

ABSTRACT
The hepatitis B virus (HBV) particle is an icosahedral nucleocapsid surrounded by a lipid envelope containing viral surface proteins. A small domain (matrix domain [MD]) in the large surface protein L and a narrow region (matrix binding domain [MBD]) including isoleucine 126 on the capsid surface have been mapped, in which point mutations such as core I126A specifically blocked nucleocapsid envelopment. It is possible that the two domains interact with each other during virion morphogenesis. By the systematic evolution of ligands by exponential enrichment (SELEX) method, we evolved DNA aptamers from an oligonucleotide library binding to purified recombinant capsids but not binding to the corresponding I126A mutant capsids. Aptamers bound to capsids were separated from unbound molecules by filtration. After 13 rounds of selections and amplifications, 16 different aptamers were found among 73 clones. The four most frequent aptamers represented more than 50% of the clones. The main aptamer, AO-01 (13 clones, 18%), showed the lowest dissociation constant (Kd) of 180 ± 82 nM for capsid binding among the four molecules. Its Kd for I126A capsids was 1,306 ± 503 nM. Cotransfection of Huh7 cells with AO-01 and an HBV genomic construct resulted in 47% inhibition of virion production at 3 days posttransfection, but there was no inhibition by cotransfection of an aptamer with a random sequence. The half-life of AO-01 in cells was 2 h, which might explain the incomplete inhibition. The results support the importance of the MBD for nucleocapsid envelopment. Inhibiting the MD-MBD interaction with a low-molecular-weight substance might represent a new approach for an antiviral therapy.
IMPORTANCE Approximately 240 million people are persistently infected with HBV. To date, antiviral therapies depend on a single target, the viral reverse transcriptase. Future additional targets could be viral protein-protein interactions. We selected a 55-base-long single-stranded DNA molecule (aptamer) which binds with relatively high affinity to a region on the HBV capsid interacting with viral envelope proteins during budding. This aptamer inhibits virion formation in cell culture. The results substantiate the current model for HBV morphogenesis and show that the capsid envelope interaction is a potential antiviral target.
INTRODUCTION
Hepatitis B virus (HBV) has infected more than 40% of the living human population and causes 240 million persistent infections (1). The treatment of chronic infections is limited to date to use of inhibitors of the viral reverse transcriptase and stimulation of the immune system by interferon. Alternative antiviral strategies are desirable.
During HBV replication, an RNA molecule (pregenome) is packaged together with the reverse transcriptase by 180 or 240 copies of the viral core protein. The assembled particle is an icosahedron with T = 3 or T = 4 symmetry and has a diameter of approximately 30 nm. The pregenome serves as a template for the synthesis of the viral DNA genome by reverse transcription occurring in the lumen of the capsid (2). This particle can then be enveloped by the three viral transmembrane surface proteins S, M, and L at an intracellular membrane, and the resulting virion is subsequently secreted from the host cell (3). Interestingly, the immature capsid containing the pregenome is not enveloped, in contrast to the case for the mature, DNA-containing capsid (4). Apparently, a structural change of the capsid surface is coupled to the synthesis of the viral DNA genome (5).
Heterologous expression of the core protein in eukaryotic cells and even in bacteria leads to capsids almost indistinguishable from authentic capsids with respect to their antigenicity and appearance by electron microscopy. The C-terminal 30-amino-acid (aa)-long region of the core protein is very rich in arginine residues and has nucleic acid binding properties. Deletion of this domain is compatible with capsid formation (6).
The budding of mature capsids is supported by cellular factors involved in multivesicular body formation (7). In addition, budding is dependent on a linear, 22-aa-long domain (matrix domain [MD]) of the surface protein L exposed at the cytoplasmic side of the cellular membrane and on a region on the capsid surface (matrix binding domain [MBD]) comprised of a ring-like groove around the base of the spike protruding from the capsid and a small area close to the pores of the capsid shell (8). Numerous single point mutations in either of the two domains block nucleocapsid envelopment (9, 10). It seems conceivable that the two domains directly interact with each other and that nucleocapsid envelopment presupposes the interaction of the MD and MBD. There is no strong biochemical evidence available for this interplay. However, the phenotypes of certain core and L protein mutants support the model: the F97L mutation of the core protein causes the envelopment of capsids containing pregenomic RNA (11, 12), and the point mutation A119F in the MD of the L protein can complement the core F97L change, restoring the selective envelopment of mature, DNA-containing capsids (13).
We intended to generate a DNA aptamer that binds to the MBD on the capsid surface. The presence of such a molecule in HBV-expressing cells was expected to block nucleocapsid envelopment and therefore virion formation. This would support the current model of HBV budding and could serve as a proof of principle that small molecules binding to the capsid surface could inhibit virion formation.
MATERIALS AND METHODS
Plasmids.
