

ABSTRACT
Interferon alpha (IFN-α) is an approved medication for chronic hepatitis B therapy. Besides acting as an immunomodulator, IFN-α elicits a pleiotropic antiviral state in hepatitis B virus (HBV)-infected hepatocytes, but whether or not IFN-α impedes the late steps of the HBV life cycle, such as HBV secretion, remains elusive. Here we report that IFN-α treatment of HepAD38 cells with established HBV replication selectively reduced HBV virion release without altering intracellular viral replication or the secretion of HBV subviral particles and nonenveloped capsids. In search of the interferon-stimulated gene(s) that is responsible for the reduction of HBV virion release, we found that tetherin, a broad-spectrum antiviral transmembrane protein that inhibits the egress of a variety of enveloped viruses, was highly induced by IFN-α in HepAD38 cells and in primary human hepatocytes. We further demonstrated that the expression of full-length tetherin, but not the C-terminal glycosylphosphatidylinositol (GPI) anchor-truncated form, inhibited HBV virion egress from HepAD38 cells. In addition, GPI anchor-truncated tetherin exhibited a dominant-negative effect and was incorporated into the liberated virions. We also found colocalization of tetherin and HBV L protein at the intracellular multivesicular body, where the budding of HBV virions takes place. In line with this, electron microscopy demonstrated that HBV virions were tethered in the lumen of the cisterna membrane under tetherin expression. Finally, knockdown of tetherin or overexpression of dominant negative tetherin attenuated the IFN-α-mediated reduction of HBV virion release. Taken together, our study suggests that IFN-α inhibits HBV virion egress from hepatocytes through the induction of tetherin.
IMPORTANCE Tetherin is a host restriction factor that blocks the egress of a variety of enveloped viruses through tethering the budding virions on the cell surface with its membrane anchor domains. Here we report that interferon directly and selectively inhibits the secretion of HBV virions, but not subviral particles or nonenveloped capsids, through the induction of tetherin in hepatocyte-derived cells. The antiviral function of tetherin requires the carboxyl-terminal GPI anchor, while the GPI anchor deletion mutant exhibits dominant negative activity and attaches to liberated HBV virions. Consistent with the fact that HBV is an intracellular budding virus, microscopy analyses demonstrated that the tethering of HBV virions occurs in the intracellular cisterna and that tetherin colocalizes with HBV virions on the multivesicular body, which is the HBV virion budding site. Our study not only expands the antiviral spectrum of tetherin but also sheds light on the mechanisms of interferon-elicited anti-HBV responses.
INTRODUCTION
Chronic hepatitis B remains a serious infectious liver disease affecting ∼350 million individuals under the risk of life-threatening cirrhosis and liver cancer (1, 2). The causative agent of hepatitis B is hepatitis B virus (HBV), which infects and propagates in the liver and secrets viral particles into the bloodstream, including infectious virions and surplus subviral particles (also known as HBV surface antigen [HBsAg]) (3, 4).
HBV is an enveloped DNA virus belonging to the Hepadnaviridae family (5). The virion contains a single copy of viral genomic DNA in a relaxed open circular (RC) form (2, 5). Upon engagement of the virion particle with the hepatocyte-specific receptor Na+-taurocholate cotransporter polypeptide (NTCP), with possibly other cofactors, the virus is endocytosed into the cell, and the viral RC DNA is transported into the nucleus to form the episomal covalently closed circular DNA (cccDNA) form of the genome (2, 5–9). By utilizing the hepatic transcription machinery, HBV cccDNA synthesizes viral mRNAs for reproduction, specifically 3.5-kb precore mRNA and pregenomic RNA (pgRNA), 2.4-kb and 2.1-kb surface antigen mRNAs, and 0.7-kb X mRNA. HBV genomic DNA replication occurs in the cytoplasmic viral nucleocapsid via reverse transcription of the encapsidated pgRNA and is catalyzed by the viral DNA polymerase. The mature nucleocapsid, which contains the newly synthesized RC DNA, is either enveloped by the viral surface antigen proteins to assemble into virions to be secreted through the cellular multivesicular body (MVB) secretory pathway or recycled back into the nucleus to amplify the cccDNA reservoir (2, 10, 11). In parallel with virion egress, the viral envelope proteins are able to self-assemble into subviral particles in the endoplasmic reticulum (ER) lumen and are secreted through the ER-Golgi constitutive secretory pathway, giving rise to the extracellular accumulation of HBsAg (10, 11).
Type I interferon (interferon alpha/beta [IFN-α/β]) is a broad-spectrum antiviral agent that plays a pivotal role in the innate defense against viral infection. In most cases, the induction of IFN-α/β serves as a hallmark for the activation of innate responses to virus infection (12–14). Although it remains debatable whether HBV induces and/or antagonizes pattern recognition receptor-mediated IFN production in hepatocytes, the virus is vulnerable to artificially activated intrahepatic innate immune responses or IFN-α treatment (15–24). In fact, IFN-α remains the only approved immunomodulatory medication for treatment of chronic hepatitis B, and it has achieved sustained virological responses in ∼30 to 40% of treated patients (25). The therapeutic effect of IFN-α against HBV functions through the coordination of the host innate and adaptive immune responses, the latter being represented by the activation of HBV-specific cytotoxic T lymphocytes (CTLs). CTL-produced cytokines, including type II interferon (IFN-γ) and tumor necrosis factor alpha (TNF-α), exert important roles in the noncytolytic clearance of HBV from hepatocytes (26). Besides acting as immunomodulators, interferons elicit an intracellular antiviral state through engagement of their specific plasma membrane receptors and in turn activate Jak-STAT pathways to induce an array of interferon-stimulated genes (ISGs), which manipulate cellular functions or directly target viral components to restrict virus infection and propagation (27). Previous studies demonstrated that interferons inhibit HBV reproduction at multiple viral replication steps in its life cycle, by downregulating viral mRNA transcription and stability (21, 28–31), blocking pgRNA encapsidation (32), reducing viral protein synthesis (21), and promoting viral nucleocapsid degradation (33), etc. In addition to these observations, a few ISGs have been identified to possess specific antiviral mechanisms against HBV replication in cell cultures. For instance, tripartite motif-containing 22 (TRIM22) suppresses HBV core promoter activity to reduce viral RNA transcription (34), host zinc finger antiviral protein (ZAP) and myeloid differentiation primary response gene 88 (MyD88) promote the decay of HBV mRNAs (29, 35), APOBEC3A deaminates and destabilizes HBV cccDNA in the nucleus (36), APOBEC3G is incorporated into the cytoplasmic HBV nucleocapsid to inhibit viral DNA replication in a deamination-independent fashion (37, 38), and IFN-γ-inducible indoleamine 2,3-dioxygenase (IDO) catalyzes tryptophan degradation to reduce HBV protein biosynthesis (21). However, since the previously observed anti-HBV activities of IFN and/or ISGs all lead to the final reduction of progeny virus, whether IFN specifically inhibits HBV particle release from hepatocytes, downstream of viral gene expression and DNA replication, is still unknown.
Blocking of virion egress would be considered the last line of defense orchestrated by IFN to restrict virus propagation and spread. Among the identified antiviral ISGs, tetherin (also known as BST2, CD317, and HM1.24) is a small transmembrane protein that physically tethers enveloped viruses at the virus budding site to prevent virion release from infected cells (39). It was first reported to be a host antiretroviral factor that impedes the departure of human immunodeficiency virus type 1 (HIV-1) progeny virions from the cell surface in the absence of the viral accessory protein Vpu (40). To date, the antiviral spectrum of tetherin has been expanded from retroviruses to a variety of enveloped viruses, including Ebola virus (41, 42), Kaposi's sarcoma-associated herpesvirus (KSHV) (43), Lassa virus (44, 45), vesicular stomatitis virus (VSV) (46), influenza virus (47–49), Sendai virus (50), human coronavirus 229E (HCoV-229E) (51), and hepatitis C virus (HCV) (52–55). Tetherin consists of a quadripartite structural domain, specifically an N-terminal cytoplasmic tail, a single transmembrane (TM) domain, a coiled-coil extracellular domain, and a C-terminal glycosylphosphatidylinositol (GPI) anchor. Upon induction by IFN, tetherin exists in a dimeric form on the cell surface through the formation of intermolecular disulfide bonds between their ectodomains, and both the terminal TM domain and GPI anchor are responsible for cross-linking the virus to the cell membrane (56). A recent mechanistic study on the inhibition of HIV-1 by tetherin favors a model in which the axially configured tetherin homodimer captures the virion by preferentially inserting a pair of GPI anchors into the viral envelope, while the other pair of TM domains remains rooted in the plasma membrane (57).
Here we report that IFN-α or IFN-γ treatment of HepAD38 cells with established HBV replication reduced virion secretion without inhibiting intracellular viral replication or the release of HBV subviral particles and naked capsids. Under such conditions, tetherin was highly induced by IFN. Expression of tetherin alone in HepAD38 cells demonstrated the same selective inhibition of HBV virion egress as did IFN. In contrast, the inactive form of tetherin with a deletion of the GPI anchor (tetherin-CΔ19) was incorporated into virions but failed to inhibit HBV virion release when expressed alone, and it abrogated the inhibitory activity of IFN-α against HBV virion secretion, suggesting that tetherin-CΔ19 exhibits dominant negative effects and the integrity of the GPI pair in the tetherin homodimer is required for its antiviral properties. In agreement with the fact that HBV is an intracellular budding virus, the intracellular colocalization of tetherin and HBV virions, as well as the intracellular tethering of HBV virions, was visualized by microscopy. Collectively, our results indicate that IFN is able to inhibit HBV virion egress through the induction of tetherin, which may shed light on the antiviral mechanisms of IFN and tetherin.
