

Abstract
Dairy industry fermentative processes mostly use Lactococcus lactis as a starter. However, some dairy L. lactis strains produce putrescine, a biogenic amine that raises food safety and spoilage concerns, via the agmatine deiminase (AGDI) pathway. The enzymatic activities responsible for putrescine biosynthesis in this bacterium are encoded by the AGDI gene cluster. The role of the catabolic genes aguB, aguD, aguA, and aguC has been studied, but knowledge regarding the role of aguR (the first gene in the cluster) remains limited. In the present work, aguR was found to be a very low level constitutively expressed gene that is essential for putrescine biosynthesis and is transcribed independently of the polycistronic mRNA encoding the catabolic genes (aguBDAC). In response to agmatine, AguR acts as a transcriptional activator of the aguB promoter (PaguB), which drives the transcription of the aguBDAC operon. Inverted sequences required for PaguB activity were identified by deletion analysis. Further work indicated that AguR is a transmembrane protein which might function as a one-component signal transduction system that senses the agmatine concentration of the medium and, accordingly, regulates the transcription of the aguBDAC operon through a C-terminal cytoplasmic DNA-binding domain typically found in LuxR-like proteins.
INTRODUCTION
Lactococcus lactis is the lactic acid bacterium (LAB) most widely used as a primary starter in the dairy industry, especially in cheese manufacturing. Despite its qualified presumption of safety (QPS) status (awarded by the European Food and Safety Authority [EFSA]) and its generally regarded as safe (GRAS) status (awarded by the Food and Drug Administration [FDA]), some L. lactis strains possess enzymatic activities that produce undesirable flavors associated with food spoilage (1). Some even produce toxic compounds, such as the biogenic amine (BA) putrescine (2). Putrescine, together with histamine and tyramine, is one of the BAs in fermented dairy products most frequently encountered at potentially unsafe levels (3–5). It has a synergistic effect on the toxicity of other BAs and can also react with nitrite to form carcinogenic nitrosamines (4, 6). In addition, the metabolism of putrescine and of its derivatives (the polyamines spermine and spermidine) plays an important role in the promotion of colorectal tumorigenesis via effects on cell proliferation and migration (7–10).
A number of putrescine-producing L. lactis strains of the subspecies L. lactis subsp. lactis and L. lactis subsp. cremoris isolated from artisanal cheeses have been shown to have a functional agmatine deiminase (AGDI) pathway. This catabolizes agmatine (a decarboxylated derivative of arginine) (11) into putrescine, yielding one molecule of ATP, one molecule of CO2, and two ammonium ions (2). The AGDI pathway increases the growth of L. lactis and causes the alkalinization of the culture medium, although it does not seem to be an acid stress resistance mechanism (12). The AGDI cluster of L. lactis is composed of five genes, aguR, aguB, aguD, aguA, and aguC, with the last four being responsible for the conversion of agmatine to putrescine (2, 13). Agmatine enters the cell via AguD (an agmatine-putrescine antiporter encoded by aguD) and is then hydrolyzed to N-carbamoylputrescine and an ammonium ion by AguA (an agmatine deiminase encoded by aguA). AguB is a putrescine carbamoyltransferase encoded by aguB that catalyzes the phosphorolysis of N-carbamoylputrescine, yielding putrescine and carbamoylphosphate. Finally, a phosphate group is transferred from carbamoylphosphate to ADP by AguC (a carbamate kinase encoded by aguC) to generate ATP, CO2, and a further ammonium ion. Putrescine is then exchanged for agmatine via the antiporter AguD (2). The protein encoded by aguR showed primary structure similarity to the AguR of Streptococcus mutans, a transcriptional activator of the agmatine deiminase system (14). The aim of the present work was to investigate whether aguR of L. lactis is involved in the transcriptional regulation of the AGDI cluster.
The strain selected for study was L. lactis subsp. cremoris CECT8666 (formerly GE2-14); this strain was originally isolated from a traditional cheese (2) and is a strong putrescine producer (13), and its genome has been completely sequenced (15). Although previously demonstrated in a L. lactis subsp. lactis putrescine-producing strain (2), it was first confirmed that the present strain's aguR was transcribed independently of the catalytic genes, which are expressed as an operon (aguBDAC). The construction of a ΔaguR knockout (KO) mutant and its subsequent analysis showed that AguR activates putrescine production. Transcriptomic studies, confirmed by independent transcriptional analysis of aguR and the aguBDAC operon, verified the involvement of AguR in the transcriptional activation of aguBDAC. Moreover, the transcriptional activation of aguBDAC was dependent on the agmatine concentration of the culture medium. In silico analysis of the topology of AguR, plus comparative studies of its structure, revealed the presence of a putative DNA-binding domain at the C terminus. It was also confirmed that AguR is located on the cell surface. Taking these results together, AguR would seem to act as a one-component signal transduction system that senses the agmatine concentration in the environment and accordingly regulates the transcription of the aguBDAC operon.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
Table 1 shows the strains and plasmids used in this study. The L. lactis subsp. cremoris strains were grown in M17 medium (Oxoid, Basingstoke, United Kingdom) supplemented with 30 mM glucose (GM17 medium) or 60 mM galactose (GalM17 medium) to prevent carbon catabolic repression (CCR) of the ADGI pathway (13). Where indicated, the media were supplemented with agmatine (Sigma-Aldrich, Barcelona, Spain) at the concentration specified below. Escherichia coli strains were grown in Luria-Bertani (LB) medium at 37°C with aeration (16). When plasmid-containing clones were grown, the medium was supplemented with the appropriate antibiotics: for L. lactis subsp. cremoris, 5 μg ml−1 of chloramphenicol (Cm) and 2 μg ml−1 of erythromycin (Em); for E. coli, 150 μg ml−1 of Em.
TABLE 1.
Strains and plasmids
| Strain or plasmid | Characteristicsa | Reference or source |
|---|---|---|
| Strains | ||
| L. lactis subsp. cremoris NZ9000 | L. lactis subsp. cremoris MG1363 containing nisRK genes, non-putrescine producer | 25 |
| L. lactis subsp. cremoris CECT8666 | Isolated from artisanal cheese, putrescine producer | 2 |
| E. coli DH11S | Life Technologies, Spain | |
| L. lactis subsp. cremoris CECT8666 ΔaguR | Strain CECT8666 lacking the aguR gene | This work |
| Plasmids | ||
| pNZ8048 | Lactococcal plasmid, Cmr | 25 |
| pCS1966 | Selection/counterselection vector, Emr | 21 |
| pNZcLIC-GFP | pNZ8048 derivative harboring gfp, Cmr | 26 |
| pNZcGFP | pNZ8048 derivative harboring gfp, Cmr | 26 |
| pAGDI | pNZ8048 derivative bearing the PaguR-aguR-PaguB-gfp fusion, Cmr | 13 |
| pIPLA1269 | pCS1966 derivative bearing a fragment of the CECT8666 ycaC-aguR genes, Emr | This work |
| pIPLA1713 | pIPLA1269 derivative bearing a fragment of the CECT8666 ycaC-aguR genes and a fragment of the aguR-aguB genes, Emr | This work |
| pAG1 | pNZ8048 derivative bearing a PaguR-gfp fusion, Cmr | This work |
| pAG2 | pNZ8048 derivative bearing a PaguB-gfp fusion, Cmr | This work |
| pAG3 | pNZcLIC-GFP derivative bearing a PnisA-aguR-gfp fusion, Cmr | This work |
| pAGDIΔ1 | pAGDI derivative with a deletion from nucleotides −209 to −179 of PaguB, Cmr | This work |
| pAGDIΔ2 | pAGDI derivative with a deletion from nucleotides −179 to −146 of PaguB, Cmr | This work |
| pAGDIΔ3 | pAGDI derivative with a deletion from nucleotides −146 to −119 of PaguB, Cmr | This work |
| pAGDIΔ4 | pAGDI derivative with a deletion from nucleotides −119 to −92 of PaguB, Cmr | This work |
Cmr, chloramphenicol resistance; Emr, erythromycin resistance; PaguR, aguR promoter; PaguB, aguB promoter; PnisA, nisA gene promoter.
Analysis of putrescine production by UHPLC.
Cultures were grown in GM17 medium in the presence of 20 mM agmatine for 24 h. The cultures were then centrifuged at 8,000 × g, and the supernatants were collected. The putrescine concentration in the supernatants (100 μl) was assessed by ultra-high-performance liquid chromatography (UHPLC) using a Waters H-class Acquity ultraperformance liquid chromatography apparatus controlled by Empower (v.2.0) software and employing a UV detection method based on derivatization with diethyl ethoxymethylene malonate (Sigma-Aldrich) (17).
DNA manipulation.