The HBV wild-type (WT) core gene was amplified by PCR from plasmid pHBV1.5 (14) (HBV genotype A) and the core gene point mutant I126A from plasmid pSVHBV1.1LE-I126A (9). The PCR primers were designed to introduce an NcoI restriction site at the initiation codon and a stop codon at triplet 149 plus a SalI restriction site. The PCR products were cleaved with NcoI and SalI and ligated into the T7 RNA polymerase-dependent bacterial expression vector pETM-13, replacing the actin binding domain (ABD) stuffer gene (vector map at http://www.helmholtz-muenchen.de/en/pepf/materials/vector-database/bacterial-expression-vectors/index.html).
In order to produce HBV virions, Huh-7 cells were transiently transfected (because of biosafety reasons) with three plasmids: (i) pSVHBV1.1LE−, containing a genotype A HBV genome with two stop codons in the surface protein open reading frame (ORF) (9); (ii) pSV45-31 (15), harboring the HBV pre-S1–pre-S2–S open reading frame for the expression of all three HBV envelope proteins (pre-S1 codons 2 to 30 were deleted in this construct because this region of the L protein is not required for virion formation [15] but has been shown to inhibit virion release in a dose-dependent fashion [16, 17]; and (iii) pSV24H (18), carrying the gene for the small HBV surface protein to optimize the ratio between the HBV envelope proteins for higher virus production.
Protein expression.
Escherichia coli BL21 Star (DE3) pRARE2 cells were transformed with the HBV core protein expression plasmids, and cultures were grown in 2× YT broth to an optical density at 600 nm (OD600) of 0.7 to 1.0. For induction of core protein expression, 200 μM isopropyl-β-d-thiogalactopyranoside was added at 20°C and left for 16 h before harvesting by centrifugation. Cells were lysed by freeze-thawing three times in lysis buffer (5 mM EDTA, 50 mM Tris HCl [pH 8.0], 2 mg/ml lysozyme), using 20 ml of lysis buffer per liter of cell culture, and then 0.1 M MgCl2 and 0.2 mg/ml DNase (end concentrations) were added, and the mixture was incubated at room temperature for 15 min before centrifugation at 15,000 rpm for 10 min at 4°C to remove cell debris. Capsids in the supernatant were precipitated by adding ammonium sulfate to an end concentration of 50% (wt/vol). The precipitate was sedimented by centrifugation at 19,000 rpm for 30 min at 4°C, and the pellet was resuspended in 10 ml of Tris-buffered saline (TBS) containing 0.1% (vol/vol) NP-40.
Protein purification.
The capsids were purified first by two subsequent steps of size exclusion chromatography using a HiPrep 26/60 Sephacryl S-500 HR (GE Healthcare) column. Fractions containing capsids were pooled, concentrated, and applied again to the column. Fractions containing capsids were detected by Coomassie blue staining of gels after SDS-PAGE or by native agarose gel electrophoresis and Western blotting with a polyclonal anti-HBc antibody (H800; kindly provided by H. Schaller, Heidelberg, Germany). Concentration of the pooled fractions was done by using a concentrator (Millipore 30000) with a cutoff of 30 kDa and centrifugation at 2,700 × g for 20 min at 4°C. A further purification step was performed by sucrose gradient ultracentrifugation at 10°C and 25,000 rpm for 24 h using an SW28 rotor and 10% to 60% (wt/wt) sucrose in TBS. Fractions containing capsids were identified, pooled, and desalted using PD-10 columns (GE Healthcare) and elution with TBS. The final protein concentration in the preparations was 1.4 mg/ml for wild-type capsids and 0.12 mg/ml for mutant capsids.
Aptamer library.
The library was obtained from Purimex (Göttingen, Germany). The oligonucleotides carried a fixed, 15-nucleotide (nt)-long sequence at both ends flanking a random sequence of 25 nt in length (5′GCGGGTCGACGTTTGN25CACATCCATGGGCGG3′). The positions of the random sequence were synthesized in the presence of an equimolar concentration of all four nucleotides A, G, C, and T. Prior to the in vitro selection, the aptamer library (10 nmol) was incubated at 85°C for 15 min, then snap-cooled on ice for 15 min, and finally equilibrated at room temperature (RT) for 15 min to induce folding of the aptamers to their 3-dimensional structures. In addition, an initial step of aptamer preselection was done by filtering the pre-snap-cooled aptamers through an alkali-pretreated Amicon Ultra 2-ml centrifugal filter (100 K) to remove aptamers binding to the matrix of the filter. Pretreatment was done with 0.5 M KOH for 20 min at room temperature followed by washing 3 times with distilled water and equilibration with binding buffer (phosphate-buffered saline [PBS]) at room temperature to reduce unspecific binding of aptamers.
In vitro selection with counterselection.
Thirteen rounds of consecutive positive and negative selections were performed. Targets of the positive in vitro selection were HBV WT capsids, while the countertargets for negative selection were the I126A mutant capsids. For binding, the aptamers and capsids were mixed in PBS (pH 7.0) and incubated at RT for various periods of time in a total volume of 100 μl. The selection of aptamers was performed by filtration using KOH-pretreated Amicon Ultra 2-ml centrifugal filters (100 K) in a swinging-bucket rotor and spinning at 4,000 × g and 25°C for 30 min. The aptamers bound to WT capsids in the positive selections, and the unbound aptamers in the negative selections were extracted with phenol-chloroform (1:1) and concentrated by using the Qiaex II kit (Qiagen). To induce selective pressure, the concentrations of aptamers, WT capsids, and mutant capsids as well as the incubation times and volumes of PBS for washing steps were adjusted during the selection process (Table 1).
