

ABSTRACT
The HIV-1 envelope (Env) glycoprotein mediates viral entry during both cell-free and cell-to-cell infection of CD4+ T cells. The highly conserved long cytoplasmic tail (CT) of Env is required in a cell type-dependent manner for optimal infectivity of cell-free virus. To probe the role of the CT in cell-to-cell infection, we tested a panel of mutations in the CT region that maintain or attenuate cell-free infection to investigate whether the functions of the CT are conserved during cell-free and cell-to-cell infection. The mutations tested included truncations of structural motifs in the gp41 CT and two point mutations in lentiviral lytic peptide 3 (LLP-3) previously described as disrupting the infectivity of cell-free virus. We found that small truncations of 28 to 43 amino acids (aa) or two LLP-3 point mutations, YW_SL and LL_RQ, severely impaired single-round cell-free infectivity 10-fold or more relative to wild-type full-length CT. These mutants showed a modest 2-fold reduction in cell-to-cell infection assays. Conversely, large truncations of 93 to 124 aa severely impaired cell-to-cell infectivity 20-fold or more while resulting in a 50% increase in infectivity of cell-free viral particles when produced in 293T cells. Intermediate truncations of 46 to 90 aa showed profound impairment of both modes of infection. Our results show that the abilities of Env to support cell-free and cell-to-cell infection are genetically distinct. These differences are cell type dependent for large-CT-truncation mutants. Additionally, point mutants in LLP-3 can maintain multiround propagation from cell-to-cell in primary CD4+ T cells.
IMPORTANCE The functions of HIV Env gp41 CT remain poorly understood despite being widely studied in the context of cell-free infection. We have identified domains of the gp41 CT responsible for striking selective deficiencies in either cell-free or cell-to-cell infectivity. These differences may reflect a different intrinsic regulatory influence of the CT on cell-associated versus particle-associated Env or differential interaction with host or viral proteins. Our findings provide novel insight into the key regulatory potential of the gp41 CT in cell-free and cell-to-cell HIV-1 infection, particularly for short-truncation mutants of ≤43 amino acids or mutants with point mutations in the LLP-3 helical domain of the CT, which are able to propagate via cell-to-cell infection in the absence of infectious cell-free virus production. These mutants may also serve as tools to further define the contributions of cell-free and cell-to-cell infection in vitro and in vivo.
INTRODUCTION
Human immunodeficiency virus (HIV-1) is an enveloped retrovirus that can enter permissive target cells through binding of its envelope glycoprotein (Env) to host CD4 and chemokine receptors. Env is a trimer of gp120-gp41 heterodimers and mediates viral attachment, fusion, and entry into CD4+ T cells during both cell-free infection and direct cell-to-cell infection via the virological synapse (VS). The gp41 cytoplasmic tail (CT) of HIV-1 Env is ∼150 amino acids (aa) long and has several functions during the viral life cycle. These include but are not limited to (i) endocytosis of cell surface Env, (ii) Env trafficking in polarized cells, (iii) Env packaging into viral particles, (iv) controlling the fusogenic potential of Env (1–6), (v) cell signaling, and (vi) regulating antibody binding (1, 7, 8) and neutralization sensitivity (8–10). Many of these functions are associated with specific sequences and/or structural motifs (11–15). The major defined motifs in the CT include a tyrosine-based endocytic signal near the membrane-spanning domain, a C-terminal dileucine endocytic motif, a highly immunogenic region (HIR) containing a neutralizing epitope called the Kennedy epitope, a nuclear factor κB (NF-κB) activation domain (16), three amphipathic alpha helices referred to as lentiviral lytic peptides (LLP), and two inhibitory sequences, is1 and is2, which inhibit Env surface expression (17). Recently, the host proteins prohibitin (18) and tail-interacting protein 47 (TIP47) (19, 20) have been implicated in binding to residues in LLP-3. Mutations in these putative protein-binding sites were found to exhibit reduced cell-free infectivity, which was cell type dependent in the case of the prohibitin binding site mutant LL_RQ. The cellular retromer complex has also recently been shown to bind to the CT (21). Disruption of retromer-CT interaction can result in the production of virus particles with enhanced Env packaging and infectivity.
A well-established approach to studying the function of the HIV-1 gp41 CT has been to examine CT deletion mutants for their fitness during cell-free infection. A mutant with a truncation of the C-terminal 144 aa (ΔCT144) exhibits a replication defect that is cell type dependent. Efficient replication of this mutant occurs in only a limited number of permissive cell lines, primarily MT-4, 293T, or HeLa cells (22, 23), but does not occur in most other T cell lines, including CEM, Jurkat, MT-2, H9, and SupT1 cells, as well as primary CD4+ T cells and monocyte-derived macrophages. The factors that define cell permissivity may relate to the efficiency of Env incorporation into budding virions. These differences in Env incorporation may be a result of differentially expressed cellular factors and/or the absence of a key binding site(s) in the mutated CT for host proteins such as prohibitin (18) or Rab11-FIP1c and Rab14 (24, 25). A number of studies have examined HIV-1 genomes carrying progressive gp41 CT truncations and observed differential impacts on Env fusogenicity (1, 3, 26), Env packaging, and/or infectivity of cell-free virus (3, 26–31). Other studies have also characterized small deletions and point mutations within the CT LLP regions or endocytic motif(s) that affect Env incorporation and/or infectivity of viral particles (2, 6, 31–35), the intracellular distribution of Env (21, 33), syncytium formation (2, 6, 34), viral fusion (6, 35), and polarized budding (36).
In the context of direct cell-to-cell infection, the role of the gp41 CT has been less well characterized. T cell-to-T cell HIV-1 infection through the VS is likely to be an important route of HIV-1 spread in vivo in lymphoid tissues where the density of lymphocytes and their ability to interact are much greater (37). This requires actin rearrangement, resulting in Env, Gag, and CD4 colocalization at the site of cell contact (38, 39), and has features that can be distinct from those of cell-free infection (40). Some of these features include resistance to neutralizing-antibody responses (9, 41–43), increased resistance to antiretroviral therapy (44–46), and the transmission of multiple viral genomes to a single cell (44, 47, 48) or to multiple cells simultaneously (49). The resistance of cell-to-cell infection to neutralizing antibodies is in part dependent upon the presence of an intact gp41 CT (9).
The role that the gp41 CT plays during in vitro cell-to-cell infection has thus far been examined with the full deletion of the CT, ΔCT144, in permissive (9, 50) and nonpermissive (51) cell types. During cell-to-cell infection, the engagement of CD4 with Env occurs at the cell surface and typically does not lead to cell-cell fusion. During VS formation, viral fusion activity of Env can be coordinated with the formation and transfer of virus particles to the target cell (52). The inhibition of fusion at the synapse may be due to the presence of fusion-inhibiting cellular factors (53, 54) or due to the presence of an immature Gag lattice that interacts with the Env CT to control viral fusogenicity (4, 5, 55). Because of the key role that the Env CT plays in Env packaging, VS formation, viral fusion, and subsequent infectivity, we were interested in understanding how different mutants in the Env CT may impact cell-to-cell transmission through the VS.
To systematically examine the domains of the Env CT required for cell-free infection in comparison to cell-to-cell infectivity we constructed a series of gp41 CT truncation mutants. We also characterized two point mutants in LLP-3, YW_SL, and LL_RQ, which have been previously described as disrupting the putative binding sites of TIP47 and prohibitin in the gp41 CT. We determined the relative levels of Env packaged into 293T-produced virus particles and expressed on the surface of Jurkat donor cells used in our cell-to-cell infectivity assays, and we measured single-round cell-free and cell-to-cell infectivity of these mutants in MT-4 cells as well as primary CD4+ T cells. We identified gp41 CT mutants with striking selective deficiencies in either cell-free or cell-to-cell infectivity, particularly for short-truncation mutants of ≤43 aa or mutants with point mutations in the LLP-3 helical domain of the CT. This provides further evidence that cell-to-cell infection is distinct from cell-free infection and can occur in isolation. Our results also indicate that there are mechanistic differences that distinguish how the CT participates in cell-free or cell-to-cell infection.
MATERIALS AND METHODS
Viral constructs.
NL-GI is based on the molecular clone pNL4-3 (56), and has the gene for green fluorescent protein (GFP) in place of nef. nef is expressed from a downstream internal ribosome entry site (IRES), as previously described (57). Gag-iCherry contains mCherry inserted into Gag, in frame between the MA and CA domains (58). NL-GI ΔCT144 (9, 26) was previously described. Δenv NL-GI and Δenv Gag-iCherry contain a frameshift mutation in the NdeI restriction site that disrupts the Env open reading frame, made by replacing the NheI-SalI fragment with the corresponding fragment from Δenv Gag-iGFP (59). All other gp41 mutants were generated by overlap extension PCR to replace the appropriate amino acid with a stop codon, or an alternate amino acid in the case of the point mutants YW_SL and LL_RQ NL-GI. Residues Y802 and W803 of HXB2 Env (19) correspond to Y800 and W801 of NL4-3 Env. Residues L799 and L800 of BH10 Env (18) correspond to L797 and L798 of NL4-3 Env. Primer sequences for several truncation mutants (3, 28) and the YW_SL (19) mutant were previously described. In this study, ΔCT118 and ΔCT43 are equivalent to ΔCT126 and ΔCT42b, described in reference 3. PCR fragments were introduced into the NL-GI backbone using the BamHI and MluI restriction sites or the NheI and MluI restriction sites. NL-GI ΔCT43 was cloned using MluI and HpaI. A similar cloning strategy was used to introduce the ΔCT100 and ΔCT28 mutations into the Gag-iCherry backbone, using the NheI and BamHI restriction sites and the BamHI and XhoI restriction sites, respectively. All mutations made by overlap extension PCR were confirmed by sequencing. The reference sources for the Env mutants are summarized in Table 1.
TABLE 1.