L. lactis subsp. cremoris genomic DNA was obtained using Kirby lytic mix following a previously described protocol (18). Genetic constructs for L. lactis subsp. cremoris were produced using L. lactis subsp. cremoris NZ9000 as an intermediate host. Plasmid DNA was isolated from L. lactis subsp. cremoris and transformed as described previously (19). Genetic constructs for E. coli were produced using E. coli DH11S (Life Technologies, Madrid, Spain) as an intermediate host. The E. coli plasmid DNA was isolated by the alkaline lysis method (16). Electroporation was performed in a Bio-Rad pulser apparatus (Bio-Rad, Barcelona, Spain) following the manufacturer's instructions. Restriction endonuclease digestions, alkaline phosphatase treatments, ligations, and other DNA manipulation procedures were performed according to standard methods (16). PCR amplifications were performed in a MyCycler thermal cycler (Bio-Rad) using Phusion high-fidelity DNA polymerase (Thermo Scientific, Barcelona, Spain) according to the manufacturer's protocol. Table 2 shows the primers used for PCR amplifications. The primers used to amplify fragments of the L. lactis subsp. cremoris CECT8666 AGDI cluster were based on its nucleotide sequence (GenBank accession no. HG317493.1). All plasmids constructed in this work were checked by nucleotide sequencing (performed by Macrogen Inc., Seoul, Republic of Korea).
TABLE 2.
Primers used
| Primer | Functiona | Nucleotide sequence (5′ to 3′) | Source or reference |
|---|---|---|---|
| AguF | Intergenic region ycaC-aguR (F) | CGAACAGACAGCGTCCCTGA | This work |
| AgurNco | Intergenic region ycaC-aguR (R) | CCCCATGGGATTAGACCTACTTATCATATTATCA | This work |
| PtcBglII | Intergenic region aguR-aguB (F) | CCCCAGATCTAAGCATATGAAAAATCAGAACTTAG | This work |
| PTC3C | Intergenic region aguR-aguB (R) | CTTTAGTGTAATCTTCTGTTGTGATG | This work |
| RT1 | Intergenic region aguB-aguD (F) | CCTTATGATTTGAAAGCACAAG | This work |
| RT2 | Intergenic region aguB-aguD (R) | GAAAGAATAGCACTAAATAGAC | This work |
| RT3 | Intergenic region aguD-aguA (F) | TTATTGGAGAACTGATTATTAAAG | This work |
| AgDI3Cre | Intergenic region aguD-aguA (R) | CTTGAGCTTCAAATTCACCGGGC | This work |
| RT4 | Intergenic region aguA-aguC (F) | GGCGGTGGAAATATTCACTG | This work |
| RT5 | Intergenic region aguA-aguC (R) | TTGTGCTGTAGGGTCACTGC | This work |
| qAguR-F | aguR expression analysis (F) | CTATCGACAGGTTAAGCAAAGCAGTT | 13 |
| qAguR-R | aguR expression analysis (R) | TCCAAAGATGATGGCCATTATGC | 13 |
| qPTC-F | aguB expression analysis (F) | ACTTGGTGGACATGAAACAATAGAAGAT | 13 |
| qPTC-R | aguB expression analysis (R) | GTCAACACGTGCCATTATGATATCG | 13 |
| rpoA-F | rpoA reference gene (F) | CACGGGCAGGTTCAACTTG | 22 |
| rpoA-R | rpoA reference gene (R) | TTCCGGCTGACGAAAATAAAG | 22 |
| qtufF | EF-Tu reference gene (F) | TCTTCATCATCAACAAGGTCTGCTT | 13 |
| qtufR | EF-Tu reference gene (R) | GAACACATCTTGCTTTCACGTCAA | 13 |
| AguRBglII | Cloning of PaguR (F) | CCCCCCAGATCTGACAAGTTTGGCTCAGATTGCTTG | This work |
| PtcNco | Cloning of PaguB (R) | CCCCCCATGGTGTTTATTCCTCCTGAATAAAATAG | This work |
| AgurlicF | Cloning of aguR (F) | ATGGGTGGTGGATTTGCTATGTTAAATTATATTTATACTACTTTTT | This work |
| AgurlicR | Cloning of aguR (R) | TTGGAAGTATAAATTTTCTTGACTAAGTTCTGATTTTTCATATG | This work |
| Gffor | Cloning of gfp (F) | GGCCATGGGTGGTGGATTTGCTCAATTC | This work |
| Gfrev | Cloning of gfp (R) | CCGCATGCCTGCATTAATGATGGTG | This work |
| KO1F | Generation of pAGDIΔ1 (F) | CACACGAATTCGAAAAAAGCACTAAACCCTCC | This work |
| KO1R | Generation of pAGDIΔ1 (R) | CACACGAATTCTCCATTCAAAAAATGGAGCT | This work |
| KO2F | Generation of pAGDIΔ2 (F) | CACACGAATTCCTCAACCCCTTGGTAGCAAAGG | This work |
| KO2R | Generation of pAGDIΔ2 (R) | CACACGAATTCAAACGCTTTCTTTTTATAAATAAA | This work |
| KO3F | Generation of pAGDIΔ3 (F) | CACACGAATTCTTGCTTTTTAAAAAGATTAAATCCT | This work |
| KO3R | Generation of pAGDIΔ3 (R) | CACACGAATTCGTCAAAAGGTTTAGGAGGGTTTAG | This work |
| KO4F | Generation of pAGDIΔ4 (F) | CACACGAATTCAGTTGATTGTTTTAAGAAATCAACC | This work |
| KO4R | Generation of pAGDIΔ4 (R) | CACACGAATTCAAAACCCTTTGCTACCAAGGG | This work |
| KO-214AguR-AF2 | Generation of pIPLA1269 (F) | CACATGACTAGTTTAGAACCTAGAAACCCAGAAAC | This work |
| KO-214AguR-AR | Generation of pIPLA1269 (R) | AACTGCAGATTTAACATCATCGGATTAGACCTAC | This work |
| KO-214AguR-BF | Generation of pIPLA1713 (F) | AACTGCAGTCAGAACTTAGTCAATAATTTAAAAG | This work |
| KO-214AguR-BR | Generation of pIPLA1713 (R) | CCATCGATAACCGCATCAACAACTTC | This work |
F, forward; R, reverse; PaguR, aguR promoter; PaguB, aguB promoter.
Reverse transcription (RT)-PCR.
Cells were grown in GM17 culture medium in the presence of 20 mM agmatine. Two milliliters of culture was collected at the end of the exponential phase of growth. Total RNA was extracted as previously described (20). cDNA was then synthesized from DNase-treated RNA samples using an iScript cDNA synthesis kit (Bio-Rad) according to the manufacturer's recommendations. The ycaC-aguR, aguR-aguB, aguB-aguD, aguD-aguA, and aguA-aguC intergenic regions (see Fig. 1A) were analyzed by PCR amplification using cDNA as a template and specific pairs of primers (Table 2). PCRs were performed using 2 μl of cDNA and 0.4 μM each gene-specific primer. Amplifications were performed for 35 cycles (94°C for 30 s, 55°C for 45 s, and 72°C for 1 min); the resulting amplicons were separated on 1.5% agarose gels in TAE (Tris-acetate-EDTA) buffer. The absence of contaminating DNA was checked via omission of reverse transcriptase in the PCR mixture; this was performed under the conditions described above, using the corresponding RNA as a template.
FIG 1.

Transcriptional analysis of the AGDI operon of L. lactis subsp. cremoris CECT8666. (A) Genetic organization of the AGDI cluster and surrounding regions. The putative aguR promoter (PaguR), the aguB promoter (PaguB), and the termination regions ( ) are indicated. (B) RT-PCR amplification of intergenic regions was conducted using total RNA extracted from cells grown in the presence of 20 mM agmatine. Five set of primers were designed to amplify the intergenic regions: ycaC-aguR (primer pair 1, lanes 1), aguR-aguB (primer pair 2, lanes 2), aguB-aguD (primer pair 3, lanes 3), aguD-aguA (primer pair 4, lanes 4), and aguA-aguC (primer pair 5, lanes 5). Negative controls were run with the same RNA samples but without reverse transcriptase. Positive controls were run with chromosomal DNA. Lanes M, DNA molecular size markers.
) are indicated. (B) RT-PCR amplification of intergenic regions was conducted using total RNA extracted from cells grown in the presence of 20 mM agmatine. Five set of primers were designed to amplify the intergenic regions: ycaC-aguR (primer pair 1, lanes 1), aguR-aguB (primer pair 2, lanes 2), aguB-aguD (primer pair 3, lanes 3), aguD-aguA (primer pair 4, lanes 4), and aguA-aguC (primer pair 5, lanes 5). Negative controls were run with the same RNA samples but without reverse transcriptase. Positive controls were run with chromosomal DNA. Lanes M, DNA molecular size markers.
Construction of an L. lactis subsp. cremoris CECT8666 ΔaguR deletion mutant.
