

ABSTRACT
Coronaviruses (CoVs) assemble by budding into the lumen of the early Golgi complex prior to exocytosis. The small CoV envelope (E) protein plays roles in assembly, virion release, and pathogenesis. CoV E has a single hydrophobic domain (HD), is targeted to Golgi complex membranes, and has cation channel activity in vitro. However, the precise functions of the CoV E protein during infection are still enigmatic. Structural data for the severe acute respiratory syndrome (SARS)-CoV E protein suggest that it assembles into a homopentamer. Specific residues in the HD regulate the ion-conducting pore formed by SARS-CoV E in artificial bilayers and the pathogenicity of the virus during infection. The E protein from the avian infectious bronchitis virus (IBV) has dramatic effects on the secretory system which require residues in the HD. Here, we use the known structural data from SARS-CoV E to infer the residues important for ion channel activity and the oligomerization of IBV E. We present biochemical data for the formation of two distinct oligomeric pools of IBV E in transfected and infected cells and the residues required for their formation. A high-order oligomer of IBV E is required for the production of virus-like particles (VLPs), implicating this form of the protein in virion assembly. Additionally, disruption of the secretory pathway by IBV E correlates with a form that is likely monomeric, suggesting that the effects on the secretory pathway are independent of E ion channel activity.
IMPORTANCE CoVs are important human pathogens with significant zoonotic potential, as demonstrated by the emergence of SARS-CoV and Middle East respiratory syndrome (MERS)-CoV. Progress has been made toward identifying potential vaccine candidates in mouse models of CoV infection, including the use of attenuated viruses that lack the CoV E protein or express E-protein mutants. However, no approved vaccines or antiviral therapeutics exist. We previously reported that the hydrophobic domain of the IBV E protein, a putative viroporin, causes disruption of the mammalian secretory pathway when exogenously expressed in cells. Understanding the mechanism of this disruption could lead to the identification of novel antiviral therapeutics. Here, we present biochemical evidence for two distinct oligomeric forms of IBV E, one essential for assembly and the other with a role in disruption of the secretory pathway. Discovery of two forms of CoV E protein will provide additional targets for antiviral therapeutics.
INTRODUCTION
Coronaviruses (CoVs) are enveloped viruses with positive-sense, single-stranded RNA genomes that infect avian and mammalian species. These viruses cause about 20% of common colds in humans. However, CoVs have presented a more serious threat to human health in recent years. The emergence of severe acute respiratory syndrome (SARS)-CoV in 2002 and the Middle East respiratory syndrome (MERS)-CoV in 2012 demonstrates the zoonotic potential of this family of viruses (1). There has been some success in the development of mouse models of SARS-CoV and MERS-CoV infection, and candidate vaccines where the envelope (E) protein is deleted or mutated have been described (2–7). However, there is still much that is unclear regarding the role of the CoV E protein in infection.
CoVs acquire their membrane envelope by budding into the endoplasmic reticulum (ER)-Golgi complex intermediate compartment (ERGIC), a characteristic that sets CoVs apart from other well-studied enveloped viruses (8). The infectious virions that bud into the ERGIC lumen must then navigate the host secretory pathway to be released from the cell. CoVs have three major structural proteins that are constituents of the virion envelope (1). The CoV S protein is the attachment and fusion protein. The CoV M protein coordinates the process of virion assembly and is the most abundant protein in the virion envelope. Lastly, the CoV E protein contains a single hydrophobic domain and is a minor component of the virion envelope. Only a small portion of the E protein expressed during infection is incorporated into the virion envelope; the majority of E remains localized to Golgi complex membranes (9–12). The E protein has been shown to be required for the robust production of virus, since recombinant CoVs lacking the E protein grow to a significantly reduced titer or are propagation incompetent (13–15).
Three roles for CoV E protein have been proposed. A role for CoV E in assembly has been suggested on the basis of the observation that CoV E, along with the M protein, can drive the production of virus-like particles (VLPs); the interaction occurs via sequences in the cytoplasmic tails (16, 17). A role in the release of infectious virus that requires the hydrophobic domain (HD) of the E protein has been reported (18, 19). Lastly, residues in the HD of SARS-CoV E have been shown to promote viral fitness and pathogenesis in a mouse-adapted model of infection (20).
Early reports on several CoVs, including infectious bronchitis virus (IBV), SARS-CoV, mouse hepatitis virus (MHV), and the human CoV 229E, demonstrated that CoV E proteins form cation-selective ion channels in planar lipid bilayers, suggesting that CoV E functions as a viroporin (21, 22). Structural data support the idea that CoV E can oligomerize and form a channel. Bacterially expressed or synthetic peptides corresponding to the SARS-CoV E protein HD form pentameric α-helical bundles in planar lipid bilayers (23–26). Solution nuclear magnetic resonance analysis of a synthetic peptide of the SARS-CoV E protein in micelles also revealed a homopentameric structure (27). Recent studies have suggested that the MERS-CoV E protein also forms pentameric ion channels in lipid bilayers (28). Furthermore, SARS-CoV E forms a proteolipidic pore in which negatively charged lipids in bilayers enhance ion conductance and cation selectivity (29, 30).
SARS-CoV E residues N15 and V25, both of which are in the HD, are necessary for ion channel activity in lipid bilayers (24, 29). N15 and V25 promote viral fitness and pathogenesis in a mouse-adapted model of SARS-CoV infection, presumably through the necessity of these residues for ion channel activity (20). Despite the plethora of in vitro evidence supporting the role of CoV E as an ion channel and the role of E as a pathogenic determinant, the precise function of E as an ion channel in infected cells and animals is unknown. The best evidence for the ion channel activity of the CoV E protein during infection comes from experiments demonstrating that the drug hexamethylene amiloride, a known channel inhibitor, reduces the titer of MHV grown in cultured cells but not that of a mutant of MHV with the entire E protein deleted (22).
We previously reported that overexpression of IBV E induces disassembly of the Golgi complex as well as reduced trafficking of cargo molecules through the Golgi complex (19). Alanine mutagenesis of the HD of IBV E revealed that a single residue, T16, is required for Golgi complex disassembly and membrane trafficking disruption (31). Given that IBV E T16 is in the position equivalent to N15 in the SARS-CoV E, we predicted that the ability of IBV E to disrupt the secretory pathway is dependent on its ion channel activity. Further, we hypothesize that the HD (and T16 specifically) is required for modification of intracellular compartments to allow the assembly and release of infectious virions.
In the study described here, we investigated how the IBV E protein and two HD mutants behave in cells. We present evidence for two distinct pools of IBV E in transfected and infected cells. The results of studies obtained with the HD mutants suggest that the Golgi complex phenotypes observed with exogenous expression are independent of IBV E ion channel activity, leading to a model in which IBV E functions as (i) a monomer potentially interacting with a cellular protein(s) to alter the host secretory machinery and (ii) a high-molecular-weight (HMW) homo-oligomer with a function in virion assembly.
MATERIALS AND METHODS
Cell culture.
HeLa and Vero cells were cultured in Dulbecco's modified Eagle medium (DMEM; Invitrogen/Gibco, Grand Island, NY) containing 10% fetal bovine serum (FBS; Atlanta Biologicals, Lawrenceville, GA) and 0.1 mg/ml Normocin (InvivoGen, San Diego, CA) at 37°C under 5% CO2.
Plasmids.
The pBluescript, pCAGGS IBV E, pCAGGS IBV E-T16A, pCAGGS IBV M, pCAGGS IBV N, and pCAGGS VSV G plasmids have been previously described (19, 31). The pCAGGS IBV E-A26F plasmid was constructed using site-directed mutagenesis of the pBluescript IBV E expression plasmid with a QuikChange site-directed mutagenesis kit (Stratagene). The IBV E-A26F-coding sequence was then subcloned into pCAGGS-MCS using the EcoRI and SacI restriction sites.
Transient transfection.
The X-tremeGENE 9 DNA transfection reagent (Roche, Indianapolis, IN) was used to transiently transfect cells according to the manufacturer's protocol. Unless otherwise noted, subconfluent HeLa cells in 35-mm dishes were transfected with the following amounts of plasmid diluted into Opti-MEM medium (Invitrogen/Gibco) with a 1:3 ratio of X-tremeGENE 9: 1.0 μg pCAGGS IBV E, 1.0 μg pCAGGS IBV E-T16A, 1.0 μg pCAGGS IBV E-A26F, and 1.0 μg pCAGGS VSV G for sucrose gradient analysis and 0.5 μg pCAGGS VSV G for endo-β-N-acetylglucosaminidase H (endo H) trafficking assay (see below). The cells were used in experiments at 16 to 22 h after transfection, unless otherwise noted.
Antibodies.
The rabbit polyclonal and rat polyclonal antibodies recognizing the C terminus of IBV E have been described previously (32). The rabbit anti-IBV M and anti-IBV N antibodies have also been described (31). The rabbit polyclonal and mouse monoclonal antibodies recognizing vesicular stomatitis virus (VSV) and VSV glycoprotein (G), respectively, have been previously described (33, 34). The mouse anti-GM130 antibody was from BD Biosciences (San Diego, CA). The horseradish peroxidase-conjugated donkey anti-rabbit immunoglobulin and the horseradish peroxidase-conjugated sheep anti-mouse immunoglobulin antibodies were from GE Healthcare (Piscataway, NJ). The horseradish peroxidase-conjugated donkey anti-rat immunoglobulin antibody was from Jackson ImmunoResearch Laboratories (Bar Harbor, ME). The Alexa Fluor 488-conjugated anti-rabbit IgG and Alexa Fluor 568-conjugated anti-mouse IgG antibodies were from Invitrogen/Molecular Probes (Eugene, OR).