TABLE 1.
Conditions during the aptamer selection processa
| Rounds | Aptamer concn (μM) | Capsid concn (nM) |
Incubation time (min) |
Vol of washing buffer (ml) |
|||
|---|---|---|---|---|---|---|---|
| WT capsids | Mutant capsids | WT capsids | Mutant capsids | Positive selection | Negative selection | ||
| 1–3 | 110 | 14 | 6 | 60 | 30 | 1 | 1 |
| 4–6 | 110 | 6 | 10 | 60 | 30 | 1 | 1 |
| 7–9 | 55 | 6 | 10 | 30 | 60 | 2 | 0.4 |
| 10–13 | 27 | 0.2 | 14 | 15 | 60 | 2 | 0.4 |
The volume of the binding reaction mixture was 100 μl.
Streptavidin-induced electrophoretic mobility shift for ssDNA preparation.
After each positive and negative selection step, the aptamer mixture was amplified by PCR. The PCR volume was 50 μl and contained aptamers, 50 μM (each) forward primer 5′GCGGGTCGACGTTTG3′ and reverse primer biotinylated 5′CCGCCCATGGATGTG3′, 1 mM each deoxynucleoside triphosphate, and 5 U Taq DNA polymerase (Promega) in Taq DNA polymerase buffer supplied by the manufacturer. After initial denaturation at 95°C for 5 min, 15 amplification cycles (denaturation at 95°C for 20 s, annealing at 51°C for 15 s, and extension at 72°C for 10 s) were performed. Afterwards, single-stranded DNA (ssDNA) plus-strand molecules were isolated from the double-stranded PCR products by using a streptavidin-induced electrophoretic mobility shift (19). The purified PCR product was suspended in streptavidin buffer and incubated with streptavidin (Thermo Scientific) at RT for 30 min (1:4 molar ratio of biotinylated strands to streptavidin). The binding mixture was then heat denatured and electrophoresed in a 10% polyacrylamide–6 M urea gel. The plus-strand ssDNA running faster in the gel than the streptavidin-bound biotinylated minus strands was then purified by passive elution from the crushed acrylamide gel and concentrated with the Qiaex II kit (Qiagen).
Kd determination.
The dissociation constants (Kd) of the aptamer-capsid binding were measured using immunoprecipitation. Different concentrations of the pre-snap-cooled aptamers (from 5 pM to 1 μM) and a fixed concentration of the HBV WT or mutant capsids (1 nM) were used. The aptamer-capsid complexes were immunoprecipitated using protein G-coupled agarose beads (Santa Cruz Biotechnologies) coated with rabbit polyclonal antibodies against the HBV core protein (H800). The bound aptamers were recovered by phenol-chloroform extraction, purified with the Qiaex II kit, and quantified by quantitative PCR (qPCR). For this PCR, the same setup was used as for the aptamer amplification during the selection process except that unbiotinylated reverse primers were used. The Kd values were estimated using the Sigma Blot 12.0 software program.
Cell transfection and virion immunoprecipitation.
Huh7 cells were transiently transfected with Fugene 6/HD/X-treme (Roche) in 6-well plates using in total 1 μg of plasmid DNA per well. When different plasmids were mixed, equal molar ratios were used. When aptamers were cotransfected, 1 μg was mixed with 1 μg of plasmids. The cell supernatants were collected at 3 days posttransfection and centrifuged at 13,000 rpm for 10 min. Virions in the supernatants were immunoprecipitated with sheep polyclonal antibodies against hepatitis B surface antigen (kindly provided by W. Gerlich, Gießen, Germany). The remnant of the plasmid DNA used for transfection was degraded by using DNase (Qiagen). The genomes of secreted virions were recovered by proteinase K digestion followed by phenol-chloroform extraction. To measure genome concentrations, an HBV genome-specific qPCR was used (9).
Aptamer secondary structure prediction.
Secondary structures of the selected aptamers were predicted by the Zuker algorithm (20), using Mfold (version 3.2) with conditions set up to 0.15 M NaCl and 25°C.
Determination of the half-life of aptamer AO-01 in cell culture.
Huh7 cells were transiently transfected using Fugene 6/HD/X-treme (Roche) in six 10-cm dishes with 1 μg (3.3 × 1013 molecules) of aptamer AO-01 per dish. After 6 h, the cells were washed 5 times with 2 ml prewarmed PBS to remove noninternalized aptamers. The aptamers were harvested at 1, 12, 24, 36, 48, and 60 h posttransfection by lysing the cells with 500 μl of lysis buffer (50 mM Tris-HCl, 100 mM NaCl, 20 mM EDTA, 0.5% [vol/vol] Nonidet P-40, pH 7.5) per dish. The aptamers were recovered by phenol-chloroform (1:1) extraction and concentrated by ethanol precipitation. The recovered aptamers were quantified by qPCR.
