

ABSTRACT
Natural-host sooty mangabeys (SM) infected with simian immunodeficiency virus (SIV) exhibit high viral loads but do not develop disease, whereas infection of rhesus macaques (RM) causes CD4+ T cell loss and AIDS. Several mechanisms have been proposed to explain these divergent outcomes, including differences in cell targeting, which have been linked to low expression of the canonical SIV entry receptor CCR5 on CD4+ T cells of SM and other natural hosts. We previously showed that infection and high-level viremia occur even in a subset of SM that genetically lack functional CCR5, which indicates that alternative entry coreceptors are used by SIV in vivo in these animals. We also showed that SM CXCR6 is a robust coreceptor for SIVsmm in vitro. Here we identify CXCR6 as a principal entry pathway for SIV in SM primary lymphocytes. We show that ex vivo SIV infection of lymphocytes from CCR5 wild-type SM is mediated by both CXCR6 and CCR5. In contrast, infection of RM lymphocytes is fully dependent on CCR5. These data raise the possibility that CXCR6-directed tropism in CCR5-low natural hosts may alter CD4+ T cell subset targeting compared with that in nonnatural hosts, enabling SIV to maintain high-level replication without leading to widespread CD4+ T cell loss.
IMPORTANCE Natural hosts of SIV, such as sooty mangabeys, sustain high viral loads but do not develop disease, while nonnatural hosts, like rhesus macaques, develop AIDS. Understanding this difference may help elucidate mechanisms of pathogenesis. Natural hosts have very low levels of the SIV entry coreceptor CCR5, suggesting that restricted entry may limit infection of certain target cells, although it is unclear how the virus replicates so robustly. Here we show that in sooty mangabey lymphocytes, infection is mediated by the alternative entry coreceptor CXCR6, as well as CCR5. In rhesus macaque lymphocytes, however, infection occurs entirely through CCR5. The use of CXCR6 for entry, combined with very low CCR5 levels, may redirect the virus to different cell targets in natural hosts. It is possible that differential targeting may favor infection of nonessential cells and limit infection of critical cells in natural hosts, thus contributing to benign outcome of infection.
INTRODUCTION
Simian immunodeficiency virus (SIV) infections of primate natural hosts, including SIVsmm infection of sooty mangabeys (SM), generally result in high viral loads without CD4+ T cell loss or progression to AIDS. In contrast, SIV infection of primate nonnatural hosts, such as SIVmac infections of rhesus macaques (RM), typically result in high viral loads, widespread loss of CD4+ T cells, and progression to AIDS. Macaques infected with SIVmac serve as the principal animal model for pathogenic infection of humans with human immunodeficiency virus type 1 (HIV-1), and understanding the mechanisms that underlie the dichotomy between natural- and nonnatural-host SIV infection may lead to a better understanding of mechanisms of disease progression and AIDS pathogenesis. Because SIVmac viruses arose from cross-species transfers of SIVsmm from SM to RM, species-matched virus-host interactions in SM and RM serve as an ideal platform for comparative analysis (1).
While both natural and nonnatural hosts exhibit high-level viremia and rapid turnover of infected cells (2), several features of natural-host SIV infection have been associated with the lack of disease progression in these primates. Both natural and nonnatural hosts experience extensive immune activation during acute infection, which resolves in natural hosts but persists during chronic infection in HIV-1-infected humans and SIVmac-infected RM and correlates with pathogenesis (3, 4). Chronic immune activation is driven, at least in part, by early damage to gut mucosal barrier and resulting microbial translocation (5). Mucosal CD4+ T cell loss is seen early in both models, but infected natural hosts exhibit preserved mucosal immunity during chronic infection and low levels of microbial translocation. TH17 cells, a key CD4+ T cell subset supporting gut barrier integrity, are depleted in pathogenic infection but preserved in natural-host infection (6, 7). Another feature distinguishing natural and nonnatural hosts is a low level of SIV infection in SM CD4+ T central memory (TCM) cells relative to that in RM CD4+ TCM cells (8), which may have an impact both directly on CD4+ T cell homeostasis and indirectly on immune activation. Similarly, CD4+ T stem cell memory (TSCM) cells are significantly protected from infection and depletion in natural hosts compared with those in nonnatural hosts (9). Thus, the factors that determine CD4+ T cell subset targeting by SIV in natural versus nonnatural hosts constitute a key issue underlying these distinct features.
HIV and SIV targeting is determined largely at entry through selective use of chemokine coreceptors, in conjunction with CD4. Established dogma holds that CCR5 is responsible for SIV entry and replication. SM and other natural hosts express exceedingly low levels of CCR5 on blood and lymph node CD4+ T cells relative to those in humans and nonnatural hosts of SIV (10), suggesting that restricted CCR5 expression may contribute to protection of important cells. Nevertheless, the levels of viremia and infected-cell turnover in natural hosts are at least equal to those in nonnatural hosts (2), raising the paradox of robust SIV replication in the face of exquisitely low CCR5 levels.
We recently identified a deletion allele of CCR5 in SM that results in abrogation of protein expression and is common among wild and captive SM (11). Approximately 8% of captive SM are homozygous for CCR5 deletion alleles and thus genetically deficient in CCR5. However, CCR5-null SM remain susceptible to SIVsmm infection and exhibit plasma viral loads that are only slightly lower than those in CCR5 wild-type animals (11), implicating non-CCR5 alternative coreceptors in infection of these animals in vivo. We subsequently showed that species-matched sooty mangabey CXCR6 (smCXCR6) supports efficient entry into transfected cells by pseudotype virus carrying SIVsmm Envs from multiple subtypes and from both CCR5 wild-type and null animals (12).
In this study, we sought to determine whether SIVsmm infection of primary SM CD4+ T cells was mediated by CXCR6, CCR5, or both. Furthermore, because SIVmac originated from cross-species transmission of SIVsmm, we also asked whether SIVmac infection of RM CD4+ T cells proceeds through similar or different pathways. Our results demonstrate that CXCR6 and CCR5 both mediate SIVsmm infection of SM primary lymphocytes ex vivo. In contrast, SIVmac infection of RM lymphocytes is entirely through CCR5. CXCR6-mediated infection in SM, when combined with restricted CCR5 expression exhibited by natural hosts, may lead to distinct CD4+ T cell targeting that contributes to the benign outcome from infection.
MATERIALS AND METHODS
MVC and CXCL16.
The CCR5 antagonist maraviroc (MVC) was obtained through the NIH AIDS Reagent Program and suspended at 20 mg/ml in dimethyl sulfoxide (DMSO). Recombinant human CXCL16 (R&D Systems) was suspended at 25 μg/ml in phosphate-buffered saline. Cells were treated 1 h preinfection with maraviroc, CXCL16, or maraviroc plus CXCL16. Maraviroc and CXCL16 were also maintained throughout infection. Final concentrations were 10 μM maraviroc and/or 500 ng/ml of CXCL16 in less than 0.03% DMSO as the vehicle. No-drug controls were treated with vehicle alone.
Viruses and viral Envs.