Sucrose gradient analysis.
HeLa cells transiently expressing wild-type (WT) or mutant IBV E or VSV G were washed with phosphate-buffered saline (PBS) at 16 h posttransfection. The cells were lysed for 10 min on ice with 100 μl of DDM lysis buffer (20 mM HEPES-KOH [pH 7.4], 100 mM NaCl, 20 mM imidazole, 1 mM CaCl2) containing 4.2% n-dodecyl-β-d-maltoside (DDM), protease inhibitor cocktail (Sigma), and 10% glycerol in all cases except for the comparison of VSV G and IBV E in Fig. 1A; qualitative analysis suggested that IBV E gradient peaks were sharper in the presence of glycerol; thus, glycerol was included in the lysis buffer and gradients for all subsequent experiments. The lysates were centrifuged at 20,000 × g for 10 min at 4°C, and the supernatants were loaded onto 5-ml 5 to 20% linear sucrose gradients consisting of DDM lysis buffer with 0.42% DDM over a 300-μl 60% sucrose cushion. The gradients were spun at 192,000 × g for 24 h at 4°C in a Beckman SW55Ti ultracentrifuge rotor. Fifteen fractions per gradient were collected using a Buchler Instruments Auto Densi-Flow II C apparatus. The fractions were then analyzed either by Western blotting or by phosphorimaging after immunoprecipitation, described below. Lysates were treated with 1% SDS prior to sucrose gradient analysis when specified.
FIG 1.
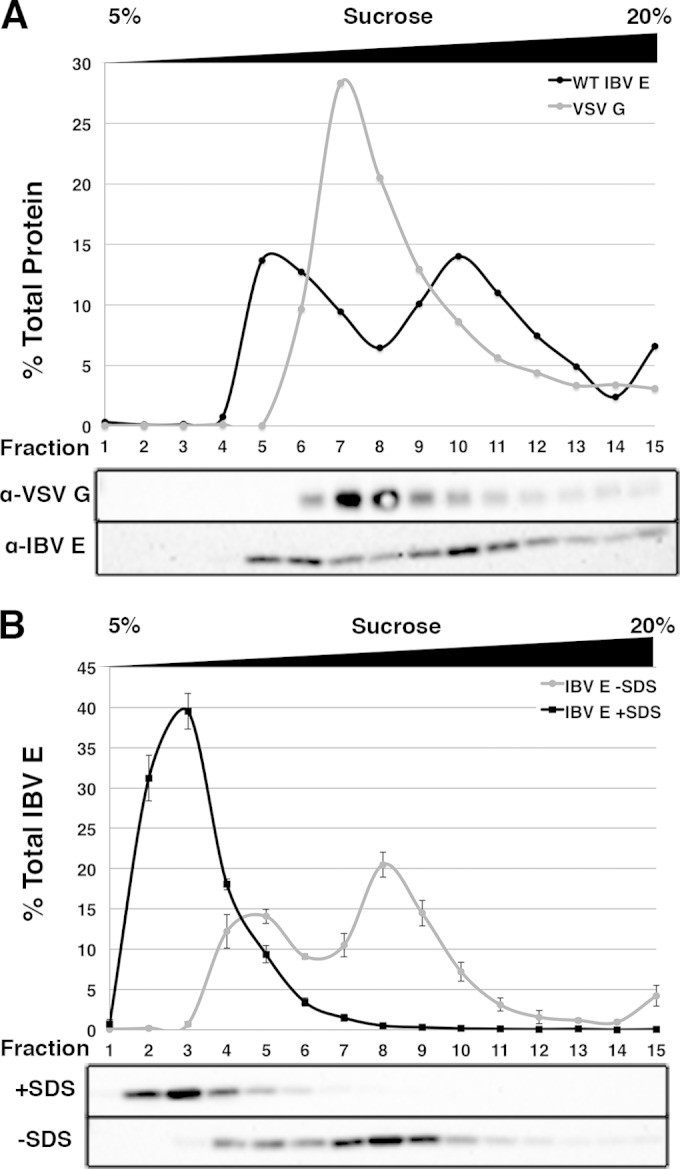
IBV E forms two pools in transfected cells. (A) HeLa cells expressing IBV E or VSV G were lysed and run on a 5 to 20% sucrose gradient, as described in Materials and Methods. Gradient fractions were collected and analyzed for the presence of IBV E or VSV G by Western blotting. Plots indicate the percentage of total protein in each fraction. (B) HeLa cells expressing IBV E were lysed and treated with SDS or not, prior to being run on a 5 to 20% sucrose gradient containing glycerol. Gradient fractions were collected and analyzed for the presence of IBV E by Western blotting. Qualitative observations suggest that sucrose gradients containing glycerol resulted in sharper gradient peaks; glycerol was thus included in all subsequent gradients. Error bars represent the SEMs from two and five independent experiments for the conditions with and without SDS, respectively.
(i) Western blot analysis.
A 4×-concentrated sample buffer (200 mM Tris-HCl [pH 6.8], 8% SDS, 60% glycerol, 0.2% bromophenol blue) was added to 15% of each fraction collected. The samples were heated at 100°C for 3 to 5 min in the presence of 2 to 5% 2-mercaptoethanol (BME), unless otherwise noted, and run on a 15% SDS-polyacrylamide gel. Gels were transferred to polyvinylidene fluoride (PVDF) Immobilon membranes (Millipore). The membranes were blocked for 1 h at room temperature in 5% milk in Tris-buffered saline (TBS) with Tween 20 (TBST; 10 mM Tris-HCl, 150 mM NaCl, 0.05% Tween 20). Proteins were detected using rabbit or rat anti-IBV E (1:10,000) in 2.5% milk in TBST overnight at 4°C. After they were washed in TBST, the membranes were then incubated in secondary horseradish peroxidase-conjugated donkey anti-rabbit or anti-rat immunoglobulin (1:10,000) for 1 h at room temperature. After washing, the membranes were incubated with the HyGlo Quick Spray chemiluminescent detection reagent (Denville Scientific Inc.). Images were collected using a Versa Doc model 5000 imaging system (Bio-Rad) and analyzed with Quantity One software.
(ii) Sucrose gradient analysis of IBV E from infected cells.
The Beaudette strain of recombinant IBV used in this study has been previously described (51, 52). Vero cells were inoculated with virus diluted in serum-free DMEM, and virus was adsorbed for 1 h with rocking. The inoculum was removed, and the cells were rinsed with DMEM containing 5% FBS and then incubated at 37°C in DMEM containing 5% FBS. For sucrose gradient analysis of IBV E during infection, Vero cells infected at a multiplicity of infection (MOI) of 1.0 were rinsed with PBS and lysed in DDM lysis buffer and protease inhibitor cocktail (Sigma) at 8 h postinfection. For sucrose gradient analysis of secreted IBV virions, Vero cells were inoculated at an MOI of 1.0 for 1 h at 37°C and treated as described above. At 24 h postinfection, the clarified cell supernatant was placed on a 4-ml 20 to 50% sucrose step gradient in TNE buffer (50 mM Tris-HCl [pH 7.4], 100 mM NaCl, 1 mM EDTA). The step gradients were spun at 198,000 × g for 90 min in an SW41Ti Beckman ultracentrifuge rotor at 4°C. The interface containing the virions was collected and diluted in TNE buffer, and the virions were pelleted with a 50-min spin as described above. The virions were resuspended in DDM lysis buffer containing protease inhibitor cocktail. Sucrose gradient analysis and Western blot analysis with the rabbit anti-IBV E antibody were performed as described above.
(iii) Cross-linking and anti-IBV E Dynabead immunoprecipitation.
HeLa cells transiently expressing IBV E, IBV E with the T16A mutation (IBV E-T16A), or IBV E with the A26F mutation (IBV E-A26F) were used at 18 h posttransfection. Lysis of transfected cells or purification of virions and sucrose gradient analysis were performed as described above. Fifteen fractions were collected for each gradient, and fractions representing the low-molecular-weight (LMW) pool (fractions 4, 5, and 6) and the high-molecular-weight pool (fractions 7, 8, and 9) were combined. The cross-linker dithiobis(succinimidyl propionate) (DSP; Pierce, Rockford, IL) was added to the combined fractions to a final concentration of 1 mM. The samples were incubated for 1 h at 4°C with rotation. The cross-linked samples were then added to 1 mg of washed rabbit anti-IBV E IgG-conjugated Dynabeads (Life Technologies AS, Oslo, Norway). The beads were prepared by conjugating 20 μg of protein G-Sepharose-purified rabbit anti-IBV E IgG per mg of Dynabeads as specified by the manufacturer's instructions. The samples were incubated for 1 h at 4°C with rotation. The Dynabeads were then washed in 1 ml of DDM lysis buffer. The Dynabeads were placed on a magnet for 1 min, the supernatant was removed, 100 μl of 1× sample buffer was added to each sample, and then the mixture was split in half. One half was treated with 5% BME to reverse the cross-links, and the other half was left unreduced. The samples were heated at 100°C for 5 min. The cross-linked samples were then evaluated on a 15% SDS-polyacrylamide gel along with various controls and analyzed by Western blotting as described above with the rat anti-IBV E polyclonal antibody.