MATERIALS AND METHODS
Cell cultures and drugs.
The HBV-inducible cell lines HepAD38 and HepDE19 were maintained in Dulbecco's modified Eagle's medium (DMEM)–F-12 medium (Mediatech) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin, and 1 μg/ml tetracycline (58, 59). To initiate HBV replication and virus particle secretion, tetracycline was withdrawn from the culture medium, and the cells were cultured for the indicated time periods. Primary human hepatocytes (PHHs) were purchased from Triangle Research Labs (TRL). Cells were maintained in hepatocyte maintenance medium (TRL) with daily medium changes. Recombinant human IFN-α, IFN-γ, and IFN-λ were purchased from PBL Biomedical Laboratories.
Generation of retroviral vectors expressing tetherin.
The nucleotide sequence that encodes N-terminally FLAG-tagged wild-type tetherin or mutant tetherin with a deletion of the C-terminal 19-amino-acid (aa) GPI anchor (tetherin-CΔ19) was amplified from plasmid pCDNA5-BST2 by PCR and cloned into the pQCXIP vector (Clontech) between the NotI and BamHI restriction sites. To prepare VSV G protein-pseudotyped retroviruses that express wild-type tetherin or tetherin-CΔ19, 1 × 106 GP2-293 cells were cotransfected with 1 μg of pCMV-VSV-G, 1.5 μg of pCDNA3-HIV1-vpu (to antagonize tetherin for the release of VSV-pseudotyped retrovirus), and 1.5 μg of the pQCXIP-derived plasmid expressing tetherin or tetherin-CΔ19 by using Lipofectamine 2000 (Life Technologies). The culture supernatant was harvested 48 and 72 h after transfection, filtered through a 0.45-μm-pore-size polyethersulfone (PES) syringe filter (Millipore), aliquoted, and stored at −80°C.
Establishment of a HepAD38 cell line stably expressing tetherin or tetherin-CΔ19 protein.
HepAD38 cells in a 35-mm dish were incubated with 2 ml of Opti-MEM containing pseudotyped control retroviruses, wild-type tetherin-expressing retroviruses, or tetherin-CΔ19-expressing retroviruses and centrifuged at room temperature for 30 min at 1,000 × g. At 48 h posttransduction, cells were cultured in DMEM–F-12 medium containing puromycin (3 μg/ml) and tetracycline (1 μg/ml) for another 2 weeks, with medium changes at 2-day intervals. The antibiotic-resistant cells were then pooled and expanded into cell lines, which were designated HepAD38-control, HepAD38-tetherin, and HepAD38-TCΔ19, respectively. During the entire procedure for HepAD38 cell transduction, 1 μg/ml of tetracycline was always included in the culture medium to prevent HBV pgRNA transcription and DNA replication.
Establishment of a HepAD38 cell line with stable knockdown of tetherin.
To stably knock down tetherin gene expression, HepAD38 cells were transduced with control or tetherin short hairpin RNA (shRNA) lentiviral particles (Santa Cruz) at 1 inclusion-forming unit (IFU) per cell for 24 h in the presence of 1 μg/ml of tetracycline. The lentivirus-transduced cells were cultured for an additional 48 h and then cultured in medium containing puromycin (3 μg/ml) and tetracycline (1 μg/ml) for 2 weeks, and antibiotic-resistant cells were pooled and expanded into cell lines, which were named HepAD38-control_K.D and HepAD38-tetherin_K.D, respectively. The knockdown level of tetherin in HepAD38-tetherin_K.D cells upon IFN-α treatment was confirmed by Western blotting.
Intracellular HBV RNA/DNA analysis.
Total cellular RNA was extracted with TRIzol reagents (Life Technologies), according to the manufacturer's instructions. Five micrograms of total RNA was resolved on a 1.5% agarose gel containing 2.2 M formaldehyde and transferred onto a Hybond-XL membrane (GE Healthcare) in 20× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate).
Intracellular viral core DNA was extracted as described previously (58). Briefly, cells from one well of a 12-well plate were lysed with 200 μl lysis buffer containing 10 mM Tris-HCl (pH 8.0), 1 mM EDTA, 1% NP-40, and 50 mM NaCl. Cell debris and nuclei were removed by centrifugation, and the supernatant was digested at 37°C for 1 h with 0.5 mg/ml pronase (Calbiochem), 0.5% sodium dodecyl sulfate (SDS), and 125 mM NaCl. The nucleic acids were extracted with phenol and precipitated with 2 volumes of ethanol with the addition of 2 μl yeast RNA as a carrier. The pellet was washed with 70% ethanol, air dried, and dissolved in TE buffer (10 mM Tris-HCl [pH 8.0], 1 mM EDTA). DNA was resolved on a 1.2% agarose gel. The gel was then subjected to denaturation in a solution containing 0.5 M NaOH and 1.5 M NaCl, followed by neutralization in a buffer containing 1 M Tris-HCl (pH 7.4) and 1.5 M NaCl. DNA was then blotted onto a Hybond-XL membrane in 20× SSC.
For the detection of HBV RNA and DNA, membranes were probed with an [α-32P]UTP (800 Ci/mmol; PerkinElmer)-labeled plus- or minus-strand-specific full-length HBV riboprobe. Hybridization was carried out by using 5 ml of Ekono hybridization buffer (G-Biosciences) with 1 h of prehybridization at 65°C and overnight hybridization at 65°C, followed by a 1-h wash with a solution containing 0.1× SSC and 0.1% SDS at 65°C. The membrane was exposed to a phosphorimager screen. Hybridization signals were visualized by using a Typhoon FLA-7000 instrument (GE Healthcare).
HBV extracellular particle gel assay.
An HBV particle gel assay was performed as previously described, with modifications (60). The secreted viral particles in culture fluid were precipitated from 1 ml of the culture supernatant by the addition of 400 μl 35% polyethylene glycol 8000 (PEG 8000) and rotated at 4°C for 2 h, followed by centrifugation at 8,000 × g for 5 min at 4°C. The precipitate was slowly dissolved in TNE buffer (10 mM Tris-HCl [pH 7.6], 150 mM NaCl, and 1 mM EDTA) at 4°C overnight. The viral particles were fractionated by electrophoresis through a nondenaturing 1% agarose gel and transferred onto a nitrocellulose filter (Whatman). To detect enveloped HBV particles and nonenveloped capsids by an enzyme immunoassay (EIA), membranes were blocked with WesternBreeze Blocker (Life Technologies) and probed with a monoclonal antibody against HBV surface antigen (catalog number N1597; Dako), with polyclonal antibodies against HBV core protein (catalog number B0586; Dako), or with tetherin antibodies (catalog number 13560-1-AP; Proteintech). Bound antibodies were revealed by using IRDye secondary antibodies (Li-Cor) and visualized by using the Li-Cor Odyssey system. For the detection of HBV DNA, the DNA-containing particles on the membrane were denatured with a solution containing 0.5 M NaOH and 1.5 M NaCl, and this step was followed by neutralization with a solution containing 1 M Tris-HCl (pH 7.4) and 1.5 M NaCl. HBV DNA was detected by hybridization with an [α-32P]UTP (800 Ci/mmol; PerkinElmer)-labeled minus-strand-specific full-length HBV riboprobe.
HBV intracellular nucleocapsid gel assay.
Cells from one well of a 12-well plate were lysed with 200 μl of 1% NP-40 lysis buffer. Cell debris and nuclei were removed by centrifugation. Twenty microliters of the clarified cell lysates was fractionated by electrophoresis through a nondenaturing 1% agarose gel and transferred onto a nitrocellulose filter (Whatman) in TNE buffer by capillary action. The membrane was then subjected to HBV capsid EIA and capsid DNA hybridization as described above.
Protein analysis.
The secreted HBV surface antigen (HBsAg) in the culture medium was detected by using HBsAg enzyme-linked immunosorbent assay (ELISA) kit (Abazyme) according to the manufacturer's instructions. For Western blot analysis, cells in one well of a 12-well plate were lysed in 150 μl of 1× Laemmli buffer and denatured at 95°C for 10 min. Fifty microliters of the cell lysate was resolved on a 12% SDS-PAGE gel, and proteins were transferred onto an Immobilon FL membrane (Millipore). The membranes were blocked with WesternBreeze Blocker and probed with antibodies against the FLAG tag (clone M2) (catalog number F3165; Sigma), tetherin (catalog number 13560-1-AP; Proteintech), HBV core (9), and β-actin (catalog number MAB1501; Millipore). Bound antibodies were revealed by using IRDye secondary antibodies and visualized by using the Li-Cor Odyssey system.
HBV virion DNA immunoprecipitation (IP)-qPCR.
One milliliter of culture fluid collected from HBV-replicating cells was incubated with 20 μl of protein A/G beads (Santa Cruz) coated with anti-HBsAg monoclonal antibody (Dako) at 4°C overnight on a rotisserie. The pelleted beads were washed three times with chilled phosphate-buffered saline (PBS) and then incubated with a solution containing 0.5 mg/ml pronase (Calbiochem), 0.5% SDS, and 125 mM NaCl for 1 h at 37°C. The virion DNAs were extracted with phenol and precipitated with 2 volumes of ethanol with the addition of 2 μl yeast RNA as a carrier. The pellet was washed with 70% ethanol, air dried, and dissolved in nuclease-free water. The quantification of virion DNA was done by quantitative PCR (qPCR) with LightCycler 480 Probes Master (Roche). The primers and probe used for qPCRs were forward primer 5′-CCGTCTGTGCCTTCTCATCTG-3′, reverse primer 5′-AGTCCAAGAGTYCTCTTATGYAAGACCTT-3′, and probe 5′–6-carboxyfluorescein (FAM)–CCGTGTGCACTTCGCTTCACCTCTGC–6-carboxytetramethylrhodamine (TAMRA)–3′. The amplification settings included 0.8 μM primers and 0.2 μM probe, annealing, and extension at 64°C for 45 cycles.