RESULTS
Selection of aptamers.
In order to generate aptamers binding to the matrix binding domain on the HBV capsid, we used the technique of systematic evolution of ligands by exponential enrichment (SELEX) (21) with positive and negative selection. A library consisting of single-stranded 55-base-long DNA molecules with 15 fixed nucleotides at each end and 25 central positions containing each of the four nucleotides in equimolar ratio served as the source for aptamers. The complexity of the library was 425 (approximately 1015). Thirty micrograms of aptamers corresponds to approximately 1015 molecules. For positive selection the library was incubated with capsids purified from E. coli (Fig. 1, lane 1). These capsids were formed by an HBV core protein lacking the 35 C-terminal arginine-rich amino acids, because this domain is dispensable for particle formation but able to bind unspecifically to nucleic acids (6). We therefore suspected that capsids formed by full-length core proteins would bind any aptamer and would not allow the selection of molecules binding to a specific region of the capsid surface. We refer to these capsids as WT capsids in this work. Aptamers attaching to WT capsids were separated from unbound molecules by filtration. The selected aptamers were extracted and then amplified by PCR using a biotinylated primer for the negative strand. Subsequently, the minus strand of each of the double-stranded oligonucleotides was removed by streptavidin-induced electrophoretic mobility shift, and the remaining positive strands were used for further rounds of selection. For negative selection, the aptamers were mixed with purified recombinant capsids also lacking the C-terminal 35 amino acids and carrying in addition the point mutation I126A (Fig. 1, lane 3). The amino acid residue isoleucine 126 is part of the matrix binding domain and is externally exposed on the capsid surface (22). The mutation to alanine strongly blocks HBV capsid envelopment (10). The residue is highly conserved; 98% of 4,043 full-length HBV genomes retrieved from the NCBI database carried a triplet coding for isoleucine at this position of the core gene. We expected that targeting residue I126 would give a higher chance for the identification of an aptamer inhibiting budding than screening for an aptamer with optimal binding just somewhere on the capsid surface. Aptamers not binding to the I126A mutant capsids were isolated by filtration, amplified by PCR, and converted to single-stranded oligomers as in the positive selection process.
FIG 1.
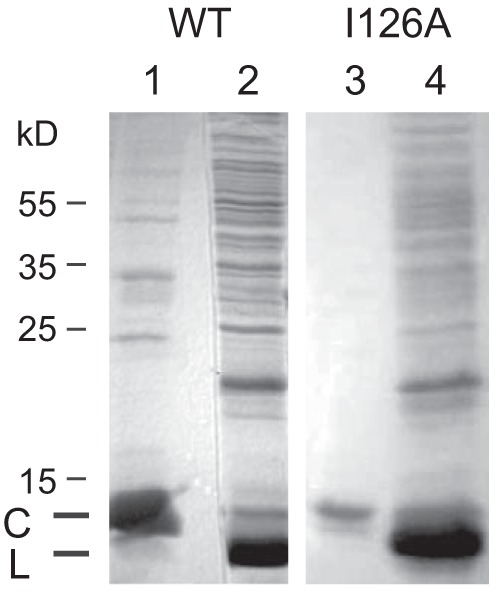
Purification of recombinant capsids consisting of C-terminally deleted core proteins. Ten microliters of purified WT and I126A mutant capsids (lanes 1 and 3, respectively) and 20 μl of cleared lysates (lanes 2 and 4, respectively) were separated by PAGE and stained with Coomassie blue. Numbers to the left indicate the positions of molecular mass marker proteins. C, C-terminally deleted core proteins; L, lysozyme.
The selection process was started with 11 nmol of aptamers, corresponding to approximately 6.6 × 1015 molecules. The concentrations of aptamers and wild type and mutant capsids, the incubation time for the aptamer-capsid binding, and the washing conditions were consecutively changed during the 13 rounds of SELEX in order to increase the selectivity during the selection process (Table 1).
At the end of the selection, double-stranded versions of the aptamers were molecularly cloned and sequenced (Table 2). Sixteen different sequences were found among 73 isolates. The most abundant sequence, AO-01, was present 13 times (18%). Ten of the 16 different aptamers contained the sequence CGCCA followed by TGTG or TGNN(N)TG. Secondary structure prediction for AO-01 shows a molecule with an imperfect 11-bp-long stem/6-base loop at the 5′ end and a 3-bp stem/9-base loop at the 3′ end, connected by 9 unpaired bases (Fig. 2). The structures of the other aptamers were also predicted. They all show a 3′ stem-loop and a 5′ stem-loop connected by a single-stranded region. The common motif (shown in bold face and underlined) is always at the 3′ end of the 5′ stem-loop and in the single-stranded linker region.
TABLE 2.