(iv) Pulse-chase analysis of IBV E on sucrose gradients.
HeLa cells expressing IBV E were analyzed at 16 h posttransfection. Cells were starved in cysteine-methionine-free DMEM for 15 min, labeled with 250 μCi of EasyTag Express protein labeling mix [35S]cysteine-methionine (PerkinElmer, Boston, MA) per dish in cysteine-methionine-free DMEM for 15 min, and chased for various times in normal growth medium. Cells were washed with PBS and lysed in 100 μl of DDM lysis buffer containing protease inhibitor cocktail (Sigma). The samples were clarified and subjected to sucrose gradient analysis as described above. Twofold-concentrated detergent solution (125 mM EDTA, 2% NP-40, 0.8% deoxycholic acid, 100 mM Tris-HCl [pH 8.0]) and SDS were added to the sucrose gradient fractions to 1× and 0.2% final concentrations, respectively. IBV E was immunoprecipitated using 2 μl of the rabbit anti-IBV E antibody per fraction with incubation at 4°C for 2 h. Immune complexes were collected using 20 μl of washed Staphylococcus aureus Pansorbin cells and washed three times with radioimmunoprecipitation assay (RIPA) buffer (10 mM Tris-HCl [pH 7.4], 0.1% SDS, 1% deoxycholic acid, 1% NP-40, 150 mM NaCl). Concentrated sample buffer was added to each sample, and the immunocomplexes were eluted at 100°C for 3 min in the presence of 2% BME and run on a 15% SDS-polyacrylamide gel. Labeled IBV E was visualized using a Molecular Imager FX phosphorimager (Bio-Rad) and quantified using Quantity One software.
VSV G endo H assay.
HeLa cells coexpressing VSV G and IBV E, IBV E-T16A, or IBV E-A26F were used at 20 h posttransfection. Cells were starved in cysteine-methionine-free DMEM for 15 min, labeled with 60 μCi of EasyTag Express protein labeling mix [35S]cysteine-methionine per dish in cysteine-methionine-free DMEM for 10 min, and chased for various times in normal growth medium. Cells were washed with PBS and lysed in 500 μl of 1× detergent solution with protease inhibitor cocktail. The samples were clarified, and SDS was added to 0.2%. The samples were immunoprecipitated with rabbit anti-VSV for 2 h at 4°C. Immune complexes were collected with 20 μl of washed S. aureus Pansorbin cells and washed three times with RIPA buffer. The immune complexes were eluted in 50 mM Tris-HCl (pH 6.8) containing 1% SDS at 100°C for 5 min. The S. aureus cells were spun out, and the supernatant was diluted with an equal amount of 150 mM Na-citrate (pH 5.5) with endo H (50 units per sample) (New England BioLabs, Beverly, MA) at 37°C overnight. Concentrated sample buffer was added, and the samples were boiled for 5 min at 100°C in the presence of 3.75% BME and run on a 10% SDS-polyacrylamide gel. Labeled VSV G was visualized using a Molecular Imager FX phosphorimager (Bio-Rad) and quantified using Quantity One software.
Indirect immunofluorescence microscopy and Golgi complex disassembly.
HeLa cells plated on glass coverslips were processed for immunofluorescence at 16 h posttransfection. Cells were washed with PBS and fixed in 3% paraformaldehyde in PBS for 10 min at 22°C. The fixative was quenched in PBS containing 10 mM glycine (PBS-Gly), and the cells were permeabilized in 0.5% Triton X-100 for 3 min. The coverslips were washed twice with PBS-Gly and incubated in primary antibody with 1% BSA for 20 min at room temperature. Rabbit anti-IBV E was used at 1:1,000, and mouse anti-GM130 was used at 1:300. The cells were then washed twice with PBS-Gly and incubated for 20 min in secondary antibody with 1% bovine serum albumin. Alexa Fluor 488-conjugated anti-rabbit IgG and Alexa Fluor 568-conjugated anti-mouse IgG were used at 1:1,000. The coverslips were washed twice in PBS-Gly and incubated with Hoechst 33285 (0.1 μg/ml) to stain the DNA, rinsed twice in PBS-Gly, and mounted on slides in glycerol containing 0.1 M N-propyl gallate. Images were captured using an Axio Scope microscope (Zeiss) equipped for epifluorescence with an Orca-03G charge-coupled-device camera (Hamamatsu, Japan). Data analysis was performed using iVision (BioVision Technologies) and Microsoft Excel software. Golgi complex disassembly was addressed in cells expressing IBV E, IBV E-T16A, or IBV E-A26F. GM130 staining of the Golgi complex was outlined. The outlined area for the Golgi complex was measured, and Golgi complexes with an area larger than 1.5 standard deviations of the average area of untransfected cells were considered disassembled. For each IBV E protein, the percentage of cells with a disassembled Golgi complex was determined by dividing the number of cells scored as disrupted by the total number of cells measured (n ≥ 100 cells).
VLP assay.
Subconfluent HeLa cells in 60-mm dishes were transfected with the following amounts of plasmids, in the combinations indicated in the figure, diluted into Opti-MEM medium (Invitrogen/Gibco) with a 1:3 ratio of X-tremeGENE 9: 0.1 μg pCAGGS IBV E, 0.1 μg pCAGGS IBV E-T16A, 0.1 μg pCAGGS IBV E-A26F, 2.0 μg pCAGGS IBV M, and 1.5 μg pCAGGS IBV N. Samples were prepared at 48 h posttransfection. The medium was clarified at 4,500 × g for 20 min. The supernatant was loaded on a 20% sucrose cushion and centrifuged at 234,000 × g in a TLA-110 rotor for 60 min. To simplify quantification of heterogeneously glycosylated IBV M, samples were deglycosylated prior to SDS-PAGE. The pellet containing VLPs was resuspended in 2× glycoprotein denaturation buffer (New England BioLabs). The cell fraction was pelleted in PBS at 4,000 × g for 2 min. The cell pellets were resuspended in detergent solution containing protease inhibitor cocktail. Glycoprotein denaturation buffer was added to a 2× final concentration. Both the VLP and cell fractions were heated to 100°C for 1 min. All samples were digested with peptide-N-glycosidase F (New England BioLabs) according to the manufacturer's protocol. A 4×-concentrated sample buffer was added to the samples, and the mixture was heated to 100°C for 3 min. Ten percent of the cell fractions and 100% of the VLP fractions were separated on 15% SDS-polyacrylamide gels. The proteins were analyzed by Western blotting as described above. Proteins were detected using rabbit anti-IBV E (1:10,000), rabbit anti-IBV M (1:5,000), and rabbit anti-IBV N (1:10,000) primary antibodies and horseradish peroxidase-conjugated donkey anti-rabbit IgG (1:10,000) secondary antibody.
RESULTS
IBV E is enriched in two pools by sucrose gradient analysis.
Structural data for SARS-CoV E suggests that its ion channel activity in artificial membranes can be attributed to homopentamer formation (23–27). To address the possibility that IBV E ion channel activity was responsible for secretory pathway disruption, we asked if IBV E forms homo-oligomers during transient expression in cells. Native lysates from HeLa cells expressing IBV E were prepared in lysis buffer containing n-dodecyl-β-d-maltoside (DDM) and separated on a 5 to 20% sucrose gradient also containing DDM. Gradient fractions were collected and analyzed by Western blotting. On a parallel control gradient, the vesicular stomatitis virus glycoprotein (VSV G), a 68-kDa membrane protein known to be 4 Svedberg units (4S) in its monomeric form on a neutral 5 to 20% sucrose gradient, was analyzed as a reference for molecular size under our assay conditions (35). IBV E was enriched in two peaks, while VSV G was enriched in one (Fig. 1A). We refer to the two IBV E peaks as the low-molecular-weight (LMW) and high-molecular-weight (HMW) pools, respectively. VSV G ran slightly further into the gradient than the LMW pool of IBV E, while the HMW pool of IBV E ran slightly further than VSV G. This led us to predict that the molecular mass of the HMW pool of IBV E was in the range of 65 to 75 kDa, potentially representing a higher-order oligomer of the protein (e.g., homopentamer).
In order to characterize the two pools of IBV E further, we tested the effect of SDS on the lysate prior to being run on a sucrose gradient (Fig. 1B). When the lysate containing IBV E was treated with SDS, IBV E was enriched in a single peak, which should reflect IBV E in its monomeric state (∼12 kDa). In the absence of SDS, IBV E was again enriched in two peaks. The LMW peak that formed in the absence of SDS ran further into the gradient than IBV E in the presence of SDS, and we concluded that this LMW peak represented a pool of IBV E that was larger than a predicted monomer. It should be noted that estimation of molecular weight of membrane proteins by sucrose gradient analysis in the presence of detergent is difficult, as various factors, including molecular size and shape, as well as the amount of bound detergent, all contribute to the migration of the protein in the gradient (36, 37). These results suggest that IBV E is present in two pools in transfected cells, possibly representing different homo-oligomeric states or the differential association of IBV E with an unknown cellular protein(s).