Immunofluorescence and confocal microscopy.
Cells were fixed with 4% paraformaldehyde for 20 min and permeabilized with 0.5% Triton X-100 in PBS for 60 min at room temperature. Fixed cells were blocked with 10% FBS and 2% bovine serum albumin (BSA) for 60 min at room temperature and then incubated with anti-HBV pre-S1 antibody (catalog number 10-H07A; Fitzgerald), anti-tetherin antibody (Proteintech), and anti-Alix antibody (catalog number sc-49268; Santa Cruz Biotechnology) diluted in PBS containing 10% FBS and 2% BSA for 2 h at room temperature. After being washed with PBS, the cells were stained with the corresponding Alexa Fluor 405 and 594 dye-conjugated secondary antibodies (Abcam) or Alexa Fluor 488 dye-conjugated secondary antibody (Life Technologies) for 60 min at room temperature. Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Finally, the cells were washed with PBS and then subjected to confocal microscopy analysis under an Olympus FV1000MPE microscope with a 60× objective. Images were processed by using FV10-ASW 3.0 Viewer software.
Transmission electron microscopy (TEM).
Cells were fixed in modified Karnovsky's fixative (2% paraformaldehyde and 2% glutaraldehyde in 0.1 M phosphate buffer), postfixed with 1% osmium tetroxide, dehydrated through a graded series of ethyl alcohol from 70% to 100%, and embedded in resin. Ultrathin sections (∼80 nm) were cut with an ultramicrotome and stained with the heavy metal uranyl acetate. Samples were viewed with a Tecnai BioTwin transmission electron microscope, and digital images were taken with an AMT charge-coupled-device (CCD) camera.
Statistical analysis.
P values were calculated by using Student's t test. A P value of <0.05 was considered significant.
RESULTS
Interferons selectively inhibit HBV virion particle release.
Previous cell culture studies have rigorously demonstrated that interferons, including type I (IFN-α/β), type II (IFN-γ), and type III (IFN-λ) interferons, inhibit intracellular HBV replication at multiple steps in the viral life cycle, eventually resulting in the reduced accumulation of secreted progeny virus particles, and thus, whether IFNs directly inhibit HBV particle release from host cells remains unclear. In our effort to characterize the antiviral activity of IFN under different cell culture and virus replication conditions, we developed a method that enables the study of the potential effect of IFN on HBV secretion. As shown in Fig. 1, a tetracycline-inducible cell line, namely, HepAD38 (59), was used to test the antiviral effect of IFN treatment on HBV DNA replication and viral particle secretion. In agreement with previously reported observations, both IFN-α and IFN-γ posttranscriptionally inhibited HBV DNA replication when the treatment was started simultaneously with tetracycline withdrawal (to induce HBV replication) (Fig. 1B, lanes 1 to 3). However, delayed IFN treatment after 6 days of HBV induction did not alter the steady-state levels of HBV RNA and DNA in HepAD38 cells (Fig. 1B, lanes 4 to 6) but significantly reduced the accumulation of HBV virion particles in the culture supernatant (Fig. 1C, middle and bottom). It has been reported that HBV also produces a considerable amount of surplus DNA-free virions with the infectious virions (61). The similar degrees of reduction of virion-associated capsid (Fig. 1C, middle) and DNA (Fig. 1C, bottom) levels with interferon treatment indicate that the secretion of both types of HBV virions is vulnerable to interferons. In marked contrast, the levels of total enveloped particles and nonenveloped (“naked”) capsids were unchanged with IFN treatment (Fig. 1C). Because enveloped HBV particles consist of virions and pleomorphic subviral HBsAg particles (the latter normally are produced in a large excess compared to virions) (62), it thus appears that the reduction in the accumulation of virions by IFN treatment did not change the EIA signal of total enveloped particles in the particle gel assay (Fig. 1C, top). In addition, the HBsAg ELISA and HBV virion DNA IP-qPCR results further confirmed that IFNs selectively inhibited HBV virion secretion but not the secretion of subviral particles (Fig. 1D).
FIG 1.

Interferon selectively inhibits HBV virion secretion in HepAD38 stable cells with established viral replication. (A) Schematic illustration of the experimental procedures. HepAD38 cells were cultured in a 6-well plate until confluent, and tetracycline (tet) was then removed from culture medium to induce HBV replication. One group of cells (group 1) was mock treated or treated with IFN-α (1,000 IU/ml) or IFN-γ (100 ng/ml) simultaneously with tetracycline withdrawal (tet−) for 6 days, and the cells and culture medium were then harvested; another group of cells (group 2) received interferon treatment after 6 days of HBV induction, and the treatment lasted 4 days before sample collection. Fresh culture media with or without IFN were replenished at 2-day intervals. (B) Cells harvested at the indicated time points were subjected to HBV RNA and core DNA analyses by Northern blotting and Southern blotting, respectively. rRNAs (28S and 18S) were used as total RNA loading controls. The positions of 3.5-kb and 2.4-kb/2.1-kb HBV RNAs and relaxed circular (RC) and single-stranded (SS) DNAs are indicated. The expression of tetherin was revealed by Western blotting, with β-actin serving as a loading control. (C) HBV particles in culture fluids harvested from group 2 cells at the endpoint of treatment (days 8 to 10) were analyzed by a particle gel assay. Enveloped HBV particles, including virions and subviral particles (HBsAg), were revealed by an EIA using HBsAb. Virion-associated capsids and nonenveloped capsids were detected by a core EIA. In situ-denatured HBV virion DNA and nonenveloped capsid DNA were detected by hybridization. (D) Culture fluid samples from group 2 were subjected to an HBsAg ELISA and HBsAg immunoprecipitation, and the immunoprecipitated samples were analyzed by HBV DNA qPCR. The relative levels of HBsAg and HBV DNA signals in each sample were plotted as a percentage of the signals from the mock-treated samples (means ± standard deviations).
As described above, we have developed an assay in which IFN fails to block intracellular HBV de novo synthesis but is still able to inhibit virion egress, making studies of the antiviral effects of IFN on HBV particle trafficking and secretion possible. We have also observed the same phenomenon in another HBV-inducible stable cell line, HepDE19 (data not shown). However, the mechanisms underlying the resistance of viral DNA replication to IFN treatment under conditions of established HBV replication await further investigations, but they might not be largely due to the blockade of IFN signaling and/or ISG production by HBV, as several reported anti-HBV ISGs were induced by IFN, such as IFN-α-induced ZAP and IFN-γ-induced IDO (data not shown). Intriguingly, in HepAD38 cells with established HBV replication, treatment with both IFN-α and IFN-γ upregulated the expression of tetherin, a particularly interesting ISG that possesses antiviral activity against a variety of enveloped viruses (Fig. 1B, bottom) (39). Consistent with this finding, it has been reported that tetherin was induced by IFN in hepatocyte-derived cells and exhibited inhibition of HCV particle release (52–54). In addition, we found that all the three types of IFN (IFN-α, -γ, and -λ) significantly upregulated tetherin expression in primary human hepatocytes by Western blotting and immunofluorescence microscopy analyses (Fig. 2), indicating that tetherin may have a physiological antiviral function in HBV-infected liver upon immune activation or immunotherapy. In this regard, we hypothesized that the IFN-mediated inhibition of HBV virion secretion is induced through the induction of tetherin. It is worth noting that while IFN-α stimulated tetherin expression at both early and late times after the induction of HBV, IFN-γ induced tetherin in HepAD38 cells only when the treatment started at 6 days post-HBV induction (Fig. 1B, bottom) and exhibited weaker antiviral activity against HBV virion release than did IFN-α (Fig. 1C and D). Such observations were also made with HepDE19 cells (data not shown); however, the underlying mechanism is unclear.
FIG 2.

Interferon induces tetherin expression in primary human hepatocytes (PHHs). PHHs were cultured in a 12-well plate and treated with IFN-α (1,000 IU/ml), IFN-γ (100 ng/ml), or IFN-λ (100 ng/ml) or left untreated (mock) for 2 days. The levels of tetherin expression were determined by a Western blot assay (A) and immunofluorescence microscopy (red) (B). β-Actin served as a Western blot loading control. Nuclei were stained with DAPI (blue).
Tetherin inhibits HBV virion egress.
To test the above-described hypothesis, we examined the antiviral effect of overexpressed tetherin in HepAD38 cells. In order to achieve overexpression of tetherin in every HBV-replicating cell, we generated cell lines stably expressing wild-type or GPI-anchor-deleted (CΔ19) tetherin through retroviral vector transduction of HepAD38 cells. Under forced expression of tetherin, HBV expression and replication were induced in HepAD38 cells by removal of tetracycline. As shown in Fig. 3A, overexpression of wild-type or CΔ19 tetherin did not affect intracellular HBV core expression, capsid formation, or DNA replication; in the supernatant, the accumulation of total enveloped HBV particles and naked capsids was unchanged with the intracellular overexpression of wild-type or mutant tetherin compared to that in control cells (Fig. 3B), but virion production was significantly reduced by wild-type tetherin and slightly augmented by tetherin-CΔ19 (TCΔ19) (Fig. 3B and C). The above-described results demonstrate that interferon-inducible tetherin mimics the observed antiviral activity of interferons against HBV virion secretion, suggesting that interferon inhibits HBV virion secretion through the induction of tetherin.
FIG 3.