SIVsmm FFv2.1 and FFv5.1 Envs were cloned from a clade 1 SIVsmm-infected sooty mangabey homozygous for the CCR5 wild-type gene (CCR5wt/wt) using single-genome amplification (SGA) as previously described (11–13). SIVsmm FPm1.1 Env was SGA cloned from a CCR5wt/wt clade 5 SIVsmm-infected sooty mangabey (12). SIVmac 239 Env was subcloned from the full-length SIVmac 239 infectious molecular clone (IMC). SIVmac 251-RZu4 1.1 Env was SGA cloned from a rhesus macaque 11 days after infection with SIVmac 251 as previously described (14). SIV E660 is a strain that originated from SIVsmm that underwent serial passage in rhesus macaques (15). The SIVsmm E660 CP3C-P-A8 and SIVsmm E660 CR54-PK-2A5 Envs were cloned by SGA from acutely SIVE660-infected rhesus macaques and were a gift from K. Barr and G. Shaw (University of Pennsylvania) (15, 16). Env pseudotypes were generated using the HIV-1 pNL-Luc-E−R+ backbone as described previously (11, 12, 17). Briefly, 293T cells were cotransfected with pNL-Luc-E−R+ and SIV Env plasmid using Fugene (Promega). The supernatant was collected 3 days later, and cell debris was removed by low-speed centrifugation. Pseudotype stocks were quantified by HIV p24 Gag enzyme-linked immunosorbent assay (ELISA; Perkin-Elmer) and treated with DNase (Roche) prior to use.
Replication-competent SIVmac 239 was generated from 293T cells transfected with the SIVmac 239 IMC, provided by J. Hoxie (University of Pennsylvania). An IMC containing the SIVmac 251-RZu4 env was generated by replacing SIVmac 239 env with the SIVmac 251-RZu4 env (14, 18). Proviral clones were transfected into 293T cells using Fugene, and the viral supernatant was harvested 3 to 4 days later and quantified by SIV p27 Gag ELISA (Perkin-Elmer). The SIVsmm E660 primary isolate was obtained through the NIH AIDS Reagent Program from V. Hirsch and P. Johnson (15). Primary isolates SIVsmm M935 and SIVsmm D215, isolated from infected CCR5wt/wt sooty mangabeys, were a gift from P. Marx (Tulane National Primate Research Center [TNPRC]).
Cell lines and PBMCs.
Human 293T cells were maintained in Dulbecco modified Eagle medium (DMEM)–10% fetal bovine serum (FBS)–1% penicillin-streptomycin–1% l-glutamine. Peripheral blood mononuclear cells (PBMCs) from uninfected rhesus macaque and sooty mangabeys were collected and Ficoll purified at the Yerkes National Primate Research Center (YNPRC) with assistance from the laboratory of F. Villinger. Sooty mangabey PBMCs were from animals with the homozygous CCR5wt/wt genotype (11): FBa1, FUu, FHz, FYz, FLa1, and FIz. Cryopreserved PBMCs were thawed and suspended in RPMI medium–10% FBS–1% penicillin-streptomycin–1% l-glutamine overnight prior to stimulation.
Coreceptors and pseudotype infections.
Rhesus macaque CCR5 (rmCCR5), rmCXCR6, and rmGPR15 were cloned from rhesus macaque PBMC genomic DNA using primers previously described for cloning of sooty mangabey coreceptors (12). The sequences of these RM coreceptor clones matched those published in GenBank. Two alleles of rmGPR15 encoding proteins with a single amino acid difference were described previously (19, 20), and all in vitro experiments were performed with the rmGPR15 M279 allele. Sooty mangabey smCCR5, smCXCR6, and smGPR15 were cloned as described previously (12).
Infections of transfected 293T cells were performed as described previously (12). Briefly, cells were transfected with CD4 and coreceptor plasmid DNA using Fugene, employing 1 μg of each plasmid unless otherwise specified. Cells were washed 24 h later and replated at 2 × 104 cells per well in 96-well plates. The next day, DNase-treated Env pseudotype viruses were added and cells were spin inoculated (2 h at 1,200 × g). Luciferase output was measured 72 h later in cell lysates based on relative light units (RLU) using a Luminoskan Ascent microplate luminometer (Thermo Scientific). Vesicular stomatitis virus G protein (VSV-G)-pseudotyped viruses served as positive controls. Env-negative pseudotypes and CD4-only no-coreceptor target cells served as negative controls.
For maraviroc titration studies, 293T cells were transfected with species-matched CD4 and CCR5, incubated for 1 h with the desired concentrations of drug, infected with pseudotype virus, and assayed for infection 3 days later based on RLU production in cell lysates. Entry in the presence of MVC was normalized to the level of entry in the absence of drug. Data were fitted to a sigmoidal dose-response curve using Prism 6 software (GraphPad, Inc.).
PBMC infection and replication experiments.
Rhesus macaque and sooty mangabey PBMCs were stimulated for 3 days with 5 μg/ml of concanavalin A (ConA) and 30 U/ml of interleukin-2 (IL-2). One hour prior to infection, cells were resuspended in medium containing IL-2 only, along with maraviroc, CXCL16, maraviroc plus CXCL16, or a vehicle alone. Cells were then plated at 1 × 106 cells/well in a 2-ml total volume in 24-well plates and infected with infectious virus using 5 ng of p27 SIVmac 239, 5 ng of p27 SIVmac RZu4, 5 ng of p27 SIVsmm E660, 100 μl of SIVsmm M935, or 100 μl of SIVsmm D215. Cells were spin inoculated and washed, and medium was collected every 2 to 3 days for SIV p27 Gag ELISA. Peak p27 production in cells with different treatment conditions was normalized to that in cells treated with the vehicle alone.
Statistics.
Statistical comparisons were made by a two-tailed unpaired t test using Welch's correction unless otherwise indicated. Significance was defined as a P value of <0.05. Statistical analyses were performed using Prism 6 software.
Ethics statement.
All animal experimentation was conducted according to guidelines established by the Animal Welfare Act and the NIH for housing and care of laboratory animals and performed in accordance with institutional regulations after review and approval by the Institutional Animal Care and Use Committees (IACUC) at the Yerkes National Primate Research Center and the Tulane National Primate Research Center. Studies were also reviewed and approved by the University of Pennsylvania IACUC.
RESULTS
Efficient use of smCXCR6 by both SIVsmm and SIVmac in vitro.
We previously reported that SIVsmm Env pseudotypes exhibit robust entry into transfected cells through smCXCR6, comparable to that with smCCR5, and modest entry through smGPR15 (12). Because SIVmac originated from cross-species transmissions of SIVsmm into rhesus macaques, we hypothesized that SIVmac and SIVsmm would exhibit similar patterns of sooty mangabey coreceptor use in vitro. We transfected 293T cells with smCD4 in combination with smCCR5, smCXCR6, and smGPR15 and infected them with luciferase-expressing Env pseudotype viruses (17). Since we previously demonstrated smCXCR6 utilization by a broad range of SIVsmm Envs, including multiple subtypes and isolates from both CCR5 wild-type and null animals (12), in this study, we used SIVsmm FFv2.1 as a representative SIVsmm, along with four Envs reflecting the two divergent lineages of SIV frequently used to induce pathogenesis in the rhesus macaque model of AIDS. The related viruses SIVmac 239 and SIVmac 251, and the two SIVsmm E660 Envs, reflect pathogenic SIV infections in rhesus macaques resulting from two independent SIVsmm cross-species transmissions (1).
