

Abstract
Caleosins are a small family of calcium-binding proteins endowed with peroxygenase activity in plants. Caleosin-like genes are present in fungi; however, their functions have not been reported yet. In this work, we identify a plant caleosin-like protein in Aspergillus flavus that is highly expressed during the early stages of spore germination. A recombinant purified 32-kDa caleosin-like protein supported peroxygenase activities, including co-oxidation reactions and reduction of polyunsaturated fatty acid hydroperoxides. Deletion of the caleosin gene prevented fungal development. Alternatively, silencing of the gene led to the increased accumulation of endogenous polyunsaturated fatty acid hydroperoxides and antioxidant activities but to a reduction of fungal growth and conidium formation. Two key genes of the aflatoxin biosynthesis pathway, aflR and aflD, were downregulated in the strains in which A. flavus PXG (AfPXG) was silenced, leading to reduced aflatoxin B1 production in vitro. Application of caleosin/peroxygenase-derived oxylipins restored the wild-type phenotype in the strains in which AfPXG was silenced. PXG-deficient A. flavus strains were severely compromised in their capacity to infect maize seeds and to produce aflatoxin. Our results uncover a new branch of the fungal oxylipin pathway and may lead to the development of novel targets for controlling fungal disease.
INTRODUCTION
Oxylipins constitute a large family of diverse oxygenated fatty acids and derivatives present in mammals, plants, algae, and fungi (1–3). While the biosynthesis and the roles of animal and plant oxylipins have been extensively studied (4–8), knowledge about fungal oxylipins remains limited. Fungal oxylipins are widespread among filamentous fungi, yeasts, and oomycetes (9–11) and were first described to be precocious sexual inducers, or psi factors (12). They are composed of a mixture of hydroxylated oxylipins derived from oleic (18:1), linoleic (18:2), and linolenic (18:3) acids under the action of psi factor-producing oxygenase (Ppo) enzymes (13–15). Linoleate diol synthase (LDS) converts linoleic acid directly to hydroxylated derivatives (8). However, most of the fungal oxylipins derive from an initial hydroperoxidation step, whereby polyunsaturated fatty acids (PUFAs) are catalyzed by lipoxygenases (LOXs) and dioxygenases (DOXs) (8, 16, 17). Whereas in plants such enzymes form essentially three types of hydroperoxides (OOHs), i.e., 9-OOH, 13-OOH, and 2-OOH, in fungi, they can introduce molecular oxygen on the carbon 8, 10, 11, or 15 of PUFA, yielding 8-OOH, 10-OOH, 11-OOH, and 15-OOH derivatives, respectively (8, 10, 11, 17, 18). Whatever their mode of formation, fatty acid hydroperoxides (FAOOHs) and their metabolites have been reported to play crucial roles in the life cycle of fungi, notably, in conidiogenesis and sclerotium formation (19). In addition, Ppo-derived psi factors produced by Aspergillus nidulans were shown to regulate both asexual and sexual spore development (12, 14, 20).
Fungal oxylipins are also involved in the regulation of secondary metabolism, involving the synthesis of mycotoxins and antibiotics. For example, deletion of Ppo enzymes yielded mutants depleted of the mycotoxin sterigmatocystin but enriched with penicillin (21). A lipoxygenase-like enzyme-deficient Aspergillus ochraceus strain producing low levels of linoleic acid-derived 13-hydroperoxyoctadecadienoic acid (13-HPOD) displayed decreased ochratoxin A production and delayed formation of conidia but increased production of sclerotia. Complementation of the culture medium with 9-HPOD and 13-HPOD enhanced the production of ochratoxin A in wild-type (WT) A. ochraceus but not in a mutant in which the LOX-like gene was deleted (19). Fungal production of aflatoxins seems to be favored by an oxidative environment. For example, the oxidative stress caused by the addition of cumene hydroperoxide and H2O2 was reported to induce aflatoxin accumulation (22). In contrast, plant-derived antioxidants diminished aflatoxin formation without affecting fungal growth (23). Besides endogenous oxylipins, several lines of evidence show that during the Aspergillus-seed interaction, the host oxylipins also play determinant roles in the biology of the fungus (24). For example, 13-HPOD affected sexual spore development in A. nidulans and Aspergillus flavus, whereas spore germination was inhibited by C6 to C12 derivatives of the phytooxylipin pathway (25). In addition, while 13-HPOD decreased the levels of production of mycotoxins, 9-HPOD did not reduce their levels of biosynthesis (26). Moreover, genetic evidence for reciprocal oxylipin cross talk during the plant-seed interaction has been reported: plant oxylipin directly or indirectly affected Aspergillus development processes, whereas plant oxylipin production, in turn, was modified during infection by the fungus (27). Intriguingly, plant fatty acid hydroperoxides and their corresponding alcohols are metabolized by fungi into trihydroxy derivatives (28). In plants, such oxylipins are derived from the hydrolysis of 15,16-epoxy-13-hydroxy-9,11-octadecenoic acid. The formation of these quite unusual epoxy alcohols is catalyzed by peroxygenase (29). In addition to α-dioxygenases and cytochrome P450 enzymes, this enzyme is known to initiate one of the branches of the phytooxylipin pathway. Such peroxygenases have been identified to be caleosins, which constitute a small family of Ca2+-binding proteins (30). They are membrane-bound hemoproteins that are strictly hydroperoxide dependent and play protective roles in response to stress (31). Although we have demonstrated that members of the plant caleosins act as peroxygenases (30, 32, 33), the enzymatic activity of fungal caleosins remains to be confirmed.
Here we identify one of the Aspergillus flavus genes to be a gene for caleosin. The corresponding protein encoded by the gene possesses peroxygenase activity, including oxylipin formation activity. The possibility that fungal caleosin could catalyze the formation of antifungal epoxy and hydroxy fatty acid (FAOH) derivatives (28) led us to ask how such compounds are physiologically relevant in fungi and how their biosynthesis is regulated during the plant-pathogen interaction. Because disruption of the caleosin gene prevents fungal development, we have used small interfering RNA (siRNA) approaches to reveal its importance in fungal growth and thus in aflatoxigenicity. Our data uncover a new oxylipin pathway in fungi.
MATERIALS AND METHODS
Materials, chemicals, host strains, and culture conditions.
Oligonucleotides were provided either by Eurofins or by Sigma-France. Aniline, thiobenzamide, cumene hydroperoxide, aflatoxin B1 (AFB1), and AFB2 were purchased from Sigma-Aldrich. [1-14C]oleic acid and [1-14C]linoleic acid (52 and 50 μCi/mmol, respectively) were purchased from PerkinElmer Life Sciences. Aspergillus flavus strain FSS63, isolated from a local farmed soil, was morphologically and biochemically identified by the Laboratory of Microbiological Enzymes of the Atomic Energy Commission of Syria (AECS). This strain was subjected to a fine molecular characterization. Using primers designed to amplify the internal transcribed spacer (ITS) region as described by White et al. (34), the ITS region was amplified and sequenced. Moreover, the identity of this strain as A. flavus was confirmed using a protocol proposed by Godet and Munaut (35) as well as by the restriction fragment length polymorphism (RFLP) method as described before (36). The primers used in this study are presented in Table 1. Stock cultures of A. flavus were maintained in slant tubes at 4°C on potato dextrose agar (PDA; Difco Laboratories, USA). For solid or liquid cultures of A. flavus, the stock culture was transferred onto petri dishes containing PDA or into a 500-ml Erlenmeyer flask containing 100 ml of potato dextrose (PD) broth, respectively, and allowed to develop for 7 days at 28°C. Escherichia coli strain TOP10 was used as the host for plasmid cloning experiments. Bacteria were grown in Luria-Bertani medium supplemented with ampicillin (100 μg ml−1) at 37°C. Saccharomyces cerevisiae Wa6 (ade his7-2 leu2-3 leu2-112 ura3-52) was used as the host for protein expression. Recombinant yeast was grown in S medium (7 g liter−1 yeast nitrogen base, 1 g liter−1 Casamino Acids, and 20 g liter−1 glucose supplemented with 50 μg ml−1 histidine, 200 μg ml−1 adenine, and 50 μg ml−1 leucine) for 2 days with shaking at 30°C.
TABLE 1.
Names and nucleotide sequences of primers used in this studya
| Name | Oligonucleotide sequence (5′–3′) |
|---|---|
| ITS1 | TCCGTAGGTGAACCTGCG |
| ITS4 | TCCTCCGCTTATTGATATGC |
| AfltF | GCACCAAATGGGTCTTTCTCGT |
| AfltR | ATCCACGGTGAAGAGGGTAAGG |
| AfafltF | CGCGCGAGATACTTCTTATACT |
| AfafltR | GAGCCCACTTCGAAAATACC |
| AfPXGqF | ATGCTCAACGGCGACGGTCC |
| AfPXGqR | GCGACGCCGGGGTTGTCTAT |
| AfPXGF | GGGGATCCATGCCTTCCAAAGTAAACATCG |
| AfPXGR | GGAAGCTTTTACGAATATATCTTTTCTCCT |
| AfPXG.NHis | GGGGATCCATGCACCACCACCACCACCACATGCCTTCCAAAGTAAACATCG |
| AfPXG.CHis | GGAAGCTTTTAGTGGTGGTGGTGGTGGTGCGAATATATCTTTTCTCCT |
| siRNAPXG1F | UGGGCCAAUAAGGUCCGGGUU |
| siRNAPXG1R | CCCGGACCUUAUUGGCCCAUU |
| siRNAPXG2F | GGCUAAGCACGGCUCGGACUU |
| siRNAPXG2R | GUCCGAGCCGUGCUUAGCCUU |
| P1 | AAGTTCGGTTAAGTATGATATCAA |
| P2 | GAGTTCAGGCTTTTTCATAAACTATTAAATAACTCA |
| P3 | CCGAGGGCAAAGAAATAGTAAAGAGGCGGTTGATAC |
| P4 | TCTTTGTACAGCGGTTACCAT |
| P5 | TGAGTTATTAATTAGTTTATGAAAAAGCCTGAACTC |
| P6 | AACTATGTATCAACCGCCCTATTTCTTTGCCCTCGG |
Underlined nucleotides indicate BamHI and HindIII restriction sites. Poly-His codons were inserted at the N terminus of primer AfPXG.NHis (6× CAC) and the C terminus of AfPXG.CHis (6× GTG).
Preparation of A. flavus subcellular fractions.
Isolation of microsomal and oleosomal fractions from fungal cells was performed essentially as described previously (37, 38) with a slight modification. Fungal mycelia taken from 7-day-old cultures were pelleted by centrifugation at 5,000 × g, and the cells were washed with 200 ml of washing buffer (50 mM Tris-HCl, pH 7.5). The pellet (10 g) was resuspended in 30 ml of the same buffer containing 0.6 M sorbitol and 1% (vol/vol) protease inhibitor cocktail (Sigma-Aldrich) and remained on ice during the preparation procedure. The fungal cells were disrupted by a high-performance disperser (Ultra-Turrax; IKA, Germany). The resulting homogeneous lysate was centrifuged once at 10,000 × g for 15 min. The supernatant was recovered and centrifuged at 100,000 × g for 90 min. After centrifugation, a fine, floating, creamy layer of lipid droplets, corresponding to oleosomes, was obtained on the surface of the preparation, while the pellet corresponded to microsomes. The crude lipid droplet fraction was carefully collected with a pipette and washed with 100 mM potassium pyrophosphate buffer that contained 0.1 M sucrose (pH 7.4). After centrifugation at 100,000 × g for 45 min, the oleosomes were taken and finally suspended in 10 mM Tris-HCl, pH 8, containing 10% (vol/vol) glycerol (buffer A). In parallel, the microsome fraction was gently homogenized and suspended in buffer A. The protein concentrations in each fraction were estimated by a Bradford assay (Bio-Rad) using bovine serum albumin as a standard (39).