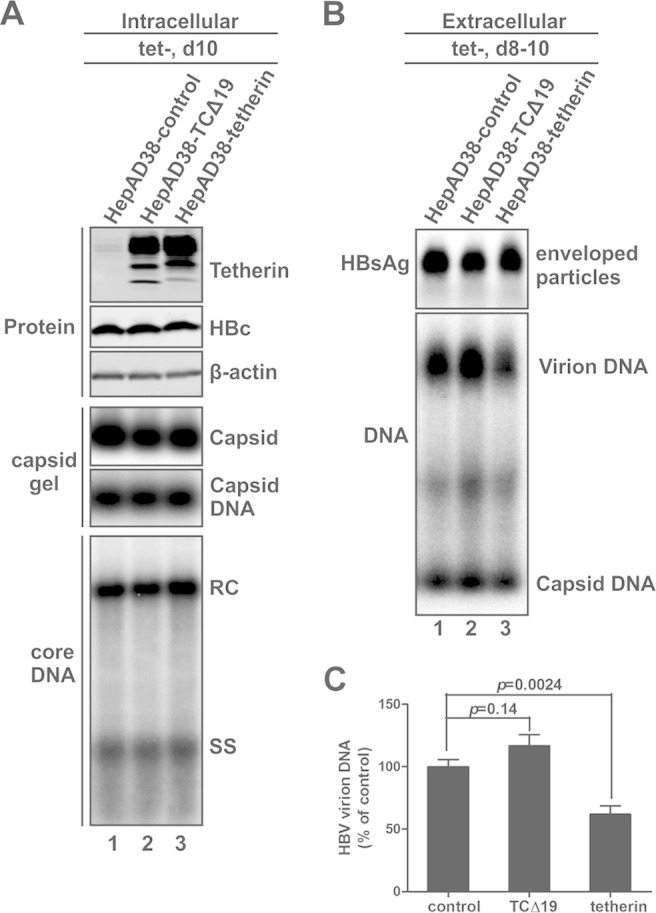
Overexpression of wild-type but not GPI-anchor-deleted tetherin inhibits HBV virion release. Retroviral vector-transduced HepAD38 cells stably expressing wild-type or mutant tetherin, namely, HepAD38-tetherin and HepAD38-TCΔ19 cells, respectively, and control retrovirus-transduced HepAD38 cells (HepAD38-control) were cultured in complete maintenance medium until they became confluent, and tetracycline was then removed from the culture medium to induce HBV DNA replication. (A) Cells were harvested at day 10 after withdrawal of tetracycline, and intracellular tetherin expression, HBV core protein expression, capsid formation, and core DNA replication were analyzed. (B and C) The extracellular accumulation of HBV enveloped particles, virions, and naked capsid DNA from days 8 to 10 post-tetracycline induction was analyzed by a particle gel assay (B) and virion DNA IP-qPCR (C).
GPI anchor deletion mutant tetherin (TCΔ19) exhibits dominant negative activity and attenuates IFN-mediated inhibition of HBV virion secretion.
It is worth noting that overexpression of TCΔ19 failed to inhibit the production of HBV virions, which is consistent with a previous report that the C-terminal 19-aa GPI anchor is functionally indispensable for the antiviral activity of tetherin against HIV-1 egress (40). More interestingly, the enhancement of HBV virion secretion by TCΔ19 expression also suggested that TCΔ19 possesses dominant negative activity. To further confirm this, we treated HepAD38-control and HepAD38-TCΔ19 cells with IFN-α as described in the legend of Fig. 4A. Consistent with the results shown in Fig. 1, IFN-α treatment induced endogenous tetherin expression in both cell lines without affecting intracellular viral replication and the release of subviral particles and naked capsids; however, IFN-α dose-dependently inhibited HBV virion secretion in control HepAD38 cells but not in TCΔ19-expressing cells, suggesting that the antiviral activity of interferon-induced tetherin was attenuated by TCΔ19 (Fig. 4B and C). Thus, in conjunction with the above-mentioned results showing that TCΔ19 marginally enhanced HBV virion secretion in the absence of IFN-α treatment (Fig. 3), we concluded that the GPI anchor deletion mutant TCΔ19 is a dominant negative form of tetherin in terms of HBV inhibition. The dominant negative effect of TCΔ19 was reported in a previous study of tetherin-mediated inhibition of HIV particle egress in the absence of Vpu (63).
FIG 4.
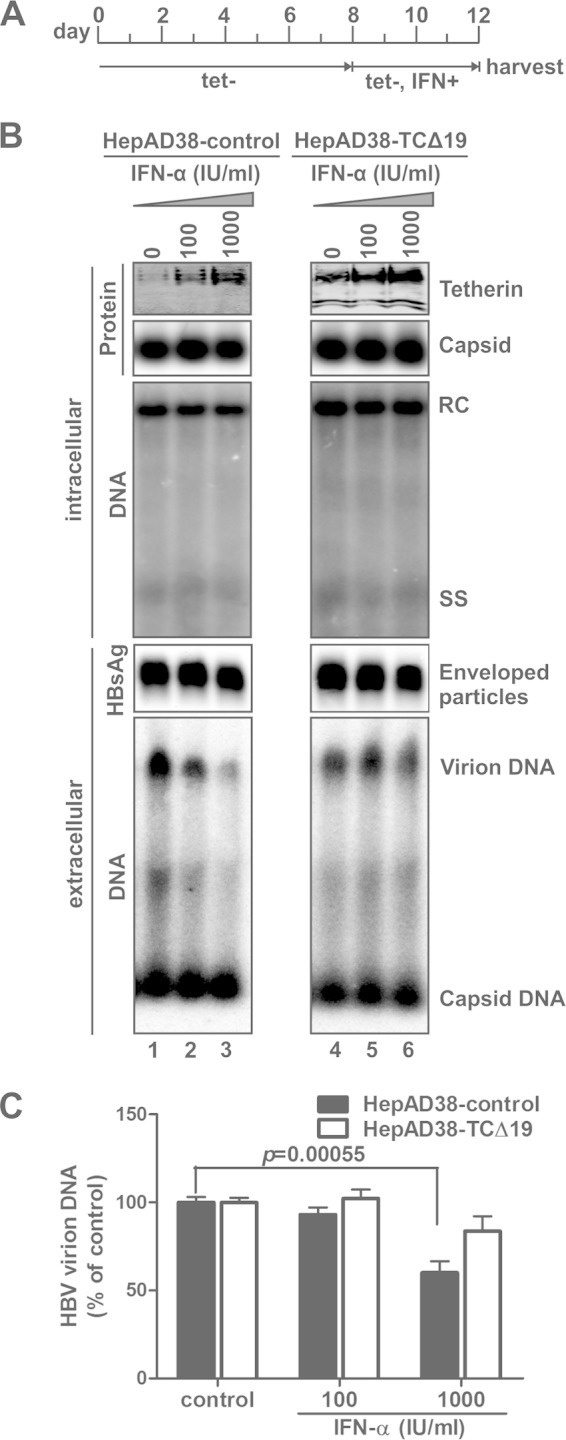
GPI-anchor-deleted tetherin (TCΔ19) rescues HBV virion egress with IFN-α treatment. (A) Experimental procedure. Confluent HepAD38-control and HepAD38-TCΔ19 cells were cultured in tetracycline-free medium for 8 days to stimulate HBV replication, and the cells were then mock treated or treated with IFN-α at the indicated concentrations for an additional 4 days, with medium changes and treatment being repeated every 2 days. (B) Harvested cell monolayers and culture fluids were subjected to intracellular and extracellular analyses, respectively, including Western blot analysis of tetherin expression and induction, a capsid gel EIA, core DNA Southern blot analysis, and a particle gel assay for enveloped HBV particles and particle-associated HBV DNA (from top to bottom). (C) The virion-associated DNA from 1 ml of the harvested supernatant was quantitated by HBsAg IP-qPCR and plotted as a percentage of the DNA signals from mock-treated cell samples (means ± standard deviations).
Evidence suggests that tetherin tethers HBV through incorporation into virions.
It is well accepted that tetherin forms homodimers in a parallel orientation by intermolecular disulfide bonds between the coiled-coil ectodomains, and this unique topology allows the tetherin dimer to trap the budding virus by inserting one pair of membrane anchors into the virus envelope while the other pair remains embedded in the cell membrane (56). The observed dominant negative effect of TCΔ19 suggested that the integrity of the GPI anchor pair is essential for tetherin to inhibit HBV virion egress. Next, we analyzed the presence of tetherin in HBV virions secreted from cells expressing wild-type or GPI anchor deletion mutant tetherin. As shown in Fig. 5, the supernatant samples prepared as described in the legend of Fig. 3 were subjected to a particle gel assay, and consistent with previous results, the three retrovirus-transduced cell lines HepAD38-control, HepAD38-TCΔ19, and HepAD38-tetherin produced the same amounts of enveloped HBV particles in the absence of tetracycline (mainly subviral particles). Interestingly, when the particle gel was stained with antibodies against tetherin, the viral particles liberated from HepAD38-TCΔ19 cells displayed a strong tetherin signal, indicating that TCΔ19 is associated with the enveloped viral particles, presumably through incorporation of the pair of N-terminal transmembrane (TM) domains into the particles (Fig. 5A, left). In addition, the physical interaction between tetherin and HBV was further confirmed by the detection of TCΔ19 in immunoprecipitated enveloped HBV particles by Western blot analysis (Fig. 5A, bottom, lanes 1 to 3).
FIG 5.