As observed previously (12), smCXCR6 supported robust SIVsmm FFv2.1 Env pseudotype infection that was similar to the level of infection mediated by smCCR5 (Fig. 1A). Notably, smCXCR6 was also a robust coreceptor for SIVmac Envs, mediating SIVmac 239, SIVmac 251-RZu4, SIVsmm E660-CP3C, and SIVsmm E660-CR54 infection at levels similar to those with CCR5. smGPR15 supported more moderate and variable entry by SIVsmm FFv2.1 and the four SIVmac Envs. Thus, smCXCR6 is an efficient alternative coreceptor in vitro for both SIVsmm and its derivative SIVmac.
FIG 1.

SIVsmm and SIVmac efficiently use sooty mangabey but not rhesus macaque CXCR6 in vitro. Target 293T cells were transfected with smCD4 plus smCCR5, smCXCR6, or smGPR15 plasmid DNA (A) or rmCD4 plus rmCCR5, rmCXCR6, or rmGPR15 plasmid DNA (B) and then infected with luciferase-expressing pseudotyped virions carrying the indicated Envs. Cells transfected with CD4 alone (“none”) served as negative controls. Infection was measured by luciferase activity 3 days postinfection, and data were normalized to 100% entry in target cells expressing species-matched CD4 plus CCR5 (n = 3, means ± SDs).
Rhesus macaque CXCR6 is a weak coreceptor of SIV in vitro.
We next assessed SIVsmm and SIVmac Env-mediated entry through coreceptors of rhesus macaque origin. Previous studies reported that SIVmac was capable of using human CXCR6 and smCXCR6 in vitro but that rmCXCR6 functioned poorly for this virus (21). In this study, we tested SIVmac 239 Env and the related SIVmac 251-RZu4 Env, the independent-lineage SIVsmm E660 Envs, and SIVsmm Env.
As shown in Fig. 1B, rmCXCR6 was a very poor entry coreceptor for all four SIVmac Envs. SIVmac 239 and SIVmac 251-RZu4 achieved a maximum of 11% of the entry mediated by rmCCR5. SIVsmm E660 Envs, which exhibited robust infection through smCXCR6, did not use rmCXCR6 at all, as infection through rmCXCR6/rmCD4 was indistinguishable from infection of cells transfected with rmCD4 alone. SIVsmm FFv2.1 Env was somewhat better than SIVmac Envs at using rmCXCR6, achieving 43% of the entry mediated by rmCCR5.
The inability of rmCXCR6 to support SIVmac infection was a striking contrast to the results with smCXCR6, which supported SIVmac entry at levels comparable to and in some cases above that mediated by CCR5 (Fig. 1A) (12). Direct comparison confirmed significantly lower levels of infection mediated by rmCD4 plus rmCXCR6 than by smCD4 plus smCXCR6 for each of the viral Env pseudotypes (SIVsmm FFv 2.1, P = 0.02; SIVmac 239, P = 0.005; SIVmac 251-RZu4, P = 0.003; and SIVsmm E660-CP3C and SIVsmm E660-CR54, P < 0.001). Of note, absolute levels of luciferase expression following infection of 293T cells expressing rmCD4 plus rmCCR5 were comparable to levels of luciferase expression following infection of 293T cells expressing smCD4 plus smCCR5 (data not shown). Thus, nonnatural host rhesus macaque CXCR6 is a poor coreceptor relative to natural host sooty mangabey CXCR6 for SIVmac and SIVsmm in vitro.
Similar to smGPR15, rmGPR15 was a moderate but variable coreceptor for SIV (Fig. 1). Entry supported by rmGPR15 ranged from 15% (SIVsmm E660-CR54) to 78% (SIVmac 251-RZu4) of that supported by rmCCR5. Thus, rmGPR15 and smGPR15 are comparable coreceptors of SIV Env pseudotypes in vitro, enabling levels of entry that are moderate but highly variable among SIV Envs. This result is in contrast to findings for CXCR6, which is a consistently robust coreceptor in its SM form but uniquely poor in its RM form.
Maraviroc is a specific inhibitor of nonhuman primate CCR5-mediated entry but is less efficient against rmCCR5 than smCCR5.
To define the relative roles of CCR5 versus other coreceptors in infection of nonhuman primate primary PBMCs ex vivo, we first examined the ability of maraviroc to specifically block smCCR5- and rmCCR5-mediated viral entry. Prior studies showed that compared with human CCR5, maraviroc bound rmCCR5 and antagonized chemokine signaling relatively poorly (22), and other CCR5 antagonists, such as SCH-C, blocked rmCCR5-mediated infection less efficiently (23). However, the relative efficiency of maraviroc in blocking rmCCR5-mediated SIV entry has not been reported directly. We therefore examined the ability of maraviroc to block infection mediated by rmCCR5 and smCCR5, as well as its potential to impact entry through relevant alternative coreceptors.
We first carried out a maraviroc dose titration using 293T cells transfected with smCCR5 and smCD4 or rmCCR5 and rmCD4 (Fig. 2A and B). Cells were incubated with various concentrations of maraviroc and then infected with SIVsmm FFv2.1, two SIVsmm E660, SIVmac 239, and SIVmac 251-RZu4 Env pseudotypes. Maraviroc was an efficient inhibitor of smCCR5-mediated entry for all viruses, with a mean 50% effective concentration (EC50) of 16 nM and essentially complete blocking at 1 μM (Fig. 2A). These results were similar to blocking of HIV-1 JRFL through human CCR5 and CD4 (data not shown). In contrast, blocking of entry through rmCCR5 was much less efficient, with a mean EC50 of 190 nM and only 80 to 85% blocking at 10 μM (Fig. 2B). Higher concentrations of maraviroc further blocked infection but induced some degree of nonspecific inhibition based on pseudotypes carrying the VSV-G Env (data not shown). Studies were also carried out with aplaviroc, vicriviroc, CMPD-167, and TAK779, which all showed similarly incomplete inhibition of SIV entry through rmCCR5 (data not shown). Therefore, 10 μM maraviroc was used to antagonize CCR5-mediated infection in primary PBMCs, recognizing that due to incomplete blocking the degree of inhibition in rmPBMCs could potentially underestimate the proportion of entry mediated by CCR5.
FIG 2.

Maraviroc is a specific inhibitor of CCR5-mediated SIV entry and is more efficient in blocking sooty mangabey than rhesus macaque CCR5 in vitro. (A and B) 293T cells were transfected with plasmids encoding smCD4 plus smCCR5 (A) or rmCD4 plus rmCCR5 (B). Cells were treated for 1 h with maraviroc (MVC) at the indicated concentrations from 0.1 nM to 10 μM, or with a vehicle alone, and then infected with luciferase-expressing SIV Env pseudotypes. Infection was measured by luciferase activity 3 days postinfection and normalized to 100% entry in the presence of vehicle alone (n = 3, means ± SDs). Data were fit to a sigmoidal dose-response curve. (C and D) 293T cells were transfected with CD4 plus CCR5, CXCR6, or GPR15 plasmid DNA cloned from a sooty mangabey (C) or rhesus macaque (D). Cells were treated or not for 1 h with maraviroc (10 μM) and then infected with SIV Env pseudotypes. Infection was measured by luciferase activity, in relative light units (RLU), 3 days postinfection (n = 3, means ± SDs).