An L. lactis subsp. cremoris CECT8666 ΔaguR deletion mutant was constructed by homologous recombination using the selection/counterselection vector pCS1966 (21). Table 2 shows the primers used to generate the ΔaguR knockout. They were designed to include the following restriction recognition sites: SpeI (in primer KO-214AguR-AF2), PstI (in primer KO-214AguR-AR), PstI (in primer KO-214AguR-BF), and ClaI (in primer KO-214AguR-BR). An 826-bp PCR fragment containing a 610-bp fragment of the 3′ end of the ycaC gene (upstream of aguR; GenBank accession no. HG317493.1), the intergenic region between ycaC and aguR, and the sequence coding for the first 5 amino acids of the aguR gene of L. lactis subsp. cremoris CECT8666 was amplified using primers KO-214AguR-AF2 and KO-214AguR-AR. The resulting fragment was digested with the SpeI and PstI restriction enzymes and cloned into the pCS1966 vector, rendering the plasmid pIPLA1269. A second 1,110-bp PCR fragment containing the last 18 bp of aguR, the intergenic region between aguR and aguB, and 856 bp of the beginning of the aguB gene was PCR amplified using primers KO-214AguR-BF and KO-214AguR-BR. The resulting fragment was digested with PstI and ClaI and cloned into the plasmid pIPLA1269, rendering the plasmid pIPLA1713. Plasmid pIPLA1713 was then transformed and integrated into L. lactis subsp. cremoris CECT8666 electrocompetent cells by homologous recombination. A previously described methodology (21) based on 5-fluoroorotate sensitivity was used to select for the loss of the plasmid (second recombination step). The resulting mutants lacking aguR (L. lactis subsp. cremoris CECT8666 ΔaguR) were confirmed by nucleotide sequence analysis of the amplicon obtained using primers KO-214AguR-AF2 and KO-214AguR-BR, which rendered the expected 1,936-bp fragment instead of the 2,894-bp fragment corresponding to the wild-type (WT) strain (data not shown).
DNA microarray experiments and data analysis.
L. lactis subsp. cremoris CECT8666 DNA microarrays (Agilent Technologies, Santa Clara, CA) were designed using the Agilent eArray (v.5.0) program according to the manufacturer's recommendations (Agilent Technologies). Each microarray (8 × 15K) was designed to contain spots of two different 60-mer oligonucleotide probes (in duplicate) specific for each of the 2,765 coding DNA sequences (CDSs) representing the protein-coding genes of the L. lactis subsp. cremoris CECT8666 genome (GenBank accession no. AZSI00000000.1) (15).
Total RNA was isolated from 10 ml of L. lactis subsp. cremoris CECT8666 and from the ΔaguR mutants, both of which were grown to late exponential phase in GalM17 medium supplemented with 20 mM agmatine. cDNA synthesis was performed using a SuperScript III reverse transcriptase kit (Life Technologies, Bleiswijk, The Netherlands) following the manufacturer's instructions. Twenty micrograms of cDNA was then labeled with Cy3/Cy5 dyes using a DyLight amine-reactive dyes kit (Thermo Scientific, Amsterdam, The Netherlands) following the manufacturer's protocol. Nine hundred nanograms of both Cy3- and Cy5-labeled cDNA was then mixed and hybridized for 17 h at 60°C in the L. lactis subsp. cremoris CECT8666 DNA microarray using an In Situ Hybridization kit Plus (Agilent Technologies) following the manufacturer's instructions. The slides were scanned using a GenePix 4200A microarray scanner (Molecular Devices, Sunnyvale, CA), and the images were analyzed using GenePix Pro (v.6.0) software. Background subtraction and locally weighted scatterplot smoothing normalization were performed using the standard routines provided by Genome2D software, available at http://server.molgenrug.nl. DNA microarray data were obtained from three independent biological replicates and two technical replicates (including a dye swap). Expression ratios were calculated from the comparison of four spots per gene per microarray (for a total of 20 measurements per gene). A gene was considered differentially expressed when a P value of at least <0.05 was obtained and the fold change in expression was at least >|0.5|.
Quantification of gene expression by RT-qPCR.
Total RNA was extracted from cultures collected at the end of the exponential phase of growth, and cDNA was synthesized by retrotranscription as described above. cDNA samples were analyzed by quantitative real-time PCR (qPCR) using an ABI Prism Fast 7500 sequence detection system (Applied Biosystems, Carlsbad, CA). The reactions were performed as previously described (20) in a 25-μl reaction volume, which included 0.9 μM each primer and Power SYBR green PCR master mix (which contains carboxy-X-rhodamine as a passive reference) (Applied Biosystems). Cycling was performed under the Applied Biosystems default settings. Amplifications were performed with previously described specific primers (13) (Table 2); primers specific for the thermounstable elongation factor (tuf) (13) and RNA polymerase alpha subunit (rpoA) (22) genes were used as references. The linearity and amplification efficiency of the reactions were tested for each primer pair at five points in a 10-fold dilution series of L. lactis subsp. cremoris CECT8666 genomic DNA. Samples with no template were included as negative controls in each run. Relative gene expression was calculated using the comparative ΔΔCT threshold cycle (CT) method as previously described (23). For each condition, RT-qPCR analysis was performed on RNA purified from three independently grown cultures. Statistical comparisons were made using the Student t test; significance was set at a P value of <0.05.
Generation of fusions with the gfp reporter gene.
A translational fusion of the aguR promoter (PaguR) attached to the gfp reporter gene (which codes for green fluorescent protein [GFP]) (PaguR-gfp) was generated. For this, the PaguR fragment was PCR amplified using primers AgurNco and AguRBglII (Table 2) and cloned into the BglII-NcoI sites of plasmid pNZ8048 (24, 25). The gfp gene was then PCR amplified from plasmid pNZcGFP (26) using primers Gffor and Gfrev and cloned into the resulting vector as an NcoI-SphI fragment, yielding the plasmid pAG1.
Similarly, a translational fusion of the promoter of aguB (PaguB) attached to the gfp reporter gene (PaguB-gfp) was generated. For this, the PaguB fragment was PCR amplified using primers PtcNco and PtcBglII (Table 2) and cloned into the BglII-NcoI sites of plasmid pNZ8048. The gfp gene was then PCR amplified from plasmid pNZcGFP using primers Gffor and Gfrev and inserted into the resulting vector as an NcoI-SphI fragment, yielding the pAG2 plasmid.
Finally, for the cellular localization of AguR, a translational fusion between aguR and gfp under the control of the nisin-inducible promoter (PnisA) was generated. For this, aguR was PCR amplified using the AgurlicF and AgurlicR primers (Table 2) and cloned into the SwaI restriction site of the pNZcLIC-GFP expression vector (26, 27), yielding the plasmid pAG3. All constructs were checked by nucleotide sequencing (performed by Macrogen Inc.).
Generation of aguB promoter deletion constructions.
Plasmids pAGDIΔ1, pAGDIΔ2, pAGDIΔ3, and pAGDIΔ4, bearing versions of PaguB with different deletions, were all derived from previously constructed plasmid pAGDI (13). Plasmid pAGDI carries the cassette PaguR-aguR-PaguB fused to the gfp reporter gene. For each construct, pAGDI was first methylated with Dam methylase and S-adenosylmethionine (New England BioLabs, Hertfordshire, United Kingdom) following the manufacturer's instructions. The whole pAGDI plasmid was amplified using divergent primers (Table 2) flanking the region of PaguB to be deleted. An EcoRI target site was included in the primers so that the amplicons obtained could be digested with EcoRI and self-ligated. The ligation mixture was digested with DpnI (in order to digest the original pAGDI plasmid used as a Dam-methylated template) before transformation into L. lactis subsp. cremoris NZ9000.
Whole-cell fluorescence measurements.
For whole-cell fluorescence measurements, equal quantities of cells were harvested, washed, and subsequently resuspended in 50 mM KPi, pH 7.2, as previously described (26). GFP emission was measured in a volume of 250 μl of cells, using a Cary Eclipse fluorescence spectrophotometer (excitation wavelength, 485 nm; emission wavelength, 530 nm; Varian Inc., Palo Alto, CA). For direct comparison, all GFP fluorescence data were normalized to the same optical density at 600 nm (OD600). Background fluorescence levels were assessed by measuring the fluorescence of nonfluorescent control cells; these values were subtracted. Statistical comparisons were made using the Student t test; significance was set at a P value of <0.05.
Fluorescence microscopy.
L. lactis subsp. cremoris NZ9000 cells containing the pAG3 plasmid carrying the PnisA-aguR-gfp translational fusion (Table 2) were grown in GM17 medium supplemented with chloramphenicol (5 μg ml−1) at 30°C until an OD600 of 0.6 was reached. The expression of aguR-gfp was then induced by the addition of 0.5 nM nisin for 2 h. Fluorescence was analyzed using a Nikon Eclipse 90i microscope (Nikon UK, Kingston, United Kingdom) running iControl software and ACT-2U camera control software, employing a ×100 objective and a B2A Nikon filter (excitation filter, 450 to 490 nm; dichroic mirror, 505 nm; emission filter, 520 nm). A minimum of 15 random fields of view were observed for each sample. Each experiment was performed in triplicate.
In silico analysis of inverted sequences of the aguB promoter.
In silico analysis of the nucleotide sequence of the putative aguB promoter (GenBank accession no. HG317493.1; nucleotides 3518 to 3726) was performed using Clone Manager (v.7) software (Scientific & Educational Software, Cary, NC).
In silico analysis of AguR.
The NCBI BLASTP program (http://blast.ncbi.nlm.nih.gov) was used to determine the similarity of the deduced amino acid sequence for AguR to sequences present in databases. Functional domains in AguR were analyzed using the Pfam database (http://pfam.xfam.org/) (28). The topology of AguR was predicted using computer-based algorithms available on the SOSUI server (http://bp.nuap.nagoya-u.ac.jp/sosui/sosui_submit.html) (29). Homology modeling was performed by searching for the most suitable template protein structure using the SWISS-MODEL work space (http://swissmodel.expasy.org) (30). Model refinement and editing were performed using Swiss-PdbViewer (v.4.0.4) software (31).