Kinetics of formation of the LMW and HMW pools of IBV E.
In an effort to better characterize the LMW and HMW pools of IBV E, we followed the newly synthesized pool of IBV E protein by metabolic labeling. HeLa cells transiently expressing IBV E were pulse-labeled for 15 min with [35S]cysteine-methionine and chased for 0 or 60 min. The cells were lysed and run over sucrose gradients as described above. IBV E was immunoprecipitated from each fraction and analyzed by SDS-PAGE. Directly after the pulse, IBV E was predominantly found in the LMW pool, with a smaller proportion being found in the HMW pool. The fraction of IBV E in the HMW pool slightly increased following a 60-min chase, but that in the LMW pool did not substantially decrease over time (Fig. 2A). To elaborate on this finding, we performed a time course of IBV E expression from 12 to 20 h posttransfection (Fig. 2B). At 12 h, more IBV E was in the LMW pool than the HMW pool; however, at 16 and 20 h posttransfection, the level of enrichment of total E in the LMW and HMW pools approached equality. Taken together, these data suggest that the LMW pool of IBV E forms more rapidly than the HMW pool and that it is unlikely to be a precursor of the HMW pool. An alternative possibility is that the LMW pool of IBV E reaches a steady state early during expression and slowly transitions into the HMW pool. Analysis of IBV E mutants (see below) makes this explanation less likely, however.
FIG 2.
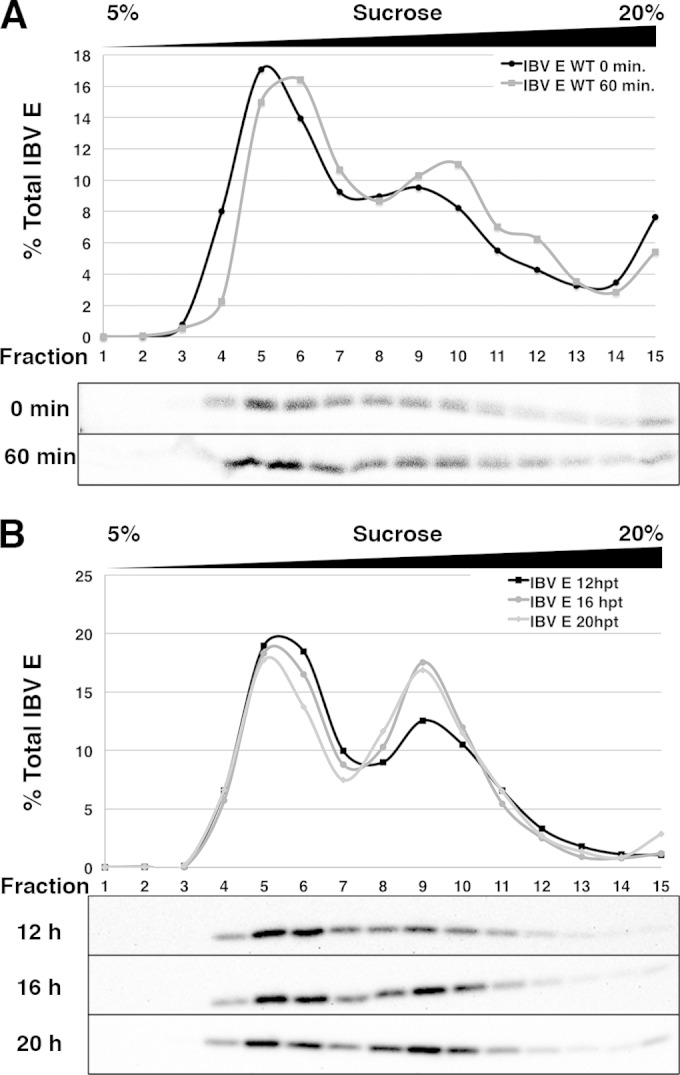
The LMW pool of IBV E persists over time, and the HMW pool increases with time. (A) HeLa cells expressing IBV E were pulse-labeled with [35S]cysteine-methionine for 15 min and lysed at 0 or 60 min of chase. The cell lysates were run on 5 to 20% sucrose gradients. Fractions were collected and immunoprecipitated with anti-IBV E antibody, analyzed by SDS-PAGE, and visualized by phosphorimaging. (B) HeLa cells expressing IBV E were lysed at 12, 16, or 20 h posttransfection (hpt). Cell lysates were separated on 5 to 20% sucrose gradients, and fractions were analyzed by Western blotting.
The LMW and HMW pools contain IBV E in different oligomeric states.
To determine the oligomeric states of IBV E in the two pools, we cross-linked the LMW and HMW gradient fractions using the reversible cross-linker DSP. IBV E was immunoprecipitated, and the eluates were treated with or without reducing agent to reverse the cross-links and analyzed by Western blotting. In the absence of reducing agent, the LMW pool, which correlates with a species of IBV E slightly larger than the monomer on the sucrose gradient, consisted primarily of monomeric IBV E, with a small amount of a second, larger species being present (Fig. 3). The HMW pool consisted of a series of six evenly spaced bands: one representing the IBV E monomer, another that appeared to be the second band observed in the LMW pool, as well as four other bands (Fig. 3). The addition of reducing agent collapsed the cross-linked species to the 12-kDa monomer. Although we cannot rule out the possibility of cross-linking of cellular proteins to IBV E, the evenly spaced cross-linked bands suggest the IBV E species represent homo-oligomers ranging from homodimers to homohexamers.
FIG 3.
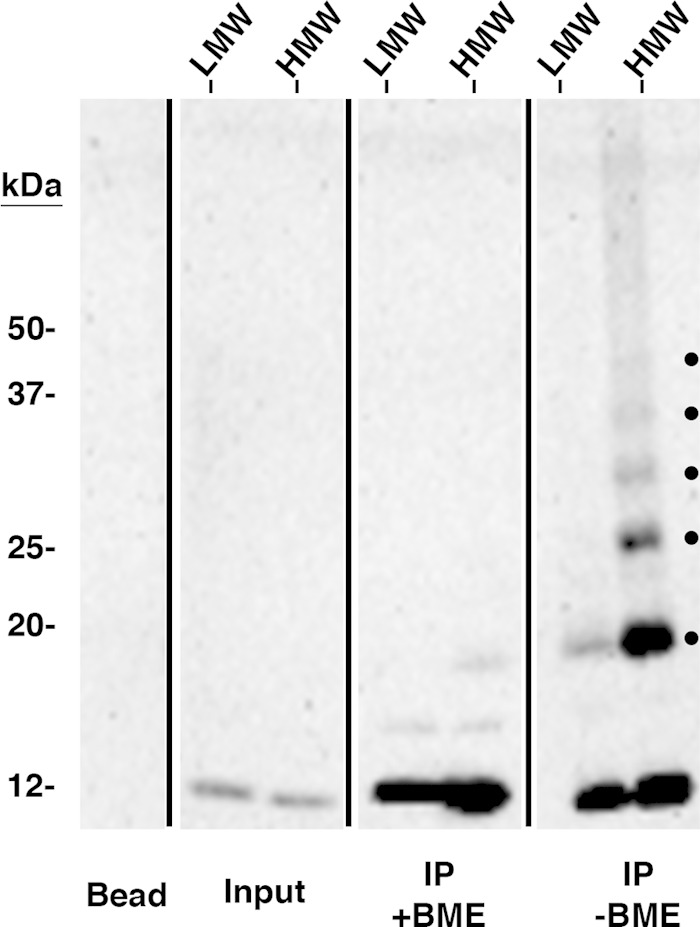
The HMW pool contains species of IBV E consistent with higher-order homo-oligomers. Pooled sucrose gradient fractions representing the LMW or HMW forms of IBV E from HeLa cells expressing IBV E were cross-linked with a reducible cross-linker (DSP), immunoprecipitated with anti-IBV E antibody, and analyzed by Western blotting. One-half of each immunoprecipitate (IP) was treated with BME to reverse the cross-linked species. Input lanes represent ∼2.0% of the preimmunoprecipitated sample to demonstrate the relative amount of IBV E in the LMW and HMW pools. The dots on the nonreduced panel indicate cross-linked species. The bead lane indicates antibody-conjugated beads that were analyzed in parallel as a negative control. This blot is representative of blots from four independent experiments with similar results.
The IBV E protein in infected cells is also present in two pools by sucrose gradient analysis.
To determine if the oligomeric states of IBV E in transfected cells were relevant during IBV infection, a native lysate from IBV-infected Vero cells was evaluated by sucrose gradient analysis. Vero cells were infected at an MOI of 1.0 for 8 h, and the cell lysate was analyzed as described above (Fig. 4A, black line). Indeed, IBV E was enriched in the same two pools observed during exogenous expression. Although the percentage of total E in the LMW pool in infected cells (∼10%) was less than that in the same pool in transfected cells (∼50%), this was consistent over multiple experiments, suggesting that the LMW form is relevant in infection. Clearly, the majority of IBV E in infected cells is in the predicted homo-oligomeric form. One possibility is that the formation of the LMW pool of IBV E could be tightly regulated during infection but not transfection. It should also be noted that more IBV E was present in fraction 15 at the bottom of the gradient in infected cells than in transfected cells.