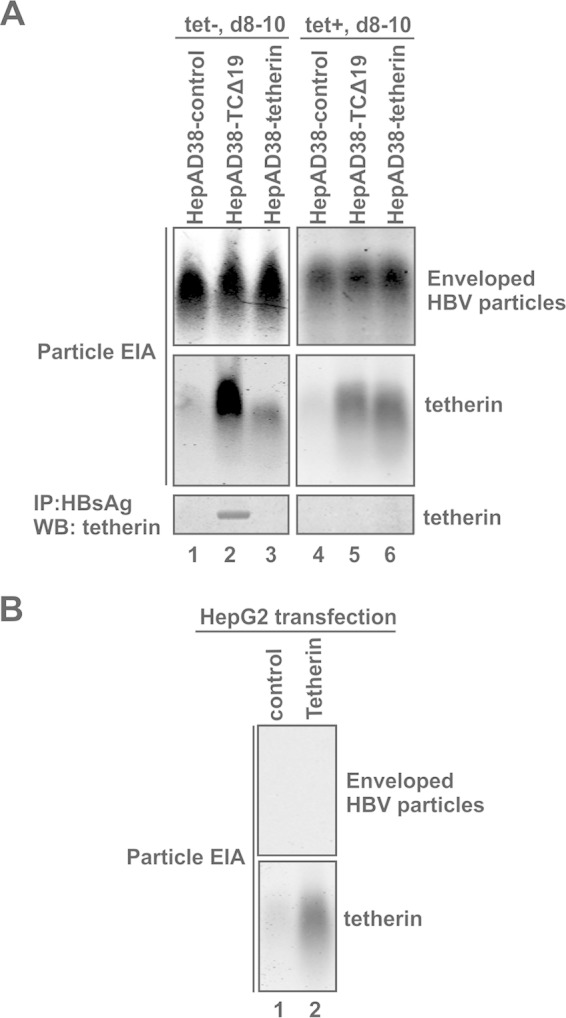
GPI-anchor-deleted tetherin is incorporated into secreted HBV virions. (A, top and middle) Culture medium of the indicated cell lines was collected between days 8 and 10 in the absence or presence of tetracycline and subjected to a particle gel assay, and the blot was stained for HBsAg and tetherin by specific antibodies. (Bottom) An aliquot of the culture fluid samples was subjected to HBsAg immunoprecipitation, followed by SDS-PAGE and Western blot analysis with tetherin antibodies. (B) HepG2 cells were transfected with the control vector or a plasmid expressing wild-type tetherin. Culture fluid was collected at day 5 posttransfection and subjected to a particle gel EIA for HBsAg and tetherin.
It is noteworthy that a weak signal for tetherin was detected on the HBV particle gel samples from HepAD38-control and HepAD38-tetherin cells at the position that overlaps the tetherin signal from the HepAD38-TCΔ19 supernatant sample (Fig. 5A, middle, lanes 1 to 3); the signal remained present in samples from the same cell lines cultured in the presence of tetracycline, in which the secreted enveloped viral particles were subviral particles (HBsAg) only, but the tetherin signals were equal between samples from HepAD38-tetherin and HepAD38-TCΔ19 cells (Fig. 5A, middle, compare lanes 5 and 6). However, when the enveloped HBV particles were purified by immunoprecipitation with HBsAg-specific antibodies and subjected to tetherin immunoblotting, the signal of tetherin vanished, except for TCΔ19 associated with HBV virions (Fig. 5A, bottom). Furthermore, we found that particle gel assays with supernatant samples from tetherin-transfected HepG2 cells also showed tetherin signals in the absence of any HBV component (Fig. 5B). Thus, we infer that the signal of full-length tetherin detected by the particle gel assay was from a host cell component(s) but not from subviral particles.
Taken together, the above-described data suggest that tetherin, per its reported antiviral mechanism, forms a dimer and inhibits HBV virion release by inserting one terminus into the virus and anchoring another terminus in the cell, and the integrity of the GPI anchor pair is required to maintain its tethering function.
Tetherin colocalizes with HBV virions intracellularly.
It has been reported that tetherin exerts a broad blocking effect against enveloped virus release, regardless of whether budding occurs at the plasma membrane or intracellular compartments (51). In fact, HBV is an intracellular budding virus whose maturation takes place in the multivesicular body (MVB) (10, 11). To investigate the tethering mechanism of tetherin-mediated restriction of HBV virion egress, we performed confocal microscopy analysis of the potential intracellular colocalization of tetherin and HBV virions. In HepAD38-tetherin cells cultured in tetracycline-free medium, the HBV virion, as represented by the viral large surface protein (L), colocalized with Alix, which is the marker for the MVB (Fig. 6A). Tetherin was found to be highly expressed on the cell membrane but also colocalized with Alix in the cytoplasm (Fig. 6B), suggesting that tetherin is associated with the MVB, which is theoretically conceivable since tetherin is a transmembrane protein. The colocalization between HBV L and tetherin was observed intracellularly but not on the cell surface (Fig. 6C). The association between tetherin and HBV virions in the MVB compartment was further confirmed by triple staining (Fig. 6D). These data indicate that tetherin inhibits HBV virion release through blocking MVB-dependent virion assembly and/or secretion, which represents a novel intracellular tethering mechanism distinct from the reported tetherin-virus interaction on the cell surface.
FIG 6.

Intracellular colocalization of tetherin with HBV virions and MVBs. HepAD38-tetherin cells were cultured in tetracycline-free medium for 12 days and subjected to immunofluorescence confocal microscopy. (A) Colocalization of HBV virions (HBV L protein served as a marker) (red) and MVBs (Alix served as a marker) (green). Nuclei were counterstained with DAPI (blue). The inset in the merged image shows the colocalization signals (yellow) at a higher magnification. (B) Association between tetherin (red) and MVBs (Alix) (green). (C) Colocalization of HBV virions (L) (red) and tetherin (green). (D) Triple immunostaining of HBV virions (L) (blue), tetherin (red), and MVBs (Alix) (green). The colocalization signal (white) is further shown in the inset with an expanded view.
To further confirm the intracellular inhibition of HBV virion release by tetherin, we conducted transmission electron microscopy (TEM) analysis of HBV virions in cells with or without tetherin overexpression. As shown in Fig. 7, consistent with data from a previously reported TEM analysis of HBV morphogenesis (64), ultrathin sections of HepAD38-control cells exhibited classical intracellular budding of mature HBV virions in cytoplasmic cisternae, presumably MVBs (Fig. 7A). We did not find any HBV virion budding through the cell surface (data not shown). In HepAD38-TCΔ19 cells expressing dominant negative tetherin, virion secretion was slightly increased, and exocytosed Dane particles were found in the extracellular space (Fig. 7B). However, in HepAD38-tetherin cells with tetherin overexpression, cisterna compartments filled with large-scale clusters of virions were observed, and the “arrested” virions were linked to each other to form an aggregate-like complex (Fig. 7C and D), which is reminiscent of previously reported observations of HIV particles entrapped by tetherin on the cell surface (40).
FIG 7.

TEM of HBV virions. The indicated cells were cultured in tetracycline-free medium for 10 days, and ultrathin sections were prepared and negatively stained for TEM analysis. (A) HepAD38-control cells; (B) HepAD38-TCΔ19 cells; (C) HepAD38-tetherin cells; (D) expanded view of the area surrounded by a dotted line in panel C. Scale bars, high voltage (HV), and direct magnification are indicated. Representative HBV virions (∼40 to 50 nm) are indicated by arrows.
Collectively, the above-described imaging data suggest that tetherin inhibits HBV virion egress through an intracellular tethering mechanism that blocks virion morphogenesis or exocytosis in the MVB compartment.
Tetherin mediates the antiviral activity of IFN-α against HBV virion egress.
By making use of the overexpression of wild-type or dominant negative tetherin, we have provided multiple pieces of evidence supporting the notion that tetherin is an end effector of interferon-mediated suppression of HBV virion secretion. In order to further confirm the physiological role of tetherin with IFN-α stimulation, we established HepAD38-based cells with stable knockdown of tetherin by lentiviral shRNA transduction. As shown in Fig. 8, after induction of HBV replication for 8 days, treatment of the control knockdown cell line HepAD38-control_K.D with IFN-α for an additional 4 days induced tetherin expression and reduced HBV virion production by ∼40% without affecting viral DNA replication (Fig. 8B, lanes 1 and 2, and C); in marked contrast, IFN-α treatment failed to upregulate tetherin expression in HepAD38-tetherin_K.D cells, and HBV DNA replication and virion secretion remained unaffected compared with those in the untreated controls (Fig. 8B, lanes 3 and 4, and C), suggesting that tetherin plays an irreplaceable role in the IFN-α-elicited antiviral response against HBV virion egress.
FIG 8.