Peroxygenase activities.
Peroxygenase activity was routinely measured during the purification procedure by use of aniline and thiobenzamide as the substrate (40). Epoxidation of [1-14C]oleic acid and [1-14C]linoleic acid was performed as described by Blee and Schuber (41). The hydroperoxide reductase activity of Aspergillus flavus PXG (AfPXG) was measured by incubation of 9-HPOD or 13-HPOD overnight at 26°C with 50 μg of purified recombinant AfPXG in a total volume of 500 μl of sodium acetate (0.1 M, pH 5.5). The reaction was stopped with a drop of HCl (4 N), and the residual substrate and products were extracted three times in 2 ml of dichloromethane-ether (1:1, vol/vol). After the extracts were dried under a nitrogen flow, the extracts were taken with 25 μl of acetonitrile-water-acetic acid (50:50:0.1, vol/vol/vol). The extracts were analyzed using a Jasco LC-2000 Plus series high-pressure liquid chromatography (HPLC) system (Jasco, USA), a UV detector (234 nm; RF-10Axl; Shimadzu), and a C18 column (150 by 4.6 mm; particle size, 5 μm; Eclipse XDB-C18; Agilent, USA). The analysis was performed using a mobile phase of acetonitrile-water-acetic acid (50:50:0.1, vol/vol/vol) at a flow rate of 0.6 ml min−1.
Primers and PCR amplification conditions.
The nucleotide sequences of the primers used in this study are presented in Table 1. The PCR amplification was performed in a 25-μl reaction final volume containing 3 mM MgSO4, 200 μM (each) the four deoxynucleoside triphosphates (dNTPs), 10 μM each primer, and 2.5 U Taq DNA polymerase. PCR conditions were 94°C for 4 min, followed by 35 cycles at 94°C for 30 s, 58°C for 30 s, and 72°C for 1 min and then a hold at 72°C for 10 min. PCR amplifications were verified by 1% agarose gel electrophoresis for 30 min at 100 V. The gel was visualized in an UV illuminator. If necessary, the PCR fragments were purified using a QIAquick PCR purification kit (Qiagen, USA).
Analysis of gene transcripts.
For studies of AfPXG gene expression, A. flavus was grown by spreading a suspension containing a known number of fungus conidia (about 1 × 106 spores ml−1) on PDA plates at 28°C for 7 days. Each developmental stage (germinated spores, mycelia, conidiophores, and conidia) was produced in triplicate. The totality of fungal growth in each stage was collected and taken for RNA isolation and cDNA synthesis. Total RNA was extracted from the fungal cells using an RNeasy kit according to the manufacturer's instructions (Qiagen, Germany). DNA traces were removed by treating RNA for 1 h at 37°C with 2 U of RNase-free RQI DNase (Promega, USA). RNAs were diluted to 50 ng μl−1 using RNase-free water and stored at −80°C. First-strand cDNA synthesis was performed using Moloney murine leukemia virus (MMLV) reverse transcriptase (RT) (Invitrogen) under the following conditions: 0.5 μg of total RNA and 0.5 μg of oligo(dT) primer were denatured for 5 min at 65°C and immediately placed on ice for 10 min. The reverse transcriptase reaction was performed in a final volume of 20 μl containing 10 mM each dNTP, 200 units of MMLV RT, and 10 units of RNase inhibitor, and the mixture was incubated for 1 h at 37°C in a Mastercycler Pro gradient PCR machine (Eppendorf, Germany). RT inactivation and RNA removal were performed by heating the reaction mixture at 70°C for 15 min, and the cDNA was stored at −80°C. Real-time PCR was carried out in 48-well plates using a StepOne cycler from Applied Biosystems, USA. The 25-μl reaction mixtures contained 0.5 μM each target and reference gene-specific primer pairs AfPXGqF/AfPXGqR (Table 1) and 18SF/18SR, 12.5 μl of SYBR green quantitative RT-PCR (qRT-PCR) mix (Bio-Rad, USA), and 2.5 μl of 10-fold-diluted cDNA. Quantitative PCR (qPCR) conditions were 3 min at 95°C for enzyme activation and 40 cycles of 15 s at 95°C, 15 s at 58°C, and 15 s at 72°C. An additional step that started at 60 and increased to 95°C (0.2°C s−1) was performed to establish the denaturation curve specific for each amplified sequence. Each point was replicated in triplicate, and the average threshold cycle (CT) value was taken. Subsequently, relative quantification (RQ) of the target gene, where RQ is equal to 2(−ΔΔCT), was calculated by the software of a StepOne cycler from Applied Biosystems, USA.
Cloning and sequence analysis of AfPXG gene.
The full-length open reading frame (ORF; 873 bp) of the AfPXG-like gene was amplified by PCR using A. flavus cDNA. For the further purification of AfPXG, primers AfPXG.NHis and AfPXG.CHis were designed to add a His tag at the N and C terminal ends of the AfPXG gene, respectively (Table 1). The purified AfPXG PCR product was ligated to the pGEM-T Easy vector (Promega, USA) following the instructions in the manufacturer's manual, resulting in pGEM-T/AfPXG. The recombinant plasmid pGEM-T/AfPXG was transferred into E. coli TOP10 competent cells. pGEM-T/AfPXG plasmids were extracted from E. coli TOP10 cells using a plasmid purification minikit (Qiagen, Germany). The presence of the AfPXG gene in the isolated plasmid was confirmed by PCR and BamHI-HindIII digestion. Both strands of at least three clones of the AfPXG gene were sequenced on an ABI 310 genetic analyzer (Applied Biosystems) using primers T7 and SP6 and a BigDye Terminator kit (Applied Biosystems). Nucleotide sequence analysis and multiple-sequence alignment of the nucleotide and deduced amino acid sequences were performed with Vector NTI Advance software (Invitrogen).
Expression of recombinant AfPXG protein.
AfPXG was subcloned into the yeast constitutive expression vector pVT102U (42) using the BamHI-HindIII site. The correct recombinant plasmid, pVT102U/AfPXG, was then introduced into the yeast Saccharomyces cerevisiae Wa6 (ade his7-2 leu2-3 leu2-112 ura3-52) (43). Expression of recombinant AfPXG in transformed yeast cells was carried out as described by Hanano et al. (30). The microsomes of the recombinant yeasts were resuspended in 10 mM potassium phosphate (pH 8) containing 10% (vol/vol) glycerol and were treated with Emulphogene (final concentration, 0.2%) for 45 min at 4°C. The mixture was centrifuged at 100,000 × g for 2 h.
Solubilization and purification of recombinant AfPXG.
All the subsequent steps were performed on ice (4°C). The oleosomes of recombinant yeasts resuspended in 10 mM potassium phosphate (pH 8) containing 10% (vol/vol) glycerol were treated separately with different detergents (CHAPS {3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate}, Triton X-100, Emulphogene; Sigma) at a final concentration of 0.2% (vol/vol) for 45 min. The mixture was then centrifuged at 100,000 × g for 45 min. After centrifugation, the supernatant was taken, the concentration of the solubilized proteins was estimated by the Bradford assay (39), and enzymatic activity was determined. The solubilized fraction was used in the protein purification procedure. The His-tagged AfPXG present in the supernatant was purified on an Ni-nitrilotriacetic acid Superflow column (Qiagen) under native (nondenaturing) conditions, according to the manufacturer's instructions. The purity of the recombinant protein was confirmed by 12% SDS-PAGE, followed by Coomassie blue staining. For Western blot experiments, proteins were fractionated by 12% SDS-PAGE and electrotransferred to Immobilon-P membranes (Millipore Corp., Bedford, MA) using a mini-Transblot transfer cell apparatus (Bio-Rad). For detection of PXG-His, a mouse monoclonal anti-His antibody and an anti-mouse immunoglobulin antibody conjugated to peroxidase were used at 1:2,000. Blots were developed using an enhanced chemiluminescence kit from Pierce.
Replacement of the PXG-like gene by an Hygr gene.
The replacement of the PXG-like gene by the hygromycin resistance (Hygr) gene was carried out as described by Ninomiya et al. (44). The 5′ and 3′ flanking DNA (≈1 kb) of the PXG-like gene was amplified by PCR using the genomic DNA of A. flavus FSS63 as a template. The primers used for the amplification of the 5′ or 3′ flanking DNA were primers P1 and P2 or primers P3 and P4, respectively (Table 1). The Hygr gene was amplified by PCR using the binary vector pCAMBIA1381 (http://www.cambia.org/daisy/cambia/585.html) (GenBank accession number AF234302) as a template and primers P5 and P6. The three PCR products were mixed and used as a template in a fusion PCR (45). The fusion PCR product was analyzed on an agarose gel (1%) and used for transformation of the protoplasts of the A. flavus FSS63 wild type.
Design of siRNAs.
Two siRNAs that targeted the mRNA sequence of the PXG-like gene of A. flavus (GenBank accession number XM_002382445), named siRNAPXG1 and siRNAPXG2,were designed online by the use of Block-iT RNAi Designer software (Life Technologies) and purchased from VBC-Biotech (Austria). The primers used to detect these siRNAs are provided in Table 1. An siRNA control (siRNACt) with no sequence homology to the A. flavus genome in any sequence database was also purchased from the same company.
Preparation of fungal protoplast and transformation.
Protoplasts were prepared from fungal conidia as described by Cheng and Belanger (46) with some modifications. Briefly, 7-day-old cultures of A. flavus were obtained on PDA plates. The expected conidia (about 1 × 108 conidia per plate) were harvested and then washed with sterile water. The spores were suspended in 20 ml of a solution of 25 mM l-mercaptoethanol and 5 mM Na2EDTA, pH 8.0, under gentle shaking for 20 min at room temperature. The treated spores were collected by centrifugation for 5 min at 5,000 rpm, and the pellets were resuspended and incubated for 2 h at 25°C in 5 ml of digestion buffer (10 mM Na2HPO4, 1.2 M MgSO4, 10 mg ml−1 lysozyme; Sigma-Aldrich). The protoplasts were purified by filtration through a small mass of cotton (about 2 cm3) and then harvested by centrifugation at 5,000 rpm for 15 min at 4°C. The resulting pellets were washed twice with 5 ml STC buffer (1 M sorbitol, 50 mM CaCl2, 50 mM Tris-HCl, pH 8). Finally, the protoplasts were resuspended in 1 ml of STC buffer and divided into 50-μl aliquots. Delivery of siRNA to the protoplast was carried out in sterile 1.5-ml microcentrifuge tubes, 10 μl of each siRNA was mixed with an equal volume of Lipofectin reagent (Invitrogen Life Technologies, United Kingdom), and the mixture was allowed to stand for 15 min at 20°C. A volume of 50 μl of protoplasts was added, and the components were gently mixed. The tubes were incubated at 20°C for 24 h to allow transfection to proceed (47). Then, the mixture was inoculated in 10 ml of PD medium with 1.2 M sorbitol for 7 days at 28°C in the dark. Different dilutions of siRNA (25, 50, 100 nM) were tested on A. flavus. A similar treatment of protoplasts without siRNA or with nonspecific siRNA was performed as a negative control. All experiments were carried out using three biological replicates. For replacement of the PXG-like gene, a solution (5 μl) containing 10 μg DNA of the fusion PCR product was mixed with 50 μl of protoplasts, and the mixture was incubated on ice for 5 min. Forty microliters of the mixture was transferred to an electroporator cell (BTX Electro Cell Manipulation 600; Genetronics) and shocked by using a charging voltage of 1.5 kV and a resistance of 186 Ω. After the electroshock, 1 ml of PD medium containing 1.2% sucrose was added and the conidia were incubated at 30°C for 2 h. The solution (200 μl) was spread on agar medium containing hygromycin B (500 μg ml−1). Colonies resistant to hygromycin were isolated and tested by PCR using primers P1 and P6. Southern blotting was used to confirm whether the colonies contained extra copies of the Hygr gene.