Sequences and frequencies of selected aptamers
| Aptamer | Sequencea | No. of appearancesb | Frequency (%)c | Length of variable region (nt) |
|---|---|---|---|---|
| A-01 | CACACGCGAGCCGCCATGTCTGGGC | 13 | 17.8 | 25 |
| A-02 | GGGACCGCAGAAGACCACATGTGCC | 11 | 15.1 | 25 |
| A-03 | GGGACGGCCCGCCATTCCGTGTGGC | 7 | 9.6 | 25 |
| A-04 | GTCGACGCGCCCATTCCGTGGGGTG | 6 | 8.2 | 25 |
| A-05 | GGCACACAACGTCGCCATGGCTGTG | 4 | 5.5 | 25 |
| A-06 | CCCACGCAACGGCGCCATGGCTGTG | 4 | 5.5 | 25 |
| A-07 | GCGTCGGCGCGCGCCATTGTGGTGC | 4 | 5.5 | 25 |
| A-08 | GGGCAGGGTCGACCGCCATGGCTGTG | 4 | 5.5 | 26 |
| A-09 | GGCACAAACGCGCCATGGCTGC | 4 | 5.5 | 22 |
| A-10 | GCCAACGACGGGCCGCCATGGTCTG | 3 | 4.1 | 25 |
| A-11 | GGCACAAACGCGGGCCATCCATGC | 3 | 4.1 | 24 |
| A-12 | GGCACCCAACGCCCCCATGGGTGTG | 2 | 2.7 | 25 |
| A-13 | GGGCAGGGTCGACCGCCATGGCTGG | 2 | 2.7 | 25 |
| A-14 | CCGAGGGGCAACGGCGCCATGGCTG | 2 | 2.7 | 25 |
| A-15 | CATAACGTTGCCCCCCATGTGTTG | 2 | 2.7 | 24 |
| A-16 | GGCAGCCTCGACCCCCCATGGC | 2 | 2.7 | 22 |
Only the sequence of the central variable region is shown. Boldface and underlining indicate the CGCCA motif and TGNTG motif, respectively.
Number of appearance of the sequence within 73 isolates.
Number of appearances × 100/73.
FIG 2.
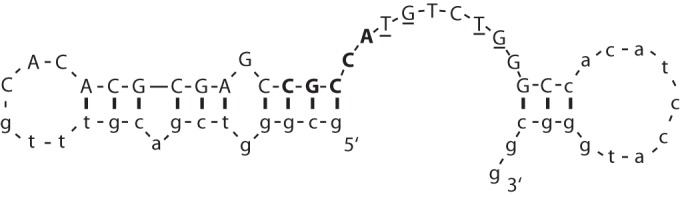
Predicted secondary structure of aptamer AO-01. The secondary structure of AO-01 was predicted by the Zuker algorithm (20). A motif common to 10 out of 16 different aptamers is shown in boldface and underlined. The fixed positions at both ends are shown in lowercase letters.
Affinity of aptamers to the matrix binding domain.
The dissociation constant Kd for the aptamer-capsid binding was measured for the four most abundant aptamers, AO-01 to AO-04, together representing 50% of the 73 isolates (Table 2). For this purpose, a constant amount of wild-type or mutant capsids was mixed with different amounts of aptamers, and bound aptamers were separated after incubation by immunoprecipitation with anti-HBc antibody from unbound molecules and quantified by qPCR (Fig. 3). The Kd values for the binding to WT capsids (Fig. 3A) correlated with the abundance of the aptamers within the 73 isolates and dropped from 180 ± 82 nM for AO-01 to 369 ± 285 nM for AO-04. In case of AO-01, the maximal value of 120 bound aptamers per capsid was observed at aptamer concentrations above 240 nM. C-terminally truncated core proteins form capsids with T = 4 icosahedral symmetry which consist of 120 core protein dimers (23). Apparently, a maximal saturation of one aptamer per dimer could not be exceeded.
FIG 3.