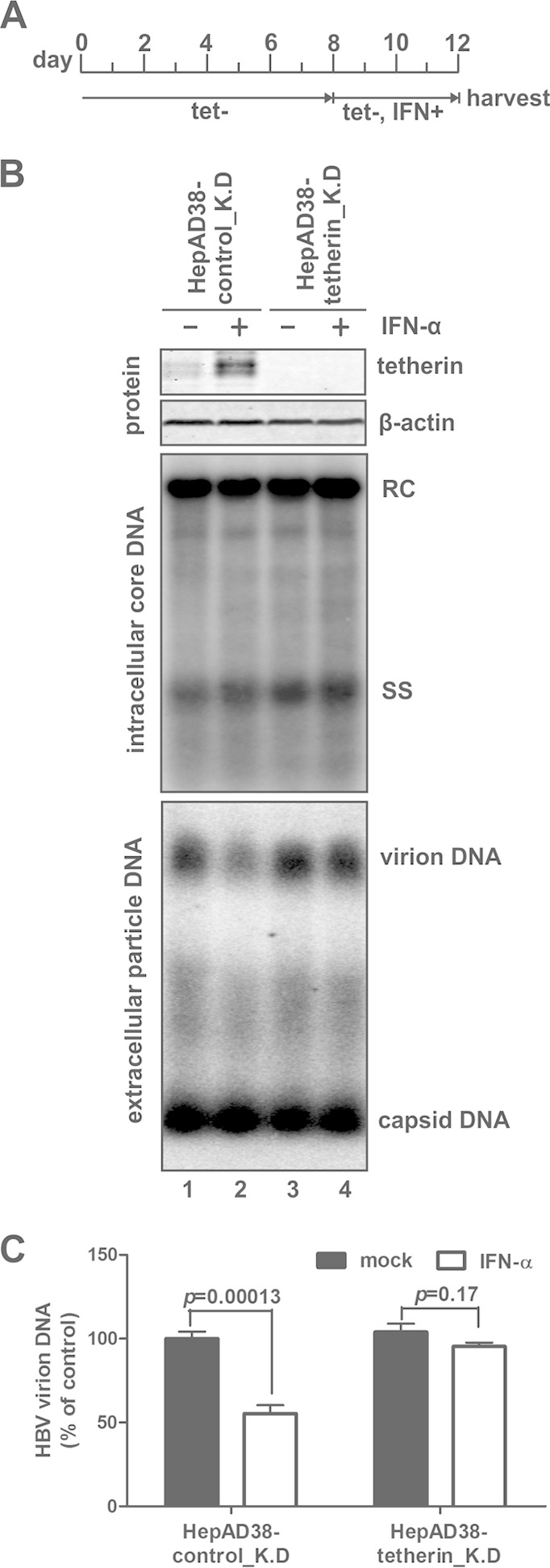
Knockdown of tetherin attenuates IFN-α-mediated suppression of HBV virion release. (A) Experimental procedure. HepAD38-control_K.D and HepAD38-tetherin_K.D cells were cultured in the presence of tetracycline until they became confluent, and the culture medium was then switched to tetracycline-free (tet−) medium to induce HBV replication. Eight days later, cells were untreated or treated with IFN-α (1,000 IU/ml) for another 4 days. During the entire culture period, fresh medium (with or without IFN-α) was replenished at 2-day intervals. (B) The harvested cell and supernatant samples were analyzed for tetherin expression, HBV core DNA replication, and extracellular accumulation of particle-associated HBV DNA. (C) The virion-associated DNA from 1 ml of the harvested supernatant was quantitated by HBsAg IP-qPCR and plotted as a percentage of the DNA signals from untreated HepAD38-control_K.D cell samples (means ± standard deviations).
DISCUSSION
Interferon-inducible tetherin is a broad-spectrum antiviral host protein that prevents the departure of progeny viruses from infected cells by directly tethering viral particles. To date, tetherin has been shown to restrict a variety of viruses from different families (see the introduction), with the only feature in common among them being that they all have a lipid envelope (39, 56). We report here that tetherin is induced in HBV-replicating cells by interferon and selectively inhibits HBV virion release, possibly through blocking intracellular virus budding, which may shed light on the antiviral mechanisms or limitations of IFN-α in the treatment of chronic hepatitis B and lead to a better understanding of tetherin biology in general.
Our previous attempts to study the potential effects of IFN and tetherin on HBV particle secretion were hampered by unsuitable experimental conditions. First, IFN-α treatment of HBV-replicating cell cultures, including transient and stable transfections and infection, normally results in reduced intracellular DNA replication and/or gene expression, which masks the potential effect of IFN on viral particle release (15, 21, 32). When we studied the antiviral function of IFN in inducible HBV stable cell lines with treatment at different time points after the initiation of viral replication, we found that intracellular HBV replication became resistant to IFN in prolonged cultures with established viral replication. Although the detailed mechanisms of viral resistance to IFN under such conditions remain unknown, the induction of tetherin by IFN seemed to indicate that IFN signaling is, at least partially, functional to induce certain ISGs (Fig. 1A and B). More importantly, such experimental conditions allow the study of IFN-mediated inhibition of HBV particle egress to become practical (Fig. 1C and D). On the other hand, we previously reported that overexpression of tetherin by transfection inhibited HBV DNA replication primarily through reducing viral RNA transcription from the transfected plasmid in cell culture (21). We reasoned that such an observation was due to a potential artifact caused by cotransfections; therefore, we established a cell system expressing tetherin and HBV from the integrated transgene template to avoid this obstacle and clearly demonstrated and characterized the antiviral activity of tetherin against HBV virion egress. Although the observations from this experimental system are conditional and may be cell type specific, this is, to the best of our knowledge, the only available cell culture system for highlighting the authentic role of tetherin in IFN-elicited antiviral responses to HBV.
HBV secretes three major types of viral particles in cell cultures, including enveloped virions, subviral particles, and nonenveloped naked capsids (Fig. 1C) (60, 61). It has been well accepted that HBV subviral particles assemble in the ER and are secreted via the ER-Golgi constitutive secretory pathway, while virion morphogenesis takes place on the membranes of the intracellular MVB, and the MVB transports virions to the plasma membrane and releases the virus through exocytosis (10, 11). The detailed noncytolytic secretion pathway for naked capsids remains elusive, but it may require the involvement of host protein Alix/AIP1 in an ESCRT-independent manner (65). While virions and subviral particles indicate HBV viremia and antigenemia in hepatitis B patients, naked capsids are absent from circulation in vivo, perhaps due to the rapid development of long-lasting anticore antibodies in patients upon HBV infection (66, 67). Nevertheless, tetherin selectively inhibits virion egress but not the release of subviral particles and naked capsids, suggesting that the antiviral specificity of tetherin relies on the composition and/or secretion pathway of different HBV particles. Considering that tetherin inhibits enveloped viruses exclusively, it is conceivable that the release of naked HBV capsid is not affected by tetherin. According to data from previous reports, most viruses restricted by tetherin acquire their envelopes by directly budding from the plasma membrane, with few exceptions, such as herpes simplex virus (HSV), HCV, and HCoV-229E, which bud at intracellular membrane structures (43, 51, 52, 68). A recent study demonstrated that tetherin and HCV core protein colocalize on lipid droplets, where the assembly and maturation of HCV particles take place (52). In agreement with the intracellular budding site of HBV, we observed that tetherin and HBV virions colocalize at the intracellular MVB by confocal microscopy and that HBV virions are entrapped in the cytoplasmic cisterna compartment under conditions of tetherin overexpression by TEM analysis. Therefore, using HBV as a model, our data further confirmed that tetherin possesses broad-spectrum antiviral activity against enveloped viruses, including both cell surface and intracellular budding viruses.
Previous studies suggest that the viral envelope structure, but not the envelope protein(s) per se, determines the physical interaction between virus and tetherin (39, 56). In our study, although tetherin colocalizes with HBV virions intracellularly at the MVB complex, we could not detect an association between tetherin and HBV envelope proteins (L/M/S) by coimmunoprecipitation assays with cell lysates prepared by a detergent-based lysis method (data not shown), which further favors the above-described notion. Interestingly, by taking advantage of the GPI anchor-truncated tetherin (TCΔ19), we demonstrated that tetherin is able to incorporate into HBV virions through its N-terminal TM domain, likely in a dimeric formation; the TCΔ19 mutant possesses dominant negative activity under basal levels or IFN-induced expression of endogenous tetherin, suggesting that TCΔ19 could form a heterogeneous dimer with endogenous tetherin; and the integrity of the GPI anchor pair is absolutely required for the tetherin dimer to fetter the HBV virion to the cell. In agreement with our findings, a previously reported study demonstrated that a GPI anchor deletion mutant retained its dimerization ability and exhibited dominant negative activity against HIV-1 secretion in the absence of Vpu (63). However, whether wild-type tetherin favors tethering of HBV virions by inserting a TM or GPI anchor domain into the virus awaits further investigation. Interestingly, we also found that the tetherin-containing host complex exists in cell culture supernatants by a particle gel assay (Fig. 5), but the nature and function of such a tetherin-associated host complex, presumably a membrane structure(s), remain obscure.
Although tetherin is capable of tethering HBV virions, the antiviral activity is not sufficient enough to completely shut down the release of progeny virions. The observed modest anti-HBV activity of tetherin is probably due to the possibility that HBV either is inherently unsusceptible to tetherin or has evolved countermeasures to antagonize tetherin. Regarding the latter, it has been reported that many viruses encode a tetherin antagonist(s) for evasion, such as the above-mentioned HIV-1 accessory protein Vpu, simian immunodeficiency virus (SIV) Nef, Ebola virus glycoprotein, KSHV K5 protein, and multiple glycoproteins of HSV-2, etc., which inhibits tetherin mainly through downregulation and/or sequestration (68, 69). However, we compared the expression levels of tetherin in HBV-transfected cells with those in mock-transfected cells and did not observe a noticeable alteration of tetherin expression by HBV (data not shown). In addition, our results demonstrated that HBV virions remained vulnerable to interferon-induced tetherin in the context of high levels of intracellular HBV expression and replication (Fig. 1 and 6). Therefore, it is unlikely that HBV actively antagonizes tetherin. On the other hand, it is also possible that hepatocyte-derived cells lack an unknown cofactor(s) for the optimal function of tetherin or express a negative regulator(s) that restricts tetherin, which may be responsible for the observed weak antiviral effect of tetherin on HBV in HepG2 cells and on HCV in Huh7 cells (52–54). In addition, compared to the 100- to 1,000-fold inhibition of production of Vpu-null HIV by tetherin in HEK 293T and HT1080 cells, overexpression of tetherin reduced Vpu-null HIV particle release by only <10-fold in Huh7 cells (40, 54).
IFN-α is an approved medication for the treatment of chronic hepatitis B (70). Acting as an ISG, tetherin single-handedly mediates the inhibition of HBV virion egress by IFN in cell cultures, which suggests that tetherin may play a specific role in the IFN-elicited antiviral response against HBV in vivo. Therefore, tetherin, or its artificially engineered mimetics (71), if with improved antiviral potency against HBV, could be potentially developed into antiviral therapies to potentiate the efficacy of IFN treatment and to prevent virus spread in hepatitis B patients receiving liver transplantation.