Biomass and conidium number measurements.
The fungal biomass of 7-day-old cultures on PDA plates was determined. The transformants or the control strain was inoculated at a single point on a cellophane membrane placed on the surface of a PDA plate. Seven days later, the membranes were removed and the corresponding mycelial mats were collected, thoroughly washed twice with distilled water, filtered through Whatman no. 4 filter papers, and dried overnight in an oven at 95°C. Mycelial dry weights were then determined as described by Rasooli and Razzaghi-Abyaneh (48). Fungal growth inhibition (in percent) was calculated according to the following formula: [(total control weight − total sample weight)/total control weight] × 100. In parallel, the total conidia were harvested from each plate and placed in 5 ml of water containing 0.01% Tween 80. The conidia were diluted to 1:10 and counted with a hemocytometer.
Extraction, cleanup, and HPLC analysis of aflatoxin.
One milliliter of the conidial suspension (1 × 106 spores ml−1) generated from A. flavus was cultivated on petri plates containing PDA. The plates were incubated at 28°C for 7 days. The total growth of the fungi was collected to extract the aflatoxins (AFs). The extraction of AFs produced by A. flavus was carried out as described by Bertuzzi et al. (49) by placing the A. flavus fungi in 100 ml of chloroform and rotating the mixture on a rotary shaker for 1 h. The cleanup of the extract was done as described previously (50) using a thin-layer chromatography (TLC) plate. Extracted AF samples were spotted onto a C18 reversed-phase TLC plate (aluminum sheets, 20 by 20 cm, 200-μm layer; Merck, Germany), and the chromatogram was developed using a solvent system of chloroform-acetone (90:10, vol/vol). After development, the spot with an Rf value similar to that of the AFB1 standard was scraped and then reextracted with chloroform and evaporated to dryness under nitrogen. The extract was resuspended with 100 μl acetonitrile and stored in an amber-colored vial under refrigeration. Extracts were analyzed using a Jasco LC-2000 Plus series HPLC system (Jasco, USA), a fluorescence detector (excitation λ, 247 nm; emission λ, 480 nm; RF-10Axl; Shimadzu), and a C18 column (150 by 4.6 mm; particle size, 5 μm; column temperature, 53°C; Eclipse XDB-C18; Agilent, USA). The analysis was performed using a mobile phase of water-methanol-acetonitrile (50:40:10, vol/vol/vol) at a flow rate of 0.8 ml min−1 and a run time of 10 min. To study the time course of aflatoxin production in the strains in which AfPXG was silenced and with different treatments, aflatoxin production was evaluated at 2, 5, 7, 9, 11, and 13 days after inoculation.
SOD and CAT enzymatic activities.
Preparation of the fungal tissues for determination of enzyme activities was carried out as described previously (51), with some modification. Briefly, 5 g of fungal tissues was homogenized with 5 ml potassium phosphate buffer (pH 7.5) containing 1 mM EDTA, 3 mM dl-dithiothreitol, and 5% (wt/vol) insoluble polyvinylpolypyrrolidone on ice. Subsequently, the homogenate was centrifuged at 12,000 rpm for 5 min, and the supernatant was used for analysis of the enzymatic activities. Superoxide dismutase (SOD) activity was assayed by measuring its ability to inhibit the photochemical reduction of nitroblue tetrazolium (NBT) as described by Beauchamp and Fridovich (52). Catalase (CAT) activity was measured by the method of Azevedo et al. (53). Activity was determined by monitoring the decrease in absorbance due to H2O2 reduction at 240 nm for 2 min.
Complementation of strains in which AfPXG was silenced with PXG pathway oxylipins.
C18:2 oxylipins derived from the LOX-PXG pathway were biosynthesized in three separate steps. In the first step, hydroperoxides 9-HPOD and 13-HPOD were prepared by incubation of C18:2 with 9-LOX and 13-LOX from tomato and soybean, respectively, as described previously (54, 55). In the second step, two possible forms of epoxide, 9,10-epoxy-12(Z)-octadecenoic acid (9,10-EOE) and 12,13-epoxy-9(Z)-octadecenoic acid (12,13-EOE), were produced from linoleic acid by incubation of radiolabeled C18:2 (50 μCi/mmol in ethanol) overnight at 26°C in the presence of 50 μg of purified recombinant AfPXG and 10 μl of cumene hydroperoxide (20 mM). Reaction, extraction, and purification of epoxides were carried out as described by Hanano et al. (30). Finally, the purified epoxides resulting from the second step were hydrolyzed to their corresponding dihydroxy compounds, 9,10-dihydroxy-12(Z)-octadecenoic acid (9,10-DHOE) and 12,13-dihydroxy-9(Z)-octadecenoic acid (12,13-DHOE). The hydrolysis of epoxides can take place chemically in acidic solution (pH 5.5). Then, the dihydroxy compounds were extracted, purified, and analyzed as described by Summerer et al. (56). An ethanol solution of 100 μM containing a mix of epoxides (mix-EOE) or dihydroxy compounds (mix-DHOE) was added to the liquid cultures of the (+) siRNAPXG1 strain in a 500-ml Erlenmeyer flask containing 100 ml of PD broth for 2 days at 28°C. To maximize any possible effects of these oxylipins on mutant growth, the mutant was first fed on PD broth containing a mix of 9-hydroxyoctadecadienoic acid (HOD) and 13-HOD (100 μM each) for 5 days at 28°C. Subsequently, a known quantity of fungal growth was then transferred onto petri dishes containing PDA supplemented with the oxylipins indicated below and allowed to develop for 5 days at 28°C. Six transformants of A. flavus were examined for their responses to each oxylipin. In parallel, the control experiment was carried out using ethanol only.
Inoculation of maize seeds with A. flavus and biomass estimation.
A quantity of 100 g of maize (Zea mays) was sterilized by emerging maize grains into 70% ethanol for 1 min. After they were dried, the grains were place in a sterile petri plate and directly inoculated with 200 μl of a liquid culture of A. flavus in PD broth. The inoculated grains were incubated at 28°C for 7 days. Fungal biomass was estimated on day 7 by careful washing of the infected grains, filtration, and then weighing of the fungal growth, expressed as the number of grams (fresh weight) per 100 g of grains.
Statistical analysis.
All data presented are expressed as means ± standard deviations (SDs). Comparisons between control and treated strains were evaluated by the t test. The difference from the control was considered significant when P was <0.05, very significant when P was <0.01, and highly significant when P was <0.001.
Nucleotide sequence accession numbers.
The sequences of the ITS region and the AfPXG gene of A. flavus were submitted to GenBank and may be found under accession numbers KC621105 and KJ668859, respectively.
RESULTS
Aspergillus flavus contains a single caleosin gene-like gene.
When the sequence of the Arabidopsis thaliana At4g26740 gene, encoding the first characterized caleosin/peroxygenase (30), was compared with the sequence of the Aspergillus flavus NNRL 3357 genome by BLAST analysis (http://blast.ncbi.nlm.nih.gov/Blast.cgi), it presented about 50% identity with the A. flavus AFLA_002850 gene. The DNA sequence of this gene, which has no biological functions known so far (57), contains an ORF of 873 nucleotides encoding a putative calcium-binding protein. Primers designed on the basis of the AFLA_002850 gene sequence (primers AfPXGF and AfPXGR; Table 1) were used to clone a homologous gene from toxigenic isolate A. flavus FSS63. An open reading frame of 873 nucleotides encoding a polypeptide of 291 amino acids (GenBank accession number KJ668859) was identified in the cloned gene. Multiple-sequence alignment of the protein sequences revealed that this fungal protein has a high degree of similarity (67%) with Arabidopsis caleosins (Fig. 1A). As expected for a caleosin, AFLA_002850 contains an EF-hand calcium-binding site motif near its N terminus and several putative phosphorylation sites in the C terminus (58). Like some plant caleosins, it lacks most of the conserved proline residues in the central hydrophobic region initially postulated to be essential for caleosin anchoring in membranes (59) (Fig. 1B), raising questions about the location of the protein in fungal cells (60). Besides the Ca2+-binding site, two histidine residues crucial for plant peroxygenase activity are present in the AFLA_002850 primary sequence, suggesting that this fungal protein might also act as an enzyme. However, compared to the sequences of Arabidopsis caleosins, both the N terminus and C terminus of AFLA_002850 are extended by additional motifs (Fig. 1A and C) that might impede substrate entrance and thus prevent peroxygenase activity.
FIG 1.

Structural features of AfPXG. (A) Multiple-sequence alignment of the deduced amino acids of the protein isolated from A. flavus (GenBank accession number KJ668859) with the A. thaliana proteins AtPXG1 (At4g26740), AtPXG2 (At5g55240), AtPXG3 (At2g33380), and AtPXG4 (At1g70670). The boxed domain (residues 62 to 97) corresponds to an EF-hand motif, and the underlined residues between residues 75 and 86 correspond to the Ca2+-binding domain. A star indicates the position of histidine 86, responsible for heme binding; an inverted triangle indicates a phosphorylation site; hyphens indicate absent residues; dark gray shading indicates similarity; light gray shading with lighter text indicates identity; light gray shading with darker text indicates a conservative residue; black text with a white background indicates nonsimilar residues. (B) Orientation and location of structural and functional domains. (C) Predicted three-dimensional structure of the putative caleosin (AFLA_00280). The virtual image was generated online using http://www.sbg.bio.ic.ac.uk/phyre2.
AFLA_002850 is a peroxygenase.
To test whether the fungal caleosin-like enzyme possesses peroxygenase activity, AFLA_002850 was expressed in yeast. As demonstrated for recombinant plant peroxygenases, crude extracts of yeast expressing AFA_002850 efficiently catalyzed the sulfoxidation of thiobenzamide (32.8 nmol min−1 mg−1 protein), the hydroxylation of aniline (24.6 nmol min−1 mg−1), and the epoxidation of oleic acid (36.2 nmol min−1 mg−1) in the presence of cumene hydroperoxide (Fig. 2A). No enzymatic activity was observed in the fractions isolated from yeast transformed with an empty vector (Fig. 2A).
FIG 2.

Biochemical characterization of recombinant AfPXG. (A) PXG activities as the sulfoxidation of thiobenzamide (light gray), the hydroxylation of aniline (dark gray), and the epoxidation of [1-14C]oleic acid (black) were measured in the crude extract, supernatant, lipid droplets, and microsomes of recombinant yeasts containing plasmid pVT102U ligated with the AfPXG insert. Results are means ± SDs (n = 3). (B) SDS-PAGE analysis of purified AfPXG. The purity of the recombinant protein was confirmed by SDS-12% PAGE, followed by Coomassie blue staining (right) and detection of PXG-His by anti-His antibody and an anti-mouse immunoglobulin antibody conjugated to peroxidase (left). Numbers to the left and right of the gels are molecular masses (in kilodaltons). (C) Specific activities of the purified recombinant AfPXG. (D) Sequential scanning of the absolute spectrum of AfPXG obtained on addition of cumene hydroperoxide over the indicated times (in minutes). (E) Correlation between the hydroxylation activity of the purified AfPXG and the disappearance of the heme content. (F) Inhibition of native AfPXG by treatment with β-mercaptoethanol (1 mM) or terbufos (3 mM). (G) Co-oxidation activities for native purified AfPXG and for the Ca2+-deionized AfPXG after extensive dialysis and for AfPXG after dialysis followed by the addition of 1 mM CaCl2 to the medium. All data are the means ± SDs (n = 3).