We next confirmed that maraviroc was specific for SM and RM CCR5 and would not block other relevant coreceptors. 293T cells were transfected with CD4 and CCR5, CXCR6, or GPR15 from sooty mangabeys and rhesus macaques, and Env pseudotype infection was measured in the presence or absence of 10 μM maraviroc (Fig. 2B and C). Maraviroc almost completely blocked smCCR5-mediated infection (98% ± 1%) but had no effect on smCXCR6- or smGPR15-mediated SIV Env infection (Fig. 2C). Maraviroc also inhibited rmCCR5-mediated pseudotype infection but had no effect on rmGPR15-mediated infection or on the limited amount of entry mediated by rmCXCR6 (Fig. 2C). In contrast to findings for smCCR5 and as expected from dose titration studies (Fig. 2B), infection of rmCCR5-transfected cells was not completely blocked by maraviroc (73% ± 2% inhibition). Therefore, maraviroc is specific for CCR5 among relevant SM and RM coreceptors, although it is less effective against rmCCR5 than smCCR5.
SIV infection of rmPBMCs is CCR5 dependent, whereas SIV infection of smPBMCs is partially CCR5 independent.
We then used maraviroc to define the role of CCR5 in SIV infection of primary sooty mangabey PBMCs (smPBMCs) and rhesus macaque PBMCs (rmPBMCs), using mPBMCs obtained from animals with the CCR5 wild-type genotype (11). For these studies, we utilized two replication-competent SIVsmm primary viral isolates (SIVsmm D215 and SIVsmm M935), two SIVmac infectious molecular clones (SIVmac 239 and SIVmac 251-RZu4), and the SIVsmm E660 viral swarm. Primary smPBMCs and rmPBMCs were stimulated with ConA and IL-2, pretreated with or without 10 μM maraviroc, and infected with virus stocks. SIV replication was measured by p27 Gag antigen in supernatant. Peak replication occurred 8 to 11 days postinfection, and the percentage of infection in maraviroc-treated cells relative to that in vehicle-treated controls was determined at the peak of infection for each virus-cell combination (Fig. 3).
FIG 3.

SIV infection of rhesus macaque PBMCs is highly CCR5 dependent, whereas sooty mangabey PBMC infection is partially CCR5 independent. Primary PBMCs from sooty mangabeys (A) and rhesus macaques (B) were stimulated with ConA and IL-2 for 3 days, treated or not for 1 h with maraviroc (10 μM), and then infected with replication-competent SIVsmm D215, SIVsmm M935, SIVsmm E660, SIVmac 239, or SIVmac 251-RZu4. Culture supernatants were collected periodically postinfection. Data represent peak supernatant p27 Gag production and are shown as the percentage of entry in maraviroc-treated PBMCs relative to that in vehicle-treated PBMCs. Each symbol represents cells from a unique sooty mangabey or rhesus macaque. SIVsmm M935 failed to replicate in rmPBMCs and is not shown in panel B.
All 5 SIV strains replicated robustly in smPBMCs, and maraviroc showed variable and incomplete inhibition of replication (Fig. 3A). The two SIVsmm isolates, D215 and M935, were reduced by approximately half (means, 53% ± 20% and 52% ± 17%, respectively) by treatment with the CCR5 antagonist maraviroc, although the extents of inhibition differed among PBMCs from different animals. Strikingly, SIVmac infection in smPBMCs was almost completely resistant to maraviroc. While the extents of blocking again differed among PBMCs from different SM, the mean peak p27 levels were reduced by averages of only 19% ± 19% for SIVmac 239, 15% ± 18% for SIVsmm E660, and 2% ± 4% for SIVmac 251-RZu4. Because maraviroc is a potent inhibitor of smCCR5-mediated SIV Env pseudotype entry into transfected 293T target cells (which overexpress CCR5 compared with primary cells [data not shown]) (Fig. 2A), these data indicate that alternative, non-CCR5 coreceptors support SIV infection in smPBMCs. We previously showed that SIVsmm replicates efficiently in CCR5-null smPBMC from animals homozygous for CCR5 deletion alleles (11). Our results here, obtained using pharmacological CCR5 blocking, are consistent with those findings and extend those data to show CCR5 independence for SIVmac in smPBMCs as well.
Even though maraviroc is a relatively poor inhibitor of rmCCR5-mediated entry and could therefore underestimate the extent to which infection is CCR5 dependent, we tested its impact on infection of rmPBMCs by the panel of SIVs (Fig. 3B). In the absence of drug, the three SIVmac isolates and SIVsmm D215 replicated efficiently in rmPBMCs, while SIVsmm M935 did not. In marked contrast to the case with smPBMCs, SIVmac and SIVsmm infections of rmPBMCs were almost completely blocked by maraviroc. Maraviroc reduced peak p27 production in rmPBMCs by averages of 97% ± 3% for the three SIVmac strains and 96% for SIVsmm D215. Given that maraviroc is an incomplete inhibitor of rmCCR5-mediated SIV pseudotype infection in 293T cells in vitro, the marked attenuation of both SIVmac and SIVsmm infections in this study indicates that entry into primary rmPBMCs is highly dependent on rmCCR5 and does not proceed through alternative entry pathways. Furthermore, the fact that both SIVmac and SIVsmm are largely CCR5 independent in smPBMCs yet are fully CCR5 dependent in rmPBMCs indicates that the restricted use of alternative coreceptors in rmPBMCs is determined mainly by the host target cell rather than by intrinsic differences in SIV Envs.
The chemokine CXCL16 is a specific but modest inhibitor of smCXCR6 coreceptor function.
The residual SIVsmm and SIVmac replication in smPBMCs treated with maraviroc confirm non-CCR5 coreceptor-mediated infection in natural host smPBMCs, in contrast to rmPBMCs. Given the broad, efficient use of smCXCR6 by SIVsmm and SIVmac strains, we asked if CXCR6 was involved in SIVmac and SIVsmm entry into smPBMCs. Conversely, rmCXCR6 is a poor coreceptor of SIVmac and SIVsmm, which is consistent with exclusive CCR5-mediated entry in rmPBMC.
Unlike for CCR5, there are no small-molecule antagonists of CXCR6-mediated SIV entry. Therefore, we tested whether entry would be inhibited by CXCL16, the chemokine ligand for CXCR6 (24). However, 293T cells transfected under standard conditions overexpress coreceptors compared with primary target cells (data not shown), and we anticipated that CXCL16 might not be a robust inhibitor if CXCR6 was overexpressed. Therefore, we first tested whether CXCL16 would inhibit infection of cells transfected with smaller amounts of coreceptor plasmid. 293T cells were transfected with CD4 in conjunction with 0.01 μg of smCXCR6, smCCR5, or smGPR15 plasmid, pretreated or not with 500 ng/ml of recombinant human CXCL16, and infected with several SIVsmm and SIVmac Env pseudotypes (Fig. 4). CXCL16 blocked 83% ± 10% of smCXCR6-mediated infection in this experiment. In contrast, CXCL16 showed no reduction of entry through smCCR5 or smGPR15. Although CXCL16 blocked the majority of smCXCR6-mediated infection under these conditions, blocking was incomplete. The effect of CXCL16 on smCXCR6-mediated SIV pseudotype infection was dependent on the amount of smCXCR6 expressed on target cells, as only a 40% reduction in pseudotype infection was seen in cells transfected with 1 μg of smCXCR6 plasmid (data not shown). Thus, CXCL16 specifically blocks smCXCR6-mediated entry, although we expect that the extent of inhibition by CXCL16 may underestimate the extent of entry mediated by CXCR6.