Microarray data accession number.
The microarray data were deposited in the Gene Expression Omnibus (GEO) database under accession no. GSE59514.
RESULTS
aguR is transcribed independently of the aguBDAC operon.
The transcriptional profiles of aguR and of the genes encoding the putrescine biosynthetic pathway (aguB, aguD, aguA, and aguC) were determined. Total RNA was isolated from L. lactis subsp. cremoris CECT8666 cells grown in GM17 medium supplemented with 20 mM agmatine and was used in RT-PCR analysis involving five sets of primers (referred to as primer sets 1 to 5 in Fig. 1A) designed to amplify the regions spanning the gene junctions (Table 2; Fig. 1A). The ycaC-aguR and aguR-aguB intergenic regions rendered no RT-PCR product (Fig. 1B, lanes 1 and 2, respectively), indicating that neither the ycaC-aguR nor the aguR-aguB intergenic region was cotranscribed. In fact, a potential transcription terminator was found in each intergenic region (ΔG = −9.4 and −10.3 kcal/mol, respectively). In contrast, RT-PCR amplifications of the aguB-aguD, aguD-aguA, and aguA-aguC intergenic regions rendered DNA fragments of the expected size (Fig. 1B, lanes 3 to 5, respectively), showing that aguB, aguD, aguA, and aguC are cotranscribed. The RT-PCRs for the negative controls failed to yield any amplification product. DNA template controls (to ensure PCR fidelity for each primer pair) uniformly yielded the PCR product of the expected size. Overall, these results indicate that the aguR gene is transcribed from its own promoter (PaguR) as a monocistronic mRNA and that its transcription is independent of both the ycaC gene located upstream of the AGDI cluster and the aguB gene. In addition, the results indicate that the aguB, aguD, aguA, and aguC genes are cotranscribed as a polycistronic mRNA (aguBDAC operon) from the PaguB promoter.
AguR is essential for putrescine biosynthesis.
To investigate the involvement of AguR in putrescine production, an L. lactis subsp. cremoris CECT8666 ΔaguR (KO) mutant was constructed as described in Materials and Methods. Both the WT and KO strains were grown in GM17 medium supplemented with increasing agmatine concentrations (0, 0.05, 0.1, 0.25, 0.5, 1, 5, 10, and 20 mM) for 24 h. Samples were collected at the end of fermentation, and putrescine production was determined by UHPLC (Fig. 2). The level of putrescine production by the WT strain strictly correlated with the initial concentration of agmatine in the medium. However, the deletion of aguR completely abolished the conversion of agmatine to putrescine; no putrescine was produced at any of the agmatine concentrations tested.
FIG 2.
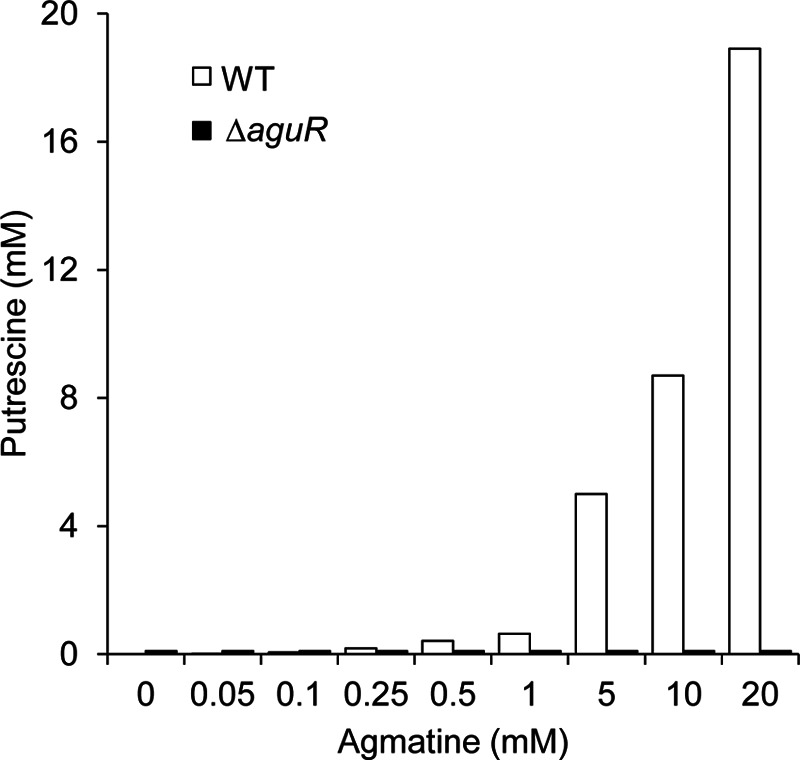
Production of putrescine by L. lactis subsp. cremoris CECT8666 (WT) and the ΔaguR deletion mutant at different agmatine concentrations. Both strains were grown in GM17 medium supplemented with 0, 0.05, 0.1, 0.25, 0.5, 1, 5, 10, or 20 mM agmatine for 24 h. Supernatants were analyzed by UHPLC to determine the putrescine concentration in the extracellular medium.
Effect of aguR deletion on transcriptomic profile.
To determine the effect of the deletion of aguR on the transcriptomic profile of L. lactis subsp. cremoris CECT8666, DNA microarray analysis was performed with the WT and the KO strains grown in the presence of 20 mM agmatine. Genes differentially expressed by the KO mutant and WT strain that fulfilled the criteria of at least a 3-fold change and a P value of <0.001, as well as the results for aguR, are shown in Table 3. The four catalytic genes (aguB, aguD, aguA, and aguC) coding for the proteins needed for the biosynthesis of putrescine were clearly downregulated in the ΔaguR strain (fold changes, −27.44, −26.27, −28.50, and −28.55, respectively). However, although the difference was statistically significant (P = 0.02), the downregulation of aguR in the KO mutant was much less (−0.73). That is, the level of expression of aguR in the WT strain was only slightly higher than that in the KO mutant, suggesting that aguR expression must be very low in the WT strain.
TABLE 3.
Genes differentially expressed in L. lactis subsp. cremoris CECT8666 ΔaguR and the WT for which the criteria for inclusion were meta
| Expression and locus tagb (gene name) | Description | Fold change | P value |
|---|---|---|---|
| Downregulated | |||
| U725_01346 (aguR) | Transcriptional regulator | −0.73 | 2.02E−2 |
| U725_01347 (aguB) | Putrescine carbamoyl transferase | −27.44 | 1.00E−4 |
| U725_01348 (aguD) | Agmatine/putrescine antiporter | −26.27 | 1.50E−4 |
| U725_01349 (aguA) | Agmatine deiminase | −28.50 | 8.00E−5 |
| U725_01350 (aguC) | Carbamate kinase | −28.55 | 8.00E−5 |
| U725_00022 | Hypothetical protein | −4.39 | 8.60E−4 |
| U725_00023 | Glycosyltransferase | −6.00 | 5.70E−4 |
| Upregulated | |||
| U725_02522 | Transposase | 5.00 | 8.00E−5 |
| U725_02523 | EpsR | 16.26 | 2.30E−4 |
| U725_02524 | EpsX | 16.00 | 7.00E−5 |
| U725_02525 | EpsA | 21.20 | 1.20E−4 |
| U725_02526 | EpsB kinase | 30.41 | 6.00E−5 |
| U725_02527 | EpsC | 16.33 | 4.20E−4 |
| U725_02528 | Undecaprenyl-phosphate galactose phosphotransferase | 18.43 | 7.00E−5 |
| U725_02529 | Putative transposase | 6.20 | 6.00E−5 |
| U725_01694 | Transposase | 6.50 | 5.00E−5 |
| U725_02271 | Putative replication protein RepA | 3.83 | 8.30E−4 |
| U725_02289 | Transposase | 3.70 | 4.10E−4 |
| U725_02472 | Transposase | 6.87 | 3.00E−5 |
| U725_02477 | EpsN protein | 25.29 | 5.00E−5 |
The criteria for inclusion were at least a 3-fold difference in expression and a P value of <0.001. aguR was also included, although the inclusion criteria were not entirely met for this gene.
Locus tags refer to those of the sequence with GenBank accession no. AZSI00000000.1.
Transcriptional regulation of aguR and aguBDAC by agmatine.
The effect of the environmental agmatine concentration on aguR and aguBDAC expression was investigated by RT-qPCR. The expression profile of aguB (the first gene of the aguBDAC operon) was analyzed as a representative of the expression of the whole aguBDAC polycistronic mRNA. Total RNA was isolated from L. lactis subsp. cremoris CECT8666 cells grown in GM17 medium as well as GM17 medium supplemented with increasing concentrations of agmatine (0, 0.05, 0.1, 0.25, 0.5, 1, 5, 10, and 20 mM). Figure 3 shows the relative aguR and aguB gene expression levels (normalized against the gene expression level of the rpoA reference gene). aguR expression was not affected by the increase in the agmatine concentration (Fig. 3A), whereas aguB expression was upregulated by concentrations of ≥0.25 mM, with the strongest overexpression (1,700-fold change) occurring in the presence of 5 mM agmatine (Fig. 3B). Agmatine concentrations of over 5 mM right up to 20 mM did not increase the level of aguB expression compared to that observed with 5 mM agmatine.