FIG 4.
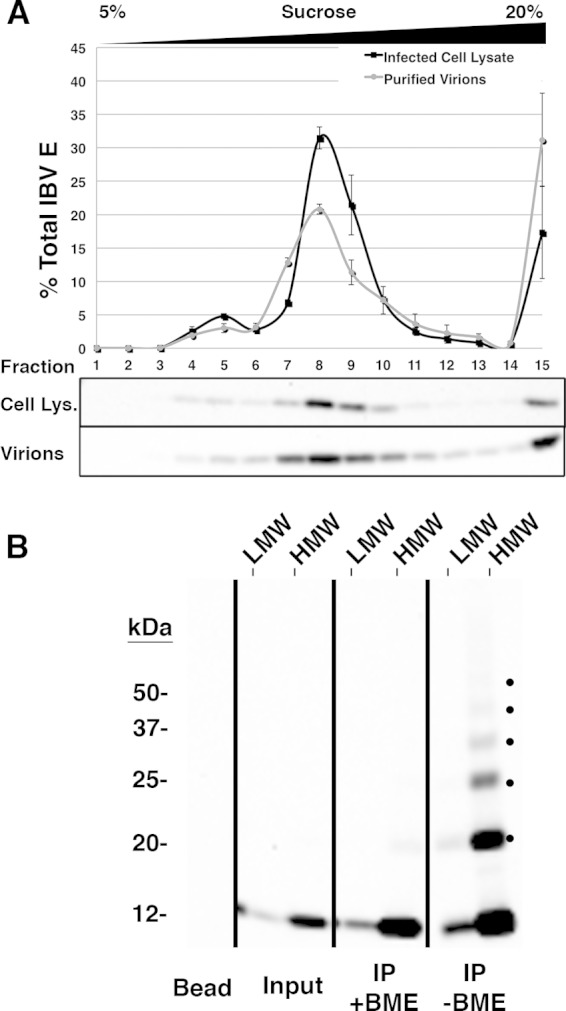
IBV E is present in two pools in infected cells and virions, with the majority being in the HMW pool. (A) Vero cells infected with IBV were lysed at 8 h postinfection and run on a 5 to 20% sucrose gradient, or virions were purified from infected cell supernatant at 24 h postinfection, resuspended in lysis buffer containing DDM, and run on a 5 to 20% sucrose gradient. Fractions were collected and analyzed by Western blotting. Error bars represent the SEMs from two independent experiments for each condition. (B) Pooled sucrose gradient fractions representing the LMW and HMW forms of IBV E from purified virions were cross-linked with DSP, immunoprecipitated with anti-IBV E antibody, and analyzed by Western blotting. One-half of each immunoprecipitate was reduced with BME to reverse the cross-linked species. Input lanes represent ∼2.0% of the preimmunoprecipitated sample to demonstrate the relative amount of IBV E in the LMW and HMW pools. The bead lane indicates antibody-conjugated beads that were analyzed in parallel as a negative control. Dots on the nonreduced panel indicate cross-linked species. This blot is representative of the blots from two independent experiments that produced similar results.
The majority of virion-associated IBV E is in the HMW pool and forms homo-oligomers.
We next addressed the forms of IBV E in the virion envelope. Vero cells were infected for 24 h at an MOI of 1.0, and virions were purified from the supernatant (see Materials and Methods). The virions were solubilized with DDM and run over a 5 to 20% sucrose gradient (Fig. 4A, gray line). The sucrose gradient profile of IBV E in secreted virions was very similar to that of IBV E from infected cells, with the majority of the E protein being in the HMW pool rather than the LMW pool. We again observed some IBV E in fraction 15. Next, we used cross-linking and immunoprecipitation of the HMW fraction from secreted virions to show that the IBV E in this pool consisted of higher-order oligomers (Fig. 4B) with a banding pattern identical to that of the HMW pool in transfected cells. Together our data from infected Vero cells and from secreted virions suggest that the majority of IBV E in both is in a higher-order homo-oligomeric state.
IBV E HD mutants have different effects on the cellular secretory pathway.
We previously reported that the wild-type (WT) IBV E protein alters the cellular secretory pathway during transient expression in HeLa cells, which requires residue T16 in the HD (19, 31). Given that both the LMW and HMW pools of IBV E are present during IBV infection, we wanted to understand the role of IBV E oligomerization in the context of secretory pathway disruption. N15 in SARS-CoV E (at the position equivalent to IBV E T16), along with V25, is necessary for ion channel activity in lipid bilayers (26, 31). Whether IBV E-T16A loses the capacity to disrupt the secretory pathway due to the loss of ion channel activity, due to improper oligomerization, or through another mechanism is not known. SARS-CoV E-V25 is located at the predicted monomer-monomer interface of the SARS-CoV E homopentamer. It has been suggested that the V25 residue in SARS-CoV E may be important for the formation of a stable homo-oligomer, given its position in the predicted structure (20). In IBV E, the residue at the position equivalent to V25 in SARS-CoV E is A26. We mutated IBV E A26 to phenylalanine to determine the effect on the cellular secretory pathway. HeLa cells expressing WT IBV E or the T16A or A26F mutant were evaluated for Golgi complex disassembly by indirect immunofluorescence microscopy (Fig. 5A and B). As previously reported, IBV E caused the disassembly of the Golgi complex, as determined by the dispersal of the cis-Golgi marker GM130, while the T16A mutant did not. The IBV E-A26F protein did elicit Golgi complex disassembly, despite the predicted role of A26 in oligomer formation.
FIG 5.

Predicted HD ion channel mutants of IBV E have different effects on the cellular secretory pathway. (A) HeLa cells expressing IBV E, IBV E-T16A, or IBV E-A26F were analyzed by indirect immunofluorescence microscopy at 16 h posttransfection. Cells were labeled with rabbit anti-IBV E and mouse anti-GM130, a cis-Golgi marker. Secondary antibodies were Alexa Fluor 488-conjugated anti-rabbit IgG and Alexa Fluor 568-conjugated anti-mouse IgG. The DNA was stained with Hoechst 33285. White arrows, disrupted Golgi complexes. (B) Quantification of the percentage of cells with Golgi complex disruption (see Materials and Methods). Data are for ≥100 cells for each condition. (C) HeLa cells coexpressing VSV G and IBV E, IBV E-T16A, IBV E-A26F, or IBV M (as a control) were pulse-labeled with [35S]cysteine-methionine for 10 min, and VSV G was immunoprecipitated at the indicated times of chase and digested with endo H. The graph shows the percentage of endo H-resistant VSV G in cells coexpressing each IBV E construct.
We then tested the effects of the IBV E-A26F mutant on the trafficking of the model cargo protein VSV G by measuring oligosaccharide processing rates. Cells expressing VSV G along with IBV E or the IBV E mutants were pulse-labeled and chased for various times, and immunoprecipitated VSV G was subjected to glycosidase digestion (Fig. 5C). As previously reported, WT IBV E dramatically reduced the rate of acquisition of endoglycosidase H resistance of VSV G, while IBV E-T16A did not. However, in accordance with the disassembly of the Golgi complex, IBV E-A26F reduced the trafficking of VSV G similarly to WT IBV E. These data suggest that two HD mutants of IBV E predicted to interfere with ion channel activity have different effects on the cellular secretory pathway.
HD mutants of IBV E are differentially enriched in LMW and HMW pools.
We next examined the oligomerization profiles of the two HD mutants of IBV E. At steady state, the IBV E-T16A protein was nearly exclusively in the HMW pool, while the IBV E-A26F protein was largely present in the LMW pool (Fig. 6A). Interestingly, we observed an increase in the amount of IBV E-T16A in fraction 15 at the bottom of the sucrose gradient compared to the amounts of the WT protein and the A26F mutant protein, an observation that we also made for IBV E expressed during infection. Preliminary data using steeper sucrose gradients suggested that IBV E-T16A in fraction 15 exists in a distinct peak and is not a heterogeneous aggregate (our unpublished data). Experiments to characterize this form of IBV E are in progress.
FIG 6.

HD mutants of IBV E are differentially enriched in the LMW and HMW pools. (A) HeLa cells expressing IBV E-T16A or IBV E-A26F were lysed and run on a 5 to 20% sucrose gradient, and fractions were analyzed by Western blotting. Error bars represent the SEMs from three independent experiments for each HD mutant. The relative positions of the LMW and HMW pools for wild-type IBV E are indicated and were determined in parallel. (B and C) HeLa cells expressing IBV E-T16A (B) or IBV E-A26F (C) were pulse-labeled for 15 min with [35S]cysteine-methionine and lysed at 0 or 60 min of chase. The cell lysates were run on 5 to 20% sucrose gradients, and fractions were immunoprecipitated and analyzed by SDS-PAGE and phosphorimaging. (D) Pooled sucrose gradient fractions representing the LMW and HMW pools of IBV E-T16A or IBV E-A26F from transfected cells were cross-linked with DSP, immunoprecipitated with anti-IBV E antibody, and analyzed by Western blotting. One-half of each immunoprecipitate was reduced with BME to reverse the cross-linked species. Input lanes represent ∼2.0% of the preimmunoprecipitated sample to demonstrate the relative amount of each IBV E HD mutant in the LMW and HMW pools. Dots on the nonreduced panel indicate cross-linked species. The bead lane indicates antibody-conjugated beads that were analyzed in parallel as a negative control. This blot is representative of the blots from two independent experiments with similar results. The blot images or phosphorimages are illustrated under the graphs for panels A, B, and C, with the image for IBV E-A26F being shown on the top and that for IBV E-T16A being shown on the bottom for panel A and with the image for the 0-min time point being shown on the top and that for the 60-min time point being shown on the bottom for panels B and C.