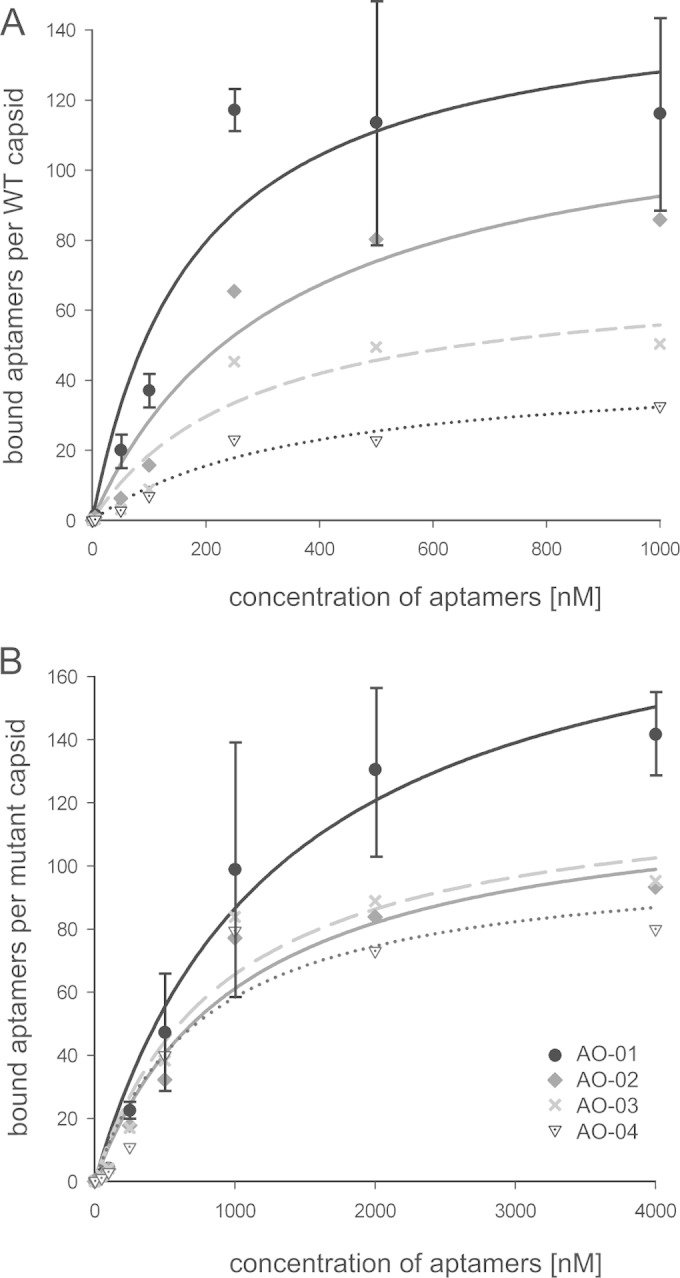
Determination of Kd values for aptamer binding to WT and mutant capsids. Capsids at a constant concentration of 1 nM were mixed with five different concentrations of the four most abundant aptamers, AO-01 to AO-04. Capsid-aptamer complexes were separated, and bound aptamers were quantified. (A) The dissociation constants of aptamer binding to WT capsid and the abundance of aptamers as shown in Table 2 are negatively correlated. A saturation of AO-01 binding was reached with 120 aptamers per capsid. (B) Binding to I126A mutant capsids. The standard deviation (n = 3) is shown only for aptamer AO-01. The standard deviations for the other aptamers were in the same range (not shown). Designation of the symbols as shown in panel B applies also to panel A.
The dissociation constants for the binding to I126A mutant capsids were 1,306 ± 503 nM for aptamer AO-01 and 2.7 to 7.3 times higher for the other characterized aptamers (Fig. 3B). This suggests that the binding of all 4 aptamers depended partially on the isoleucine residue at position 126. Aptamer AO-01 also reached a saturation of one aptamer per mutant dimer but only at concentrations above 2 μM.
Inhibition of virus formation by aptamer AO-01.
We transiently cotransfected Huh7 cells with an envelope-negative but replication-competent genomic HBV construct and expression plasmids for the synthesis of all three viral envelope proteins (9) as a positive control. At 3 days posttransfection, the amount of secreted virions corresponded to 1.6 × 106 viral DNA genome copies/ml culture supernatant as measured by a PCR-based method (Fig. 4, left bar) (9). When the plasmid for envelope protein expression was omitted, no virions could be formed (14), but the assay resulted in a signal of 2 × 105 viral DNA genomes/ml culture supernatant (bar B). This background is mainly due to residual amounts of the genomic plasmid used for transfection, which was amplified by the PCR. Cotransfection of both plasmids together with aptamer AO-01 resulted in a 50% lower virus concentration in the culture supernatant (bar A-01) than for the transfection without AO-01, whereas cotransfection of a random aptamer (bar A-N) showed no significant change relative to the positive control. Aptamers AO-02, -03, and -04 showed weaker effects (between 18% and 13% inhibition) than aptamer AO-01 (data not shown).
FIG 4.
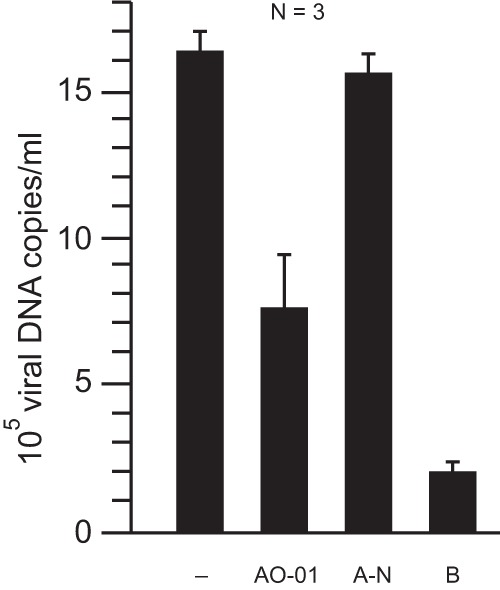
Inhibition of virus release by aptamer AO-01. Huh7 cells were transiently transfected with plasmids initiating HBV formation, and the concentration of viral genomes in the culture supernatant was measured at 3 days posttransfection. Leaving out the plasmid for HBV envelope protein expression prevented virion formation (14) and resulted in a background signal (bar B). Cotransfection of the plasmids with the aptamer AO-01 reduced the amount of virus in the culture supernatant by approximately 50% (bar AO-01) whereas cotransfection of an aptamer with a random sequence had no effect (bar A-N).