When yeast crude extracts were subfractionated by differential centrifugations, only membrane fractions, i.e., microsomes and lipid droplets, showed catalytic activity, while the supernatant fraction did not show activity (Fig. 2A). Since addition of the His tag to the N terminus or the C terminus of the protein did not modify its oxidative ability (not shown), we attempted to purify a His-tagged protein. Lipid droplet proteins were first solubilized (about 45%) with 0.2% Emulphogene, and then the solubilized enzymatic fraction (representing 85% of the total activity present in the lipid droplets) was purified by affinity chromatography on an Ni2+ column with a purification factor of 9.2 (Table 2). When analyzed by SDS-PAGE, the purified fraction showed one major single band at 32 kDa with a degree of purity higher than 98% when estimated by scanning densitometry (Fig. 2B). The purified fraction catalyzed the oxidation of thiobenzamide (1.55 μmol min−1 mg−1 of protein), aniline (1.24 μmol min−1 mg−1 of protein), and oleic acid (2.1 μmol min−1 mg−1 of protein) in the presence of cumene hydroperoxide as a cosubstrate. The heat-inactivated fraction was unable to catalyze such reactions. These data strongly suggest that peroxygenase activity is associated with the 32-kDa membrane-bound protein (Fig. 2C).
TABLE 2.
Representative purification of oleosomal recombinant AfPXGa
| Fraction | Vol (ml) | Total activity (nmol min−1) | Amt of protein (mg [%]) | Sp act (nmol min−1 mg−1) | Purification factor |
|---|---|---|---|---|---|
| Oleosomes | 4 | 655.6 | 4.8 (100) | 136.5 | 1 |
| Solubilized fraction | 8 | 558.4 | 2.1 (43.7) | 265.9 | 1.9 |
| Purified AfPXG | 2 | 2,016.8 | 1.6 (33.3) | 1,260.5 | 9.2 |
Recombinant AfPXG was solubilized in the presence of 0.2% Emulphogene for 45 min at 4°C. His-tagged AfPXG was purified by affinity chromatography on an Ni2+ column. The activity was measured with 1 mM aniline in the presence of 1 mM cumene hydroperoxide at 310 nm.
In Arabidopsis, such peroxygenase activity was assigned to a heme prosthetic group presumably bound to a histidine residue of the active site of the caleosin (30). The preservation of this amino acid in the primary structure of AFLA_002850 suggested that the fungal protein may also contain a heme group (Fig. 1A). Analysis of the light absorption spectra revealed a peak at 407 nm representative of the Soret band of hemoproteins. Addition of 1 mM cumene hydroperoxide resulted in a gradual decrease of the Soret band (Fig. 2D) that was correlated with a decline in the amount of hydroperoxide-supported oxidation by the fungal caleosin-like protein. Both the decrease in the absorbance at 407 nm and enzyme inactivation followed pseudo-first-order kinetics (Fig. 2E), in agreement with the presence of heme in AFLA_002850 acting as a prosthetic group. Moreover, the oxidative ability of AFLA_002850 was totally abolished by the addition of 1 mM β-mercaptoethanol and 3 mM terbufos, which act as competitive and suicide inhibitors of plant PXGs, respectively (30) (Fig. 2F). Taken together, these results indicate that AFLA_002850 has several features similar to those of plant peroxygenases. The enzymatic activity of Arabidopsis thaliana PXG1 (AtPXG1) requires calcium. To verify that A. flavus caleosin also requires the presence of Ca2+ to function as a peroxygenase, we performed extensive dialysis of this protein against the chelating agent EDTA to remove any trace of metal. This treatment completely abolished the co-oxidative properties of AFLA_002850, which were restored (up to 78.5%) by adding 1 mM CaCl2 to the medium (Fig. 2G). Thus, calcium appears to be crucial for the structure and/or the activity of the A. flavus caleosin-like protein. Together these results favor a peroxygenase identity for AFLA_002850, and we have proposed to name this gene AfPXG (GenBank accession number KJ668859).
AfPXG efficiently metabolizes linoleic acid and its 13-hydroperoxide derivative.
AfPXG catalyzed the oxidation of monounsaturated fatty acids (Fig. 3A). However, most of the oxylipins so far identified in pathogenic fungi derive from linoleic acid (24). To examine whether polyunsaturated fatty acids are also substrates, 14C-labeled C18 fatty acids with one to three double bonds were incubated with recombinant AfPXG in the presence of cumene hydroperoxide, H2O2, 9-HPOD, and 13-HPOD, and their metabolization was followed by TLC coupled with radiodetection. Linoleic acid was efficiently epoxidized by AfPXG followed by linolenic acid, whereas in comparison, only weak epoxidation of oleic acid was observed regardless of the hydroperoxide used. The most active cosubstrates were the fatty acid hydroperoxides followed by H2O2. In contrast, cumene hydroperoxide poorly promoted the epoxidation of unsaturated fatty acids (Fig. 3A).
FIG 3.

Peroxygenase activities of the purified recombinant AfPXG. (A) Co-oxidation of radiolabeled polyunsaturated fatty acids in the presence of cumene hydroperoxide (CuOOH), hydrogen peroxide (H2O2), 9-hydroperoxy-10,12-octadecadienoic acid (9-HPOD), or 13-hydroperoxy-9,11-octadecadienoic acid (13-HPOD). 14C-labeled substrates metabolized by purified recombinant AfPXG were separated by TLC and analyzed by radiodetection. C18:1, oleic acid; C18:2, linoleic acid; C18:3, linolenic acid. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Different lowercase letters indicate significant differences (P < 0.05) for the fatty acids used. (B and C) UV-HPLC analysis of the metabolization of fatty acid hydroperoxides by purified AfPXG. (I) Products formed after 2 h of incubation at 27°C of 13-HPOD or 9-HPOD by AfPXG; (II) 13-HOD or 9-HOD standards; (III) 13-HPOD or 13-HPOD incubated with heat-inactivated AfPXG. 9-HPOD and 13-HPOD did not differ in their ability to co-oxidize the reactions when the difference was analyzed by the t test. This FAOOH reduction capacity was significantly different from that measured in the presence of H2O2 and cumene hydroperoxide when analyzed by the t test.
Next, the metabolism of fatty acid hydroperoxides in the presence of the purified recombinant AfPXG was analyzed by HPLC (with UV detection). After 2 h of incubation, only 20% of the 13-HPOD and 42% of the 9-HPOD remained intact (peaks 1 and 2, respectively, in Fig. 3B and C, panels I). Peak 3 and peak 4 were identified to be the alcohol products 13-HOD and 9-HOD, respectively, by coelution with standards (Fig. 3B and C, panels II). No metabolites were detected from incubations of 9-HPOD and 13-HPOD with heat-inactivated AfPXG (Fig. 3B and C, panels III). These data indicate that recombinant AfPXG may preferably metabolize linoleic acid and its 13-hydroperoxide.
AfPXG is highly expressed in the early stage of spore germination.
Plant caleosins/peroxygenases are present in seeds but also in vegetative tissues, where they play distinct physiological roles. To examine whether AfPXG was constitutively expressed throughout the fungal life cycle or whether it was induced during particular phases of development, we evaluated AfPXG expression and AfPXG enzymatic activity in conidia, germinated spores, vegetative mycelium, and the conidiophore stage. The AfPXG transcription level was first evaluated by RT-PCR, and it level of expression was compared to that of the 18S rRNA gene, which was used as a reference. The accumulation of the AfPXG transcript was undetectable in the mature (nongerminated) conidia of A. flavus (time zero; Fig. 4A). The transcription of AfPXG was activated in the early stage of conidial germination (1 day after inoculation), and the level of the AfPXG transcript reached its maximal level after 2 days. The accumulation of AfPXG transcripts was reduced in the vegetative mycelium stage (days 4 and 5). Only low levels of AfPXG accumulated in the conidiophore stage (days 6 and 7). Variations in the expression of AfPXG during development were confirmed by quantitative real-time RT-PCR analysis (Fig. 4B). AfPXG enzymatic activity was also assessed by measuring its capacity to oxidize the aniline of microsomal fractions isolated from the different developmental stages of A. flavus. Maximal peroxygenase activity was measured (168.2 nmol min−1 mg−1 of protein) with microsomes isolated from germinated spores (day 2). Lower levels of enzymatic activity were found in microsomes isolated from vegetative mycelium and conidiophores (Fig. 4B). In summary, these results indicate that both the activation of AfPXG expression and the peroxygenase activity of the AfPXG-encoded protein were maximal in the early stages of conidium germination of A. flavus.
FIG 4.
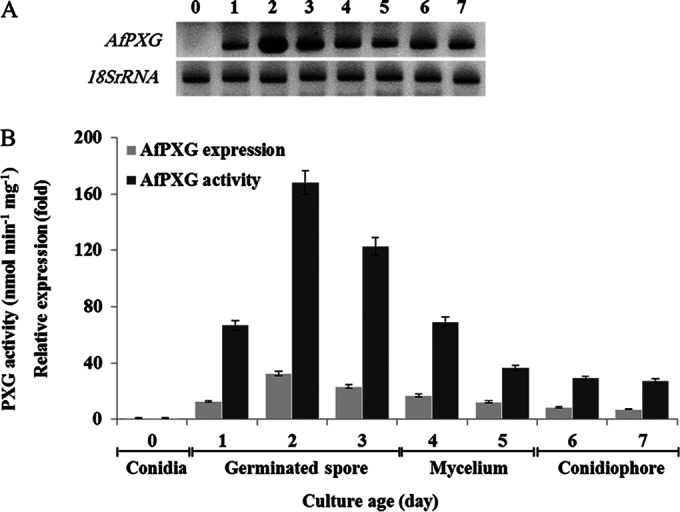
The transcriptional level of AfPXG and the enzymatic activity of PXG of A. flavus are dependent on A. flavus development. (A) AfPXG transcriptional level detected by RT-PCR at different stages of fungal development (the 18S rRNA gene was used as a reference). The numbers at the top are days of culture. (B) AfPXG expression and the AfPXG-specific hydroxylation activity of aniline in the microsomal fraction prepared from conidia, germinated spores, mycelium, and conidiophores. The measurements were done in triplicate. Values are the means ± SDs (n = 3).
The replacement of PXG by Hygr dramatically affects A. flavus development.
Using fusion PCR, we deleted the PXG-like gene by creating DNA fragments carrying Hygr with ≈1-kb 5′ and 3′ flanking sequences from the A. flavus PXG-like gene. The fusion PCR products were introduced into the protoplast of A. flavus FSS63, and hygromycin-resistant colonies were isolated. The presence of the Hygr gene or the absence of the AfPXG gene in the genomic DNA of the transformant was confirmed by PCR (data not shown). In parallel, Hygr transformants were screened by Southern hybridizations, and approximately 65% of the transformants showed a pattern diagnostic for the replacement of PXG by Hygr (data not shown). We therefore use the abbreviation AfPXGΔ to indicate this replacement. We examined the AfPXGΔ strain for growth, sporulation, and aflatoxin production. The growth of the AfPXGΔ strain was dramatically decreased, its biomass did not exceed 20% of that of the wild-type strain, and it failed to produce any remarkable spores after 7 days of culture. No expression of AfPXG was detected by qPCR, and no AfPXG activity was measured in the AfPXGΔ strain. These results for biomass and sporulation did not change after 14 days of culture (data not shown). Due to the dramatic effects on fungal development caused by the deletion of AfPXG, we next silenced AfPXG via a small interfering RNA (siRNA) approach to obtain putative intermediate phenotypes.
AfPXG expression is silenced by siRNA.