Cell-free and cell-to-cell infectivity of gp41 CT mutants in MT-4 cells
| gp41 CT mutation |
No. of amino acid changes in Rev | Infectivitya |
||
|---|---|---|---|---|
| Description | Mutant name | Cell free | Cell to cell | |
| None (full-length gp41) | None (WT) | 0 | 100 | 100 |
| No gp41 | Δenv mutant | 0 | 1.42 ± 0.37 | 0.09 ± 0.04 |
| No gp41 CT; no YSPL motif | ΔCT144 | 0 | 68.16 ± 11.85 | 1.90 ± 0.37 |
| Large gp41 CT truncation; no YSPL motif | ΔCT124FSb | 0 | 38.67 ± 14.07 | 0.75 ± 0.22 |
| Large gp41 CT truncation | ΔCT124c | 1 | 177.78i | 0.12 ± 0.03 |
| ΔCT118d | 3 | 149.07i | 1.05 ± 0.29 | |
| ΔCT100e | 0 | 261.20 ± 13.49 | 2.63 ± 1.18 | |
| ΔCT93f | 1 | 185.71 ± 2.93 | 4.96 ± 2.04 | |
| Intermediate gp41 CT truncation | ΔCT90d | 2 | 9.18 ± 2.58 | 0.20 ± 0.08 |
| ΔCT83f | 0 | 2.87 ± 0.23 | 0.16 ± 0.01 | |
| ΔCT67e | 0 | 1.50 ± 0.92 | 0.71 ± 0.45 | |
| ΔCT66d | 2 | 2.42 ± 1.56 | 0.38 ± 0.15 | |
| ΔCT60e | 0 | 2.88 ± 0.57 | 0.79 ± 0.35 | |
| ΔCT59d | 3 | 3.24 ± 1.95 | 1.16 ± 0.74 | |
| ΔCT56f | 1 | 4.97 ± 0.58 | 2.15 ± 0.17 | |
| ΔCT49d | 3 | 18.25 ± 6.20 | 3.95 ± 2.99 | |
| ΔCT46e | 0 | 3.32 ± 0.32 | 17.13 ± 6.00 | |
| Short gp41 truncation | ΔCT43d | 0 | 10.91 ± 1.89 | 52.51 ± 10.38 |
| ΔCT28d | 0 | 5.69 ± 1.98 | 45.36 ± 12.22 | |
| Full-length gp41 CT; two missense mutations in LLP-3 | YW_SLg | 0 | 8.87 ± 2.96 | 60.39 ± 10.11 |
| LL_RQh | 0 | 9.55 ± 2.89 | 55.46 ± 13.40 | |
Values were normalized to the infectivity of WT NL-GI and are means ± SEMs from at least two experiments performed in duplicate.
Primers for this mutant were based on reference 8.
Primers for this mutant were based on reference 1.
Primers for this mutant were from reference 3.
Primers for this mutant were designed specifically for this study.
Primers for this mutant were from reference 28.
Primers for this mutant were based on reference 19.
Primers for this mutant were based on reference 18.
Single experiment performed in duplicate.
Cells and cell culture.
Cells of the human cell lines Jurkat clone E6-1 and MT-4 were obtained through the NIH AIDS Reagent Program (ARP), Division of AIDS, NIAID, NIH, from Arthur Weiss and Douglass Richman, respectively. Primary CD4+ T cells were isolated from human peripheral blood obtained from deidentified HIV-negative blood donors through the New York Blood Center. All primary CD4+ T cells and T cell lines were cultured as described in reference 9. 293T cells (American Type Culture Association) were maintained in Dulbecco's modification of Eagle's medium supplemented with 10% Cosmic Calf serum (HyClone), 100 U/ml penicillin, 100 μg/ml streptomycin, and 200 μM l-glutamine.
Cell-free infection assays.
Cell-free virus particles were made by transfection of 293T cells using the calcium phosphate transfection method (60) or by nucleofection of Jurkat cells (Amaxa Biosystems) with wild-type (WT) or mutant NL-GI plasmid DNA. Virus particles were harvested 48 h later from 293T cells or 72 h later from Jurkat cells. Vesicular stomatitis virus envelope G protein (VSV-G) pseudotyped viruses were generated by cotransfection of WT or mutant NL-GI plasmid with plasmid pMD2.G, which encodes the VSV envelope G protein (Addgene), at a ratio of 1:1. Virus was quantified by p24 enzyme-linked immunosorbent assay (ELISA). Virus stocks were normalized for p24 and used to infect 1.25 × 105 MT-4 cells in a 96-well round bottom plate. For single-round infection assays, medium was replaced with medium containing 10 μM zidovudine (AZT; ARP) approximately 18 h after infection. At 40 h after infection, cells were treated with trypsin-EDTA, washed with phosphate-buffered saline (PBS), and fixed in 2% paraformaldehyde before detection by flow cytometry with a BD LSRII flow cytometer (BD Biosciences) and analysis using FlowJo software (Tree Star, Inc.). To spinoculate activated primary CD4+ T cells, 5 × 105 cells per well were spun at 1,200 × g for 99 min with 293T-produced virus in a 96-well flat-bottom plate. After 3 h of incubation at 37°C, 100 μl medium containing 50 U/ml interleukin 2 was added to each well. At 24 h after spinoculation, cells were trypsinized, fixed, and used for flow cytometry, as described above.
Cell-to-cell infection assays.
Donor Jurkat cells were nucleofected (Amaxa Biosystems) with WT or mutant NL-GI plasmid DNA, cultured overnight in antibiotic-free medium, and purified by Ficoll-Hypaque density gradient centrifugation to remove dead cells (9, 59). The amount of DNA used for nucleofection was adjusted to give approximately the same GFP expression levels at the time of coculture with target cells. Donor Jurkat cells and target MT-4 or activated primary CD4+ T cells were labeled with a 4 μM concentration of the cell proliferation dye eFluor 670 or a 6 μM concentration of the cell proliferation dye eFluor 450 (eBioscience), respectively. Approximately 1.25 × 105 Jurkat cells were cocultured with 1.25 × 105 MT-4 cells or activated primary CD4+ T cells per well in a 96-well round bottom plate. For single-round infection assays, culture medium was replaced with medium containing 10 μM AZT approximately 18 h after coculture. At 40 h after coculture, cells were trypsinized and fixed, as described for cell-free infection.
Cell-to-cell transfer assays.
Donor Jurkat cells were nucleofected with WT or mutant Gag-iCherry plasmid DNA, cultured overnight, purified by Ficoll-Hypaque density gradient centrifugation to remove dead cells, and dye labeled as described above. Resting primary CD4+ T cells were labeled with 4 μM eFluor 450. Approximately 1.25 × 105 Jurkat cells were cocultured with 1.25 × 105 resting primary CD4+ T cells per well in a 96-well round-bottom plate for 3 h, trypsinized, and fixed as described for cell-free infection.
Cell-to-cell viral membrane fusion assays.
Donor Jurkat cells were nucleofected with WT or mutant NL4-3 plasmid DNA, as well as the plasmid pMM310 (61, 62), which encodes the β-lactamase (BlaM)–Vpr fusion protein (ARP; catalog no. 11444 from Michael Miller), at a ratio of 3:1. Cells were cultured overnight and purified by Ficoll-Hypaque density gradient centrifugation. Approximately 2 × 105 unlabeled Jurkat cells were cocultured with 2 × 105 eFluor 670-labeled activated primary CD4+ T cells per well in a 96-well round-bottom plate for 3 to 5 h. Cells were loaded with loading solution containing CCF2-AM (Pan Vera) for 1 to 1.5 h and developed for 16 h in development solution as previously described (63). A total of 4 × 105 Jurkat donor cells were also fixed at the time of coculture and stained for intracellular p24 using the Fix & Perm cell permeabilization kit (Invitrogen) and the anti-p24 monoclonal antibody KC57-51 (Beckman Coulter).
Western blotting.
HIV-1-transfected 293T cells were lysed with 1% Triton lysis buffer containing complete protease inhibitor cocktail (Roche) and centrifuged for 10 min at 5,000 rpm at 4°C to remove nuclei. Total protein concentration was measured with a Coomassie Plus (Bradford) assay kit (Thermo Scientific) according to the manufacturer's protocol. Medium from transfected 293T cells containing virus particles was quantified by p24 enzyme-linked immunosorbent assay (ELISA), concentrated by sucrose gradient centrifugation through 20% sucrose and 1 mM EDTA, and resuspended in 1% Triton lysis buffer. Concentrated virus lysates were quantified by p24 ELISA. 293T cell lysates and virus particle lysates were normalized for protein concentration and p24 concentration, respectively. Reduced, denatured samples were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were transferred to a polyvinylidene difluoride (PVDF) membrane (GE Healthcare) and probed with the monoclonal antibody 2F5 (Hermann Katinger, ARP) to detect gp41. Membranes were then stripped, cut, and reprobed with sheep antiserum to HIV-1 gp120 (Michael Phelan, ARP) to detect gp120 or HIV immunoglobulin (HIV-IG) (Luiz Barbosa, ARP) to detect all HIV proteins. Primary antibodies were detected with horseradish peroxidase (HRP)-conjugated anti-human or anti-sheep secondary antibodies (Jackson ImmunoResearch Laboratories, Inc.). Western blots were visualized with the FluorChem E imaging system (ProteinSimple) and the signal intensity of protein bands was determined using Image Studio Lite software (LiCor Biosciences).
Detection of cell surface Env by flow cytometry.
Jurkat cells were nucleofected with WT or mutant NL-GI plasmid DNA, cultured overnight, and purified as described for cell-to-cell infection assays. A total of 2 × 105 Jurkat cells per well were stained with 15 μG/ml HIV-IG (Luiz Barbosa, ARP), followed by Alexa Fluor 647 goat anti-human IgG (Life Technologies). The average geometric mean fluorescence intensity (MFI) of cell surface Env was calculated from three independent experiments and used in a two-tailed unpaired t test to determine statistical significance between the geometric MFI of Jurkat cells expressing the Δenv mutant and Jurkat cells expressing either the WT protein or a given gp41 CT truncation mutant.
Calculations.