FIG 4.
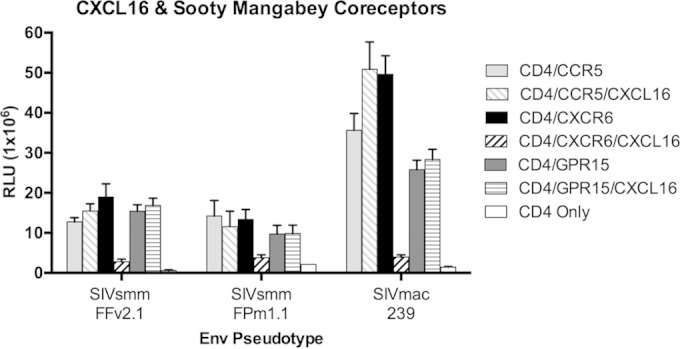
CXCL16 specifically inhibits smCXCR6-mediated infection in vitro. 293T cells were transfected with sooty mangabey smCD4 (1 μg plasmid DNA) in conjunction with smCCR5, smCXCR6, smGPR15, or pcDNA3.1 vector control (0.01 μg of plasmid DNA). Cells were treated or not for 1 h with 500 ng/ml of recombinant human CXCL16 and infected with luciferase-expressing SIV Env pseudotypes. Infection was measured by luciferase activity, in RLU, 3 days postinfection (n = 3, means ± SDs).
Sooty mangabey CXCR6 mediates SIV infection in primary smPBMCs.
To determine whether CXCR6 plays a role in SIV entry into SM primary cells, we stimulated smPBMCs with ConA and IL-2 and infected them with SIVsmm and SIVmac in the presence or absence of maraviroc, CXCL16, or both maraviroc and CXCL16. Supernatant p27 Gag was monitored, and peak viral production in treated cells was normalized to that in cells of the same animal in the absence of inhibitors (Fig. 5).
FIG 5.

CXCR6 mediates CCR5-independent SIV infection of sooty mangabey PBMCs. Primary smPBMCs were stimulated with ConA and IL-2 for 3 days and then treated for 1 h with maraviroc (10 μM), CXCL16 (500 ng/ml), maraviroc and CXCL16 combined, or a vehicle alone. Cells were then infected with replication-competent SIVsmm D215 (A), SIVsmm M935 (B), SIVsmm E660 (C), and SIVmac 239 (D). Culture supernatants were collected periodically postinfection. Data represent peak supernatant p27 Gag production and are shown as the percentage of entry in treated PBMCs relative to that in vehicle-treated PBMCs (means ± SDs). Each symbol represents cells from a unique sooty mangabey. *, P < 0.05; **, P < 0.005 (two-tailed paired t test). Peak p27 antigen levels for viruses in the absence of blocking (means with ranges in parentheses, in nanograms per milliliter) were as follows: SIVsmm D215, 36.8 (21.6 to 49.9); SIVsmm M935, 12.7 (5.7 to 15.3); SIVsmm E660, 48.9 (15.2 to 76.6); and SIVmac 239, 17.4 (8.4 to 33.8).
As shown in Fig. 5, the levels of inhibition with each agent alone and in combination varied among individual animals. The overall pattern indicated that both CXCR6 and CCR5 mediate SIV infection of primary smPBMCs. CXCL16 alone and maraviroc alone significantly reduced SIVsmm D215 infection relative to that of vehicle-treated control cells (53% ± 20% for MVC and 57% ± 21% for CXCL16), indicating use of both entry pathways (Fig. 5A). The combined blocking further reduced infection (78% ± 15% inhibition) relative to that with maraviroc alone, further indicating that CXCR6 supports SIVsmm D215 infection of smPBMC. For SIVsmm M935, only maraviroc blocking reached statistical significance, although a nonsignificant reduction of infection was seen with CXCL16 alone and combined blocking. Both SIVsmm E660 and SIVmac 239 showed nonsignificant reductions in infection with CXCL16 alone and maraviroc alone. However, CXCL16 and maraviroc together significantly blocked infection in comparison to infection of both untreated cells and maraviroc-treated PBMCs (71% ± 20% and 55% ± 19% reduction relative to infection of untreated cells for SIVsmm E660 and SIVmac 239, respectively), indicating CXCR6 utilization in smPBMCs.
Taken together, these data indicate that both CXCR6 and CCR5 mediate infection of primary smPBMCs by SIVsmm D215, SIVsmm E660, and SIVmac 239, although less clear conclusions can be drawn for SIVsmm M935. Notably, in no case was infection completely blocked by the use of both inhibitors together, indicating either incomplete CXCR6 blockade or the use of other coreceptors in addition to CXCR6 and CCR5. Nevertheless, our data demonstrate dualtropic CXCR6 and CCR5 SIV entry in primary smPBMCs, which stands in marked contrast CCR5-dependent entry in primary rmPBMCs.
DISCUSSION
In this study, we showed that SIVsmm/SIVmac infection of primary sooty mangabey PBMCs is mediated by both CXCR6 and CCR5, whereas infection of rhesus macaque PBMCs is mediated exclusively through CCR5. This reveals a striking dichotomy in coreceptor tropism between the two principal hosts for the SIVsmm and SIVmac virus family, which corresponds with natural hosts that exhibit a benign course of infection versus nonnatural hosts that develop AIDS.
SM and other African monkey species that are natural hosts for endemic SIVs exhibit extremely low levels of CCR5 on CD4+ T lymphocytes in blood and lymphoid tissues (10). However, plasma viral loads in natural hosts are similar to those in SIV-infected macaques and HIV-infected humans, and viral replication is sustained by short-lived cells in both pathogenic and nonpathogenic infections (2). Thus, the mechanism by which such high-level replication occurs in the face of exquisitely low CCR5+ CD4+ cell targets in the natural host SM, and why rapid cell turnover of infected cells does not result in pathogenesis, has remained an enigma. Involvement of non-CCR5 alternative entry pathways was implicated by our previous observation that some SM genetically lacked CCR5 yet were susceptible to infection and exhibited high plasma viremia (11). Our demonstration here that CXCR6 mediates SIV infection of primary SM lymphocytes provides an explanation, at least in part, for robust SIVsmm replication in the complete absence of CCR5 in CCR5-null SM and in the presence of extremely low CCR5 levels in the broader SM population.