ACKNOWLEDGMENTS
We thank Christoph Seeger (Fox Chase Cancer Center) for providing the HepAD38 cell line and Andrea Cuconati for critical readings of the manuscript. We also thank the confocal microscopy facility and electron microscopy facility at Indiana University School of Medicine for technical support.
This study was supported by the National Institutes of Health (grants R21AI103838 and R01AI094474) and by the Hepatitis B Foundation through an appropriation from the Commonwealth of Pennsylvania.
REFERENCES
- 1.Liang TJ. 2009. Hepatitis B: the virus and disease. Hepatology 49:S13–S21. doi: 10.1002/hep.22881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Block TM, Guo H, Guo JT. 2007. Molecular virology of hepatitis B virus for clinicians. Clin Liver Dis 11:685–706, vii. doi: 10.1016/j.cld.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deny P, Zoulim F. 2010. Hepatitis B virus: from diagnosis to treatment. Pathol Biol (Paris) 58:245–253. doi: 10.1016/j.patbio.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 4.Trepo C, Chan HL, Lok A. 2014. Hepatitis B virus infection. Lancet 384:2053–2063. doi: 10.1016/S0140-6736(14)60220-8. [DOI] [PubMed] [Google Scholar]
- 5.Seeger C, Mason WS. 2000. Hepatitis B virus biology. Microbiol Mol Biol Rev 64:51–68. doi: 10.1128/MMBR.64.1.51-68.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ni Y, Lempp FA, Mehrle S, Nkongolo S, Kaufman C, Falth M, Stindt J, Koniger C, Nassal M, Kubitz R, Sultmann H, Urban S. 2014. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 146:1070–1083. doi: 10.1053/j.gastro.2013.12.024. [DOI] [PubMed] [Google Scholar]
- 7.Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, Huang Y, Qi Y, Peng B, Wang H, Fu L, Song M, Chen P, Gao W, Ren B, Sun Y, Cai T, Feng X, Sui J, Li W. 2012. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 1:e00049. doi: 10.7554/eLife.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang HC, Chen CC, Chang WC, Tao MH, Huang C. 2012. Entry of hepatitis B virus into immortalized human primary hepatocytes by clathrin-dependent endocytosis. J Virol 86:9443–9453. doi: 10.1128/JVI.00873-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo H, Mao R, Block TM, Guo JT. 2010. Production and function of the cytoplasmic deproteinized relaxed circular DNA of hepadnaviruses. J Virol 84:387–396. doi: 10.1128/JVI.01921-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Patient R, Hourioux C, Roingeard P. 2009. Morphogenesis of hepatitis B virus and its subviral envelope particles. Cell Microbiol 11:1561–1570. doi: 10.1111/j.1462-5822.2009.01363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prange R. 2012. Host factors involved in hepatitis B virus maturation, assembly, and egress. Med Microbiol Immunol 201:449–461. doi: 10.1007/s00430-012-0267-9. [DOI] [PubMed] [Google Scholar]
- 12.Cai X, Chiu YH, Chen ZJ. 2014. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol Cell 54:289–296. doi: 10.1016/j.molcel.2014.03.040. [DOI] [PubMed] [Google Scholar]
- 13.Kawai T, Akira S. 2006. Innate immune recognition of viral infection. Nat Immunol 7:131–137. doi: 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- 14.Sadler AJ, Williams BR. 2008. Interferon-inducible antiviral effectors. Nat Rev Immunol 8:559–568. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robek MD, Boyd BS, Chisari FV. 2005. Lambda interferon inhibits hepatitis B and C virus replication. J Virol 79:3851–3854. doi: 10.1128/JVI.79.6.3851-3854.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Isogawa M, Robek MD, Furuichi Y, Chisari FV. 2005. Toll-like receptor signaling inhibits hepatitis B virus replication in vivo. J Virol 79:7269–7272. doi: 10.1128/JVI.79.11.7269-7272.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wieland SF, Chisari FV. 2005. Stealth and cunning: hepatitis B and hepatitis C viruses. J Virol 79:9369–9380. doi: 10.1128/JVI.79.15.9369-9380.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang H, Ryu WS. 2010. Hepatitis B virus polymerase blocks pattern recognition receptor signaling via interaction with DDX3: implications for immune evasion. PLoS Pathog 6:e1000986. doi: 10.1371/journal.ppat.1000986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lang T, Lo C, Skinner N, Locarnini S, Visvanathan K, Mansell A. 2011. The hepatitis B e antigen (HBeAg) targets and suppresses activation of the Toll-like receptor signaling pathway. J Hepatol 55:762–769. doi: 10.1016/j.jhep.2010.12.042. [DOI] [PubMed] [Google Scholar]
- 20.Liu Y, Li J, Chen J, Li Y, Wang W, Du X, Song W, Zhang W, Lin L, Yuan Z. 2015. Hepatitis B virus polymerase disrupts K63-linked ubiquitination of STING to block innate cytosolic DNA-sensing pathways. J Virol 89:2287–2300. doi: 10.1128/JVI.02760-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mao R, Zhang J, Jiang D, Cai D, Levy JM, Cuconati A, Block TM, Guo JT, Guo H. 2011. Indoleamine 2,3-dioxygenase mediates the antiviral effect of gamma interferon against hepatitis B virus in human hepatocyte-derived cells. J Virol 85:1048–1057. doi: 10.1128/JVI.01998-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo H, Jiang D, Ma D, Chang J, Dougherty AM, Cuconati A, Block TM, Guo JT. 2009. Activation of pattern recognition receptor-mediated innate immunity inhibits the replication of hepatitis B virus in human hepatocyte-derived cells. J Virol 83:847–858. doi: 10.1128/JVI.02008-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo F, Han Y, Zhao X, Wang J, Liu F, Xu C, Wei L, Jiang JD, Block TM, Guo JT, Chang J. 2015. STING agonists induce an innate antiviral immune response against hepatitis B virus. Antimicrob Agents Chemother 59:1273–1281. doi: 10.1128/AAC.04321-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang J, Block TM, Guo JT. 2012. The innate immune response to hepatitis B virus infection: implications for pathogenesis and therapy. Antiviral Res 96:405–413. doi: 10.1016/j.antiviral.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 25.Scaglione SJ, Lok AS. 2012. Effectiveness of hepatitis B treatment in clinical practice. Gastroenterology 142:1360–1368.e1361. doi: 10.1053/j.gastro.2012.01.044. [DOI] [PubMed] [Google Scholar]
- 26.Chisari FV, Isogawa M, Wieland SF. 2010. Pathogenesis of hepatitis B virus infection. Pathol Biol (Paris) 58:258–266. doi: 10.1016/j.patbio.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schneider WM, Chevillotte MD, Rice CM. 2014. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol 32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Uprichard SL, Wieland SF, Althage A, Chisari FV. 2003. Transcriptional and posttranscriptional control of hepatitis B virus gene expression. Proc Natl Acad Sci U S A 100:1310–1315. doi: 10.1073/pnas.252773599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mao R, Nie H, Cai D, Zhang J, Liu H, Yan R, Cuconati A, Block TM, Guo JT, Guo H. 2013. Inhibition of hepatitis B virus replication by the host zinc finger antiviral protein. PLoS Pathog 9:e1003494. doi: 10.1371/journal.ppat.1003494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu F, Campagna M, Qi Y, Zhao X, Guo F, Xu C, Li S, Li W, Block TM, Chang J, Guo JT. 2013. Alpha-interferon suppresses hepadnavirus transcription by altering epigenetic modification of cccDNA minichromosomes. PLoS Pathog 9:e1003613. doi: 10.1371/journal.ppat.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Belloni L, Allweiss L, Guerrieri F, Pediconi N, Volz T, Pollicino T, Petersen J, Raimondo G, Dandri M, Levrero M. 2012. IFN-alpha inhibits HBV transcription and replication in cell culture and in humanized mice by targeting the epigenetic regulation of the nuclear cccDNA minichromosome. J Clin Invest 122:529–537. doi: 10.1172/JCI58847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wieland SF, Eustaquio A, Whitten-Bauer C, Boyd B, Chisari FV. 2005. Interferon prevents formation of replication-competent hepatitis B virus RNA-containing nucleocapsids. Proc Natl Acad Sci U S A 102:9913–9917. doi: 10.1073/pnas.0504273102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu C, Guo H, Pan XB, Mao R, Yu W, Xu X, Wei L, Chang J, Block TM, Guo JT. 2010. Interferons accelerate decay of replication-competent nucleocapsids of hepatitis B virus. J Virol 84:9332–9340. doi: 10.1128/JVI.00918-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao B, Duan Z, Xu W, Xiong S. 2009. Tripartite motif-containing 22 inhibits the activity of hepatitis B virus core promoter, which is dependent on nuclear-located RING domain. Hepatology 50:424–433. doi: 10.1002/hep.23011. [DOI] [PubMed] [Google Scholar]