Next, we observed a window of IBV E expression for the two HD mutants by [35S]cysteine-methionine metabolic labeling, as described above for WT IBV E. The pulse-chase analysis revealed that the majority of IBV E-T16A was present in the HMW pool even at 0 min of chase, and the percentage of total IBV E-T16A at the bottom of the gradient in fraction 15 increased with the chase (Fig. 6B). On the other hand, IBV E-A26F was predominantly in the LMW pool, remained in the LMW pool over time, and did not accumulate in fraction 15 (Fig. 6C).
We performed cross-linking followed by immunoprecipitation of the pooled LMW and HMW fractions for each mutant. IBV E-T16A robustly formed the predicted HMW homo-oligomer, while IBV E-A26F did not (Fig. 6D), consistent with the migration of these proteins on sucrose gradients. These results indicate that the T16 residue, predicted to be required for ion channel activity and secretory pathway phenotypes, is not required for the HMW oligomerization of IBV E but is required for stable formation of the LMW pool. The A26F mutation, which was predicted to affect ion channel activity and oligomerization of the E protein, did markedly reduce the predicted oligomerization in the HMW pool. These results suggest that the effects of IBV E on the cellular secretory pathway correlate with the LMW pool and thus are likely to occur by a mechanism distinct from the proposed ion channel activity of this protein.
VLP production requires the HMW pool.
We previously reported that cells expressing IBV E and IBV E-T16A produce VLPs to a similar extent when coexpressed with IBV M and IBV N, suggesting that T16 in IBV E is required for secretory pathway disruption but not for virion assembly (31). We evaluated VLP production from HeLa cells expressing IBV E-A26F compared to that from HeLa cells expressing WT IBV E and IBV E-T16A (Fig. 7). Cell lysates and supernatants were collected, and VLPs were concentrated from the supernatants by centrifugation through a 20% sucrose cushion. VLP release was determined by comparing the percentage of expressed IBV M released into the supernatant, as quantified by Western blotting. As expected, VLPs were released into the supernatants of cells expressing IBV E, M, and N. Surprisingly, cells expressing IBV E-T16A along with IBV M and N released ∼4-fold more IBV M into the supernatant than the WT protein. We believe that the difference from our previous study (31) can be attributed, at least in part, to differences in the transfection protocol. Intriguingly, cells expressing IBV E-A26F were unable to produce VLPs. These results suggest that the LMW pool of IBV E present during expression of IBV E-A26F does not support VLP production. On the other hand, the higher-order oligomer, present in the HMW pool, supports the robust production of VLPs.
FIG 7.
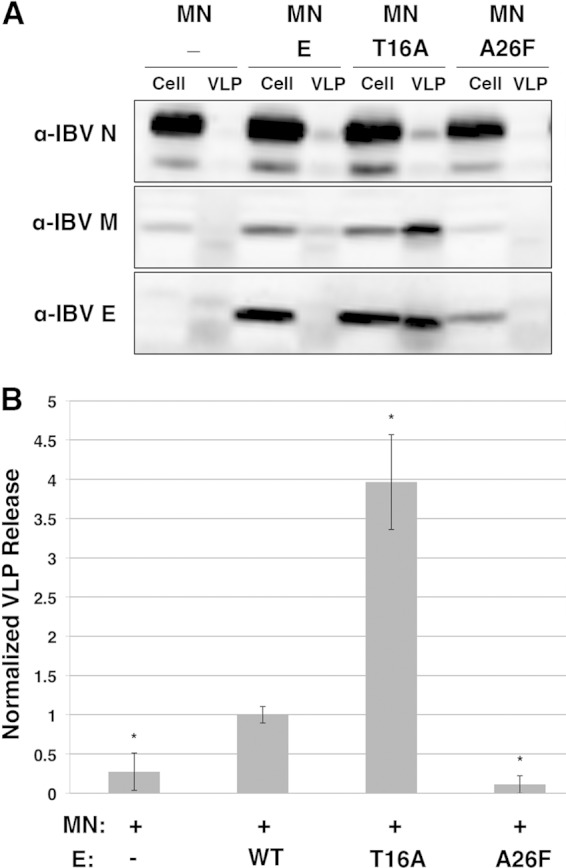
IBV E-T16A supports VLP production, while IBV E-A26F does not. (A) A representative immunoblot showing the amount of IBV N, M, and E coexpressed in 10% of HeLa cell fractions and 100% of VLP fractions. (B) Quantification of the amount of IBV M released with no E, WT E, E-T16A, or E-A26F. The amount of M released with WT E was set equal to 1. Error bars represent the SEMs from three independent experiments. *, a significant change in VLP level compared to that obtained with WT E by Student's t test (P < 0.05). MN, M and N proteins.
DISCUSSION
Two distinct pools of IBV E.
Our sucrose gradient analysis of IBV E indicates that the protein exists in two populations during both transient expression and infection. Pulse-chase and steady-state analyses led us to conclude that the LMW and HMW pools of IBV E represent distinct populations of the protein and that the LMW pool is unlikely to be a precursor of the HMW pool. Rather, the LMW pool of IBV E appears to represent a persistent population of the protein, potentially possessing its own function.
We determined that the IBV E in the LMW pool is slightly larger than the predicted monomer present on the gradient in the presence of SDS and that the IBV E in the HMW pool is slightly larger than the 68-kDa VSV G protein. Though we could not calculate the precise size of the HMW form of IBV E by sucrose gradient analysis, it is similar to the predicted size of an IBV E homopentamer. Cross-linking and subsequent immunoprecipitation of the gradient fractions containing the two pools of IBV E suggested that the LMW pool contains a monomer or dimer, whereas the HMW pool contains a homo-oligomer up to a hexamer. It is thus possible that IBV E may be different from both SARS-CoV E and MERS-CoV E, both of which have been shown to form homopentamers in artificial membranes (23–28). However, our experiments do not rule out the possibility that some bands could contain IBV E cross-linked to host cell proteins, in the case of transfection, and host or other viral proteins, in the case of infection. Preliminary mass spectroscopy data suggest that immunoprecipitates of IBV E from the LMW pool from transfected cell lysates contain several host proteins, whereas immunoprecipitates of IBV E from the HMW pool do not (our unpublished data). These preliminary data suggest that a small host protein interacting with IBV E could explain the migration of the LMW pool further into the gradient than the E monomer. Additionally, the sole presence of IBV E in the immunoprecipitates of the HMW pool would lend support to the interpretation that IBV E forms a homohexamer. However, if an interacting host protein is present, it is possible that IBV E does indeed form a homopentamer in the HMW pool. We are currently pursuing these preliminary results. Our data suggesting that mutation of IBV E-A26 inhibits homo-oligomer formation would lend support to a model for homopentamer formation, given that it is the residue analogous to SARS-CoV E-V25. Additional evidence for a model of homopentamer formation by IBV E has been reported by Parthasarathy et al., who demonstrated by perfluoro-octanoic acid-PAGE and analytical ultracentrifugation that IBV E purified from bacteria can form an oligomer consistent with a homopentamer (38).
IBV E mutants support distinct functions for the two pools.
IBV E has been proposed to play roles both in virion assembly and in the release of infectious virions from the host cell (8). However, the mechanism(s) by which the protein executes these roles remains unknown. Based on the persistence of the two pools of IBV E in pulse-chase experiments, we hypothesize that the LMW and HMW pools may represent populations of IBV E with distinct functions. The sucrose gradient profiles of the IBV E-T16A and -A26F mutants support this idea and revealed several interesting points. First, the T16A mutant, predicted to be deficient in ion channel activity, formed very little of the LMW pool. Second, the sucrose gradient profile for the IBV E-A26F mutant, which we predicted would prevent homo-oligomerization, behaved as expected, with the formation of the HMW pool being severely inhibited. Cross-linking and immunoprecipitation of the IBV E-T16A and A26F LMW and HMW fractions corroborated our interpretation of the sucrose gradient profiles. Additionally, analysis of the IBV E mutants by pulse-chase labeling followed by sucrose gradient analysis showed that IBV E-T16A formed very little of the LMW pool even at 0 min of chase. Alternatively, the IBV E-A26F protein remained in the LMW pool from 0 to 60 min of chase. The findings of the pulse-chase analyses of the HD mutants correlate with the findings for the WT protein, suggesting the existence of distinct populations of IBV E. In addition, we conclude that A26 is required for the robust formation of the higher-order homo-oligomers observed in the HMW pool of IBV E and T16 is required for the formation of the LMW pool. The fact that the sucrose gradient profiles for the T16A mutant during transfection and WT E during infection are similar has led us to hypothesize that formation of the LMW pool of IBV E during infection may be a regulated event, which would suggest that T16 not only is required for putative ion channel activity but also could take part in this regulatory event during infection, possibly through its interaction with a host cell protein(s). We predict that this regulated interaction is crucial for modification of the host cell environment, leading to proper navigation of the host secretory pathway by undamaged, infectious virions.