For cell-free infection assays, the percent infection relative to WT NL-GI was determined for each experiment by calculating the average percent infection in target cells of duplicate wells for WT NL-GI and gp41 mutant A NL-GI, followed by the formula (% infection by gp41 mutant A/% infection by WT) × 100, where mutant A is a gp41 ΔCT truncation mutant or LLP-3 mutant. For cell-to-cell infection assays, the average percent infection in target cells of duplicate wells for WT NL-GI and gp41 mutant A NL-GI was calculated and then adjusted for differences in donor cell transfection levels at the time of coculture, i.e., time zero, using the formula (% transfection of WT NL-GI donor cells at time zero/% transfection of gp41 mutant A NL-GI donor cells at time zero) × % infection of target cells by gp41 mutant A NL-GI. This value was then used to calculate the percent infection relative to WT NL-GI, as for cell-free infection assays.
For the comparison of cell surface Env expression levels, the relative MFI was calculated by examining the population expressing high levels of GFP, which corresponds to HIV-1-infected cells expressing both early and late HIV-1 genes, including Env. The geometric MFI of Env-AF647 of this population was expressed as a ratio over the geometric MFI for Δenv NL-GI using the formula: geometric MFI ratio of mutant A = geometric MFI for mutant A/geometric MFI for the Δenv mutant.
To calculate the relative MFI, each ratio was then expressed relative to the ratio for WT NL-GI using the formula: relative MFI = (geometric MFI ratio of mutant A − 1)/(geometric MFI ratio of WT − 1).
RESULTS
Characterization of Env expression by gp41 CT truncation mutants.
To assess the impact of gp41 CT mutations on cell-to-cell infection, we first constructed a series of truncation mutants that were studied previously for Env incorporation and fusion (3). This mutant series systematically removes the amphipathic alpha helices LLP-1, LLP-3, and LLP-2 or their intervening sequences (Fig. 1). These were compared to WT NL-GI and the well-characterized Env mutant ΔCT144 (26), which contains a stop codon that disrupts the membrane-proximal endocytosis motif and truncates the entire gp41 CT. We also constructed ΔCT124FS and ΔCT124 in NL4-3 Env, based on characterization studies reported in references 8 and 1, respectively. ΔCT124FS contains a frameshift mutation upstream of the YSPL endocytosis motif that results in a stop codon proximal to LLP-2 that removes 124 aa of the gp41 CT (ΔCT124FS). ΔCT124 contains a stop codon in the same position but has an intact open reading frame and YSPL motif.
FIG 1.
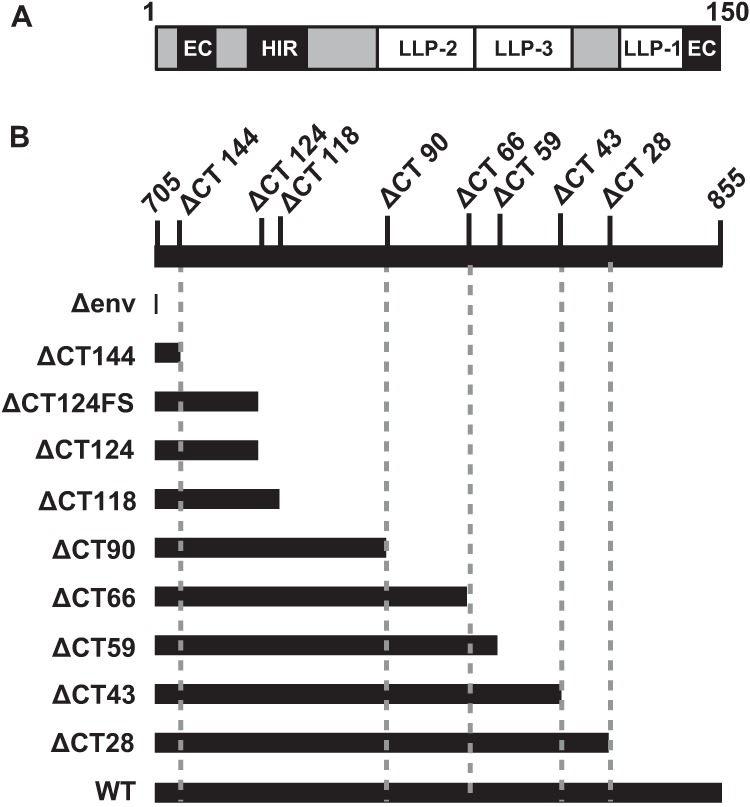
Schematic of gp41 cytoplasmic tail (CT) truncation mutants. (A) Schematic of the gp41 CT showing major structural and functional domains removed in truncation mutants. EC, endocytosis signal; HIR, highly immunogenic region; LLP, lentiviral lytic peptide. The schematic is based on reference 14. (B) Series of gp41 CT truncation mutants constructed by overlap PCR in relation to the full-length, wild-type (WT) NL4-3 gp41 CT residues 705 to 855. A stop codon was introduced to remove the indicated number of amino acids from the C terminus of the gp41 CT, based on reference 3.
When generated in 293T cells, the mutant viral clones produced Env precursor gp160 and gp41 proteins with the predicted decreases in size of the CT (Fig. 2A, top and middle). Anti-HIV antisera detected similar levels of p24 capsid expression in these cells (Fig. 2A, bottom). Most of the CT truncation mutants expressed higher levels of gp120 in 293T lysates than the WT (ΔCT144 to ΔCT59); however, ΔCT43 expressed gp120 levels similar to those of the WT, and ΔCT28 expressed lower levels than the WT (Fig. 2A; quantified in Fig. 2C). Env incorporation into virus particles was assessed by performing Western blotting on virus particles normalized for p24 input after concentration by sucrose gradient centrifugation. The large-truncation mutants, ΔCT144, ΔCT124, and ΔCT118, incorporated levels of gp120 into virus particles similar to those of the WT, while the remaining mutants incorporated 50% or less gp120 (Fig. 2B and C). The relative expression pattern of gp41 in the cell lysates versus the virus lysates was similar to what was observed for gp120 (data not shown). These findings are consistent with the Env expression and incorporation levels obtained by Jiang and Aiken using the same mutants in pNL4-3 (3). While the Western blots of virus lysates were normalized for p24 input (Fig. 2B, bottom), these mutants released various levels of viral particles to the medium, as assessed by p24 ELISA (Fig. 2D). We noted that the p24 production of ΔCT124 and ΔCT118 was consistently low across independent 293T cell transfections, and this may be explained by missense mutations in Rev (Table 1), whose gene overlaps in open reading frame with that of the Env CT.
FIG 2.

HIV-1 envelope expression by gp41 CT truncation mutants. (A) Western blot of 293T cell lysates after transfection with WT NL-GI and gp41 CT truncation mutants. (B) Western blot of virus particle lysates of WT NL-GI and gp41 CT truncation mutants purified from transfected 293T supernatants. (C) Quantification of gp120 levels expressed in 293T cell lysates and virus particle lysates. Env expression was calculated as a ratio of gp120 to p24 and expressed relative to that of the WT. Error bars represent the standard errors of the means (SEMs) from at least two independent experiments. #, cell lysate gp120/p24 values from a single experiment. (D) HIV-1 p24 produced by transfection of 293T cells, as measured by p24 ELISA. Error bars represent the SEMs from at least three individual ELISAs, each performed in duplicate using virus from independently transfected 293T cells for each ELISA. (E) Representative histograms of cell surface HIV-1 Env on Jurkat cells expressing WT NL-GI or gp41 CT truncation mutants (black line) compared to the Δenv mutant (filled gray curve). Env was detected on cells expressing high levels of HIV-1 infection using pooled polyclonal HIV-IG antibody. P values represent statistically significant differences in the average geometric MFI of cell surface Env compared to the Δenv mutant for three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (F) Relative MFI of cell surface HIV-1 Env for gp41 CT truncation mutants compared to WT NL-GI. Error bars represent the SEMs from three experiments, each performed with independently transfected Jurkat cells.
We also examined the surface Env expression of the mutants by staining nucleofected Jurkat cells with pooled HIV-1 patient immunoglobulin (HIV-IG) (Fig. 2E and F). All CT mutants expressed cell surface Env, as measured by positive staining on the population expressing high levels of GFP. The level of surface Env staining of the mutants ΔCT28, ΔCT43, ΔCT59, and ΔCT66 was similar to the surface staining of cells expressing WT Env. As previously reported for ΔCT144, Env staining was markedly higher than that of the WT due to a lack of the YSPL endocytosis motif. ΔCT124, ΔCT118, and ΔCT90 expressed the smallest amount of cell surface Env relative to WT; however, this was still higher than that of the Δenv negative control (Fig. 2E).
gp41 CT truncation mutants show selective cell-free and cell-to-cell infectivity.
We tested the single-round infectivity of 293T cell-derived virus particles in MT-4 target cells, a T cell line known to be highly permissive for HIV-1 infection, and initiated infections with virus supernatants containing the same amount of p24 antigen. We found that ΔCT144 was 68% as infectious as the WT (Fig. 3B; Table 1). Similar results were observed for ΔCT124FS (Table 1). Interestingly, the truncation mutants ΔCT124 and ΔCT118 yielded higher levels of cell-free infectivity than the WT, up to ∼180% of WT levels. Mutants ΔCT90, ΔCT66, ΔCT59, ΔCT43, and ΔCT28, which have progressively smaller portions of the CT removed, showed 10-fold or greater loss of infectivity relative to WT (Fig. 3A and B; Table 1).
FIG 3.

Cell-free and cell-to-cell single-round infectivity in MT-4 cells. (A) Representative flow cytometry plots of cell-free infection of MT-4 cells by the WT, ΔCT118, or ΔCT28 NL-GI. (B) Cell-free infection levels of MT-4 cells by gp41 CT truncation mutants, expressed as a percentage of WT NL-GI. Error bars represent the SEMs from at least three experiments, each performed in duplicate using independently produced virus to infect target cells. #, single experiment performed in duplicate. (C) Representative flow cytometry plots of cell-to-cell infection of MT-4 target cells by Jurkat donor cells transfected with the WT, ΔCT118, or ΔCT28 NL-GI. (D) Cell-to-cell infection levels of MT-4 cells after coculture with Jurkat donor cells expressing gp41 CT truncation mutants, expressed as a percentage of WT NL-GI. Error bars represent the SEMs from at least three experiments, each performed in duplicate with independently transfected Jurkat donor cells.