Modifying the expression of a given gene is a powerful strategy to access biological functions. We therefore attempted to reduce AfPXG expression by the siRNA approach. The success of this strategy was evaluated by quantitative RT-PCR analysis and confirmed by measuring the enzymatic activity of AfPXG in the microsomal fractions isolated from strains in which AfPXG was silenced. The primers designed to be specific for the two siRNAs siRNAPXG1 and siRNAPXG2 differed in efficacy, with the primer specific for siRNAPXG1 being the most effective (Table 1). For example, compared to the results for the control strain, only 8% of the AfPXG transcript accumulated after the addition of 100 nM the primer specific for siRNAPXG1, whereas double that amount of gene expression was retained after the addition of the same concentration of the primer specific for siRNAPXG2 (Fig. 5A). Similarly, compared to the peroxygenase activity in the control strain, peroxygenase activities were more affected in the (+) siRNAPXG1 strain than in the (+) siRNAPXG2 strain (Fig. 5B). Thus, the siRNA strategy resulted in low levels of AfPXG peroxygenase activity linked to the efficient reduction of AfPXG expression in the treated strains.
FIG 5.
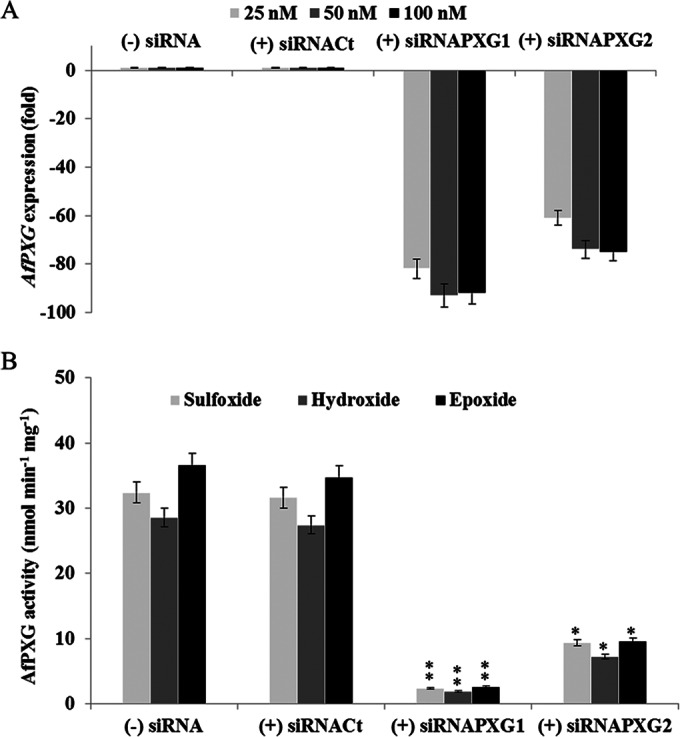
Efficiency of silencing of AfPXG at the gene transcript and enzymatic activity levels. (A) Two siRNAs specific for PXG, siRNAPXG1 and siRNAPXG2 (Table 1), were delivered to the protoplast via the Lipofectin reagent at various concentrations. Treatment of the protoplasts without (−) siRNA or with nonspecific control siRNA, (+) siRNACt, used as a negative control, was performed. AfPXG transcripts were quantified by qRT-PCR. (B) AfPXG activities were measured by sulfoxidation of thiobenzamide (light gray), hydroxylation of aniline (dark gray), and epoxidation of oleic acid (black). Values are means ± SDs (n = 3). The activities of lines in which AfPXG was silenced differed significantly from those of the control when analyzed by the t test (*, P < 0.05; **, P < 0.01).
AfPXG silencing leads to fatty acid hydroperoxide accumulation.
To examine whether AfPXG metabolized fatty acid hydroperoxides in the fungus like the recombinant protein did in vitro (see above), we first quantified the total hydroperoxide (ROOH) content present in the tissues of fungal strains in which AfPXG was silenced and the control strain. Hydroperoxide levels tripled and doubled in the (+) siRNAPXG1 and (+) siRNAPXG2 strains, respectively, compared to the level in the control (data not shown). The rise in the amount of ROOH was mostly due to the accumulation of fatty acid hydroperoxides, with the levels of 13-HPOD being higher than those of 9-HPOD in the two strains in which AfPXG was silenced (Fig. 6). Notably, the corresponding alcohol, 13-HOD, was present in WT A. flavus but was not be detected in the lines carrying siRNAPXG. These results are in agreement with a functional fatty acid hydroperoxide reductase activity supported by AfPXG in A. flavus.
FIG 6.
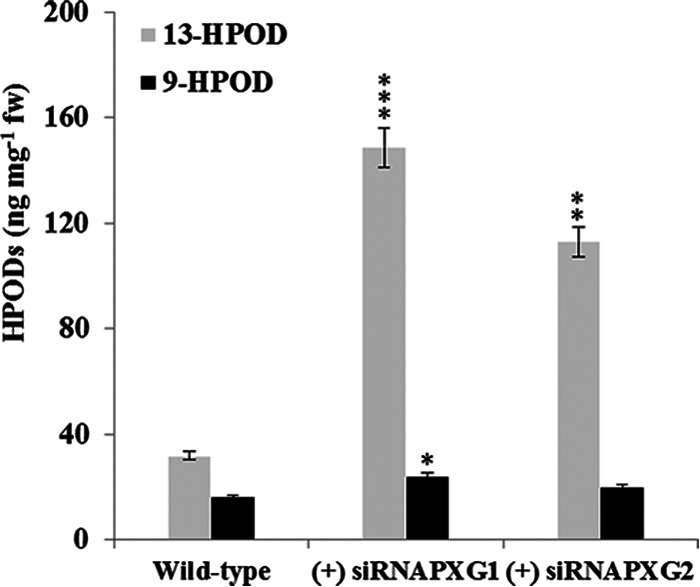
Repression of AfPXG expression resulted in hydroperoxide accumulation. The 9-HPOD and 13-HPOD contents in the tissues of the WT strain and strains in which AfPXG was silenced were analyzed. Each point represents the result of triplicate measurements. Values are means ± SDs (n = 3). The result for each line in which AfPXG was silenced was significantly different from that for the WT (*, P < 0.05; **, P < 0.01; ***, P < 0.001). fw, fresh weight.
Silencing of AfPXG reduces fungal biomass and leads to inhibition of conidial formation.
The most striking visual effect of silencing of AfPXG was the poor fitness of the resulting strains compared to the fitness of the control. For example, only thin mycelia developed from the (+) siRNAPXG1 strain, with the mycelium weight representing about 30% of the mycelium weight of the control (Fig. 7A and B). Moreover, development of this strain was blocked at the reproductive stage because it failed to form conidia. The phenotypes were also attenuated in the (+) siRNAPXG2 strain. Its mycelial weight was still reduced compared to that of the control (it had about half of the mycelium weight of the control), but this strain was able to form conidia, although the conidia were 7-fold less numerous than they were in the control (Fig. 7B; Table 3). Thus, silencing of AfPXG resulted in a dramatic decrease of the growth of the transformed strains and a significantly reduced capacity to undergo conidiogenesis.
FIG 7.

Phenotypes of strains in which AfPXG was silenced. (A) Phenotypes of the (+) siRNAfPXG1 and (+) siRNAfPXG2 strains compared with the phenotype of the (+) siRNACt control strain on PDA cultures. Cultures were carried out at 28°C for 7 days. (B) Evaluation of number of conidia and biomass in strains in which AfPXG was silenced and the control strain. The measurements were done in triplicate. Values are means ± SDs (n = 3). The number of conidia from each line in which AfPXG was silenced was significantly different from that for the WT when analyzed by the t test (*, P < 0.05; **, P < 0.01).
TABLE 3.
Dry weight of A. flavus mycelium in relation to AfPXG silencing before and after treatment with exogenous oxylipins
| A. flavus strain or strain and treatment | Mean ± SD mycelium wt (g DW per plate)a | CVb |
|---|---|---|
| Strain | ||
| (+) siRNACt | 3.61 ± 0.034 | 0.96 |
| (+) siRNAPXG1 | 1.13 ± 0.014c | 1.23 |
| (+) siRNAPXG2 | 1.43 ± 0.026c | 1.81 |
| Strain and treatment | ||
| WT without oxylipins | 3.73 ± 0.042 | 1.12 |
| (+) siRNAPXG1 without oxylipins | 1.15 ± 0.023 | 2.00 |
| (+) siRNAPXG1 with 9-HOD | 1.86 ± 0.024d | 1.29 |
| (+) siRNAPXG1 with 13-HOD | 2.07 ± 0.020d | 0.96 |
| (+) siRNAPXG1 with mix-EOE | 1.31 ± 0.032 | 2.44 |
| (+) siRNAPXG1 with mix-HOE | 1.24 ± 0.030 | 2.41 |
Mean weight for fungal growth on six separate plates (n = 6) on day 7 after inoculation. DW, dry weight.
CV, coefficient of variation, which was calculated as (standard deviation of the weight/mean weight) × 100.
The mycelium weight for each silenced strain was significantly different from that for the WT (P < 0.05).
The mycelium weight for treated strain (+) siRNAPXG1 was significantly different from that for the WT (P < 0.05).
Silencing of AfPXG leads to poor production of AFB1.
We next examined the effect of silencing of AfPXG on the production of aflatoxin B1 (AFB1). A. flavus FSS63 generated a unique peak coinciding with that of the AFB1 standard when analyzed by HPLC coupled with fluorescence detection (Fig. 8A, peak 2). By using a standard curve established with known concentrations of AFB1, about 13 μg ml−1 of AFB1 was recovered from cultures of the control strain, siRNACt (Fig. 8A, inset). Silencing of AfPXG decreased the concentration of AFB1 by a factor of about 6 on day 7 after inoculation (Fig. 8A), suggesting a role of AfPXG in aflatoxin accumulation. The result for this one time point was confirmed by evaluating the time course of AFB1 production by the line in which AfPXG was silenced in comparison with that by the wild type (Fig. 8B).
FIG 8.

Aflatoxigenicity was affected in strains in which AfPXG was silenced. (A) HPLC analysis of the aflatoxin B1 produced by wild-type A. flavus and A. flavus strains in which AfPXG was silenced (peaks 1, 3, 4, and 6, 25, 10, 5, and 1 ng ml−1 of standard AFB2, respectively). (Inset) Quantification of AFB1 in the (+) siRNAfPXG2 and (+) siRNAfPXG1 (open circles) strains compared to AFB1 standard (closed circles). (B) Time course of AFB1 production in strains in which AfPXG was silenced. AFB1 was extracted and analyzed on days 2, 5, 7, 9, 11, and 13 after inoculation, as described in Materials and Methods. (C) Silencing of AfPXG downregulated aflR and aflD expression. Each point represents the result of triplicate measurements. Values are means ± SDs (n = 3). Asterisks indicate significant differences in gene expression between strains in which AfPXG was silenced and the control strain (*, P < 0.05; **, P < 0.01).
To determine at what stage of the aflatoxin biosynthetic chain AfPXG was acting, we analyzed the expression of two crucial genes. The first one, aflD (alternatively named nor-1), intervenes in the early steps of aflatoxin biosynthesis when the first stable aflatoxin norsolorinic acid intermediate is converted into averantin. The second gene, aflR, is a regulatory gene involved in transcriptional activation of most of the structural genes. Expression of both genes was strongly repressed (by up to a factor of 51) in both strains in which AfPXG was silenced and the control strain (Fig. 8C). Thus, silencing of AfPXG contributes to a reduction in the expression of aflatoxin biosynthetic genes and, therefore, toxin accumulation.
Silencing of AfPXG enhances ROS-degrading enzyme activities.
Aflatoxin biosynthesis is stimulated under stress oxidative conditions that provoke reactive oxygen species (ROS) accumulation (61). To evaluate whether silencing of AfPXG modified the fungal ROS status, we measured A. flavus superoxide dismutase (SOD) and catalase (CAT) activities. These two enzymes control O2− and H2O2 accumulation, respectively, in cells. SOD activity was stimulated by factors of 3 and 2 in strains (+) siRNAPXG1 and (+) siRNAPXG2, respectively, compared to its activity in the WT. CAT activity was similarly induced and doubled in the two strains, respectively, compared to its activity in the WT (Fig. 9). These data suggest that silencing of AfPXG leads to enhanced ROS scavenging activities that probably result in reduced ROS accumulation in the transformed strains.