Golgi complex phenotypes in transfected cells correlate with the LMW pool.
We previously reported that, when exogenously expressed, IBV E has significant effects on the host cell secretory pathway, including the disassembly of the Golgi complex as well as a reduction in the rate of protein trafficking through the secretory pathway. T16 in the HD is required for these phenotypes (31). In this study, we show that IBV E-T16A migrates predominantly in the HMW pool in sucrose gradients, whereas the WT protein has a relatively equal distribution in the LMW and HMW pools. In contrast, the A26F mutation shifted the majority of IBV E into the LMW pool. Since expression of A26F disrupted the Golgi complex while that of T16A did not, we suggest that the secretory pathway phenotypes are induced by the fraction of IBV E in the LMW pool. Thus, secretory pathway disruption is unlikely to be associated with the ion channel activity of the E protein, which we assume requires higher-order oligomerization, as is found in the HMW pool.
Assembly of VLPs correlates with the HMW pool.
VLP analysis, together with the sucrose gradient profiles and oligomerization data from our cross-linking studies, suggests that the HMW pool of IBV E has a role in the production of VLPs, while the LMW pool does not. The lack of VLP production by the IBV E-A26F mutant suggests that higher-order oligomerization is required for VLP production; these data thus implicate the HMW pool of IBV E in the process of virion assembly. The fact that the IBV E-T16A mutant released 4-fold more IBV M into the supernatant of IBV E-T16A-expressing cells in our VLP assay is intriguing. Perhaps VLPs are released at a higher rate when T16 is mutated, as a consequence of losing the LMW pool and secretory pathway disruption. If so, virions with the T16A mutation would also travel more quickly through the secretory pathway, leading to damage from host proteases and reduced infectivity. Future studies will address this possibility, as well as whether the higher-order oligomer of IBV E found in the HMW pool is required for induction of membrane curvature, membrane scission during budding, or efficient exocytosis of virions.
Precedent for multiple functions of the CoV E protein.
SARS-CoV E interacts with the tight junction protein PALS1 and was suggested to play a role in pathogenesis by disassembling tight junctions in lung epithelium, although this interaction has not been characterized further (39). More recently, it has been suggested that SARS-CoV E interacts with syntenin, resulting in the activation of p38 mitogen-activated protein kinase signaling during infection (40). Together with the fact that the ion channel activity of SARS-CoV E has been shown to be a pathogenic determinant in a mouse infection model (20), these examples set a strong precedence for our hypothesis suggesting multiple functions for IBV E. Other viroporins have been shown to have interactions with host and other viral proteins (41). The HIV-1 Vpu1 viroporin has been shown to interact with and induce the degradation of CD4, tetherin, and the tetraspanin protein CD81 (42–44). The influenza A virus M2 viroporin has roles in assembly, virus release, and entry, and the hepatitis C virus p7 viroporin is required for the assembly and release of infectious virus (41, 45–50). While the ion channel activity of various viroporins has been implicated in certain roles, it is likely that some functions are ion channel independent, similar to our findings presented here.
In summary, this study has established that IBV E exists in two populations when expressed in mammalian cells and that it forms oligomers consistent with a homopentamer or homohexamer. Importantly, the secretory pathway phenotypes induced by the WT IBV E protein in transfected cells are likely to be independent of virus ion channel activity. We suggest that T16 in the HD of IBV E not only is required for the secretory pathway phenotypes but also is required for the formation of the LMW pool of IBV E, which correlates with the secretory pathway phenotypes. This could occur through a regulated interaction with a host cell protein. Additionally, we demonstrate that the HMW pool of IBV E present during IBV E-T16A expression is capable of robust VLP production, thus suggesting a role for a higher-order oligomer of IBV E in the process of assembly. Understanding the dynamics of oligomerization, secretory pathway disruption, ion channel activity, and E-protein interactors during infection will help resolve some of the interesting questions posed by this study.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grant R01 GM42522 and the Biochemistry, Cellular and Molecular Biology Graduate Program (T32 GM007445).
We thank Travis Ruch, Catherine Gilbert, David Zuckerman, and Jason Berk for helpful discussions and critical readings of the manuscript. We also thank Travis Ruch for the development of some of the tools used in this study.
REFERENCES
- 1.Hilgenfeld R, Peiris M. 2013. From SARS to MERS: 10 years of research on highly pathogenic human coronaviruses. Antiviral Res 100:286–295. doi: 10.1016/j.antiviral.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dediego ML, Pewe L, Alvarez E, Rejas MT, Perlman S, Enjuanes L. 2008. Pathogenicity of severe acute respiratory coronavirus deletion mutants in hACE-2 transgenic mice. Virology 376:379–389. doi: 10.1016/j.virol.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Netland J, DeDiego ML, Zhao J, Fett C, Alvarez E, Nieto-Torres JL, Enjuanes L, Perlman S. 2010. Immunization with an attenuated severe acute respiratory syndrome coronavirus deleted in E protein protects against lethal respiratory disease. Virology 399:120–128. doi: 10.1016/j.virol.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McCray PB, Pewe L, Wohlford-Lenane C, Hickey M, Manzel L, Shi L, Netland J, Jia HP, Halabi C, Sigmund CD, Meyerholz DK, Kirby P, Look DC, Perlman S. 2007. Lethal infection of K18-hACE2 mice infected with severe acute respiratory syndrome coronavirus. J Virol 81:813–821. doi: 10.1128/JVI.02012-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Regla-Nava JA, Nieto-Torres JL, Jimenez-Guardeno JM, Fernandez-Delgado R, Fett C, Castano-Rodriguez C, Perlman S, Enjuanes L, DeDiego ML. 2015. Severe acute respiratory syndrome coronaviruses with mutations in the E protein are attenuated and promising vaccine candidates. J Virol 89:3870–3887. doi: 10.1128/JVI.03566-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao J, Li K, Wohlford-Lenane C, Agnihothram SS, Fett C, Zhao J, Gale MJJ, Baric RS, Enjuanes L, Gallagher T, McCray PBJ, Perlman S. 2014. Rapid generation of a mouse model for Middle East respiratory syndrome. Proc Natl Acad Sci U S A 111:4970–4975. doi: 10.1073/pnas.1323279111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo X, Deng Y, Chen H, Lan J, Wang W, Zou X, Hung T, Lu Z, Tan W. 12 March 2015. Systemic and mucosal immunity in mice elicited by a single immunisation with human adenovirus type 5 or 41 vector-based vaccines carrying the spike protein of Middle East respiratory syndrome coronavirus (MERS-CoV). Immunology doi: 10.1111/imm.12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hogue BG, Machamer CE. 2008. Coronavirus structural proteins and virus assembly, p 179–200. In Perlman S, Gallagher T, Snijder EJ (ed), Nidoviruses. ASM Press, Washington, DC. [Google Scholar]
- 9.Yu X, Bi W, Weiss SR, Leibowitz JL. 1994. Mouse hepatitis virus gene 5b protein is a new virion envelope protein. Virology 202:1018–1023. doi: 10.1006/viro.1994.1430. [DOI] [PubMed] [Google Scholar]
- 10.Liu DX, Inglis SC. 1991. Association of the infectious bronchitis virus 3c protein with the virion envelope. Virology 185:911–917. doi: 10.1016/0042-6822(91)90572-S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Godet M, L'Haridon R, Vautherot JF, Laude H. 1992. TGEV corona virus ORF4 encodes a membrane protein that is incorporated into virions. Virology 188:666–675. doi: 10.1016/0042-6822(92)90521-P. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Venkatagopalan P, Daskalova SM, Lopez LA, Dolezal KA, Hogue BG. 2015. Coronavirus envelope (E) protein remains at the site of assembly. Virology 478C:75–85. doi: 10.1016/j.virol.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuo L, Masters PS. 2003. The small envelope protein E is not essential for murine coronavirus replication. J Virol 77:4597–4608. doi: 10.1128/JVI.77.8.4597-4608.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ortego J, Ceriani JE, Patino C, Plana J, Enjuanes L. 2007. Absence of E protein arrests transmissible gastroenteritis coronavirus maturation in the secretory pathway. Virology 368:296–308. doi: 10.1016/j.virol.2007.