The inhibitory effect of aptamer AO-01 on virion formation is evident but relatively weak. This may be due to a short half-live of the aptamer within transfected cells. We measured the residual amount of aptamer AO-01 in cell cultures at 0 to 60 h after transfection (Fig. 5). The aptamer was exponentially degraded with a half-life (t1/2) of log10(2)/0.147 h ≈ 2.05 h. Therefore, an inhibitory effect of the aptamer on virus production can be expected only during the early period of the cotransfection.
FIG 5.
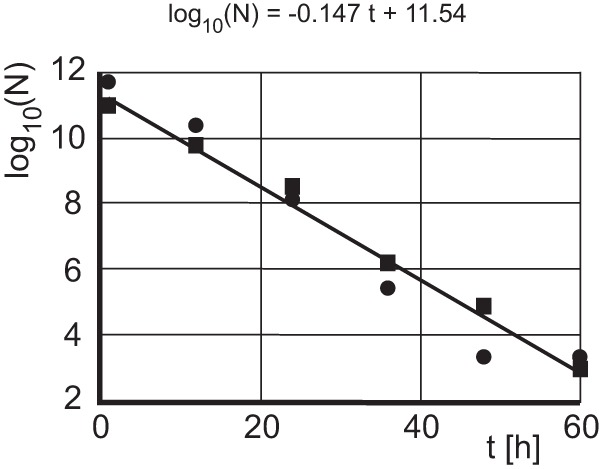
Determination of the half-life of aptamer AO-01 in Huh7 cells. Huh7 cells were transiently transfected with aptamer AO-01. The aptamer in the culture was quantified every 12 h by PCR. The amount of aptamers dropped exponentially. The half-life was approximately 2 h. The experiment was performed twice (squares and circles).
DISCUSSION
The formation of HBV particles is not well characterized. For example, models for the structural basis of the maturation signal coupling viral DNA genome synthesis in the lumen of the capsid to the competence of the particle for envelopment (4, 24) have been proposed (5, 25, 26) but have not been verified. Also, controversial data regarding the contact sites between capsid and envelope proteins have been reported. One model proposes that the tips of the spikes protruding from the capsid interact with the envelope proteins (27–29). Another model suggests that a domain (MBD) at the base of the spikes and lateral regions interacts with the large envelope protein (8, 30).
We generated an aptamer (31) that binds to or close to the MBD on HBV capsids expressed in bacteria, with a dissociation constant of 180 nM, by a positive/negative selection process. The I126A mutant capsids were chosen for negative selection because all tested amino acid substitutions at this position (A, V, L, G, W, F, Y, S, T, C, Q, and N), with the exceptions of methionine and proline, allowed capsid formation but strongly blocked capsid envelopment in transfected human hepatoma cells (10). The fact that the aptamer AO-01 bound to I126A mutant capsids with a 7-fold-higher dissociation constant than to WT capsids argues for a binding site including this residue. An alternative explanation, however, is that the I126A mutation induces a change at a different site of the capsid which is then involved in AO-01 binding. To date, we cannot rule out the second possibility. Our measurements revealed that a maximum of 120 AO-01 aptamers can be bound by one capsid. Apparently, two core proteins contribute to one binding site.
The aptamer AO-01 inhibited HBV secretion in Huh7 cells cotransfected with a genomic HBV construct. Although the half-life of AO-01 was only 2 h, a clear inhibition of virus secretion of approximately 50% during a 3-day period posttransfection could be observed. The half-life of AO-01 was measured in cells not expressing HBV capsids. Potentially, the half-life is extended by binding to capsids. It seems possible that the inhibitory effect of AO-01 was relatively strong during the early phase after the transfection, while later during the 3-day period, virus production may be more and more undisturbed. A DNA aptamer against the NSB5 protein of hepatitis C virus has a dissociation constant of 132 nM, which is similar to the Kd of aptamer AO-01, and reduced viral mRNA levels by 90% in a cell culture system (32). Another possible explanation besides the short half-life for the relatively weak inhibition of budding by aptamer AO-01 is that the MBD of mature HBV capsids competent for budding may have a structure different from that of the MBD of recombinant capsids lacking the C-terminal domain. To date, we cannot rule out this possibility. However, empty capsids apparently can be enveloped (26, 33), and it therefore seems possible that the MBD of recombinant capsids from bacteria mimics the MBD on mature capsids.
Which step in the viral life cycle was blocked by the aptamer AO-01 is not clear. The most straightforward model would be that the aptamer binds to or close to the MBD and thereby inhibits the interaction of the capsid with the matrix domain of the L protein. This explanation would support the model that the MD-MBD interaction is crucial for envelopment. It is not clear how many MD-MBD interactions would be necessary for complete envelopment of the capsid. If, e.g., close to 120 interactions per particle are required for this morphogenesis step, a few bound aptamers might be able to inhibit virion formation. However, if a few MD-MBD interactions per particle are sufficient, then an almost complete coverage of MBD sites by aptamers might be necessary for efficient inhibition. However, alternative models are well possible. For example, it is conceivable that aptamer binding blocks the generation of the maturation signal of the capsid or that it blocks the transport of the capsid to budding sites.