To determine the single-round infectivity of these mutants in cell-to-cell infection, we nucleofected Jurkat donor cells with these mutant clones and cocultured these cells with MT-4 target cells. Unlike the cell-free viral infection, truncation mutants ΔCT144, ΔCT124, ΔCT118, ΔCT90, ΔCT66, and ΔCT59 were severely attenuated in cell-to-cell infection by 20-fold or more relative to WT (Fig. 3C and D; Table 1). In contrast, ΔCT43 and ΔCT28 showed a modest 2-fold reduction in infectivity relative to WT.
We next examined whether similar infection phenotypes are also observed in a more physiologically relevant cell type, primary activated CD4+ T cells. Similar to what was observed in the MT-4 cell-free infectivity assay, we observed the highest relative levels of cell-free infection with ΔCT124 and ΔCT118 (>100%), ∼40% infection with ΔCT144, and <10% infectivity for the remaining mutants 24 h after spinoculation (Fig. 4A and B). We then performed cell-to-cell infection experiments using nucleofected Jurkat cells as donors and primary CD4+ T cells as target cells. In these studies, the nucleofection efficiency of the Jurkat cells was similar among the different viral constructs. Studies with these donor cells also indicated that ΔCT43 and ΔCT28 were the fittest mutants with regard to cell-to-cell infection, while they were very poorly infectious by cell-free routes (Fig. 4C and D).
FIG 4.

Cell-free and cell-to-cell single-round infectivity in primary CD4+ T cells. (A) Representative flow cytometry plots of cell-free infection of primary CD4+ T cells by spinoculation with the WT, ΔCT118, or ΔCT28 NL-GI. (B) Cell-free infection levels of primary CD4+ T cells after spinoculation with gp41 CT truncation mutants, expressed as a percentage of WT NL-GI. Error bars represent the SEMs for two CD4+ T cell donors each performed in duplicate. (C) Representative flow cytometry plots of cell-to-cell infection of primary CD4+ T cells by Jurkat donor cells transfected with the WT, ΔCT118, or ΔCT28 NL-GI. (D) Cell-to-cell infection levels of primary CD4+ T cells after coculture with Jurkat donor cells expressing gp41 CT truncation mutants, expressed as a percentage of WT NL-GI. Error bars represent the SEMs from three experiments performed in duplicate with independently transfected Jurkat donor cells and CD4+ T cells from a total of three blood donors.
The lower viral production observed for some of the truncation mutants (Fig. 2D) may be attributed to missense mutations in Rev, generated when stop codons were introduced into the overlapping reading frame of Env (summarized in Table 1). To exclude the possibility that differences in cell-free and cell-to-cell infectivity were due to defects in Rev and to further define the regions of the CT responsible for infectivity differences, we generated and tested several additional mutants. ΔCT100, ΔCT83, ΔCT67, and ΔCT60 do not contain changes in the Rev amino acid sequence after introduction of a stop codon in the gp41 open reading frame. ΔCT93 contains 1 amino acid change in Rev. We also tested ΔCT56 (1 amino acid change in Rev), ΔCT49 (3 amino acid changes in Rev), and ΔCT46 (no amino acid changes in Rev), to further define the region responsible for conferring the difference in cell-to-cell infectivity seen between ΔCT59 and ΔCT43. We observed that these additional mutants produced WT levels of virus, with the exception of ΔCT49, which contained 3 amino acid mutations in Rev (Fig. 5A). Compared to the WT, the large-truncation mutants ΔCT100 and ΔCT93 showed ≥150% cell-free infectivity, similar to ΔCT118 and ΔCT124 (Fig. 4B; Table 1). ΔCT49 showed some cell-free infectivity (18%), while the remaining mutants were severely attenuated (≤5%). These additional mutants all showed ≤5% infectivity compared to WT during cell-to-cell infection, with the exception of ΔCT46 (17%) (Fig. 5C; Table 1). Overall, robust preferential cell-free infection was observed for four mutants with large truncations (ΔCT124, ΔCT118, ΔCT100, and ΔCT93), while preferential cell-to-cell infection was observed for two mutants with small truncations (ΔCT43 and ΔCT28).
FIG 5.
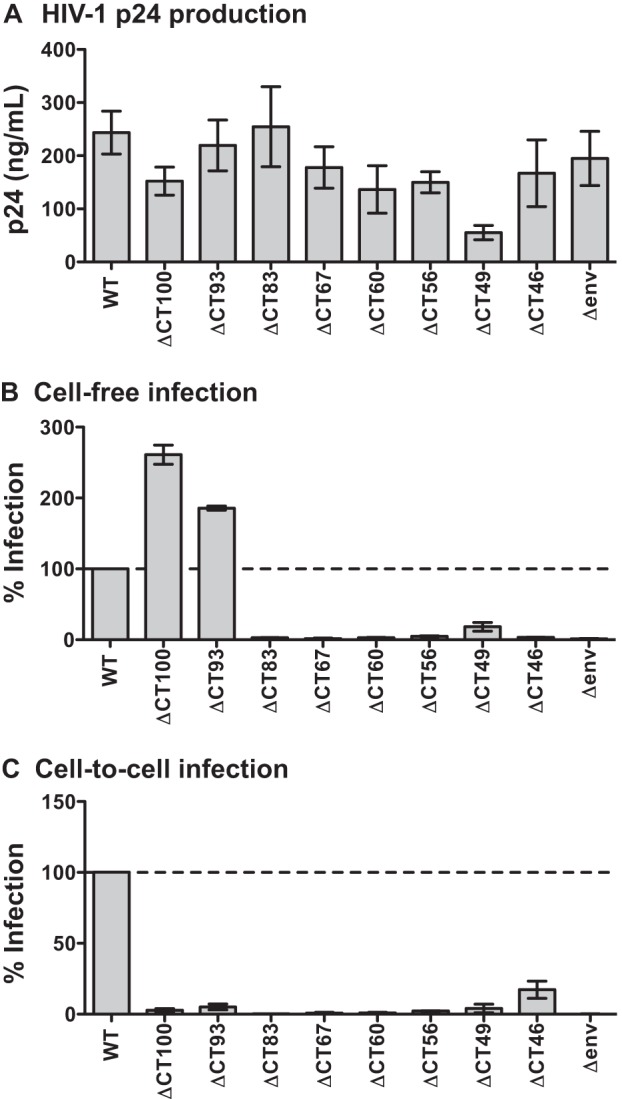
Characterization of additional gp41 truncation mutants. (A) HIV-1 p24 produced by transfection of 293T cells with WT NL-GI and additional gp41 CT truncation mutants, as measured by p24 ELISA. Error bars represent the SEMs from at least three individual ELISAs, each performed in duplicate using virus from independently transfected 293T cells for each ELISA. (B) Cell-free infection levels of MT-4 cells by additional gp41 CT truncation mutants, expressed as a percentage of WT NL-GI. Error bars represent the SEMs from at least two experiments, each performed in duplicate using independently produced virus to infect target cells. (C) Cell-to-cell infection levels of MT-4 cells after coculture with Jurkat donor cells expressing additional gp41 CT truncation mutants, expressed as a percentage of WT NL-GI. Error bars represent the SEMs from at least two experiments, each performed in duplicate with independently transfected Jurkat donor cells.
LLP-3 point mutants show selective attenuation of cell-free infectivity.
In order to better understand the contribution of the LLP-3 region of gp41 to the CT mutant phenotypes, we constructed two mutants with point mutations in LLP-3, YW_SL and LL_RQ, which were previously described as detrimental mutations for cell-free virus infectivity. Figure 6A shows the positions of these substitutions in LLP-3, relative to the nearest truncation mutation we generated, the ΔCT59 mutation. The resulting mutants did not show any significant differences in p24 expression in producer 293T cells (Fig. 6B). Similar amounts of p24 were also detected in the supernatants of transfected 293T cells (data not shown). However, gp120 and gp41 levels were reduced in the virus particles compared to WT levels for both mutants in Western blots loaded with the same amount of p24 (Fig. 6C and D). This suggests a defect in Env packaging, as 293T lysates contained amounts of gp120 and gp41 similar to or greater than those seen with the WT.
FIG 6.

HIV-1 envelope expression and single-round infectivity of gp41 LLP-3 mutants YW_SL and LL_RQ in MT-4 cells. (A) Amino acid sequence alignment of gp41 LLP-3 mutants LL_RQ and YW_SL in relation to the full-length, wild-type (WT) NL4-3 gp41 CT residues 792 to 805, as well as the nearest truncation mutant, ΔCT59. Amino acid identity (-), the stop codon (*), and substitutions are shown. (B) Western blot of 293T cell lysates after transfection with WT NL-GI or gp41 LLP-3 mutants. (C) Western blot of virus particle lysates of WT NL-GI or gp41 LLP-3 mutants purified from transfected 293T supernatants. (D) Quantification of gp120 levels expressed in 293T cell lysates and virus particle lysates. Env expression was calculated as a ratio of gp120/p24 and expressed relative to WT. Error bars represent the SEMs from at least two independent experiments. (E) Cell-free infection levels of MT-4 cells by gp41 LLP-3 mutants, expressed as a percentage of WT NL-GI. Error bars represent the SEMs from three experiments, each performed in duplicate using independently produced virus to infect target cells. (F) Cell-to-cell infection levels of MT-4 cells after coculture with Jurkat donor cells expressing gp41 LLP-3 mutants, expressed as a percentage of WT NL-GI. Error bars represent the SEMs from four experiments, each performed in duplicate with independently transfected Jurkat donor cells.
Both LLP-3 point mutants were strongly attenuated in cell-free infectivity (∼10% infectivity compared to WT) (Fig. 6E; Table 1). During cell-to-cell infection, YW_SL and LL_RQ showed 60% and 55% infectivity, respectively (Fig. 6F; Table 1). Table 1 summarizes the relative infectivity of all of the truncation and point mutants tested for single-round infectivity in MT-4 cells using 293T-produced cell-free virus particles for cell-free infection or infected Jurkat donor cells for cell-to-cell infection.