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeDiego ML, Alvarez E, Almazan F, Rejas MT, Lamirande E, Roberts A, Shieh W-J, Zaki SR, Subbarao K, Enjuanes L. 2007. A severe acute respiratory syndrome coronavirus that lacks the E gene is attenuated in vitro and in vivo. J Virol 81:1701–1713. doi: 10.1128/JVI.01467-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vennema H, Godeke GJ, Rossen JW, Voorhout WF, Horzinek MC, Opstelten DJ, Rottier PJ. 1996. Nucleocapsid-independent assembly of coronavirus-like particles by co-expression of viral envelope protein genes. EMBO J 15:2020–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Corse E, Machamer CE. 2003. The cytoplasmic tails of infectious bronchitis virus E and M proteins mediate their interaction. Virology 312:25–34. doi: 10.1016/S0042-6822(03)00175-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ye Y, Hogue BG. 2007. Role of the coronavirus E viroporin protein transmembrane domain in virus assembly. J Virol 81:3597–3607. doi: 10.1128/JVI.01472-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruch TR, Machamer CE. 2011. The hydrophobic domain of infectious bronchitis virus E protein alters the host secretory pathway and is important for release of infectious virus. J Virol 85:675–685. doi: 10.1128/JVI.01570-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nieto-Torres JL, DeDiego ML, Verdia-Baguena C, Jimenez-Guardeno JM, Regla-Nava JA, Fernandez-Delgado R, Castano-Rodriguez C, Alcaraz A, Torres J, Aguilella VM, Enjuanes L. 2014. Severe acute respiratory syndrome coronavirus envelope protein ion channel activity promotes virus fitness and pathogenesis. PLoS Pathog 10:e1004077. doi: 10.1371/journal.ppat.1004077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilson L, McKinlay C, Gage P, Ewart G. 2004. SARS coronavirus E protein forms cation-selective ion channels. Virology 330:322–331. doi: 10.1016/j.virol.2004.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilson L, Gage P, Ewart G. 2006. Hexamethylene amiloride blocks E protein ion channels and inhibits coronavirus replication. Virology 353:294–306. doi: 10.1016/j.virol.2006.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Torres J, Wang J, Parthasarathy K, Liu DX. 2005. The transmembrane oligomers of coronavirus protein E. Biophys J 88:1283–1290. doi: 10.1529/biophysj.104.051730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Torres J, Maheswari U, Parthasarathy K, Ng L, Liu DX, Gong X. 2007. Conductance and amantadine binding of a pore formed by a lysine-flanked transmembrane domain of SARS coronavirus envelope protein. Protein Sci 16:2065–2071. doi: 10.1110/ps.062730007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Torres J, Parthasarathy K, Lin X, Saravanan R, Kukol A, Liu DX. 2006. Model of a putative pore: the pentameric alpha-helical bundle of SARS coronavirus E protein in lipid bilayers. Biophys J 91:938–947. doi: 10.1529/biophysj.105.080119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parthasarathy K, Ng L, Lin X, Liu DX, Pervushin K, Gong X, Torres J. 2008. Structural flexibility of the pentameric SARS coronavirus envelope protein ion channel. Biophys J 95:L39–L41. doi: 10.1529/biophysj.108.133041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pervushin K, Tan E, Parthasarathy K, Lin X, Jiang FL, Yu D, Vararattanavech A, Soong TW, Liu DX, Torres J. 2009. Structure and inhibition of the SARS coronavirus envelope protein ion channel. PLoS Pathog 5:e1000511. doi: 10.1371/journal.ppat.1000511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Surya W, Li Y, Verdia-Baguena C, Aguilella VM, Torres J. 2015. MERS coronavirus envelope protein has a single transmembrane domain that forms pentameric ion channels. Virus Res 201:61–66. doi: 10.1016/j.virusres.2015.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Verdia-Baguena C, Nieto-Torres JL, Alcaraz A, DeDiego ML, Torres J, Aguilella VM, Enjuanes L. 2012. Coronavirus E protein forms ion channels with functionally and structurally-involved membrane lipids. Virology 432:485–494. doi: 10.1016/j.virol.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Verdia-Baguena C, Nieto-Torres JL, Alcaraz A, Dediego ML, Enjuanes L, Aguilella VM. 2013. Analysis of SARS-CoV E protein ion channel activity by tuning the protein and lipid charge. Biochim Biophys Acta 1828:2026–2031. doi: 10.1016/j.bbamem.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ruch TR, Machamer CE. 2012. A single polar residue and distinct membrane topologies impact the function of the infectious bronchitis coronavirus E protein. PLoS Pathog 8:e1002674. doi: 10.1371/journal.ppat.1002674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Corse E, Machamer CE. 2000. Infectious bronchitis virus E protein is targeted to the Golgi complex and directs release of virus-like particles. J Virol 74:4319–4326. doi: 10.1128/JVI.74.9.4319-4326.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weisz OA, Swift AM, Machamer CE. 1993. Oligomerization of a membrane protein correlates with its retention in the Golgi complex. J Cell Biol 122:1185–1196. doi: 10.1083/jcb.122.6.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kreis TE. 1986. Microinjected antibodies against the cytoplasmic domain of vesicular stomatitis virus glycoprotein block its transport to the cell surface. EMBO J 5:931–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doms RW, Keller DS, Helenius A, Balch WE. 1987. Role for adenosine triphosphate in regulating the assembly and transport of vesicular stomatitis virus G protein trimers. J Cell Biol 105:1957–1969. doi: 10.1083/jcb.105.5.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Helenius A, Simons K. 1975. Solubilization of membranes by detergents. Biochim Biophys Acta 415:29–79. doi: 10.1016/0304-4157(75)90016-7. [DOI] [PubMed] [Google Scholar]
- 37.Marks MS. 1998. Determination of molecular size by zonal sedimentation analysis on sucrose density gradients. Curr Protoc Cell Biol Chapter 5:Unit 5.3. doi: 10.1002/0471143030.cb0503s00. [DOI] [PubMed] [Google Scholar]
- 38.Parthasarathy K, Lu H, Surya W, Vararattanavech A, Pervushin K, Torres J. 2012. Expression and purification of coronavirus envelope proteins using a modified beta-barrel construct. Protein Expr Purif 85:133–141. doi: 10.1016/j.pep.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Teoh K-T, Siu Y-L, Chan W-L, Schluter MA, Liu C-J, Peiris JSM, Bruzzone R, Margolis B, Nal B. 2010. The SARS coronavirus E protein interacts with PALS1 and alters tight junction formation and epithelial morphogenesis. Mol Biol Cell 21:3838–3852. doi: 10.1091/mbc.E10-04-0338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jimenez-Guardeno JM, Nieto-Torres JL, DeDiego ML, Regla-Nava JA, Fernandez-Delgado R, Castano-Rodriguez C, Enjuanes L. 2014. The PDZ-binding motif of severe acute respiratory syndrome coronavirus envelope protein is a determinant of viral pathogenesis. PLoS Pathog 10:e1004320. doi: 10.1371/journal.ppat.1004320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nieva JL, Madan V, Carrasco L. 2012. Viroporins: structure and biological functions. Nat Rev Microbiol 10:563–574. doi: 10.1038/nrmicro2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lambele M, Koppensteiner H, Symeonides M, Roy NH, Chan J, Schindler M, Thali M. 2015. Vpu is the main determinant for tetraspanin downregulation in HIV-1-infected cells. J Virol 89:3247–3255. doi: 10.1128/JVI.03719-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dubé M, Bego MG, Paquay C, Cohen ÉA. 2010. Modulation of HIV-1-host interaction: role of the Vpu accessory protein. Retrovirology 7:114. doi: 10.1186/1742-4690-7-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andrew A, Strebel K. 2010. HIV-1 Vpu targets cell surface markers CD4 and BST-2 through distinct mechanisms. Mol Aspects Med 31:407–417. doi: 10.1016/j.mam.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCown MF, Pekosz A. 2005. The influenza A virus M2 cytoplasmic tail is required for infectious virus production and efficient genome packaging. J Virol 79:3595–3605. doi: 10.1128/JVI.79.6.3595-3605.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang J, Qiu JX, Soto C, DeGrado WF. 2011. Structural and dynamic mechanisms for the function and inhibition of the M2 proton channel from influenza A virus. Curr Opin Struct Biol 21:68–80. doi: 10.1016/j.sbi.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pielak RM, Chou JJ. 2011. Influenza M2 proton channels. Biochim Biophys Acta 1808:522–529. doi: 10.1016/j.bbamem.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jones CT, Murray CL, Eastman DK, Tassello J, Rice CM. 2007. Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J Virol 81:8374–8383. doi: 10.1128/JVI.00690-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Steinmann E, Penin F, Kallis S, Patel AH, Bartenschlager R, Pietschmann T. 2007. Hepatitis C virus p7 protein is crucial for assembly and release of infectious virions. PLoS Pathog 3:e103. doi: 10.1371/journal.ppat.0030103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brohm C, Steinmann E, Friesland M, Lorenz IC, Patel A, Penin F, Bartenschlager R, Pietschmann T. 2009. Characterization of determinants important for hepatitis C virus p7 function in morphogenesis by using trans-complementation. J Virol 83:11682–11693. doi: 10.1128/JVI.00691-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Youn S, Collisson EW, Carolyn E, Machamer CE. 2005. Contribution of trafficking signals in the cytoplasmic tail of the infectious bronchitis virus spike protein to virus infection. J Virol 79:13209–13217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Machamer CE, Youn S. 2006. The transmembrane domain of the infectious bronchitis virus E protein is required for efficient virus release. Adv Exp Med Biol 581:193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]