FIG 3.

Influence of agmatine concentration on the expression of aguR and aguBDAC as determined by RT-qPCR. Cell cultures were supplemented with 0, 0.05, 0.1, 0.25, 0.5, 1, 5, 10, or 20 mM agmatine, and samples were collected at the end of the exponential phase of growth. The relative expression of aguR (A) and aguB, representing the whole aguBDAC operon (B), relative to the transcript level for samples grown in the absence of agmatine was calculated. Data were normalized to the total RNA content using rpoA and tuf as reference genes. The values shown are the means from three replicates; the standard deviations are indicated by bars. *, P < 0.05; **, P < 0.001.
In response to agmatine, AguR acts as a transcriptional activator of PaguB.
To study the activity of the PaguR and PaguB promoters, PaguR-gfp and PaguB-gfp fusions were constructed by substituting the aguR gene or aguBDAC genes for the gfp reporter gene and comparing the activity with that of the PaguR-aguR-PaguB-gfp fusion (pAGDI; Table 3). The constructs were assayed in L. lactis subsp. cremoris NZ9000, a strain without the AGDI cluster, grown in GM17 medium in the presence (20 mM) or absence of agmatine by measurement of whole-cell fluorescence (Fig. 4). Interestingly, when fused independently, neither PaguR nor PaguB was associated with any detectable activity. However, PaguB activity was recorded in assays involving the PaguR-aguR-PaguB-gfp construct, but, strikingly, activity was recorded only under the agmatine supplementation conditions. The fact that PaguB showed activity only when aguR (driven by its own promoter) was included in the genetic cassette supports that suggestion that AguR is a transcriptional activator of PaguB.
FIG 4.

Cloning and assay of PaguR and PaguB activity, reported as GFP fluorescence, in the presence and absence of 20 mM agmatine. The genetic fusions PaguR-gfp, PaguB-gfp, and PaguR-aguR-PaguB-gfp were transformed into L. lactis subsp. cremoris NZ9000 cells, and promoter activity was determined by measuring whole-cell fluorescence (250 μl of cells) at a similar OD600. The values shown are the means from three replicates; standard deviations are indicated by bars. a.u., arbitrary units.
To further determine the dose-dependent activator effect of agmatine on the promoter activity, the gfp fusion constructs were assayed in L. lactis subsp. cremoris NZ9000 grown in GM17 medium supplemented with increasing amounts of agmatine. Figure 5 shows the whole-cell fluorescence results obtained. Once again, no activity was detected for the PaguR-gfp or PaguB-gfp construct (at any agmatine concentration tested) (Fig. 5A and B, respectively), while a dose-dependent activation of the PaguR-aguR-PaguB-gfp fusion was seen, with maximum activity being recorded for 0.1 mM agmatine (6 fluorescent arbitrary units). Agmatine concentrations above 0.1 mM did not significantly increase the intensity of fluorescence compared to that achieved with 0.1 mM agmatine (Fig. 5C).
FIG 5.

Effect of the agmatine concentration on the transcriptional activity of the AGDI cluster promoters, measured by whole-cell fluorescence. L. lactis subsp. cremoris NZ9000 cells harboring either the PaguR-gfp (A), the PaguB-gfp (B), or the PaguR-aguR-PaguB-gfp (C) genetic fusion were grown in GM17 medium supplemented with 0, 10−6, 10−5, 10−4, 10−3, 10−2, 0.1, 0.5, 1, 2, 5, 10, or 20 mM agmatine for 7 h, after which GFP fluorescence was monitored. The values shown are the means from three replicates; standard deviations are indicated by bars. a.u.: arbitrary units.
Functional analysis of inverted sequences of the aguB promoter region.
Clone software analysis of the nucleotide sequence upstream of the putative −35 region of the aguB promoter revealed the presence of one direct and three reversed sequences (Fig. 6A). To determine whether these inverted sequences are necessary for transcriptional activity, a series of deletions in the pAGDI plasmid (from nucleotides −209 to −179 [pAGDIΔ1 plasmid], from nucleotides −179 to −146 [plasmid pAGDIΔ2], from nucleotides −146 to −119 [plasmid pAGDIΔ3], and from nucleotides −119 to −92 [plasmid pAGDIΔ4]; Fig. 6B) was generated. gfp was used as the reporter gene, and L. lactis subsp. cremoris NZ9000 was used as the host. Plasmid pAGDI containing the complete aguB promoter was used as a control. NZ9000 cells were transformed with either pAGDI, pAGDIΔ1, pAGDIΔ2, pAGDIΔ3, or pAGDIΔ4 and grown in GM17 medium supplemented with 20 mM agmatine. At the end of the exponential phase, cells were collected and the activity of the promoters was examined via whole-cell fluorescence. The obtained transcriptional activities were expressed as percentages relative to the activity obtained with pAGDI (100% activity) (Fig. 6B). The deletion of the fragment from nucleotides −209 to −179 did not affect the activity of the promoter, which was equal to that shown by the control. However, the deletion of the fragments located downstream of this region did prevent expression. This indicates that these sequences are required for PaguB activity.
FIG 6.

(A) Sequence of the aguB promoter region. The putative −10 and −35 regions, the ribosome binding site (RBS), and the aguB start codon are shown in bold. Direct and reversed sequences are indicated by arrows. The deletions generated in this study (Δ1, Δ2, Δ3, and Δ4) are indicated by dashed lines. Asterisks indicate matches with the palindromic consensus sequence of the DosR binding site. (B) Effect of sequential deletions within the aguB promoter region. Plasmids pAGΔ1, pAGΔ2, pAGΔ3, and pAGΔ4 were constructed from pAGDI (PaguR-aguR-PaguB-gfp fusion). The dashed lines indicate the fragments deleted. The corresponding GFP fluorescence in L. lactis subsp. cremoris NZ9000 cells grown in GM17 medium supplemented with 20 mM agmatine was measured. The activities associated with the deletion constructs are expressed as percentages relative to the activity of pAGDI (which was considered to show 100% activity). The values shown are the means from three replicates; standard deviations are indicated by bars. *, P < 0.05.
AguR is a transmembrane protein.
As described above, the PaguR-aguR-PaguB-gfp fusion became active in response to the extracellular agmatine concentration in L. lactis subsp. cremoris NZ9000, a strain lacking the AGDI cluster, in which aguD codes for the agmatine/putrescine antiporter. Database checks were made to confirm that the genome of the L. lactis subsp. cremoris NZ9000 strain (GenBank accession no. CP002094.1) (32) is defective in predicted agmatine transporters. The ability of L. lactis subsp. cremoris NZ9000 to internalize agmatine in vivo was therefore assessed. The strain was grown in GM17 medium plus 20 mM agmatine, but after 24 h the concentration of extracellular agmatine in the supernatant was the same (20 mM), indicating that L. lactis subsp. cremoris NZ9000 likely lacks a system for agmatine internalization (data not shown). Thus, agmatine in the extracellular medium might trigger the induction of aguBDAC transcription.
To gain insight into the mechanism of the response to the extracellular agmatine concentration, the subcellular localization of the AguR protein was predicted by in silico topology analysis using the computer-based algorithms provided by the SOSUI server. The topology revealed a membrane protein secondary structure with seven predicted transmembrane-spanning segments, with the N-terminal being outside the cell and a long (105-amino-acid-residue) C-terminal domain being inside (Fig. 7A). The localization of AguR was also examined experimentally using the PnisA-aguR-gfp translational fusion (pAG3 plasmid) in which the aguR gene fused to the gfp gene is under the control of the nisA promoter. This fusion was assayed in L. lactis subsp. cremoris NZ9000 with induction by nisin, and the cells were examined by fluorescence microscopy. As shown in Fig. 7, panel B1, the AguR-GFP fusion protein was evenly distributed on the periphery of the cell, confirming the predicted transmembrane nature of AguR in L. lactis subsp. cremoris. A control assay with a cytoplasmic GFP showing a fluorescent pattern that contrasted with the fluorescent pattern of the AguR-GFP product was carried out in parallel (Fig. 7, panel B2).
FIG 7.

Cellular localization of AguR. (A) Predicted secondary structure and topology of AguR obtained via analysis of the amino acid sequence (performed using the SOSUI server). Seven transmembrane domains were predicted (gray shading). (B1) In vivo membrane localization of AguR in L. lactis subsp. cremoris NZ9000 cells overexpressing the AguR-GFP translational fusion protein imaged by fluorescence microscopy. (B2) Control image showing the fluorescent pattern of the same cells overexpressing a cytoplasmic GFP.
AguR has a LuxR_C-like domain.