To examine the reported cell type-specific infectivity defect of cell-free viruses with large tail truncations, we compared the relative infectivity of virus particles produced in Jurkat cells (nonpermissive for full tail truncation) to the 293T-produced virus used in Fig. 3 and 4. We produced WT, ΔCT100, ΔCT28, YW_SL, and LL_RQ virus particles in Jurkat cells and performed cell-free infectivity assays after normalizing for p24 input. Compared to WT, the ΔCT100 mutant showed ≤10% infectivity in MT-4 cells, similar to ΔCT28 (∼15%), while YW_SL and LL_RQ showed ∼25 to 30% infectivity (data not shown). The cell type-dependent cell-free infectivity we observed for ΔCT100 is consistent with previous cell-free infectivity studies of mutants with large truncations of the gp41 CT (22, 23, 27–30).
VSV-G pseudotyping rescues defect in cell-free infectivity of mutants with small gp41 CT truncation and LLP-3 point mutants.
To determine whether the defect in cell-free infection observed for the short-truncation mutants and the LLP-3 point mutants was due to viral entry, we generated virus particles in 293T cells pseudotyped with VSV-G and tested their single-round infectivity in MT-4 cells. The infectivity of the short-truncation mutant ΔCT28 as well as the LLP-3 point mutants YW_SL and LL_RQ was greatly enhanced when these mutants were pseudotyped with VSV-G, compared to the infectivity of nonpseudotyped mutants (Fig. 7). All VSV-G-pseudotyped viruses (WT, gp41 CT mutants, and Δenv mutants) were able to infect MT-4 cells to similar levels. This indicates that the viruses with defects in cell-free infection are all due to an intrinsic deficiency in entry rather than any other steps of viral infection.
FIG 7.
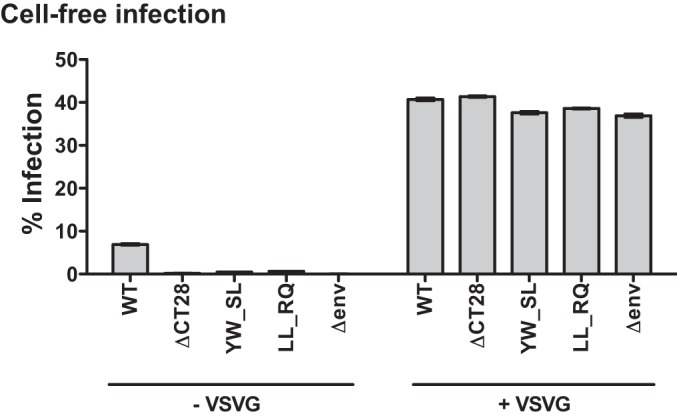
Cell-free single-round infectivity of select VSV-G pseudotyped gp41 CT mutants in MT-4 cells. Cell-free infection levels of MT-4 cells by 3.5 ng/well of WT, ΔCT 28, the gp41 LLP-3 mutants YW_SL and LL_RQ, or Δenv NL-GI (left), compared to 3.5 ng/well of virus pseudotyped with VSV-G (right). Error bars represent the SEMs for duplicate wells.
A defect in cell-to-cell infection of mutants with large gp41 CT truncations occurs after cell-to-cell transfer and viral membrane fusion.
To determine if the selective block in cell-to-cell infection of the large-truncation mutants ΔCT124, ΔCT118, ΔCT100, and ΔCT93 NL-GI (Fig. 3D and 5C) was due to a defect in cell-to-cell transfer, we examined the efficiency of transfer of fluorescent HIV particles from cell to cell. We compared ΔCT100 Gag-iCherry to WT Gag-iCherry as well as ΔCT28 Gag-iCherry, a mutant that exhibited a preferential block to cell-free over cell-to-cell infection. Transfer was initiated with nucleofected Jurkat donor cells. After 3 h coculture with resting primary CD4+ T cells, there was no significant difference in the level of transfer between ΔCT100 Gag-iCherry and ΔCT28 Gag-iCherry (Fig. 8A and B), which were 59% and 54% of WT Gag-iCherry, respectively. This indicates that the ΔCT100 Env can initiate VS formation and mediate the transfer of virus particles from donor to target cell. The block in cell-to-cell infection by the large-truncation mutants, including ΔCT100 NL-GI (Fig. 3D and 5C), therefore likely occurs after transfer of virus particles.
FIG 8.

Cell-to-cell transfer and cell-to-cell viral membrane fusion of selected gp41 CT truncation mutants into primary CD4+ T cells. (A) Representative flow cytometry plots of cell-to-cell transfer to primary CD4+ T cells by Jurkat donor cells transfected with WT, ΔCT100, ΔCT28, or Δenv Gag-iCherry. (B) Cell-to-cell transfer levels of primary CD4+ T cells after coculture with Jurkat donor cells expressing WT, ΔCT100, ΔCT28, or Δenv Gag-iCherry, expressed as a percentage of WT Gag-iCherry. Error bars represent the SEMs from two experiments performed in duplicate, using independently transfected donor cells for each experiment and CD4+ T cells from a total of three blood donors. (C) Representative flow cytometry plots of cell-to-cell viral membrane fusion with primary CD4+ T cells by Jurkat donor cells transfected with WT, ΔCT100, ΔCT28, or Δenv NL4-3 coexpressing BlaM-Vpr. (D) Cell-to-cell viral membrane fusion levels of primary CD4+ T cells after coculture with Jurkat donor cells coexpressing WT, ΔCT100, ΔCT28, or Δenv NL4-3 and BlaM-Vpr, expressed as a percentage of WT NL4-3 BlaM-Vpr. Error bars represent the SEMs from two experiments performed in duplicate, using independently transfected Jurkat donor cells and CD4+ T cells from a total of three blood donors.
We next examined whether the ability of virus particles to fuse after transfer across the VS may be impaired in the ΔCT100 mutant. We performed a cell-to-cell viral membrane fusion assay using Jurkat donor cells cotransfected with either WT, ΔCT100, or ΔCT28 NL4-3 and a BlaM-Vpr expression plasmid. This variation of the BlaM-Vpr viral fusion assay (62) measures fusion of the viral and cellular membranes after cell-to-cell transfer of HIV-1. After coculture with activated primary CD4+ T cells, we observed similar levels of viral membrane fusion mediated by WT, ΔCT100, and ΔCT28 after cell-to-cell contact (Fig. 8C and D). This indicates that viral membrane fusion following cell-to-cell contact is intact with the ΔCT100 mutant.
Selective attenuation of viral spread in MT-4 or primary CD4+ T cell cultures initiated with cell-free virus or with infected cells.
ΔCT100 NL-GI undergoes single-round cell-free infection but is deficient in cell-to-cell infection (Fig. 5), while ΔCT28, YW_SL, and LL_RQ NL-GI show attenuated single-round cell-free infection but are capable of single-round cell-to-cell infection (Fig. 3 and 6). We therefore examined ΔCT100 and ΔCT28 as representative truncation mutants to test for the ability to undergo multiround infection in MT-4 cells. We also tested the LLP-3 point mutants YW_SL and LL_RQ. We determined the number of infected cells at each time point by detecting GFP expression by flow cytometry. When the infection was initiated with cell-free virus made in 293T cells, ΔCT100 was similar in infectivity to WT HIV-1 throughout the 5-day course of infection, while ΔCT28 showed infection similar to that of the Δenv control (Fig. 9A). When infection was initiated with infected Jurkat donor cells, ΔCT28 was more infectious than ΔCT100, while ΔCT100 was only slightly more infectious than the Δenv control (Fig. 9B). The ΔCT28 mutant continued to spread in culture between day 1 and day 3, indicating that cell-to-cell infection allows this virus to spread. The LLP-3 mutants YW_SL and LL_RQ, like ΔCT28, showed very low levels of cell-free infection at 1 day postinfection (Fig. 9C). While these viruses were able to spread in culture, the infection was initially low and was unable to attain the level seen with WT virus. In contrast, when infection with the mutants YW_SL and LL_RQ was initiated with infected Jurkat donor cells, these mutants showed levels of replication very similar to those of the WT through a 5-day time course (Fig. 9D). Overall, these results indicate that viruses with a significant defect in cell-free viral infectivity (ΔCT28, YW_SL, and LL_RQ) can replicate with kinetics similar to WT HIV-1 when the infection is initiated with a cell-to-cell inoculum. Likewise, a virus with a defect in cell-to-cell infectivity (ΔCT100) can replicate similarly to WT when the infection is initiated with cell-free virus produced in 293T cells. When viral spread was initiated in primary activated CD4+ T cells as target cells, our observations were similar to those for the cell line MT-4, particularly between day 1 and day 3 of the infection (Fig. 9E and F).
FIG 9.

Multiround infectivity of gp41 CT mutants in MT-4 and primary CD4+ T cells. (A) Multiround infection of MT-4 cells initiated with 293T-produced cell-free WT NL-GI or the indicated truncation mutants. (B) Multiround infection of MT-4 cells initiated by coculture with Jurkat donor cells expressing WT NL-GI or the indicated truncation mutants. (C) Multiround infection of MT-4 cells initiated with 293T-produced cell-free WT NL-GI or the indicated mutants. (D) Multiround infection of MT-4 cells initiated by coculture with Jurkat donor cells expressing WT NL-GI or the indicated mutants. For panels A to D, error bars represent the SEMs for duplicate wells. (E) Multiround infection of primary CD4+ T cells initiated with 293T-produced cell-free WT NL-GI or the indicated mutants. (F) Multiround infection of primary CD4+ T cells initiated by coculture with Jurkat donor cells expressing WT NL-GI or the indicated mutants. For panels E and F, error bars represent the SEMs for two CD4+ T cell donors each performed in duplicate. All cell-free infections were initiated at day 0 with the same amount of virus p24. All cell-to-cell infections were initiated at day 0 by coculture with Jurkat donor cells transfected to similar levels with WT or mutant NL-GI.
Overall, these results indicate that viruses with a significant defect in cell-free viral infectivity (ΔCT28, YW_SL, and LL_RQ) can replicate with kinetics similar to those of WT HIV-1 when the infection is initiated with infected Jurkat donor cells. Likewise, a virus with a defect in cell-to-cell infectivity (ΔCT100) can replicate similar to WT when the infection is initiated with cell-free virus produced in 293T cells.