FIG 9.
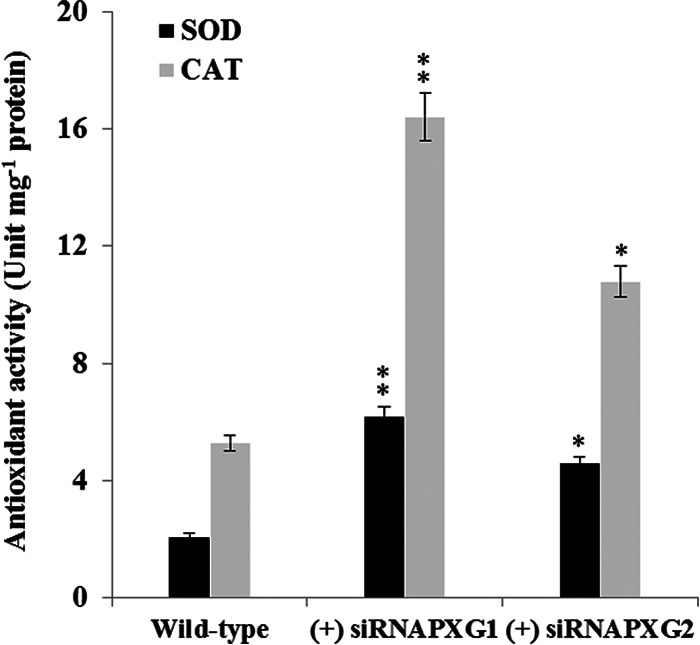
Silencing of AfPXG enhanced antioxidant activities. SOD and CAT activities were measured in the strainsin which AfPXG was silenced and the WT strain. SOD activity was assayed by measuring its ability to inhibit the photochemical reduction of nitroblue tetrazolium (NBT). CAT activity was measured by monitoring the decrease in absorbance due to H2O2 reduction at 240 nm for 2 min. Each point represents the result of triplicate measurements. Values are means ± SDs (n = 3). Asterisks indicate significant differences between the enzymatic activities of each line in which AfPXG was silenced and the control (*, P < 0.05; **, P < 0.01).
Oxylipins derived from the AfPXG pathway restore phenotypes in which AfPXG is silenced.
To investigate whether phenotypes resulting from silencing of AfPXG were specifically due to AfPXG peroxygenase activity, we tried to complement strains in which AfPXG was silenced chemically using oxylipins derived from AfPXG metabolization of C18:2 and C18:2 fatty acid hydroperoxides. Thus, the (+) siRNAPXG1 strain was cultivated in the presence of 100 μM 9-HOD, 13-HOD, mix-EOE [9,10-epoxy-12(Z)-octadecenoic acid and 12,13-epoxy-9(Z)-octadecenoic acids], or mix-HOE [9,10-dihydroxy-12(Z)-octadecenoic acid, 12,13-dihydroxy-9(Z)-octadecenoic acid, 9,12,13-trihydroxy-11-octadecenoic acid, and 9,10,13-trihydroxy-12-octadecenoic acid]. In particular, the level of restoration of the mycelium growth of strain (+) siRNAPXG1 was about 50% of that of the control when 9-HOD and 13-HOD were added (Fig. 10A and B; Table 3). In addition, conidiation was significantly reestablished (up to 60% compared to that in the wild type) in the strains in which AfPXG was silenced if they were grown in the presence of these fatty acid alcohols but far less (about 20%) when they were grown in the presence of mixtures of epoxides or di- and trihydroxylated derivatives (Fig. 10C). Moreover, oxylipins generated by AfPXG restored aflatoxigenicity in strain (+) siRNAPXG1. Compared to the level of transcription in the WT, addition of 13-HOD resulted in the recovery of up to 90% of the level of aflD transcription and 80% of the level of aflR transcription. 9-HOD, mix-EOE, and mix-HOE were less effective (Fig. 10D and E). As a consequence, AFB1 production was stimulated in the strains in which AfPXG was silenced and that were grown in the presence of 13-HOD (up to 70% compared to the level of AFB1 production in the wild type), and AFB1 production was stimulated to a lesser extent by the other oxylipins (Fig. 10F). The restoration of AFB1 production in strains in which AfPXG was silenced reached a peak on day 7 after inoculation, and its maximal level was observed when they were complemented with 13-HOD compared to the levels achieved when they were complemented with other oxylipins (Fig. 10G). Of note, we observed that feeding of the AfPXGΔ strain with a mixture of 9- and 13-HOD similarly restored the growth, conidiation, and AFB1 production of the null mutant (data not shown). Together, these results indicate that the oxylipins generated by AfPXG are able to complement phenotypes resulting from alteration of the expression of AfPXG.
FIG 10.

The phenotypes of strains in which AfPXG was silenced were due to peroxygenase activity. Addition of 100 μM 9-HOD, 13-HOD, mix-EOE (mono- or diepoxides), or mix-HOE (di- or trihydroxy compounds) to the (+) siRNAPXG1 strain restored fungal development. (A) Morphology and appearance of fungal growth on PDA medium; (B) mycelium dry weight; (C) conidium formation; (D and E) relative expression of aflD (D) and aflR (E) expression. (F) Aflatoxin B1 production. Controls (the WT strain and strains in which AfPXG was silenced) were treated with ethanol. (G) The production of AFB1 in the treatments over time (2, 5, 7, 9, 11, and 13 days [d] after inoculation). Treatments and measurements were done in triplicate. Values are means ± SDs (n = 9). Asterisks indicate significant differences between treatment with each exogenous oxylipin and the control treatment (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
Silencing of AfPXG limits maize seed infection by A. flavus.
Finally, we investigated whether strains in which AfPXG was silenced were still able to colonize maize seeds. Compared to the level of plant colonization by the WT A. flavus strain grown on maize seeds for 7 days, the (+) siRNAPXG1 strain exhibited poor plant colonization (Fig. 11A). Both mycelial mass and aflatoxin accumulation were reduced by a factor of 40 compared to the mycelial mass of and the level of aflatoxin accumulation by the WT (Fig. 11B). These data strongly suggest that AfPXG is crucial for the successful infection of maize seeds by A. flavus.
FIG 11.
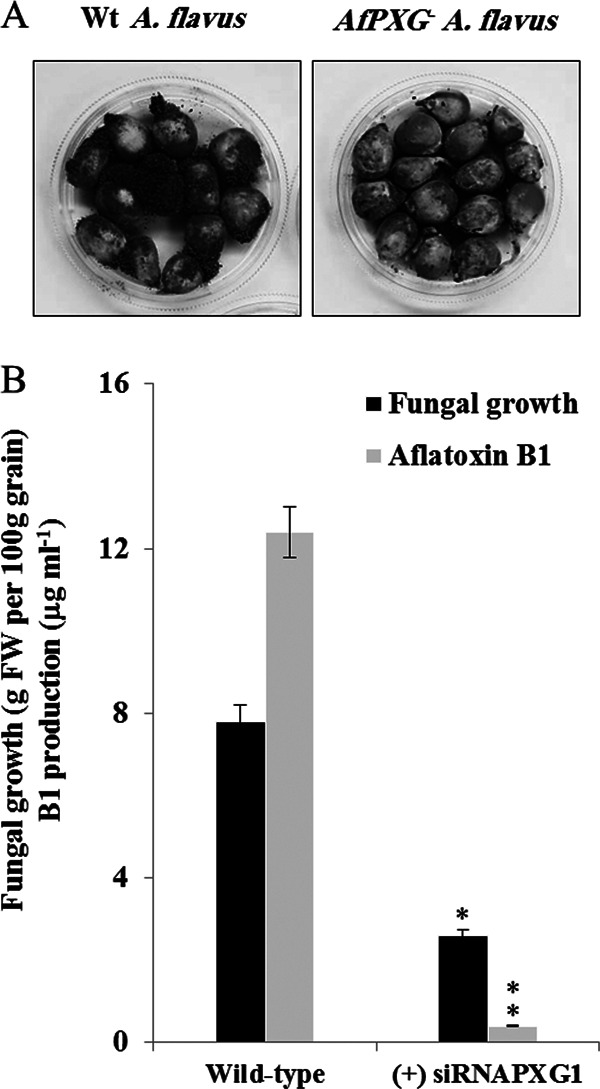
Impact of silencing of AfPXG on maize seed colonization by A. flavus. (A) Maize seeds infected with a wild-type A. flavus strain (left) or strain (+) siRNAPXG1 (right). After sterilization, grains were place in a sterile petri plate and directly inoculated with a 200 μl of a liquid culture of A. flavus in PD broth. The photograph was taken at 7 days after infection. (B) Estimation of the mycelium weight and level of aflatoxin B1 accumulation in the (+) siRNAPXG1 and WT strains on day 7 after infection of maize seeds. Asterisks indicate significant differences between the strains in which AfPXG was silenced and the wild type (**, P < 0.01).
DISCUSSION
Characteristics of AfPXG.
We identified and characterized a fungal caleosin-like protein as a peroxygenase and consequently ascribe a new branch to the oxylipin pathway present in Aspergillus flavus. Similarly to plant caleosins, the enzymatically active AfPXG was found to be bound to the endoplasmic reticulum and lipid droplets when expressed in yeast. Ppo enzymes have also been detected in lipid droplets (62), suggesting that this organelle might be a major site for oxylipin biosynthesis in fungi. However, the mode of anchorage of AfPXG remains to be clarified because, like class II caleosins, it does not possess the canonical proline knot presumed to be crucial for targeting to lipid droplets (33).
AfPXG activity is pivotal for fungal development.
Arabidopsis possesses at least seven isoforms of caleosins (58). Each of them fulfills a distinct function linked to its catalytic activity (30, 33). In contrast, AfPXG is a monocopy gene in A. flavus and might play multiple roles in fungal biology. Silencing of this gene severely affected and reduced fungal growth, conidium production, and aflatoxin accumulation. The restrained vegetative growth of the strains in which AfPXG was silenced could partly result from a defect in spore germination, a developmental stage where the gene expression and enzymatic activity of the fungal caleosin were at their highest levels in wild-type A. flavus. Because the caleosin ATS1 (63) was shown to control the germination of seeds by affecting the degradation of their lipid reserves (64), it can be postulated that AfPXG might similarly promote spore germination by stimulating the metabolism of some spore reserve in the fungus. Alternatively, most of the A. flavus phenotypes observed when we deleted or silenced AfPXG might result from the alteration of the expression of other oxylipin biosynthetic enzymes, such as lipoxygenase or Ppo (62). In favor of this hypothesis, similar phenotypes for growth, conidiogenesis, and aflatoxin biosynthesis have been reported for the A. flavus lox1 (Aflox1) mutant devoid of an Mn-dependent lipoxygenase (65). However, whether these symptoms were due to the action of FAOOHs per se or that of oxylipin products derived from further FAOOH degradation was not determined. In disagreement with this hypothesis is the restoration of the wild-type phenotypes in the strains altered in AfPXG expression by AfPXG-derived oxylipins, which are not substrates of LOXs or Ppo. Moreover, deletion of AfPXG resulted in the arrest of fungal growth at as early as 2 days when the induction of Ppo was not accomplished (17).