A C-terminal DNA-binding domain that is typically found in LuxR-like proteins (the LuxR_C-like domain) and that contains a helix-turn-helix (HTH) DNA-binding functional motif (14, 33) was found in AguR (C terminus residues 265 to 323; Fig. 7A). Structure-based multiple-sequence alignment between the predicted LuxR_C-like domain sequence of AguR and the orthologous domains of LuxR member proteins with known structures was performed: DosR from Mycobacterium tuberculosis (34), GerE from Bacillus subtilis (35), StyR from Pseudomonas fluorescens (36), CviR from Chromobacterium violaceum (37), and VraR from Staphylococcus aureus (38) (Fig. 8A). A remarkably strong similarity between the accepted four-α-helix motif distribution model of the solved LuxR domains and the predicted α-helix motifs within the AguR LuxR_C-like domain was found (Fig. 8A). Half of the total residues involved within the LuxR_C-like domain were conserved or conservatively substituted across all the structures compared, indicating strong sequence similarity. Moreover, 9 out of the 13 residues described to act as DNA-binding residues in the LuxR_C-like domain of the DosR regulator of M. tuberculosis were conserved in AguR (Fig. 8A). Since the LuxR_C-like domain of DosR binds to DNA as a homodimer, it is remarkable that when we replaced the LuxR_C-like domain of chain A of AguR in the solved DosR model (34), a perfect fit was shown (Fig. 8B). Further, the largest inverted repeated sequence found in the aguR promoter region (Fig. 6) showed a 13-bp match with the 20-bp palindromic consensus sequence of DosR binding sites (39).
FIG 8.

In silico structural analysis of the C-terminal LuxR_C-like domain of AguR. (A) Structural alignment of the LuxR_C-like domain of AguR with homologue domains retrieved from the Protein Data Bank (PDB). Residues identical in the majority of the proteins are indicated by capital letters in the consensus sequence, while c indicates conservative substitutions. The shadowed residues are those involved in α helices within the domain. The α helices in the AguR sequence are derived from a structural alignment performed with the DosR LuxR_C-like domain as a template. Arrows indicate those residues from the DosR domain that interact with DNA, while asterisks indicate those involved in DosR dimerization. The sequences of DosR from Mycobacterium tuberculosis (PDB accession no. 1ZLK), GerE from Bacillus subtilis (PDB accession no. 1FSE), VraR from Staphylococcus aureus (PDB accession no. 2RNJ), StyR from Pseudomonas fluorescens (PDB accession no. 1YIO), CviR from Chromobacterium violaceum (PDB accession no. 3QP6), and an unknown protein from Bacteroides thetaiotaomicron (PDB accession no. 3CLO) were aligned. (B) Homology modeling analysis between LuxR_C-like domains from DosR and AguR. A DosR dimer (chains A and B) bound to DNA was used as the template. Modeling was performed by substituting the LuxR_C-like domain from DosR chain A for the LuxR_C-like domain from AguR (red). DosR chain B is shown in yellow; the DNA helix is gray. The residues involved in DosR dimerization within the α10 helix are shown in dark blue, the AguR putative DNA-interacting residues are shown in green, and the putative dimerization residues of AguR are shown in light blue.
DISCUSSION
Food safety is a major social concern in developed countries, in part stemming from the worldwide recorded incidence of foodborne illnesses. A great deal of effort has therefore been invested in the development of processing methods and techniques that prevent contaminants such as BAs from entering foodstuffs. Fermented foods, particularly cheese, are of special concern in this respect (40–42). Putrescine is one of the BAs most commonly detected in dairy products (2, 3, 43, 44). Prompted by the increasing awareness of the risks associated with the dietary intake of high BA loads and the importance of L. lactis as a primary starter in the dairy industry, the aim of this work was to decipher the genetic regulation of the putrescine biosynthesis cluster of L. lactis subsp. cremoris CECT8666.
The transcriptional studies performed detected an mRNA spanning the intergenic regions of aguB, aguD, aguA, and aguC, thereby confirming that these genes are cotranscribed from the PaguB promoter as a single aguBDAC polycistronic mRNA. In fact, no terminator-like sequences were identified in the aguBDAC intergenic regions. The transcription of the catabolic genes of the AGDI cluster as a single mRNA molecule has previously been reported for Pseudomonas aeruginosa PAO1 (45), S. mutans UA159 (46), L. lactis subsp. lactis CHCC7244 (2), and Enterococcus faecalis JH2-2 (47). The similar degree of downregulation between the ΔaguR mutant and WT strains seen in the DNA microarray comparisons in the present study (Table 3) supports the idea that these genes are cotranscribed. In addition, the present data reveal that the expression of the adjacent upstream aguR gene occurs via an independent mRNA transcribed from the PaguR promoter.
The role of aguR in the AGDI operon has been described to be a positive regulator in S. mutans (14), E. faecalis JH2-2 (47), and E. faecalis V583 (48) and a TetR-family repressor in P. aeruginosa (45). In the present work, the deletion of aguR in putrescine-producing L. lactis subsp. cremoris CECT8666 fully impaired expression of the catalytic AGDI genes and thereby the catabolism of agmatine to putrescine (Fig. 2), indicating that this gene behaves as a positive regulator. Moreover, when fused to gfp, the promoter regions of aguR and aguBDAC showed no activity in cells of strain NZ9000 (which does not carry the AGDI cluster) regardless of the agmatine concentration (Fig. 4 and 5). In contrast, when aguR was present in the gfp fusion, an agmatine-specific induction of PaguB activity was observed. Together, these data reveal the dual role of AguR in L. lactis subsp. cremoris: not only is it required for sensing the agmatine concentration, but it is also involved in the transcriptional activation of putrescine biosynthesis. However, the transcription of aguR was revealed to be completely independent of the agmatine concentration, suggesting that AguR is constantly present in the cells, although its expression must be at a very low level since very small differences in aguR transcription were observed between the WT and KO strains in transcriptomic analyses.
As previously reported, the transcription of the aguBDAC operon is regulated by carbon catabolic repression (CCR), mediated by the catabolite control protein CcpA. However, the expression level of aguR is independent of the glucose concentration (13). CcpA would control the expression of the aguBDAC operon, whose promoter, PaguB, in fact has a cre site (13), and would not control aguR expression. Therefore, our data suggest that CCR and AguR activation would work as two independent systems exerting a parallel control on the PaguB promoter.
The in silico analysis of the amino acid sequence of AguR revealed the presence of seven transmembrane domains, a short extracytoplasmic N terminus, and a longer cytoplasmic C terminus (Fig. 7A). A membrane localization of AguR has also been predicted on the basis of the localization of its orthologous protein present in S. mutans (14), although the authors of the latter work proposed a four-transmembrane-domain model and determined that the N terminus lies in the cytosol. In the present work, fluorescence microscopy analysis of NZ9000 cells expressing aguR fused to gfp showed that AguR localizes at the bacterial surface, where it is evenly distributed (Fig. 7B). Moreover, agmatine concentration sensing was maintained when aguR was coexpressed with PaguB-gfp in L. lactis subsp. cremoris NZ9000, an AGDI-defective strain (lacking aguR) unable to internalize agmatine. This confirms that extracellular agmatine activates the AGDI system without being internalized. However, NZ9000 cells containing the PaguB-gfp construct but lacking AguR were unable to transduce the agmatine signal to the inside of the cell and activate PaguB. These results strongly suggest that AguR is a transmembrane protein that behaves both as a sensor of the extracellular agmatine concentration and as a signal transducer demanding that the transcription of the aguBDAC genes be initiated. It should be noted that the non-AGDI cluster genes, which, in the present transcriptomic studies, showed different degrees of expression in the KO and WT strains (Table 3), are not present in the genome of L. lactis subsp. cremoris NZ9000 (except for the glycosyltransferase and transposase genes). They do not, therefore, seem to be required by the proposed regulation model.
BLAST analysis of the amino acid sequence of AguR showed that this protein belongs to the transcriptional regulators of the LuxR family, as described for its orthologs in L. lactis subsp. lactis CHCC7244 (2), S. mutans UA159 (14, 46), and E. faecalis JH2-2 (47). The members of the LuxR family of DNA-binding proteins are transcription factors involved in quorum sensing via the detection of autoinducers, such as oligopeptide-signaling molecules (in Gram-positive bacteria) (49, 50) or acylated homoserine lactones (in Gram-negative bacteria) (51). These proteins have two functional domains: an amino-terminal domain involved in the binding of the signaling molecule and a LuxR_C-like transcription regulation domain at the C terminus of the protein which includes a helix-turn-helix (HTH) DNA-binding motif (33). LuxR transcription factors can therefore behave as regulators (transcriptional activators) by binding a cognate extracellular inducer and targeting specific gene promoters (52). A high degree of structural homology was noted when the AguR intracellular LuxR_C-like domain was compared to the LuxR_C-like domains of LuxR family members with solved structures (Fig. 8); indeed, the characteristic four-α-helix secondary structure for this domain was shared (53). Moreover, half of the amino acid residues of the LuxR_C-like domain (34 of 62 residues) were strongly conserved across all the compared structures, indicating a high degree of sequence conservation. In fact, 9 out of 13 DNA-binding residues in the LuxR_C-like domain of DosR in M. tuberculosis (34) were conserved in the AguR LuxR_C-like domain, as were the 3 residues involved in dimerization within the α10 helix of DosR monomers (34). The similarity between the two proteins is such that a DosR chain could be perfectly replaced by one from AguR. Since the LuxR_C-like domain of DosR binds to DNA as a homodimer, the LuxR_C-like domain of AguR should be able to bind to DNA, probably with a dimeric structure.