DISCUSSION
The HIV-1 gp41 CT has been the subject of numerous studies using systematic mutation to pinpoint the role of specific CT sequences in Env packaging, fusion, intracellular localization, and overall infectivity of cell-free virus. However, in the context of cell-to-cell infection, the CT has mainly been studied using the truncation mutant ΔCT144. Here, we determined that individual mutations of the CT can have a dramatically different impact on cell-free or cell-to-cell infection. Specifically, large truncations of the CT that permit cell-free infection of 293T-produced virus drastically attenuate cell-to-cell infection. Small truncations or substitution mutations in LLP-3 can result in the reciprocal phenotype when cells are cocultured with infected Jurkat cells. We conclude that the CT plays unique roles in supporting infection during these two modes of infection.
In agreement with other studies that generated virus particles in HeLa or 293T cells (3, 28), we found reduced levels of gp120 in virus particles for mutants ΔCT90 to ΔCT28 (Fig. 2B and C), correlating with a severe reduction in cell-free infectivity compared to WT (Fig. 3B and 4B; Table 1). This is consistent with the role of LLP-1 (2) or inhibitory sequences (17) in envelope packaging. It is important to note that while Env incorporation appeared to be important for cell-free infectivity, this phenotype did not correlate with cell-to-cell infectivity. The ΔCT28 and ΔCT43 mutants exhibited decreases in Env incorporation but still showed relatively robust infection in cell-to-cell infection assays. This indicates that cell-to-cell infection can overcome deficiencies of viral Env incorporation. Further, our multiround infectivity assays with ΔCT28 (Fig. 9A and B) show that such viruses can propagate over time via cell-to-cell infection in permissive cell types such as MT-4 cells. This phenotype is similar to a 20-aa truncation isolated from an HIV-1-infected patient (64). This study by Beaumont et al. provides evidence that clones with low cell-free infectivity can persist in vivo, although these mutations may be rare (64). However, traditional methods for the detection and isolation of replication-competent viral clones are based on cell-free assays and therefore would not detect mutants that are defective by cell-free infection but still capable of cell-to-cell spread. The proportion of such clones in vivo could be underestimated by these detection methods.
In our assays, the LLP-3 substitution mutants YW_SL and LL_RQ are also compromised in cell-free but not cell-to-cell infectivity (single-round infection [Fig. 6E and F] and multiround infection [Fig. 9]). These viruses packaged moderately reduced levels of Env on virus particles (Fig. 6C and D), similar to prior findings for YW_SL (19, 20). Env packaging was at least 2-fold reduced in these mutants; however, cell-free infectivity was ∼10-fold lower than that of the WT. While the presence of Env is necessary for cell-free infection, the structure of the tail or its ability to recruit a trans-acting factor(s) may influence infection levels of cell-free but not cell-to-cell HIV-1. A recent study by Checkley et al. found that knockdown of TIP47 in Jurkat cells did not alter envelope incorporation, virus production, or infectivity of cell-free HIV particles (65), indicating that TIP47 may not be critical to the originally described phenotype of reduced Env incorporation and cell-free infectivity. While the role of TIP47 as a trans-acting factor involved in Env packaging has been called into question, our results are consistent with studies describing these LLP-3 amino acid residues as important for infection (18–20). Although these specific LLP-3 residues may serve as binding sites for other uncharacterized trans-acting factors, it seems more likely that the residues in this region may be critical for maintaining a specific conformation of LLP-3 that is required for efficient Env packing/function.
An interaction between the host trafficking factor Rab11-FIP1C and the YW795 motif in LLP-3 of the gp41 CT has recently been shown to regulate Env incorporation into virus particles in nonpermissive cells (24, 25). Truncation of the CT, demonstrated using ΔCT144, or mutation of YW795 alters Rab11-FIP1C localization in infected cells, Env-Gag colocalization, and Env incorporation in virus particles produced in nonpermissive cells. Disruption of this interaction may account for the lack of cell-free or cell-to-cell infectivity of the ΔCT100 and other large-truncation mutants when nonpermissive Jurkat cells are used either to produce cell-free virus particles or as donor cells during cell-to-cell infection. The LLP-3 point mutants YW_SL and LL_RQ have disruptions in the putative binding sites of the host factors TIP47 and prohibitin in the same region of the CT as Rab11-FIP1C. These mutants are capable of cell-to-cell spread in primary CD4+ T cells at levels similar to that of the WT but remain attenuated when cell-free inocula are used to initiate the infection, although the YW795 motif remains intact (Fig. 9E and F), suggesting that the differences in gp41 CT-mediated infectivity for these mutants may occur independently of Rab11-FIP1C interaction.
The large-truncation mutants ΔCT124, ΔCT118, ΔCT100, and ΔCT93 showed 2- to 3-fold-higher infectivity than the WT, although the same amount of p24 was used for all infections. In addition, these mutants are selectively defective in cell-to-cell infection (Fig. 3D, 4D, and 5C). In previous studies of cell-to-cell infection via the VS, we observed that fusion of the viral and cellular membranes occurs after transfer of virus into intracellular compartments within the target cell (52). Here, our surface staining experiments confirm that Env is present on the surface of all Jurkat donor cells expressing the truncation series (Fig. 2E and F). The levels of cell surface Env are sufficient to mediate transfer of Gag into the target cell (Fig. 8A and B) and also support viral fusion (Fig. 8C and D); however, the large-truncation mutant ΔCT100 is defective in cell-to-cell infection (Fig. 3D, 4D, and 5C). This may indicate that there are functions of the CT that go beyond cell adhesion and induction of fusion and may impact cell-to-cell infection at the early postentry stage of infection.
Interestingly, during spreading infection in both permissive MT-4 cells and nonpermissive primary CD4+ T cells, the selective mutants do not bypass the initial defect in either cell-free or cell-to-cell infection and thus never replicate to WT levels (Fig. 9). Rather, the type of initial inoculum determines the ability of the mutant to spread over time. One possible explanation for this is that if the initial inoculum is too weak to establish a critical number of infected cells, this can have a prolonged impact on successive rounds of replication even if clones can replicate well when introduced through a different route. For example, although the initial infection is low for ΔCT28, it is still sufficient to establish a critical number of infected cells and initiate efficient replication from cell-to-cell. However, the initial number of cells infected by cell-free inocula may be below this critical threshold, and therefore too low to effectively initiate spreading infection. This is less apparent with the LLP-3 mutants YW_SL and LL_RQ, which are somewhat more infectious than ΔCT28 in our cell-free infection assays.
Model for mutants with diminished cell-free but not cell-to-cell infectivity (ΔCT28, ΔCT43, YW_SL, and LL_RQ).
Efficient Env incorporation into cell-free virus likely requires direct or indirect interactions with Gag that are needed for Env packaging and/or subsequent productive cell-free infection (11, 28, 64) but may be less stringent for cell-to-cell infection. In the context of HIV-1 NL4-3, we found that these mutants are capable of cell-free infection when pseudotyped with VSV-G (Fig. 7), showing that their defect in cell-free infectivity is due to a defect in Env at the step of viral entry. Truncation up to the last 43 aa of the CT or point mutations in the LLP-3 region may disrupt CT conformation or alter the intracellular interactions that occur at the CT during assembly and budding at the plasma membrane. This may change the ectodomain conformation of gp120 such that it is no longer functional during cell-free infection. A recent report describes direct mutations in the V1V2 loop of the gp120 ectodomain that prevent cell-free infection while permitting cell-to-cell infection (66), similar to mutants we describe here. In an alternate but not mutually exclusive model, the C-terminal region of the CT may be critical for proper subcellular localization of Env (including targeting of Env to sites of virus budding) or to activate the recruitment of Gag to plasma membrane-localized Env during cell-free viral assembly. A different, more proximal CT domain may mediate this function during cell-to-cell infection (see below), allowing cell-to-cell infection to continue even in the presence of these mutations.
Infectivity of these mutants is rescued during cell-to-cell infection, implying that there is something different about the function of Env during VS formation versus during cell-free infection. During cell-to-cell infection, Env engagement with CD4 and formation of the VS precede viral release, as opposed to cell-free infection, where viral release precedes engagement of CD4 on an uninfected target cell. Because some of these mutants that exhibit packaging defects in cell-free virions are still infectious when introduced as cell-associated virus, our data imply that the formation of the VS can overcome packaging or other defects, perhaps by concentrating Env at the site of cell-cell contact.
Model for mutants with diminished cell-to-cell but not cell-free infectivity (ΔCT124, ΔCT118, ΔCT100, and ΔCT93).
The putative interaction between Gag and Env that is required for cell-free infection may be overcome by large truncations in the tail, which may permit passive incorporation of Env into virus particles made in 293T cells, consistent with previous reports (27, 28). In contrast, in nonpermissive cells such as Jurkat or primary CD4+ T cells, both cell-free and cell-to-cell infectivity may be mediated by domains in the region of LLP-2/3, as large CT truncations that delete this region are not infectious. For cell-free infection, this effect may be attributed to altered Env trafficking and packaging caused by mutation of the Rab11-FIP1C binding site in this region of the CT (25); however, surprisingly, we found that these mutants are fully capable of being transferred from donor cell to target cell (Fig. 8A and B). We also measured the release of a BlaM-Vpr fusion protein into the target cells following cell-cell coculture, which is also not affected by large CT truncations (Fig. 8C and D). Taken together, our data are indicative of an early block in infection of the incoming virus that occurs after fusion. During cell-to-cell transmission, CD4 engagement, virus assembly, and transfer to the target cell are all tightly coordinated. Several viral and cellular factors are required for the coordinated assembly of infectious virus particles in the infected donor cell for VS formation, for example, components of the T cell signaling machinery such as the kinase Zap70 (67). It is plausible that other factors may be required in the infected donor cell that impact steps post-VS formation and viral transfer, to affect virus infectivity once transferred to the target cell. Large CT truncations may fail to recruit key viral or cellular factors necessary to specifically maintain cell-to-cell infectivity. Alternatively, the timing of viral fusion with respect to viral assembly may result in the transfer of virus into the target cell but inappropriate intracellular localization of fusion.