Our data rather suggested that the physiological deficiencies of lines in which AfPXG was silenced and probably those of the Aflox1 mutant resulted from the lack of production of oxylipins by AfPXG. Among them, FAOHs were more active in restoring the phenotypes of strains in which AfPXG was silenced than fatty acid epoxides and/or the di- and trihydroxylated derivatives. Such FAOHs were recently found to modulate reactive oxygen species accumulation in plants (32). For example, deletion of an FAOH-producing caleosin in Arabidopsis thaliana resulted in the accumulation of the corresponding hydroperoxide precursors, which was correlated with a low cellular oxidative status under physiological conditions. It is tempting to make a parallel with the fungal situation. We showed here that the alteration of AfPXG expression resulted in the accumulation of linoleic acid-derived hydroperoxides and to an increase in the activities of antioxidative enzymes, leading to low levels of ROS production. These data would explain, at least in part, the functioning of the fungal caleosin in aflatoxin production. Indeed, it was previously reported that 13-HPOD represses aflatoxin biosynthesis when added to Aspergillus cultures, whereas 9-HPOD lengthens the time during which aflatoxin transcripts accumulate (26). Thus, endogenous accumulation of both 13-HPOD and 9-HPOD in strains in which AfPXG was silenced might account for the poor accumulation of AFB1. On the other hand, the biosynthesis of aflatoxin is favored under conditions with high levels of ROS and silencing of AfPXG minimized ROS accumulation in fungal tissues. Thus, the combined effects of low levels of ROS and high levels of FAOOH production might account for the reduced production of aflatoxin in lines altered in AfPXG expression. This result would be in accordance with the findings of previous studies suggesting that the fungal cell responds to incomplete scavenging of reactive oxygen species at the intracellular level by producing toxins (61). Alternatively, restrained conidium and aflatoxin production in lines in which AfPXG was silenced might also result from the limited growth of the transformed fungus.
Aspergillus species produce aflatoxins at a time that coincides with spore development (66). In A. nidulans, these processes are regulated by G-protein signaling pathway components (67). Likewise, deletion of one β subunit of the heterotrimeric G-protein complex in A. flavus yielded an aconidial, aflatoxin-null phenotype for the resulting strain (66). Plant caleosin has been found to link to the G-protein component (68). Thus, at present, we cannot exclude the possibility that fungal caleosin and the oxylipins derived from it act by modulating the G-protein signaling pathway, resulting in an alteration of fungal development and, therefore, mycotoxin production.
Implication of AfPXG pathway in interactions between A. flavus and plant seeds.
The incapacity of lines in which AfPXG was silenced to develop on maize seeds contrasts with the findings for Aflox1 strains, which, surprisingly, recovered the ability to form conidia and aflatoxin when inoculated onto viable corn kernels (65). It was thus presumed for the Aflox1 mutant that the oxylipins released by seeds induce the activation of secondary metabolite synthesis and fungal morphological changes (27, 65). Accordingly, it was earlier shown that the addition of 13-HPOD and 9-HPOD caused an increase in conidial development in A. flavus (24). Because the seed oxylipins closely resemble psi factors in structure, it was hypothesized that the sporogenic effect of the seed oxylipins could take place through interference and/or mimicking of psi factors (66).
Our results, identifying a fungal caleosin using plant fatty acid hydroperoxides as the substrates, make the story more complex. Accordingly, the lack of recovery of conidiogenesis and aflatoxin production of the strains in which AfPXG was silenced in the presence of maize seeds might be due to their inability to generate active fungal FAOHs from the fatty acid hydroperoxides released by the maize seed lipoxygenase. This assumption would explain why conidiation and the production of aflatoxin were drastically reduced in Zmlox3 mutant fungi with a 9-LOX deletion infecting kernels (69). Similarly, this hypothesis would also explain how oxylipins generated by ZmLOX3 cloned in an aconidial ΔppoAC mutant of A. nidulans are able to restore the capacity of this mutant to produce conidia (27). Intriguingly, the growth of strains with AfPXG (Fig. 11) and Aflox1 (65) was not rescued when they infected maize kernels. These results point to a specificity of fungal oxylipins for the control of A. flavus mass that act independently of the pathogenic process. In conclusion, by identifying a novel oxylipin enzyme, our data underline the importance of plant-like hydroxy fatty acid derivatives in fungal development and pathogenesis possibly through the control of oxidative status.
ACKNOWLEDGMENTS
We thank I. Othman, director general of AECS, and N. MirAli, head of the Molecular Biology and Biotechnology Department, for their support. We also kindly thank John Mansfield, Imperial College of London, and Peter Lamont from the Ecology Department of Scottish Marine Institute, United Kingdom, for their precious help in critical reading of the manuscript.
REFERENCES
- 1.Andreou A, Brodhun F, Feussner I. 2009. Biosynthesis of oxylipins in non-mammals. Prog Lipid Res 48:148–170. doi: 10.1016/j.plipres.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 2.Blee E. 1998. Phytooxylipins and plant defense reactions. Prog Lipid Res 37:33–72. doi: 10.1016/S0163-7827(98)00004-6. [DOI] [PubMed] [Google Scholar]
- 3.Funk CD. 2001. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science 294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 4.Shah J. 2005. Lipids, lipases, and lipid-modifying enzymes in plant disease resistance. Annu Rev Phytopathol 43:229–260. doi: 10.1146/annurev.phyto.43.040204.135951. [DOI] [PubMed] [Google Scholar]
- 5.Trienens M, Rohlf M. 2012. Insect-fungus interference competition—the potential role of global secondary metabolite regulation, pathway-specific mycotoxin expression and formation of oxylipins. Fungal Ecol 5:191–199. [Google Scholar]
- 6.Shea JM, Del Poeta M. 2006. Lipid signaling in pathogenic fungi. Curr Opin Microbiol 9:352–358. doi: 10.1016/j.mib.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 7.Sakuradani E, Ando A, Ogawa J, Shimizu S. 2009. Improved production of various polyunsaturated fatty acids through filamentous fungus Mortierella alpina breeding. Appl Microbiol Biotechnol 84:1–10. doi: 10.1007/s00253-009-2076-7. [DOI] [PubMed] [Google Scholar]
- 8.Su C, Oliw EH. 1996. Purification and characterization of linoleate 8-dioxygenase from the fungus Gaeumannomyces graminis as a novel hemoprotein. J Biol Chem 271:14112–14118. doi: 10.1074/jbc.271.24.14112. [DOI] [PubMed] [Google Scholar]
- 9.Alem MA, Douglas LJ. 2005. Prostaglandin production during growth of Candida albicans biofilms. J Med Microbiol 54:1001–1005. doi: 10.1099/jmm.0.46172-0. [DOI] [PubMed] [Google Scholar]
- 10.Hamberg M. 1986. Isolation and structures of lipoxygenase products from Saprolegnia parasitica. Biochim Biophys Acta 876:688–692. doi: 10.1016/0005-2760(86)90059-7. [DOI] [Google Scholar]
- 11.Matsuda Y, Satho T, Beppu T, Arima K. 1976. Purification and properties of Co2+ requiring heme protein having lipoxygenase activity from Fusarium oxysporum. Agric Biol Chem 40:963–976. doi: 10.1271/bbb1961.40.963. [DOI] [Google Scholar]
- 12.Champe SP, el-Zayat AA. 1989. Isolation of a sexual sporulation hormone from Aspergillus nidulans. J Bacteriol 171:3982–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mazur P, Meyers HV, Nakanishi K, El-Zayat AAE, Champe SP. 1990. Structural elucidation of sporogenic fatty acid metabolites from Aspergillus nidulans. Tetrahedron Lett 31:3837–3840. doi: 10.1016/S0040-4039(00)97482-3. [DOI] [Google Scholar]
- 14.Mazur P, Nakanishi K, Elzayat AAE, Champe SP. 1991. Structure and synthesis of sporogenic psi factors from Aspergillus nidulans. J Chem Soc Chem Commun 20:1486–1487. [Google Scholar]
- 15.Tsitsigiannis DI, Kowieski TM, Zarnowski R, Keller NP. 2004. Endogenous lipogenic regulators of spore balance in Aspergillus nidulans. Eukaryot Cell 3:1398–1411. doi: 10.1128/EC.3.6.1398-1411.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hornsten L, Su C, Osbourn AE, Garosi P, Hellman U, Wernstedt C, Oliw EH. 1999. Cloning of linoleate diol synthase reveals homology with prostaglandin H synthases. J Biol Chem 274:28219–28224. doi: 10.1074/jbc.274.40.28219. [DOI] [PubMed] [Google Scholar]
- 17.Tsitsigiannis DI, Kowieski TM, Zarnowski R, Keller NP. 2005. Three putative oxylipin biosynthetic genes integrate sexual and asexual development in Aspergillus nidulans. Microbiology 151:1809–1821. doi: 10.1099/mic.0.27880-0. [DOI] [PubMed] [Google Scholar]
- 18.Tsitsigiannis DI, Kunze S, Willis DK, Feussner I, Keller NP. 2005. Aspergillus infection inhibits the expression of peanut 13S-HPODE-forming seed lipoxygenases. Mol Plant Microbe Interact 18:1081–1089. doi: 10.1094/MPMI-18-1081. [DOI] [PubMed] [Google Scholar]
- 19.Reverberi M, Punelli F, Scarpari M, Camera E, Zjalic S, Ricelli A, Fanelli C, Fabbri AA. 2010. Lipoperoxidation affects ochratoxin A biosynthesis in Aspergillus ochraceus and its interaction with wheat seeds. Appl Microbiol Biotechnol 85:1935–1946. doi: 10.1007/s00253-009-2220-4. [DOI] [PubMed] [Google Scholar]
- 20.Calvo AM, Gardner HW, Keller NP. 2001. Genetic connection between fatty acid metabolism and sporulation in Aspergillus nidulans. J Biol Chem 276:25766–25774. doi: 10.1074/jbc.M100732200. [DOI] [PubMed] [Google Scholar]
- 21.Tsitsigiannis DI, Keller NP. 2006. Oxylipins act as determinants of natural product biosynthesis and seed colonization in Aspergillus nidulans. Mol Microbiol 59:882–892. doi: 10.1111/j.1365-2958.2005.05000.x. [DOI] [PubMed] [Google Scholar]
- 22.Jayashree T, Subramanyam C. 2000. Oxidative stress as a prerequisite for aflatoxin production by Aspergillus parasiticus. Free Radic Biol Med 29:981–985. doi: 10.1016/S0891-5849(00)00398-1. [DOI] [PubMed] [Google Scholar]
- 23.Mahoney N, Molyneux RJ. 2010. Rapid analytical method for the determination of aflatoxins in plant-derived dietary supplement and cosmetic oils. J Agric Food Chem 58:4065–4070. doi: 10.1021/jf9039028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Calvo AM, Hinze LL, Gardner HW, Keller NP. 1999. Sporogenic effect of polyunsaturated fatty acids on development of Aspergillus spp. Appl Environ Microbiol 65:3668–3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doehlert DC, Wicklow DT, Gardner HW. 1993. Evidence implicating the lipoxygenase pathway in providing resistance to soybeans against Aspergillus flavus. Phytopathology 83:1473–1477. doi: 10.1094/Phyto-83-1473. [DOI] [Google Scholar]