The target DNA-binding sites (Lux-type boxes) of many LuxR-type proteins have a dyad symmetry structure (51) and are often located just upstream of the −35 region of the regulated promoters. Such is the case of a direct repeat element essential for transcription from the PaguB promoter (Fig. 6A), which shows a 13-bp match with the 20-bp DosR binding site consensus sequence (39). This similarity between the proteins and their DNA-binding sites is even more remarkable considering the taxonomic distance between the GC content of M. tuberculosis (65%) and L. lactis (35%).
The results reveal the role of the regulatory protein AguR as both an agmatine sensor and a transcriptional activator of the AGDI genes (aguB, aguD, aguA, and aguC). In other lactic acid bacteria with the AGDI pathway, such as S. mutans (14, 46) and E. faecalis (47), the role of AguR would be the same. However, the system seems to be slightly different in Lactobacillus brevis, since the AGDI cluster does not contain an aguR gene. L. brevis has a putative transcription regulator gene adjacent to the AGDI cluster that belongs to the RpiR family, which is distantly related to AguR (54) and lacks transmembrane domains. A mechanism similar to the one proposed here for AguR has been described in E. coli for the biosynthesis operon of cadaverine, another BA: the transmembrane protein CadC binds lysine outside the cell, and the signal is then transduced to the N-terminal cytoplasmic portion of the protein, which contains the HTH domain (55–58). Nevertheless, further analyses are needed to determine the precise mechanism by which AguR senses the agmatine concentration and transduces the activation signal to the promoter of the aguBDAC genes.
ACKNOWLEDGMENTS
This work was performed with the financial support of the Spanish Ministry of Economy and Competitiveness (AGL2013-45431-R) and the Plan for Science, Technology and Innovation 2013–2017, funded by the European Regional Development Fund and the Principality of Asturias (GRUPIN14-137). D.M.L. and B.D.R. were the beneficiaries of JAE DOC contracts (CSIC).
We are grateful to Bert Poolman for providing the GFP-based cloning vectors and Adrian Burton for linguistic assistance. Strain L. lactis subsp. cremoris NZ9000 and plasmid pNZ8048 were kindly provided by NIZO Food Research.
REFERENCES
- 1.Smit G, Smit BA, Engels WJ. 2005. Flavour formation by lactic acid bacteria and biochemical flavour profiling of cheese products. FEMS Microbiol Rev 29:591–610. doi: 10.1016/j.fmrre.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 2.Ladero V, Rattray FP, Mayo B, Martin MC, Fernandez M, Alvarez MA. 2011. Sequencing and transcriptional analysis of the biosynthesis gene cluster of putrescine-producing Lactococcus lactis. Appl Environ Microbiol 77:6409–6418. doi: 10.1128/AEM.05507-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fernandez M, Linares DM, Del Rio B, Ladero V, Alvarez MA. 2007. HPLC quantification of biogenic amines in cheeses: correlation with PCR-detection of tyramine-producing microorganisms. J Dairy Res 74:276–282. doi: 10.1017/S0022029907002488. [DOI] [PubMed] [Google Scholar]
- 4.Ladero V, Calles-Enríquez M, Fernández M, Álvarez MA. 2010. Toxicological effects of dietary biogenic amines. Curr Nutr Food Sci 6:145–156. doi: 10.2174/157340110791233256. [DOI] [Google Scholar]
- 5.Linares DM, Fernández M, Del-Río B, Ladero V, Martín MC, Alvarez MA. 2012. The tyrosyl-tRNA synthetase like gene located in the tyramine biosynthesis cluster of Enterococcus durans is transcriptionally regulated by tyrosine concentration and extracellular pH. BMC Microbiol 12:23. doi: 10.1186/1471-2180-12-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ten Brink B, Damink C, Joosten HM, Huis in't Veld JH. 1990. Occurrence and formation of biologically active amines in foods. Int J Food Microbiol 11:73–84. doi: 10.1016/0168-1605(90)90040-C. [DOI] [PubMed] [Google Scholar]
- 7.Linsalata M, Russo F. 2008. Nutritional factors and polyamine metabolism in colorectal cancer. Nutrition 24:382–389. doi: 10.1016/j.nut.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 8.Moffatt J, Hashimoto M, Kojima A, Kennedy DO, Murakami Koshimizu K, Ohigashi H, Matsui-Yuasa I. 2000. Apoptosis induced by 1′-acetoxychavicol acetate in Ehrlich ascites tumor cells is associated with modulation of polyamine metabolism and caspase-3 activation. Carcinogenesis 21:2151–2157. doi: 10.1093/carcin/21.12.2151. [DOI] [PubMed] [Google Scholar]
- 9.Shah P, Swiatlo E. 2008. A multifaceted role for polyamines in bacterial pathogens. Mol Microbiol 68:4–16. doi: 10.1111/j.1365-2958.2008.06126.x. [DOI] [PubMed] [Google Scholar]
- 10.Casero RA, Marton LJ. 2007. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat Rev Drug Discov 6:373–390. doi: 10.1038/nrd2243. [DOI] [PubMed] [Google Scholar]
- 11.Simon JP, Stalon V. 1982. Enzymes of agmatine degradation and the control of their synthesis in Streptococcus faecalis. J Bacteriol 152:676–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.del Rio B, Linares DM, Ladero V, Redruello B, Fernández M, Martin MC, Alvarez MA. 2015. Putrescine production via the agmatine deiminase pathway increases the growth of Lactococcus lactis and causes the alkalinization of the culture medium. Appl Microbiol Biotechnol 99:897–905. doi: 10.1007/s00253-014-6130-8. [DOI] [PubMed] [Google Scholar]
- 13.Linares DM, Del Rio B, Ladero V, Redruello B, Martin MC, Fernandez M, Alvarez MA. 2013. The putrescine biosynthesis pathway in Lactococcus lactis is transcriptionally regulated by carbon catabolic repression, mediated by CcpA. Int J Food Microbiol 165:43–50. doi: 10.1016/j.ijfoodmicro.2013.04.021. [DOI] [PubMed] [Google Scholar]
- 14.Liu YL, Zeng L, Burne RA. 2009. AguR is required for induction of the Streptococcus mutans agmatine deiminase system by low pH and agmatine. Appl Environ Microbiol 75:2629–2637. doi: 10.1128/AEM.02145-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ladero V, del Rio B, Linares DM, Fernandez M, Mayo B, Martin MC, Alvarez MA. 2014. Genome sequence analysis of the biogenic amine-producing strain Lactococcus lactis subsp. cremoris CECT 8666 (formerly GE2-14). Genome Announc 2(5):e01088-14. doi: 10.1128/genomeA.01088-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Green MR, Sambrook J. 2012. Molecular cloning: a laboratory manual, 4th ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 17.Redruello B, Ladero V, Cuesta I, Ávarez-Buylla JR, Martín MC, Fernández M, Alvarez MA. 2013. A fast, reliable, ultra high performance liquid chromatography method for the simultaneous determination of amino acids, biogenic amines and ammonium ions in cheese, using diethyl ethoxymethylenemalonate as a derivatising agent. Food Chem 139:1029–1035. doi: 10.1016/j.foodchem.2013.01.071. [DOI] [PubMed] [Google Scholar]
- 18.Hopwood DA, Bibb MJ, Chater KF, Kieser T, Bruton CJ, Kieser HM, Lydiate DJ, Smith CP, Ward JM, Schrempf H. 1985. Genetic manipulation of Streptomyces: a laboratory manual. The John Innes Foundation, Norwich, United Kingdom, and Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 19.de Vos WM, Vos P, Dehaard H, Boerritger I. 1989. Cloning and expression of the Lactococcus lactis subsp. cremoris SK11 gene encoding an extracellular serine proteinase. Gene 85:169–176. doi: 10.1016/0378-1119(89)90477-0. [DOI] [PubMed] [Google Scholar]
- 20.Linares DM, Fernández M, Martín MC, Alvarez MA. 2009. Tyramine biosynthesis in Enterococcus durans is transcriptionally regulated by the extracellular pH and tyrosine concentration. Microb Biotechnol 2:625–633. doi: 10.1111/j.1751-7915.2009.00117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Solem C, Defoor E, Jensen PR, Martinussen J. 2008. Plasmid pCS1966, a new selection/counterselection tool for lactic acid bacterium strain construction based on the oroP gene, encoding an orotate transporter from Lactococcus lactis. Appl Environ Microbiol 74:4772–4775. doi: 10.1128/AEM.00134-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taibi A, Dabour N, Lamoureux M, Roy D, LaPointe G. 2011. Comparative transcriptome analysis of Lactococcus lactis subsp. cremoris strains under conditions simulating cheddar cheese manufacture. Int J Food Microbiol 146:263–275. doi: 10.1016/j.ijfoodmicro.2011.02.034. [DOI] [PubMed] [Google Scholar]