It is increasingly clear that subset-specific targeting within the CD4+ T cell population plays a critical role in outcome from infection. SIV-infected RM have high levels of viral DNA in both CD4+ central memory T (TCM) cells and effector memory (TEM) cells, while infected SM have high viral DNA levels in TEM cells but much lower levels in TCM cells (8). Relative sparing of TCM cells in natural hosts may favor the preservation of CD4+ T cell homeostasis (8, 25). Other subsets that are differentially affected in natural and nonnatural hosts that may affect T cell homeostasis, immune competence, or other functions include CD4+ T stem cell memory (TSCM) cells and T follicular helper (TFH) cells, as well as distinct localization patterns within lymphoid tissues (9, 26). Furthermore, sustained immune activation in chronically infected RM as well as in HIV-infected humans, but not natural hosts, has been linked to differential infection and depletion of CD4+ TH17 cells, which play a critical role in mucosal barrier integrity (6, 7, 27). Thus, understanding the targeting of specific cell subsets among CD4+ T cells in natural hosts is critical in unraveling AIDS pathogenesis. Relative sparing of SM TCM cells in SM compared with RM was associated with particularly low CCR5 levels in SM TCM cells, but even SM TEM cells had little CCR5 compared with RM TEM cells despite high viral DNA levels (8). Our data suggest that alternative coreceptors, such as CXCR6, may direct SIV in infected SM to cells that have less impact on T cell homeostasis or other facets of pathogenesis.
An important next step will be to define the expression patterns of CXCR6 in SM CD4+ T cells. In humans, CXCR6 is found on CD4+ Teff, TH1, and TH17 cells and intestinal lamina propria lymphocytes (28–31). However, commercially available antibodies to human CXCR6 do not cross-react with smCXCR6 or rmCXCR6 (data not shown), so delineating target cell populations in SM natural hosts that enable high-level replication without inducing disease will require new reagents.
The ability of SIV Envs to use alternative coreceptors in vitro has been recognized for nearly 2 decades, but largely in artificial systems employing human coreceptors, and standard dogma has been that SIV infection in vivo is mediated by CCR5. One long-recognized exception is the red-capped mangabey (RCM), which carries a common deletion allele of CCR5 (32). As a result, most RCMs are CCR5 null, and SIVrcm was found to use human CCR2b but not CCR5 for entry (33). Our prior observation that 8% of SM are CCR5 null yet susceptible to SIVsmm infection identified a second natural host species demonstrating in vivo infection through non-CCR5 pathways (11), which we now show is mediated by smCXCR6. While observations in CCR5-null animals provided the first clue that alternative coreceptors support robust SIV infection in vivo, our data here demonstrate that non-CCR5 pathways mediate PBMC infection even in CCR5 wild-type animals. Since highly restricted CCR5 expression is common to all natural host species examined (10), we hypothesize that non-CCR5 entry pathways may play a role in SIV infection in primate natural hosts in vivo more generally. Thus, it will be important to determine whether efficient primary cell entry by species-matched CXCR6 (or other non-CCR5 coreceptors) is a feature shared broadly by other SIV–natural-host combinations.
Although we have confirmed the use of CXCR6 in SIVsmm infection of smPBMC, we cannot be certain that it is the only CCR5-independent coreceptor used. Blocking of SIVsmm infection of smPBMCs by combined CXCL16 and maraviroc was incomplete (Fig. 5). This likely reflects the fact that CXCL16 is only a partial inhibitor of CXCR6-mediated entry, even at low levels of coreceptor expression (Fig. 4). However, we cannot exclude the possibility that additional non-CCR5 pathways, such as GPR15, contribute to smPBMC infection as well. Unfortunately, neither small-molecule antagonists nor a chemokine ligand is available for GPR15. However, in contrast to the case with smPBMCs, the near-complete inhibition of rmPBMC infection by maraviroc clearly demonstrates that GPR15 is not a major pathway for SIVmac in rmPBMCs, despite the moderate albeit variable use of rmGPR15 by Env pseudotypes in transfected cells. This result is consistent with in vivo studies showing that abrogation of GPR15 use by mutating the SIVmac 239 Env had no effect on replication and disease in rhesus macaques (34). In addition, SIVmac 239 does not infect CD4+ GPR15+ human PBMCs from CCR5-null human donors (35). On the other hand, use of human GPR15 is suggested by the ability of SIVmac 239 and SIVmac 251 to replicate in human CEMx174 cells that express high levels of GPR15 but not CCR5 (35, 36). It is not clear why GPR15 might enable infection in a cell line but not in primary rhesus macaque cells.
In contrast to smCXCR6, rmCXCR6 was a poor coreceptor for both of the independent SIVmac lineages studied, which is consistent with in vitro data published previously (21) and in vivo studies showing that short-term administration of CCR5 antagonists to SIVmac-infected RM leads to a rapid drop in plasma viremia of up to 2 logs (37, 38). The inability of SIVmac to use rmCXCR6 results from a host coreceptor-dependent restriction, as SIVmac Envs were fully capable of using smCXCR6. The defect in rmCXCR6 entry coreceptor function has been mapped to amino acid 31 in the N-terminal domain of the molecule, where smCXCR6 contains a serine and rmCXCR6 carries an arginine (21). The poorly functional R31 sequence is also shared by CXCR6 of cynomolgus and pigtail macaques, which develop AIDS following SIV infection, whereas the functional S31 sequence is also present in African green monkeys, which are natural hosts for SIVagm and do not develop disease. Human CXCR6 has the functional serine amino acid at this position and is efficiently used by SIV. However, HIV-1 does not typically use CXCR6, a restriction that is determined by the viral Env glycoprotein.
We speculate that in their natural hosts, coevolution has led to two processes that together maintain virus-host equilibrium. In response to SIV infection, natural hosts over time may have downregulated CCR5 to preserve critical cells from infection and avoid pathogenesis (10); concomitantly, viral evolution under pressure to maintain efficient replication may have led SIV to acquire expanded coreceptor use. Whether by additional host adaptation also restricting alternative coreceptor expression on critical cells or by happenstance that targeted a coreceptor distributed in a benign manner, these two processes together may have led to coexistence enabling high-level infection of nonessential cell types. The fact that efficient alternative coreceptor use is a feature shared broadly by essentially all SIV strains examined suggests that this process may have been a common mechanism for virus-host coadaptation in natural host relationships. In contrast, host evolution would not occur in the time scale or current context of nonnatural host infection, and abundant CCR5 in nonnatural hosts would offer no selective pressure for virus to utilize species-matched alternative coreceptors.
In summary, we have identified CXCR6 as a principal coreceptor for SIVsmm infection of primary SM lymphocytes. In contrast, SIVmac infection of RM lymphocytes is restricted to CCR5. CXCR6/CCR5 dual tropism of SIVsmm, combined with low levels of CCR5 expression, may enable differential cell targeting in nonpathogenic natural-host infection compared with pathogenic infection of humans with HIV-1 and RM with SIVmac.
ACKNOWLEDGMENTS
We thank K. Bar, G. Shaw, J. Hoxie, and P. Marx for SIV clones and isolates and F. Villinger for assistance with sooty mangabey primary cells.
This work was supported by NIH grants R56-AI091516 and R01-MH061139 (R.G.C.), R01-AI058706 (C.A.D.), and R37-AI066998 (G.S.). S.T.C.E. was supported by NIH grant T32-AI007632. Extensive assistance was provided by the Viral/Molecular, Immunology, and Nonhuman Primate Cores of the Penn Center for AIDS Research (P30-AI045008). Work was also enabled by NIH/Office of Research Infrastructure Programs support for the Yerkes National Primate Research Center and its Comparative AIDS Core (OD P51OD011132).