It is important to note that the studies presented here were performed exclusively with Env from the HIV-1 laboratory isolate NL4-3. We have yet to determine if CT truncations in other strains generate similar infectivity phenotypes. There is at least one example of a ΔCT20 mutant identified in a patient infected with a B-clade virus. This mutant was tested in vitro in the R5-tropic virus backbone NL(AD8) and was shown to selectively replicate by cell-to-cell infection (64). Further studies are needed to determine if additional CT truncations or LLP-3 mutations exhibit similar phenotypes when created in R5-tropic primary-isolate Envs. If broadly applicable, these primary isolate Env mutants may be tested in humanized mouse models to examine the contribution of cell-free versus cell-to-cell infection in an in vivo system, which remains a major open question in the HIV-1 field.
The data presented here point to the multiple roles of the gp41 CT in differentially modulating cell-free and cell-to-cell HIV-1 infection. Further study of the distinct mechanisms involved in cell-to-cell transmission is needed to elucidate how these differential phenotypes impact Env conformations, interactions with Gag and the incoming viral core, and/or signaling events that may facilitate infection of the target cell.
ACKNOWLEDGMENTS
We thank members of the B. K. Chen lab for helpful comments and the Flow Cytometry Shared Resource Facility, Icahn School of Medicine at Mount Sinai, New York, NY.
This work was supported by grants to B.K.C. from the NIH, AI074420 and GM113885.
We have no conflicting financial interests.
REFERENCES
- 1.Wyss S, Dimitrov AS, Baribaud F, Edwards TG, Blumenthal R, Hoxie JA. 2005. Regulation of human immunodeficiency virus type 1 envelope glycoprotein fusion by a membrane-interactive domain in the gp41 cytoplasmic tail. J Virol 79:12231–12241. doi: 10.1128/JVI.79.19.12231-12241.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kalia V, Sarkar S, Gupta P, Montelaro RC. 2003. Rational site-directed mutations of the LLP-1 and LLP-2 lentivirus lytic peptide domains in the intracytoplasmic tail of human immunodeficiency virus type 1 gp41 indicate common functions in cell-cell fusion but distinct roles in virion envelope incorporation. J Virol 77:3634–3646. doi: 10.1128/JVI.77.6.3634-3646.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiang J, Aiken C. 2007. Maturation-dependent human immunodeficiency virus type 1 particle fusion requires a carboxyl-terminal region of the gp41 cytoplasmic tail. J Virol 81:9999–10008. doi: 10.1128/JVI.00592-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wyma DJ, Jiang J, Shi J, Zhou J, Lineberger JE, Miller MD, Aiken C. 2004. Coupling of human immunodeficiency virus type 1 fusion to virion maturation: a novel role of the gp41 cytoplasmic tail. J Virol 78:3429–3435. doi: 10.1128/JVI.78.7.3429-3435.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murakami T, Ablan S, Freed EO, Tanaka Y. 2004. Regulation of human immunodeficiency virus type 1 Env-mediated membrane fusion by viral protease activity. J Virol 78:1026–1031. doi: 10.1128/JVI.78.2.1026-1031.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhakta SJ, Shang L, Prince JL, Claiborne DT, Hunter E. 2011. Mutagenesis of tyrosine and di-leucine motifs in the HIV-1 envelope cytoplasmic domain results in a loss of Env-mediated fusion and infectivity. Retrovirology 8:37. doi: 10.1186/1742-4690-8-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Joyner AS, Willis JR, Crowe JE Jr, Aiken C. 2011. Maturation-induced cloaking of neutralization epitopes on HIV-1 particles. PLoS Pathog 7:e1002234. doi: 10.1371/journal.ppat.1002234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Edwards TG, Wyss S, Reeves JD, Zolla-Pazner S, Hoxie JA, Doms RW, Baribaud F. 2002. Truncation of the cytoplasmic domain induces exposure of conserved regions in the ectodomain of human immunodeficiency virus type 1 envelope protein. J Virol 76:2683–2691. doi: 10.1128/JVI.76.6.2683-2691.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Durham ND, Yewdall AW, Chen P, Lee R, Zony C, Robinson JE, Chen BK. 2012. Neutralization resistance of virological synapse-mediated HIV-1 Infection is regulated by the gp41 cytoplasmic tail. J Virol 86:7484–7495. doi: 10.1128/JVI.00230-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kalia V, Sarkar S, Gupta P, Montelaro RC. 2005. Antibody neutralization escape mediated by point mutations in the intracytoplasmic tail of human immunodeficiency virus type 1 gp41. J Virol 79:2097–2107. doi: 10.1128/JVI.79.4.2097-2107.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Checkley MA, Luttge BG, Freed EO. 2011. HIV-1 envelope glycoprotein biosynthesis, trafficking, and incorporation. J Mol Biol 410:582–608. doi: 10.1016/j.jmb.2011.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steckbeck JD, Kuhlmann AS, Montelaro RC. 2013. C-terminal tail of human immunodeficiency virus gp41: functionally rich and structurally enigmatic. J Gen Virol 94:1–19. doi: 10.1099/vir.0.046508-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murakami T. 2012. Retroviral env glycoprotein trafficking and incorporation into virions. Mol Biol Int 2012:682850. doi: 10.1155/2012/682850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Postler TS, Desrosiers RC. 2013. The tale of the long tail: the cytoplasmic domain of HIV-1 gp41. J Virol 87:2–15. doi: 10.1128/JVI.02053-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Santos da Silva E, Mulinge M, Perez Bercoff D. 2013. The frantic play of the concealed HIV envelope cytoplasmic tail. Retrovirology 10:54. doi: 10.1186/1742-4690-10-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Postler TS, Desrosiers RC. 2012. The cytoplasmic domain of the HIV-1 glycoprotein gp41 induces NF-kappaB activation through TGF-beta-activated kinase 1. Cell Host Microbe 11:181–193. doi: 10.1016/j.chom.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bultmann A, Muranyi W, Seed B, Haas J. 2001. Identification of two sequences in the cytoplasmic tail of the human immunodeficiency virus type 1 envelope glycoprotein that inhibit cell surface expression. J Virol 75:5263–5276. doi: 10.1128/JVI.75.11.5263-5276.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Emerson V, Holtkotte D, Pfeiffer T, Wang IH, Schnolzer M, Kempf T, Bosch V. 2010. Identification of the cellular prohibitin 1/prohibitin 2 heterodimer as an interaction partner of the C-terminal cytoplasmic domain of the HIV-1 glycoprotein. J Virol 84:1355–1365. doi: 10.1128/JVI.01641-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blot G, Janvier K, Le Panse S, Benarous R, Berlioz-Torrent C. 2003. Targeting of the human immunodeficiency virus type 1 envelope to the trans-Golgi network through binding to TIP47 is required for env incorporation into virions and infectivity. J Virol 77:6931–6945. doi: 10.1128/JVI.77.12.6931-6945.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lopez-Verges S, Camus G, Blot G, Beauvoir R, Benarous R, Berlioz-Torrent C. 2006. Tail-interacting protein TIP47 is a connector between Gag and Env and is required for Env incorporation into HIV-1 virions. Proc Natl Acad Sci U S A 103:14947–14952. doi: 10.1073/pnas.0602941103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Groppelli E, Len AC, Granger LA, Jolly C. 2014. Retromer regulates HIV-1 envelope glycoprotein trafficking and incorporation into virions. PLoS Pathog 10:e1004518. doi: 10.1371/journal.ppat.1004518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akari H, Fukumori T, Adachi A. 2000. Cell-dependent requirement of human immunodeficiency virus type 1 gp41 cytoplasmic tail for Env incorporation into virions. J Virol 74:4891–4893. doi: 10.1128/JVI.74.10.4891-4893.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murakami T, Freed EO. 2000. The long cytoplasmic tail of gp41 is required in a cell type-dependent manner for HIV-1 envelope glycoprotein incorporation into virions. Proc Natl Acad Sci U S A 97:343–348. doi: 10.1073/pnas.97.1.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qi M, Williams JA, Chu H, Chen X, Wang JJ, Ding L, Akhirome E, Wen X, Lapierre LA, Goldenring JR, Spearman P. 2013. Rab11-FIP1C and Rab14 direct plasma membrane sorting and particle incorporation of the HIV-1 envelope glycoprotein complex. PLoS Pathog 9:e1003278. doi: 10.1371/journal.ppat.1003278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qi M, Chu H, Chen X, Choi J, Wen X, Hammonds J, Ding L, Hunter E, Spearman P. 2015. A tyrosine-based motif in the HIV-1 envelope glycoprotein tail mediates cell-type- and Rab11-FIP1C-dependent incorporation into virions. Proc Natl Acad Sci U S A 112:7575–7580. doi: 10.1073/pnas.1504174112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilk T, Pfeiffer T, Bosch V. 1992. Retained in vitro infectivity and cytopathogenicity of HIV-1 despite truncation of the C-terminal tail of the env gene product. Virology 189:167–177. doi: 10.1016/0042-6822(92)90692-I. [DOI] [PubMed] [Google Scholar]