- 23.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−delta delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 24.Kuipers OP, de Ruyter PG, Kleerebezem M, de Vos WM. 1997. Controlled overproduction of proteins by lactic acid bacteria. Trends Biotechnol 15:135–140. doi: 10.1016/S0167-7799(97)01029-9. [DOI] [PubMed] [Google Scholar]
- 25.Kuipers OP, de Ruyter PG, Kleerebezem M, de Vos WM. 1998. Quorum sensing-controlled gene expression in lactic acid bacteria. J Biotechnol 64:15–21. doi: 10.1016/S0168-1656(98)00100-X. [DOI] [Google Scholar]
- 26.Linares DM, Geertsma ER, Poolman B. 2010. Evolved Lactococcus lactis strains for enhanced expression of recombinant membrane proteins. J Mol Biol 401:45–55. doi: 10.1016/j.jmb.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 27.Geertsma ER, Groeneveld M, Slotboom DJ, Poolman B. 2008. Quality control of overexpressed membrane proteins. Proc Natl Acad Sci U S A 105:5722–5727. doi: 10.1073/pnas.0802190105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer EL, Eddy SR, Bateman A, Finn RD. 2012. The Pfam protein families database. Nucleic Acids Res 40:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hirokawa T, Boon-Chieng S, Mitaku S. 1998. SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics 14:378–379. doi: 10.1093/bioinformatics/14.4.378. [DOI] [PubMed] [Google Scholar]
- 30.Arnold K, Bordoli L, Kopp J, Schwede T. 2006. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 31.Guex N, Peitsch MC. 1997. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 32.Linares DM, Kok J, Poolman B. 2010. Genome sequences of Lactococcus lactis MG1363 (revised) and NZ9000 and comparative physiological studies. J Bacteriol 192:5806–5812. doi: 10.1128/JB.00533-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nasser W, Reverchon S. 2007. New insights into the regulatory mechanisms of the LuxR family of quorum sensing regulators. Anal Bioanal Chem 387:381–390. doi: 10.1007/s00216-006-0702-0. [DOI] [PubMed] [Google Scholar]
- 34.Wisedchaisri G, Wu MT, Rice AE, Roberts DM, Sherman DR, Hol WG. 2005. Structures of Mycobacterium tuberculosis DosR and DosR-DNA complex involved in gene activation during adaptation to hypoxic latency. J Mol Biol 354:630–641. doi: 10.1016/j.jmb.2005.09.048. [DOI] [PubMed] [Google Scholar]
- 35.Ducros VM, Lewis RJ, Verma CS, Dodson EJ, Leonard G, Turkenburg JP, Murshudov GN, Wilkinson AJ, Brannigan JA. 2001. Crystal structure of GerE, the ultimate transcriptional regulator of spore formation in Bacillus subtilis. J Mol Biol 306:759–771. doi: 10.1006/jmbi.2001.4443. [DOI] [PubMed] [Google Scholar]
- 36.Milani M, Leoni L, Rampioni G, Zennaro E, Ascenzi P, Bolognesi M. 2005. An active-like structure in the unphosphorylated StyR response regulator suggests a phosphorylation-dependent allosteric activation mechanism. Structure 13:1289–1297. doi: 10.1016/j.str.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 37.Chen G, Swem LR, Swem DL, Stauff DL, O'Loughlin CT, Jeffrey PD, Bassler BL, Hughson FM. 2011. A strategy for antagonizing quorum sensing. Mol Cell 42:199–209. doi: 10.1016/j.molcel.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leonard PG, Golemi-Kotra D, Stock AM. 2013. Phosphorylation-dependent conformational changes and domain rearrangements in Staphylococcus aureus VraR activation. Proc Natl Acad Sci U S A 110:8525–8530. doi: 10.1073/pnas.1302819110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park H, Guinn KM, Harrell MI, Liao R, Voskuil ML, Tompa M, Schoolnik GK, Sherman DR. 2003. Rv3133c/dosR is a transcription factor that mediates the hypoxic response of Mycobacterium tuberculosis. Mol Microbiol 48:833–843. doi: 10.1046/j.1365-2958.2003.03474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Linares DM, Del-Río B, Ladero V, Martinez N, Fernández M, Martín MC, Alvarez MA. 2012. Factors influencing biogenic amines accumulation in dairy products. Front Microbiol 3:180. doi: 10.3389/fmicb.2012.00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sumner SS, Speckhard MW, Somers EB, Taylor SL. 1985. Isolation of histamine-producing Lactobacillus buchneri from Swiss cheese implicated in a food poisoning outbreak. Appl Environ Microbiol 50:1094–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taylor SL, Keefe TJ, Windham ES, Howell JF. 1982. Outbreak of histamine poisoning associated with consumption of Swiss cheese. J Food Prot 45:455–457. [DOI] [PubMed] [Google Scholar]
- 43.Bunkova L, Bunka F, Mantlova G, Cablova A, Sedlacek I, Svec P, Pachlova V, Kracmar S. 2010. The effect of ripening and storage conditions on the distribution of tyramine, putrescine and cadaverine in Edam-cheese. Food Microbiol 27:880–888. doi: 10.1016/j.fm.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 44.Pircher A, Bauer F, Paulsen P. 2007. Formation of cadaverine, histamine, putrescine and tyramine by bacteria isolated from meat, fermented sausages and cheeses. Eur Food Res Technol 226:225–231. doi: 10.1007/s00217-006-0530-7. [DOI] [Google Scholar]
- 45.Nakada Y, Jiang Y, Nishijyo T, Itoh Y, Lu CD. 2001. Molecular characterization and regulation of the aguBA operon, responsible for agmatine utilization in Pseudomonas aeruginosa PAO1. J Bacteriol 183:6517–6524. doi: 10.1128/JB.183.22.6517-6524.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Griswold AR, Chen YY, Burne RA. 2004. Analysis of an agmatine deiminase gene cluster in Streptococcus mutans UA159. J Bacteriol 186:1902–1904. doi: 10.1128/JB.186.6.1902-1904.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suarez C, Espariz M, Blancato VS, Magni C. 2013. Expression of the agmatine deiminase pathway in Enterococcus faecalis is activated by the AguR regulator and repressed by CcpA and PTS(Man) systems. PLoS One 8:e76170. doi: 10.1371/journal.pone.0076170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Linares DM, Perez M, Ladero V, del Rio B, Redruello B, Martin MC, Fernandez M, Alvarez MA. 2014. An agmatine-inducible system for the expression of recombinant proteins in Enterococcus faecalis. Microb Cell Fact 13:169. doi: 10.1186/s12934-014-0169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lyon GJ, Novick RP. 2004. Peptide signaling in Staphylococcus aureus and other Gram-positive bacteria. Peptides 25:1389–1403. doi: 10.1016/j.peptides.2003.11.026. [DOI] [PubMed] [Google Scholar]
- 50.Miller MB, Bassler BL. 2001. Quorum sensing in bacteria. Annu Rev Microbiol 55:165–199. doi: 10.1146/annurev.micro.55.1.165. [DOI] [PubMed] [Google Scholar]
- 51.Fuqua C, Parsek MR, Greenberg EP. 2001. Regulation of gene expression by cell-to-cell communication: acyl-homoserine lactone quorum sensing. Annu Rev Genet 35:439–468. doi: 10.1146/annurev.genet.35.102401.090913. [DOI] [PubMed] [Google Scholar]
- 52.Gobbetti M, De Angelis M, Di Cagno R, Minervini F, Limitone A. 2007. Cell-cell communication in food related bacteria. Int J Food Microbiol 120:34–45. doi: 10.1016/j.ijfoodmicro.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 53.Santos CL, Correia-Neves M, Moradas-Ferreira P, Mendes MV. 2012. A walk into the LuxR regulators of actinobacteria: phylogenomic distribution and functional diversity. PLoS One 7:e46758. doi: 10.1371/journal.pone.0046758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lucas PM, Blancato VS, Claisse O, Magni C, Lolkema JS, Lonvaud-Funel A. 2007. Agmatine deiminase pathway genes in Lactobacillus brevis are linked to the tyrosine decarboxylation operon in a putative acid resistance locus. Microbiology 153:2221–2230. doi: 10.1099/mic.0.2007/006320-0. [DOI] [PubMed] [Google Scholar]
- 55.Neely MN, Dell CL, Olson ER. 1994. Roles of LysP and CadC in mediating the lysine requirement for acid induction of the Escherichia coli Cad operon. J Bacteriol 176:3278–3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rauschmeier M, Schüppel V, Tetsch L, Jung K. 2014. New insights into the interplay between the lysine transporter LysP and the pH sensor CadC in Escherichia coli. J Mol Biol 426:215–229. doi: 10.1016/j.jmb.2013.09.017. [DOI] [PubMed] [Google Scholar]
- 57.Tetsch L, Koller C, Dönhöfer A, Jung K. 2011. Detection and function of an intramolecular disulfide bond in the pH-responsive CadC of Escherichia coli. BMC Microbiol 11:74. doi: 10.1186/1471-2180-11-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tetsch L, Koller C, Haneburger I, Jung K. 2008. The membrane-integrated transcriptional activator CadC of Escherichia coli senses lysine indirectly via the interaction with the lysine permease LysP. Mol Microbiol 67:570–583. doi: 10.1111/j.1365-2958.2007.06070.x. [DOI] [PubMed] [Google Scholar]