REFERENCES
- 1.Hatziioannou T, Evans DT. 2012. Animal models for HIV/AIDS research. Nat Rev Microbiol 10:852–867. doi: 10.1038/nrmicro2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gordon SN, Dunham RM, Engram JC, Estes J, Wang Z, Klatt NR, Paiardini M, Pandrea IV, Apetrei C, Sodora DL, Lee HY, Haase AT, Miller MD, Kaur A, Staprans SI, Perelson AS, Feinberg MB, Silvestri G. 2008. Short-lived infected cells support virus replication in sooty mangabeys naturally infected with simian immunodeficiency virus: implications for AIDS pathogenesis. J Virol 82:3725–3735. doi: 10.1128/JVI.02408-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Silvestri G, Sodora DL, Koup RA, Paiardini M, O'Neil SP, McClure HM, Staprans SI, Feinberg MB. 2003. Nonpathogenic SIV infection of sooty mangabeys is characterized by limited bystander immunopathology despite chronic high-level viremia. Immunity 18:441–452. doi: 10.1016/S1074-7613(03)00060-8. [DOI] [PubMed] [Google Scholar]
- 4.Meythaler M, Martinot A, Wang Z, Pryputniewicz S, Kasheta M, Ling B, Marx PA, O'Neil S, Kaur A. 2009. Differential CD4+ T-lymphocyte apoptosis and bystander T-cell activation in rhesus macaques and sooty mangabeys during acute simian immunodeficiency virus infection. J Virol 83:572–583. doi: 10.1128/JVI.01715-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann D, Blazar BR, Rodriguez B, Teixeira-Johnson L, Landay A, Martin JN, Hecht FM, Picker LJ, Lederman MM, Deeks SG, Douek DC. 2006. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med 12:1365–1371. [DOI] [PubMed] [Google Scholar]
- 6.Brenchley JM, Paiardini M, Knox KS, Asher AI, Cervasi B, Asher TE, Scheinberg P, Price DA, Hage CA, Kholi LM, Khoruts A, Frank I, Else J, Schacker T, Silvestri G, Douek DC. 2008. Differential Th17 CD4 T-cell depletion in pathogenic and nonpathogenic lentiviral infections. Blood 112:2826–2835. doi: 10.1182/blood-2008-05-159301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Favre D, Lederer S, Kanwar B, Ma ZM, Proll S, Kasakow Z, Mold J, Swainson L, Barbour JD, Baskin CR, Palermo R, Pandrea I, Miller CJ, Katze MG, McCune JM. 2009. Critical loss of the balance between Th17 and T regulatory cell populations in pathogenic SIV infection. PLoS Pathog 5:e1000295. doi: 10.1371/journal.ppat.1000295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paiardini M, Cervasi B, Reyes-Aviles E, Micci L, Ortiz AM, Chahroudi A, Vinton C, Gordon SN, Bosinger SE, Francella N, Hallberg PL, Cramer E, Schlub T, Chan ML, Riddick NE, Collman RG, Apetrei C, Pandrea I, Else J, Munch J, Kirchhoff F, Davenport MP, Brenchley JM, Silvestri G. 2011. Low levels of SIV infection in sooty mangabey central memory CD4(+) T cells are associated with limited CCR5 expression. Nat Med 17:830–836. doi: 10.1038/nm.2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cartwright EK, McGary CS, Cervasi B, Micci L, Lawson B, Elliott ST, Collman RG, Bosinger SE, Paiardini M, Vanderford TH, Chahroudi A, Silvestri G. 2014. Divergent CD4+ T memory stem cell dynamics in pathogenic and nonpathogenic simian immunodeficiency virus infections. J Immunol 192:4666–4673. doi: 10.4049/jimmunol.1303193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pandrea I, Apetrei C, Gordon S, Barbercheck J, Dufour J, Bohm R, Sumpter B, Roques P, Marx PA, Hirsch VM, Kaur A, Lackner AA, Veazey RS, Silvestri G. 2007. Paucity of CD4+CCR5+ T cells is a typical feature of natural SIV hosts. Blood 109:1069–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Riddick NE, Hermann EA, Loftin LM, Elliott ST, Wey WC, Cervasi B, Taaffe J, Engram JC, Li B, Else JG, Li Y, Hahn BH, Derdeyn CA, Sodora DL, Apetrei C, Paiardini M, Silvestri G, Collman RG. 2010. A novel CCR5 mutation common in sooty mangabeys reveals SIVsmm infection of CCR5-null natural hosts and efficient alternative coreceptor use in vivo. PLoS Pathog 6:e1001064. doi: 10.1371/journal.ppat.1001064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elliott ST, Riddick NE, Francella N, Paiardini M, Vanderford TH, Li B, Apetrei C, Sodora DL, Derdeyn CA, Silvestri G, Collman RG. 2012. Cloning and analysis of sooty mangabey alternative coreceptors that support simian immunodeficiency virus SIVsmm entry independently of CCR5. J Virol 86:898–908. doi: 10.1128/JVI.06415-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li B, Stefano-Cole K, Kuhrt DM, Gordon SN, Else JG, Mulenga J, Allen S, Sodora DL, Silvestri G, Derdeyn CA. 2010. Nonpathogenic simian immunodeficiency virus infection of sooty mangabeys is not associated with high levels of autologous neutralizing antibodies. J Virol 84:6248–6253. doi: 10.1128/JVI.00295-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Francella N, Gwyn SE, Yi Y, Li B, Xiao P, Elliott ST, Ortiz AM, Hoxie JA, Paiardini M, Silvestri G, Derdeyn CA, Collman RG. 2013. CD4+ T cells support production of simian immunodeficiency virus Env antibodies that enforce CD4-dependent entry and shape tropism in vivo. J Virol 87:9719–9732. doi: 10.1128/JVI.01254-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirsch VM, Johnson PR. 1994. Pathogenic diversity of simian immunodeficiency viruses. Virus Res 32:183–203. doi: 10.1016/0168-1702(94)90041-8. [DOI] [PubMed] [Google Scholar]
- 16.Keele BF, Li H, Learn GH, Hraber P, Giorgi EE, Grayson T, Sun C, Chen Y, Yeh WW, Letvin NL, Mascola JR, Nabel GJ, Haynes BF, Bhattacharya T, Perelson AS, Korber BT, Hahn BH, Shaw GM. 2009. Low-dose rectal inoculation of rhesus macaques by SIVsmE660 or SIVmac251 recapitulates human mucosal infection by HIV-1. J Exp Med 206:1117–1134. doi: 10.1084/jem.20082831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Connor RI, Chen BK, Choe S, Landau NR. 1995. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 206:935–944. doi: 10.1006/viro.1995.1016. [DOI] [PubMed] [Google Scholar]
- 18.Francella N, Elliott ST, Yi Y, Gwyn SE, Ortiz AM, Li B, Silvestri G, Paiardini M, Derdeyn CA, Collman RG. 2013. Decreased plasticity of coreceptor use by CD4-independent SIV Envs that emerge in vivo. Retrovirology 10:133. doi: 10.1186/1742-4690-10-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weiler A, May GE, Qi Y, Wilson N, Watkins DI. 2006. Polymorphisms in eight host genes associated with control of HIV replication do not mediate elite control of viral replication in SIV-infected Indian rhesus macaques. Immunogenetics 58:1003–1009. doi: 10.1007/s00251-006-0166-6. [DOI] [PubMed] [Google Scholar]