- 27.Freed EO, Martin MA. 1995. Virion incorporation of envelope glycoproteins with long but not short cytoplasmic tails is blocked by specific, single amino acid substitutions in the human immunodeficiency virus type 1 matrix. J Virol 69:1984–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Freed EO, Martin MA. 1996. Domains of the human immunodeficiency virus type 1 matrix and gp41 cytoplasmic tail required for envelope incorporation into virions. J Virol 70:341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu X, Yuan X, McLane MF, Lee TH, Essex M. 1993. Mutations in the cytoplasmic domain of human immunodeficiency virus type 1 transmembrane protein impair the incorporation of Env proteins into mature virions. J Virol 67:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dubay JW, Roberts SJ, Hahn BH, Hunter E. 1992. Truncation of the human immunodeficiency virus type 1 transmembrane glycoprotein cytoplasmic domain blocks virus infectivity. J Virol 66:6616–6625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Piller SC, Dubay JW, Derdeyn CA, Hunter E. 2000. Mutational analysis of conserved domains within the cytoplasmic tail of gp41 from human immunodeficiency virus type 1: effects on glycoprotein incorporation and infectivity. J Virol 74:11717–11723. doi: 10.1128/JVI.74.24.11717-11723.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murakami T, Freed EO. 2000. Genetic evidence for an interaction between human immunodeficiency virus type 1 matrix and alpha-helix 2 of the gp41 cytoplasmic tail. J Virol 74:3548–3554. doi: 10.1128/JVI.74.8.3548-3554.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lambele M, Labrosse B, Roch E, Moreau A, Verrier B, Barin F, Roingeard P, Mammano F, Brand D. 2007. Impact of natural polymorphism within the gp41 cytoplasmic tail of human immunodeficiency virus type 1 on the intracellular distribution of envelope glycoproteins and viral assembly. J Virol 81:125–140. doi: 10.1128/JVI.01659-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Newman JT, Sturgeon TJ, Gupta P, Montelaro RC. 2007. Differential functional phenotypes of two primary HIV-1 strains resulting from homologous point mutations in the LLP domains of the envelope gp41 intracytoplasmic domain. Virology 367:102–116. doi: 10.1016/j.virol.2007.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Day JR, Munk C, Guatelli JC. 2004. The membrane-proximal tyrosine-based sorting signal of human immunodeficiency virus type 1 gp41 is required for optimal viral infectivity. J Virol 78:1069–1079. doi: 10.1128/JVI.78.3.1069-1079.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deschambeault J, Lalonde JP, Cervantes-Acosta G, Lodge R, Cohen EA, Lemay G. 1999. Polarized human immunodeficiency virus budding in lymphocytes involves a tyrosine-based signal and favors cell-to-cell viral transmission. J Virol 73:5010–5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murooka TT, Deruaz M, Marangoni F, Vrbanac VD, Seung E, von Andrian UH, Tager AM, Luster AD, Mempel TR. 2012. HIV-infected T cells are migratory vehicles for viral dissemination. Nature 490:283–287. doi: 10.1038/nature11398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jolly C, Mitar I, Sattentau QJ. 2007. Requirement for an intact T-cell actin and tubulin cytoskeleton for efficient assembly and spread of human immunodeficiency virus type 1. J Virol 81:5547–5560. doi: 10.1128/JVI.01469-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jolly C, Kashefi K, Hollinshead M, Sattentau QJ. 2004. HIV-1 cell to cell transfer across an Env-induced, actin-dependent synapse. J Exp Med 199:283–293. doi: 10.1084/jem.20030648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alvarez RA, Barria MI, Chen BK. 2014. Unique features of HIV-1 spread through T cell virological synapses. PLoS Pathog 10:e1004513. doi: 10.1371/journal.ppat.1004513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abela IA, Berlinger L, Schanz M, Reynell L, Gunthard HF, Rusert P, Trkola A. 2012. Cell-cell transmission enables HIV-1 to evade inhibition by potent CD4bs directed antibodies. PLoS Pathog 8:e1002634. doi: 10.1371/journal.ppat.1002634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martin N, Welsch S, Jolly C, Briggs JA, Vaux D, Sattentau QJ. 2010. Virological synapse-mediated spread of human immunodeficiency virus type 1 between T cells is sensitive to entry inhibition. J Virol 84:3516–3527. doi: 10.1128/JVI.02651-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Malbec M, Porrot F, Rua R, Horwitz J, Klein F, Halper-Stromberg A, Scheid JF, Eden C, Mouquet H, Nussenzweig MC, Schwartz O. 2013. Broadly neutralizing antibodies that inhibit HIV-1 cell to cell transmission. J Exp Med 210:2813–2821. doi: 10.1084/jem.20131244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sigal A, Kim JT, Balazs AB, Dekel E, Mayo A, Milo R, Baltimore D. 2011. Cell-to-cell spread of HIV permits ongoing replication despite antiretroviral therapy. Nature 477:95–98. doi: 10.1038/nature10347. [DOI] [PubMed] [Google Scholar]
- 45.Titanji BK, Aasa-Chapman M, Pillay D, Jolly C. 2013. Protease inhibitors effectively block cell-to-cell spread of HIV-1 between T cells. Retrovirology 10:161. doi: 10.1186/1742-4690-10-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Agosto LM, Zhong P, Munro J, Mothes W. 2014. Highly active antiretroviral therapies are effective against HIV-1 cell-to-cell transmission. PLoS Pathog 10:e1003982. doi: 10.1371/journal.ppat.1003982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Del Portillo A, Tripodi J, Najfeld V, Wodarz D, Levy DN, Chen BK. 2011. Multiploid inheritance of HIV-1 during cell-to-cell infection. J Virol 85:7169–7176. doi: 10.1128/JVI.00231-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jung A, Maier R, Vartanian JP, Bocharov G, Jung V, Fischer U, Meese E, Wain-Hobson S, Meyerhans A. 2002. Recombination: Multiply infected spleen cells in HIV patients. Nature 418:144. doi: 10.1038/418144a. [DOI] [PubMed] [Google Scholar]
- 49.Rudnicka D, Feldmann J, Porrot F, Wietgrefe S, Guadagnini S, Prevost MC, Estaquier J, Haase AT, Sol-Foulon N, Schwartz O. 2009. Simultaneous cell-to-cell transmission of human immunodeficiency virus to multiple targets through polysynapses. J Virol 83:6234–6246. doi: 10.1128/JVI.00282-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Monel B, Beaumont E, Vendrame D, Schwartz O, Brand D, Mammano F. 2012. HIV cell-to-cell transmission requires the production of infectious virus particles and does not proceed through env-mediated fusion pores. J Virol 86:3924–3933. doi: 10.1128/JVI.06478-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Emerson V, Haller C, Pfeiffer T, Fackler OT, Bosch V. 2010. Role of the C-terminal domain of the HIV-1 glycoprotein in cell-to-cell viral transmission between T lymphocytes. Retrovirology 7:43. doi: 10.1186/1742-4690-7-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dale BM, McNerney GP, Thompson DL, Hubner W, de Los Reyes K, Chuang FY, Huser T, Chen BK. 2011. Cell-to-Cell Transfer of HIV-1 via Virological Synapses Leads to Endosomal Virion Maturation that Activates Viral Membrane Fusion. Cell Host Microbe 10:551–562. doi: 10.1016/j.chom.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roy NH, Lambele M, Chan J, Symeonides M, Thali M. 2014. Ezrin is a component of the HIV-1 virological presynapse and contributes to the inhibition of cell-cell fusion. J Virol 88:7645–7658. doi: 10.1128/JVI.00550-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weng J, Krementsov DN, Khurana S, Roy NH, Thali M. 2009. Formation of syncytia is repressed by tetraspanins in human immunodeficiency virus type 1-producing cells. J Virol 83:7467–7474. doi: 10.1128/JVI.00163-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pang HB, Hevroni L, Kol N, Eckert DM, Tsvitov M, Kay MS, Rousso I. 2013. Virion stiffness regulates immature HIV-1 entry. Retrovirology 10:4. doi: 10.1186/1742-4690-10-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol 59:284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cohen GB, Gandhi RT, Davis DM, Mandelboim O, Chen BK, Strominger JL, Baltimore D. 1999. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity 10:661–671. doi: 10.1016/S1074-7613(00)80065-5. [DOI] [PubMed] [Google Scholar]
- 58.Hubner W, Chen P, Del Portillo A, Liu Y, Gordon RE, Chen BK. 2007. Sequence of human immunodeficiency virus type 1 (HIV-1) Gag localization and oligomerization monitored with live confocal imaging of a replication-competent, fluorescently tagged HIV-1. J Virol 81:12596–12607. doi: 10.1128/JVI.01088-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen P, Hubner W, Spinelli MA, Chen BK. 2007. Predominant mode of human immunodeficiency virus transfer between T cells is mediated by sustained Env-dependent neutralization-resistant virological synapses. J Virol 81:12582–12595. doi: 10.1128/JVI.00381-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pear WS, Nolan GP, Scott ML, Baltimore D. 1993. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci U S A 90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tobiume M, Lineberger JE, Lundquist CA, Miller MD, Aiken C. 2003. Nef does not affect the efficiency of human immunodeficiency virus type 1 fusion with target cells. J Virol 77:10645–10650. doi: 10.1128/JVI.77.19.10645-10650.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cavrois M, De Noronha C, Greene WC. 2002. A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary T lymphocytes. Nat Biotechnol 20:1151–1154. doi: 10.1038/nbt745. [DOI] [PubMed] [Google Scholar]
- 63.Cavrois M, Neidleman J, Bigos M, Greene WC. 2004. Fluorescence resonance energy transfer-based HIV-1 virion fusion assay. Methods Mol Biol 263:333–344. [DOI] [PubMed] [Google Scholar]
- 64.Beaumont E, Vendrame D, Verrier B, Roch E, Biron F, Barin F, Mammano F, Brand D. 2009. Matrix and envelope coevolution revealed in a patient monitored since primary infection with human immunodeficiency virus type 1. J Virol 83:9875–9889. doi: 10.1128/JVI.01213-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Checkley MA, Luttge BG, Mercredi PY, Kyere SK, Donlan J, Murakami T, Summers MF, Cocklin S, Freed EO. 2013. Reevaluation of the requirement for TIP47 in human immunodeficiency virus type 1 envelope glycoprotein incorporation. J Virol 87:3561–3570. doi: 10.1128/JVI.03299-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brandenberg OF, Rusert P, Magnus C, Weber J, Boni J, Gunthard HF, Regoes RR, Trkola A. 2014. Partial rescue of V1V2 mutant infectivity by HIV-1 cell-cell transmission supports the domain's exceptional capacity for sequence variation. Retrovirology 11:75. doi: 10.1186/s12977-014-0075-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sol-Foulon N, Sourisseau M, Porrot F, Thoulouze MI, Trouillet C, Nobile C, Blanchet F, di Bartolo V, Noraz N, Taylor N, Alcover A, Hivroz C, Schwartz O. 2007. ZAP-70 kinase regulates HIV cell-to-cell spread and virological synapse formation. EMBO J 26:516–526. doi: 10.1038/sj.emboj.7601509. [DOI] [PMC free article] [PubMed] [Google Scholar]