- 35.Li J, Lin S, Chen Q, Peng L, Zhai J, Liu Y, Yuan Z. 2010. Inhibition of hepatitis B virus replication by MyD88 involves accelerated degradation of pregenomic RNA and nuclear retention of pre-S/S RNAs. J Virol 84:6387–6399. doi: 10.1128/JVI.00236-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lucifora J, Xia Y, Reisinger F, Zhang K, Stadler D, Cheng X, Sprinzl MF, Koppensteiner H, Makowska Z, Volz T, Remouchamps C, Chou WM, Thasler WE, Huser N, Durantel D, Liang TJ, Munk C, Heim MH, Browning JL, Dejardin E, Dandri M, Schindler M, Heikenwalder M, Protzer U. 2014. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 343:1221–1228. doi: 10.1126/science.1243462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nguyen DH, Hu J. 2008. Reverse transcriptase- and RNA packaging signal-dependent incorporation of APOBEC3G into hepatitis B virus nucleocapsids. J Virol 82:6852–6861. doi: 10.1128/JVI.00465-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nguyen DH, Gummuluru S, Hu J. 2007. Deamination-independent inhibition of hepatitis B virus reverse transcription by APOBEC3G. J Virol 81:4465–4472. doi: 10.1128/JVI.02510-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neil SJ. 2013. The antiviral activities of tetherin. Curr Top Microbiol Immunol 371:67–104. doi: 10.1007/978-3-642-37765-5_3. [DOI] [PubMed] [Google Scholar]
- 40.Neil SJ, Zang T, Bieniasz PD. 2008. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451:425–430. doi: 10.1038/nature06553. [DOI] [PubMed] [Google Scholar]
- 41.Jouvenet N, Neil SJ, Zhadina M, Zang T, Kratovac Z, Lee Y, McNatt M, Hatziioannou T, Bieniasz PD. 2009. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J Virol 83:1837–1844. doi: 10.1128/JVI.02211-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaletsky RL, Francica JR, Agrawal-Gamse C, Bates P. 2009. Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc Natl Acad Sci U S A 106:2886–2891. doi: 10.1073/pnas.0811014106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mansouri M, Viswanathan K, Douglas JL, Hines J, Gustin J, Moses AV, Fruh K. 2009. Molecular mechanism of BST2/tetherin downregulation by K5/MIR2 of Kaposi's sarcoma-associated herpesvirus. J Virol 83:9672–9681. doi: 10.1128/JVI.00597-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sakuma T, Noda T, Urata S, Kawaoka Y, Yasuda J. 2009. Inhibition of Lassa and Marburg virus production by tetherin. J Virol 83:2382–2385. doi: 10.1128/JVI.01607-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Radoshitzky SR, Dong L, Chi X, Clester JC, Retterer C, Spurgers K, Kuhn JH, Sandwick S, Ruthel G, Kota K, Boltz D, Warren T, Kranzusch PJ, Whelan SP, Bavari S. 2010. Infectious Lassa virus, but not filoviruses, is restricted by BST-2/tetherin. J Virol 84:10569–10580. doi: 10.1128/JVI.00103-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weidner JM, Jiang D, Pan XB, Chang J, Block TM, Guo JT. 2010. Interferon-induced cell membrane proteins, IFITM3 and tetherin, inhibit vesicular stomatitis virus infection via distinct mechanisms. J Virol 84:12646–12657. doi: 10.1128/JVI.01328-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yondola MA, Fernandes F, Belicha-Villanueva A, Uccelini M, Gao Q, Carter C, Palese P. 2011. Budding capability of the influenza virus neuraminidase can be modulated by tetherin. J Virol 85:2480–2491. doi: 10.1128/JVI.02188-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Watanabe R, Leser GP, Lamb RA. 2011. Influenza virus is not restricted by tetherin whereas influenza VLP production is restricted by tetherin. Virology 417:50–56. doi: 10.1016/j.virol.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mangeat B, Cavagliotti L, Lehmann M, Gers-Huber G, Kaur I, Thomas Y, Kaiser L, Piguet V. 2012. Influenza virus partially counteracts restriction imposed by tetherin/BST-2. J Biol Chem 287:22015–22029. doi: 10.1074/jbc.M111.319996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bampi C, Rasga L, Roux L. 2013. Antagonism to human BST-2/tetherin by Sendai virus glycoproteins. J Gen Virol 94:1211–1219. doi: 10.1099/vir.0.051771-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang SM, Huang KJ, Wang CT. 2014. BST2/CD317 counteracts human coronavirus 229E productive infection by tethering virions at the cell surface. Virology 449:287–296. doi: 10.1016/j.virol.2013.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Amet T, Byrd D, Hu N, Sun Q, Li F, Zhao Y, Hu S, Grantham A, Yu Q. 2014. BST-2 expression in human hepatocytes is inducible by all three types of interferons and restricts production of hepatitis C virus. Curr Mol Med 14:349–360. doi: 10.2174/1566524013666131118111719. [DOI] [PubMed] [Google Scholar]
- 53.Pan XB, Qu XW, Jiang D, Zhao XL, Han JC, Wei L. 2013. BST2/tetherin inhibits hepatitis C virus production in human hepatoma cells. Antiviral Res 98:54–60. doi: 10.1016/j.antiviral.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 54.Ye L, Wang X, Li J, Liu J, Ramirez SH, Wu J, Ho W. 2012. Tetherin has negligible activity in restricting hepatitis C virus in hepatocytes. Innate Immun 18:398–405. doi: 10.1177/1753425911412984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dafa-Berger A, Kuzmina A, Fassler M, Yitzhak-Asraf H, Shemer-Avni Y, Taube R. 2012. Modulation of hepatitis C virus release by the interferon-induced protein BST-2/tetherin. Virology 428:98–111. doi: 10.1016/j.virol.2012.03.011. [DOI] [PubMed] [Google Scholar]
- 56.Swiecki M, Omattage NS, Brett TJ. 2013. BST-2/tetherin: structural biology, viral antagonism, and immunobiology of a potent host antiviral factor. Mol Immunol 54:132–139. doi: 10.1016/j.molimm.2012.11.008. [DOI] [PubMed] [Google Scholar]
- 57.Venkatesh S, Bieniasz PD. 2013. Mechanism of HIV-1 virion entrapment by tetherin. PLoS Pathog 9:e1003483. doi: 10.1371/journal.ppat.1003483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guo H, Jiang D, Zhou T, Cuconati A, Block TM, Guo JT. 2007. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J Virol 81:12472–12484. doi: 10.1128/JVI.01123-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ladner SK, Otto MJ, Barker CS, Zaifert K, Wang GH, Guo JT, Seeger C, King RW. 1997. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob Agents Chemother 41:1715–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guo H, Pan X, Mao R, Zhang X, Wang L, Lu X, Chang J, Guo JT, Passic S, Krebs FC, Wigdahl B, Warren TK, Retterer CJ, Bavari S, Xu X, Cuconati A, Block TM. 2011. Alkylated porphyrins have broad antiviral activity against hepadnaviruses, flaviviruses, filoviruses, and arenaviruses. Antimicrob Agents Chemother 55:478–486. doi: 10.1128/AAC.00989-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ning X, Nguyen D, Mentzer L, Adams C, Lee H, Ashley R, Hafenstein S, Hu J. 2011. Secretion of genome-free hepatitis B virus—single strand blocking model for virion morphogenesis of para-retrovirus. PLoS Pathog 7:e1002255. doi: 10.1371/journal.ppat.1002255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gerlich WH, Glebe D, Schuttler CG. 2007. Deficiencies in the standardization and sensitivity of diagnostic tests for hepatitis B virus. J Viral Hepat 14(Suppl 1):16–21. doi: 10.1111/j.1365-2893.2007.00912.x. [DOI] [PubMed] [Google Scholar]
- 63.Lv M, Wang J, Zhu Y, Wang X, Zuo T, Liu D, Zhang J, Wu J, Zhang H, Kong W, Yu X. 2013. Overexpression of inactive tetherin delGPI mutant inhibits HIV-1 Vpu-mediated antagonism of endogenous tetherin. FEBS Lett 587:37–43. doi: 10.1016/j.febslet.2012.11.022. [DOI] [PubMed] [Google Scholar]
- 64.Roingeard P, Sureau C. 1998. Ultrastructural analysis of hepatitis B virus in HepG2-transfected cells with special emphasis on subviral filament morphogenesis. Hepatology 28:1128–1133. doi: 10.1002/hep.510280431. [DOI] [PubMed] [Google Scholar]
- 65.Bardens A, Doring T, Stieler J, Prange R. 2011. Alix regulates egress of hepatitis B virus naked capsid particles in an ESCRT-independent manner. Cell Microbiol 13:602–619. doi: 10.1111/j.1462-5822.2010.01557.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hoofnagle JH, Gerety RJ, Ni LY, Barker LF. 1974. Antibody to hepatitis B core antigen. A sensitive indicator of hepatitis B virus replication. N Engl J Med 290:1336–1340. [DOI] [PubMed] [Google Scholar]
- 67.Hoofnagle JH, Gerety RJ, Barker LF. 1973. Antibody to hepatitis-B-virus core in man. Lancet ii:869–873. [DOI] [PubMed] [Google Scholar]
- 68.Liu Y, Luo S, He S, Zhang M, Wang P, Li C, Huang W, Hu B, Griffin GE, Shattock RJ, Hu Q. 2015. Tetherin restricts HSV-2 release and is counteracted by multiple viral glycoproteins. Virology 475:96–109. doi: 10.1016/j.virol.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 69.Douglas JL, Gustin JK, Viswanathan K, Mansouri M, Moses AV, Fruh K. 2010. The great escape: viral strategies to counter BST-2/tetherin. PLoS Pathog 6:e1000913. doi: 10.1371/journal.ppat.1000913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hoofnagle JH, Doo E, Liang TJ, Fleischer R, Lok AS. 2007. Management of hepatitis B: summary of a clinical research workshop. Hepatology 45:1056–1075. doi: 10.1002/hep.21627. [DOI] [PubMed] [Google Scholar]
- 71.Perez-Caballero D, Zang T, Ebrahimi A, McNatt MW, Gregory DA, Johnson MC, Bieniasz PD. 2009. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell 139:499–511. doi: 10.1016/j.cell.2009.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]