- 26.Burow GB, Nesbitt TC, Dunlap J, Keller NP. 1997. Seed lipoxygenase products modulate Aspergillus mycotoxin biosynthesis. Mol Plant Microbe Interact 10:8. [Google Scholar]
- 27.Brodhagen M, Tsitsigiannis DI, Hornung E, Goebel C, Feussner I, Keller NP. 2008. Reciprocal oxylipin-mediated cross-talk in the Aspergillus-seed pathosystem. Mol Microbiol 67:378–391. doi: 10.1111/j.1365-2958.2007.06045.x. [DOI] [PubMed] [Google Scholar]
- 28.Prost I, Dhondt S, Rothe G, Vicente J, Rodriguez MJ, Kift N, Carbonne F, Griffiths G, Esquerre-Tugaye MT, Rosahl S, Castresana C, Hamberg M, Fournier J. 2005. Evaluation of the antimicrobial activities of plant oxylipins supports their involvement in defense against pathogens. Plant Physiol 139:1902–1913. doi: 10.1104/pp.105.066274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blee E, Wilcox AL, Marnett LJ, Schuber F. 1993. Mechanism of reaction of fatty acid hydroperoxides with soybean peroxygenase. J Biol Chem 268:1708–1715. [PubMed] [Google Scholar]
- 30.Hanano A, Burcklen M, Flenet M, Ivancich A, Louwagie M, Garin J, Blee E. 2006. Plant seed peroxygenase is an original heme-oxygenase with an EF-hand calcium binding motif. J Biol Chem 281:33140–33151. doi: 10.1074/jbc.M605395200. [DOI] [PubMed] [Google Scholar]
- 31.Blee E. 2002. Impact of phyto-oxylipins in plant defense. Trends Plant Sci 7:315–322. doi: 10.1016/S1360-1385(02)02290-2. [DOI] [PubMed] [Google Scholar]
- 32.Blee E, Boachon B, Burcklen M, Le Guedard M, Hanano A, Heintz D, Ehlting J, Herrfurth C, Feussner I, Bessoule JJ. 2014. The reductase activity of the Arabidopsis caleosin responsive to dessication20 mediates gibberellin-dependent flowering time, abscisic acid sensitivity, and tolerance to oxidative stress. Plant Physiol 166:109–124. doi: 10.1104/pp.114.245316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blee E, Flenet M, Boachon B, Fauconnier ML. 2012. A non-canonical caleosin from Arabidopsis efficiently epoxidizes physiological unsaturated fatty acids with complete stereoselectivity. FEBS J 279:3981–3995. doi: 10.1111/j.1742-4658.2012.08757.x. [DOI] [PubMed] [Google Scholar]
- 34.White TJ, Bruns T, Lee S, Taylor JW. 1990. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics, p 315–322. In PCR protocols: a guide to methods and applications. Academic Press, Inc., New York, NY. [Google Scholar]
- 35.Godet M, Munaut F. 2010. Molecular strategy for identification in Aspergillus section Flavi. FEMS Microbiol Lett 304:157–168. doi: 10.1111/j.1574-6968.2009.01890.x. [DOI] [PubMed] [Google Scholar]
- 36.Klich MA, Mullaney EJ. 1987. DNA restriction enzyme fragment polymorphism as a tool for rapid differentiation of Aspergillus flavus from Aspergillus oryzae. Exp Mycol 11:170–175. doi: 10.1016/0147-5975(87)90002-8. [DOI] [Google Scholar]
- 37.Ferreira de Oliveira JM, van Passel MW, Schaap PJ, de Graaff LH. 2010. Shotgun proteomics of Aspergillus niger microsomes upon d-xylose induction. Appl Environ Microbiol 76:4421–4429. doi: 10.1128/AEM.00482-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Record E, Asther M, Moukha S, Marion D, Burlat V, Ruel K. 1998. Localization of a phosphatidylglycerol/phosphatidylinositol transfer protein in Aspergillus oryzae. Can J Microbiol 44:945–953. doi: 10.1139/w98-092. [DOI] [PubMed] [Google Scholar]
- 39.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 40.Blee E, Durst F. 1987. Hydroperoxide-dependent sulfoxidation catalyzed by soybean microsomes. Arch Biochem Biophys 254:43–52. doi: 10.1016/0003-9861(87)90079-8. [DOI] [PubMed] [Google Scholar]
- 41.Blee E, Schuber F. 1990. Efficient epoxidation of unsaturated fatty acids by a hydroperoxide-dependent oxygenase. J Biol Chem 265:12887–12894. [PubMed] [Google Scholar]
- 42.Vernet T, Dignard D, Thomas DY. 1987. A family of yeast expression vectors containing the phage f1 intergenic region. Gene 52:225–233. doi: 10.1016/0378-1119(87)90049-7. [DOI] [PubMed] [Google Scholar]
- 43.Schiestl RH, Gietz RD. 1989. High efficiency transformation of intact yeast cells using single stranded nucleic acids as a carrier. Curr Genet 16:339–346. doi: 10.1007/BF00340712. [DOI] [PubMed] [Google Scholar]
- 44.Ninomiya Y, Suzuki K, Ishii C, Inoue H. 2004. Highly efficient gene replacements in Neurospora strains deficient for nonhomologous end-joining. Proc Natl Acad Sci U S A 101:12248–12253. doi: 10.1073/pnas.0402780101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuwayama H, Obara S, Morio T, Katoh M, Urushihara H, Tanaka Y. 2002. PCR-mediated generation of a gene disruption construct without the use of DNA ligase and plasmid vectors. Nucleic Acids Res 30:E2. doi: 10.1093/nar/30.2.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheng Y, Belanger RR. 2000. Protoplast preparation and regeneration from spores of the biocontrol fungus Pseudozyma flocculosa. FEMS Microbiol Lett 190:287–291. doi: 10.1111/j.1574-6968.2000.tb09300.x. [DOI] [PubMed] [Google Scholar]
- 47.Whisson SC, Avrova AO, Van West P, Jones JT. 2005. A method for double-stranded RNA-mediated transient gene silencing in Phytophthora infestans. Mol Plant Pathol 6:153–163. doi: 10.1111/j.1364-3703.2005.00272.x. [DOI] [PubMed] [Google Scholar]
- 48.Rasooli I, Razzaghi-Abyaneh M. 2004. Inhibitory effects of thyme oils on growth and aflatoxin production by Aspergillus parasiticus. Food Control 15:479–483. doi: 10.1016/j.foodcont.2003.07.002. [DOI] [Google Scholar]
- 49.Bertuzzi T, Rastelli S, Mulazzi A, Pietri A. 2011. Evaluation and improvement of extraction methods for the analysis of aflatoxins B1, B2, G1 and G2 from naturally contaminated maize. Food Anal Methods 5:512–519. [Google Scholar]
- 50.Shannon GM, Shotwell OL, Kwolek WF. 1983. Extraction and thin layer chromatography of aflatoxin B1 in mixed feeds. J Assoc Off Anal Chem 66:582–586. [PubMed] [Google Scholar]
- 51.Zhang B, Zhang H, Jin J, Ni Y, Chen J. 2012. PCDD/Fs-induced oxidative damage and antioxidant system responses in tobacco cell suspension cultures. Chemosphere 88:798–805. doi: 10.1016/j.chemosphere.2012.03.085. [DOI] [PubMed] [Google Scholar]
- 52.Beauchamp C, Fridovich I. 1971. Superoxide dismutase: improved assays and an assay applicable to acrylamide gels. Anal Biochem 44:276–287. doi: 10.1016/0003-2697(71)90370-8. [DOI] [PubMed] [Google Scholar]
- 53.Azevedo RA, Alas RM, Smith RJ, Lea PJ. 1998. Response of antioxidant enzymes to transfer from elevated carbon dioxide to air and ozone fumigation, in the leaves and roots of wild-type and a catalase-deficient mutant of barley. Physiol Plant 104:280–292. doi: 10.1034/j.1399-3054.1998.1040217.x. [DOI] [Google Scholar]
- 54.Gardner HW, Grove MJ, Keller NP. 1998. Soybean lipoxygenase is active on nonaqueous media at low moisture: a constraint to xerophilic fungi and aflatoxins? J Am Oil Chem Soc 75:1801–1808. doi: 10.1007/s11746-998-0334-y. [DOI] [Google Scholar]
- 55.Lie M, Pasha K. 1998. Fatty acids, fatty acid analogues and their derivatives. Nat Prod Rep 15:607–629. doi: 10.1039/a815607y. [DOI] [Google Scholar]
- 56.Summerer S, Hanano A, Utsumi S, Arand M, Schuber F, Blee E. 2002. Stereochemical features of the hydrolysis of 9,10-epoxystearic acid catalysed by plant and mammalian epoxide hydrolases. Biochem J 366:471–480. doi: 10.1042/BJ20011778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Payne WGA, Dean RA, Nierman WC, Amedeo P, Caler EGA, Fedorova ND, Maiti R, Joardar V, Inman J, Galinsky KJ, Yu J, Bhatnagar D, Cleveland TE. 2007. Genome sequence of Aspergillus flavus strain NRRL 3357. Submitted (04-SEP-2007) to the INSDC. J. Craig Venter Institute, Rockville, MD. [Google Scholar]
- 58.Murphy DJ. 2001. The biogenesis and functions of lipid bodies in animals, plants and microorganisms. Prog Lipid Res 40:325–438. doi: 10.1016/S0163-7827(01)00013-3. [DOI] [PubMed] [Google Scholar]
- 59.Kim YY, Jung KW, Yoo KS, Jeung JU, Shin JS. 2011. A stress-responsive caleosin-like protein, AtCLO4, acts as a negative regulator of ABA responses in Arabidopsis. Plant Cell Physiol 52:874–884. doi: 10.1093/pcp/pcr039. [DOI] [PubMed] [Google Scholar]
- 60.Abell BM, Holbrook LA, Abenes M, Murphy DJ, Hills MJ, Moloney MM. 1997. Role of the proline knot motif in oleosin endoplasmic reticulum topology and oil body targeting. Plant Cell 9:1481–1493. doi: 10.1105/tpc.9.8.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reverberi M, Zjalic S, Ricelli A, Punelli F, Camera E, Fabbri C, Picardo M, Fanelli C, Fabbri AA. 2008. Modulation of antioxidant defense in Aspergillus parasiticus is involved in aflatoxin biosynthesis: a role for the ApyapA gene. Eukaryot Cell 7:988–1000. doi: 10.1128/EC.00228-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tsitsigiannis DI, Zarnowski R, Keller NP. 2004. The lipid body protein, PpoA, coordinates sexual and asexual sporulation in Aspergillus nidulans. J Biol Chem 279:10. [DOI] [PubMed] [Google Scholar]
- 63.Nuccio ML, Thomas TL. 1999. ATS1 and ATS3: two novel embryo-specific genes in Arabidopsis thaliana. Plant Mol Biol 39:1153–1163. doi: 10.1023/A:1006101404867. [DOI] [PubMed] [Google Scholar]
- 64.Poxleitner M, Rogers SW, Lacey Samuels A, Browse J, Rogers JC. 2006. A role for caleosin in degradation of oil-body storage lipid during seed germination. Plant J 47:917–933. doi: 10.1111/j.1365-313X.2006.02845.x. [DOI] [PubMed] [Google Scholar]
- 65.Scarpari M, Punelli M, Scala V, Zaccaria M, Nobili C, Ludovici M, Camera E, Fabbri AA, Reverberi M, Fanelli C. 2014. Lipids in Aspergillus flavus-maize interaction. Front Microbiol 5:74. doi: 10.3389/fmicb.2014.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Calvo AM, Wilson RA, Bok JW, Keller NP. 2002. Relationship between secondary metabolism and fungal development. Microbiol Mol Biol Rev 66:447–459. doi: 10.1128/MMBR.66.3.447-459.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Affeldt KJ, Brodhagen M, Keller NP. 2012. Aspergillus oxylipin signaling and quorum sensing pathways depend on G protein-coupled receptors. Toxins 4:695–717. doi: 10.3390/toxins4090695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Khalil HB, Wang Z, Wright JA, Ralevski A, Donayo AO, Gulick PJ. 2011. Heterotrimeric Gα subunit from wheat (Triticum aestivum), GA3, interacts with the calcium-binding protein, Clo3, and the phosphoinositide-specific phospholipase C, PI-PLC1. Plant Mol Biol 77:145–158. doi: 10.1007/s11103-011-9801-1. [DOI] [PubMed] [Google Scholar]
- 69.Gao X, Shim WB, Göbel C, Kunze S, Feussner I, Meeley R, Balint-Kurti P, Kolomiets M. 2007. Disruption of a maize 9-lipoxygenase results in increased resistance to fungal pathogens and reduced levels of contamination with mycotoxin fumonisin. Mol Plant Microbe Interact 20:922–933. doi: 10.1094/MPMI-20-8-0922. [DOI] [PubMed] [Google Scholar]