- 20.Prétet JL, Brussel A, Guillet JG, Butor C. 1999. Two alleles for rhesus macaque GPR15 (BOB). AIDS Res Hum Retroviruses 15:945–947. doi: 10.1089/088922299310665. [DOI] [PubMed] [Google Scholar]
- 21.Pöhlmann S, Lee B, Meister S, Krumbiegel M, Leslie G, Doms RW, Kirchhoff F. 2000. Simian immunodeficiency virus utilizes human and sooty mangabey but not rhesus macaque STRL33 for efficient entry. J Virol 74:5075–5082. doi: 10.1128/JVI.74.11.5075-5082.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Napier C, Sale H, Mosley M, Rickett G, Dorr P, Mansfield R, Holbrook M. 2005. Molecular cloning and radioligand binding characterization of the chemokine receptor CCR5 from rhesus macaque and human. Biochem Pharmacol 71:163–172. doi: 10.1016/j.bcp.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 23.Billick E, Seibert C, Pugach P, Ketas T, Trkola A, Endres MJ, Murgolo NJ, Coates E, Reyes GR, Baroudy BM, Sakmar TP, Moore JP, Kuhmann SE. 2004. The differential sensitivity of human and rhesus macaque CCR5 to small-molecule inhibitors of human immunodeficiency virus type 1 entry is explained by a single amino acid difference and suggests a mechanism of action for these inhibitors. J Virol 78:4134–4144. doi: 10.1128/JVI.78.8.4134-4144.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matloubian M, David A, Engel S, Ryan JE, Cyster JG. 2000. A transmembrane CXC chemokine is a ligand for HIV-coreceptor Bonzo. Nat Immunol 1:298–304. doi: 10.1038/79738. [DOI] [PubMed] [Google Scholar]
- 25.McGary CS, Cervasi B, Chahroudi A, Micci L, Taaffe J, Meeker T, Silvestri G, Davenport MP, Paiardini M. 2014. Increased stability and limited proliferation of CD4+ central memory T cells differentiate nonprogressive simian immunodeficiency virus (SIV) infection of sooty mangabeys from progressive SIV infection of rhesus macaques. J Virol 88:4533–4542. doi: 10.1128/JVI.03515-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brenchley JM, Vinton C, Tabb B, Hao XP, Connick E, Paiardini M, Lifson JD, Silvestri G, Estes JD. 2012. Differential infection patterns of CD4+ T cells and lymphoid tissue viral burden distinguish progressive and nonprogressive lentiviral infections. Blood 120:4172–4181. doi: 10.1182/blood-2012-06-437608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nigam P, Kwa S, Velu V, Amara RR. 2011. Loss of IL-17-producing CD8 T cells during late chronic stage of pathogenic simian immunodeficiency virus infection. J Immunol 186:745–753. doi: 10.4049/jimmunol.1002807. [DOI] [PubMed] [Google Scholar]
- 28.Kim CH, Kunkel EJ, Boisvert J, Johnston B, Campbell JJ, Genovese MC, Greenberg HB, Butcher EC. 2001. Bonzo/CXCR6 expression defines type 1-polarized T-cell subsets with extralymphoid tissue homing potential. J Clin Invest 107:595–601. doi: 10.1172/JCI11902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lim HW, Lee J, Hillsamer P, Kim CH. 2008. Human Th17 cells share major trafficking receptors with both polarized effector T cells and FOXP3+ regulatory T cells. J Immunol 180:122–129. doi: 10.4049/jimmunol.180.1.122. [DOI] [PubMed] [Google Scholar]
- 30.Sharron M, Pohlmann S, Price K, Lolis E, Tsang M, Kirchhoff F, Doms RW, Lee B. 2000. Expression and coreceptor activity of STRL33/Bonzo on primary peripheral blood lymphocytes. Blood 96:41–49. [PubMed] [Google Scholar]
- 31.Unutmaz D, Xiang W, Sunshine MJ, Campbell J, Butcher E, Littman DR. 2000. The primate lentiviral receptor Bonzo/STRL33 is coordinately regulated with CCR5 and its expression pattern is conserved between human and mouse. J Immunol 165:3284–3292. doi: 10.4049/jimmunol.165.6.3284. [DOI] [PubMed] [Google Scholar]
- 32.Chen Z, Kwon D, Jin Z, Monard S, Telfer P, Jones MS, Lu CY, Aguilar RF, Ho DD, Marx PA. 1998. Natural infection of a homozygous delta24 CCR5 red-capped mangabey with an R2b-tropic simian immunodeficiency virus. J Exp Med 188:2057–2065. doi: 10.1084/jem.188.11.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gautam R, Gaufin T, Butler I, Gautam A, Barnes M, Mandell D, Pattison M, Tatum C, Macfarland J, Monjure C, Marx PA, Pandrea I, Apetrei C. 2009. Simian immunodeficiency virus SIVrcm, a unique CCR2-tropic virus, selectively depletes memory CD4+ T cells in pigtailed macaques through expanded coreceptor usage in vivo. J Virol 83:7894–7908. doi: 10.1128/JVI.00444-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pöhlmann S, Stolte N, Munch J, Ten Haaft P, Heeney JL, Stahl-Hennig C, Kirchhoff F. 1999. Coreceptor usage of BOB/GPR15 in addition to CCR5 has no significant effect on replication of simian immunodeficiency virus in vivo. J Infect Dis 180:1494–1502. doi: 10.1086/315097. [DOI] [PubMed] [Google Scholar]
- 35.Kiene M, Marzi A, Urbanczyk A, Bertram S, Fisch T, Nehlmeier I, Gnirss K, Karsten CB, Palesch D, Munch J, Chiodi F, Pohlmann S, Steffen I. 2012. The role of the alternative coreceptor GPR15 in SIV tropism for human cells. Virology 433:73–84. doi: 10.1016/j.virol.2012.07.012. [DOI] [PubMed] [Google Scholar]
- 36.Chen Z, Zhou P, Ho DD, Landau NR, Marx PA. 1997. Genetically divergent strains of simian immunodeficiency virus use CCR5 as a coreceptor for entry. J Virol 71:2705–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wolinsky SM, Veazey RS, Kunstman KJ, Klasse PJ, Dufour J, Marozsan AJ, Springer MS, Moore JP. 2004. Effect of a CCR5 inhibitor on viral loads in macaques dual-infected with R5 and X4 primate immunodeficiency viruses. Virology 328:19–29. doi: 10.1016/j.virol.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 38.Veazey RS, Klasse PJ, Ketas TJ, Reeves JD, Piatak M Jr, Kunstman K, Kuhmann SE, Marx PA, Lifson JD, Dufour J, Mefford M, Pandrea I, Wolinsky SM, Doms RW, DeMartino JA, Siciliano SJ, Lyons K, Springer MS, Moore JP. 2003. Use of a small molecule CCR5 inhibitor in macaques to treat simian immunodeficiency virus infection or prevent simian-human immunodeficiency virus infection. J Exp Med 198:1551–1562. doi: 10.1084/jem.20031266. [DOI] [PMC free article] [PubMed] [Google Scholar]