Other attempts have been made to inhibit the generation of HBV particles by interfering with the envelope-capsid interaction using peptides or chemicals (29, 34, 35). For example, cell-permeative peptides containing the matrix domain added at low micromolar concentrations to HBV-producing cells reduced the production of extracellular HBV DNA by 50% (34), and an oxazolidine derivative found during an in vitro screen for molecules inhibiting the matrix domain-capsid interaction reduced HBV DNA released from transiently transfected cells by a factor of 4 at a concentration of 20 μM (35). Our study supports the notion that molecules binding to the MBD on HBV capsids can suppress hepatitis B virion formation. Whether an aptamer-derived molecule can be applied in humans is open (36). However, in the future this target may expand therapeutic options for the treatment of chronic hepatitis B.
ACKNOWLEDGMENTS
The anti-HBc antibody H800 was provided by Heinz Schaller (Heidelberg, Germany). We thank Gabriele Möller (Institute for Experimental Genetics, Helmholtz Zentrum München, Germany) for assistance with Kd measurements.
A.O. was supported by the Deutsche Akademische Austauschdienst (DAAD) in cooperation with the Egyptian Ministry of Higher Education and Scientific Research. We thank Ulrike Protzer for financial support.
REFERENCES
- 1.Ott JJ, Stevens GA, Groeger J, Wiersma ST. 2012. Global epidemiology of hepatitis B virus infection: new estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine 30:2212–2219. doi: 10.1016/j.vaccine.2011.12.116. [DOI] [PubMed] [Google Scholar]
- 2.Beck J, Nassal M. 2007. Hepatitis B virus replication. World J Gastroenterol 13:48–64. doi: 10.3748/wjg.v13.i1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bruss V. 2007. Hepatitis B virus morphogenesis. World J Gastroenterol 13:65–73. doi: 10.3748/wjg.v13.i1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gerelsaikhan T, Tavis JE, Bruss V. 1996. Hepatitis B virus nucleocapsid envelopment does not occur without genomic DNA synthesis. J Virol 70:4269–4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roseman AM, Berriman JA, Wynne SA, Butler PJ, Crowther RA. 2005. A structural model for maturation of the hepatitis B virus core. Proc Natl Acad Sci U S A 102:15821–15826. doi: 10.1073/pnas.0504874102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gallina A, Bonelli F, Zentilin L, Rindi G, Muttini M, Milanesi G. 1989. A recombinant hepatitis B core antigen polypeptide with the protamine-like domain deleted self-assembles into capsid particles but fails to bind Nucleic acids. J Virol 63:4645–4652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stieler JT, Prange R. 2014. Involvement of ESCRT-II in hepatitis B virus morphogenesis. PLoS One 9:e91279. doi: 10.1371/journal.pone.0091279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ponsel D, Bruss V. 2003. Mapping of amino acid side chains on the surface of hepatitis B virus capsids required for envelopment and virion formation. J Virol 77:416–422. doi: 10.1128/JVI.77.1.416-422.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schittl B, Bruss V. 2014. Mutational profiling of the variability of individual amino acid positions in the hepatitis B virus matrix domain. Virology 458-459:183–189. doi: 10.1016/j.virol.2014.04.030. [DOI] [PubMed] [Google Scholar]
- 10.Pairan A, Bruss V. 2009. Functional surfaces of the hepatitis B virus capsid. J Virol 83:11616–11623. doi: 10.1128/JVI.01178-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yuan TT, Sahu GK, Whitehead WE, Greenberg R, Shih C. 1999. The mechanism of an immature secretion phenotype of a highly frequent naturally occurring missense mutation at codon 97 of human hepatitis B virus core antigen. J Virol 73:5731–5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yuan TT, Tai PC, Shih C. 1999. Subtype-independent immature secretion and subtype-dependent replication deficiency of a highly frequent, naturally occurring mutation of human hepatitis B virus core antigen. J Virol 73:10122–10128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Le Pogam S, Shih C. 2002. Influence of a putative intermolecular interaction between core and the pre-S1 domain of the large envelope protein on hepatitis B virus secretion. J Virol 76:6510–6517. doi: 10.1128/JVI.76.13.6510-6517.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bruss V, Ganem D. 1991. The role of envelope proteins in hepatitis B virus assembly. Proc Natl Acad Sci U S A 88:1059–1063. doi: 10.1073/pnas.88.3.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bruss V, Thomssen R. 1994. Mapping a region of the large envelope protein required for hepatitis B virion maturation. J Virol 68:1643–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuroki K, Russnak R, Ganem D. 1989. Novel N-terminal amino acid sequence required for retention of a hepatitis B virus glycoprotein in the endoplasmic reticulum. Mol Cell Biol 9:4459–4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prange R, Clemen A, Streeck RE. 1991. Myristylation is involved in intracellular retention of hepatitis B virus envelope proteins. J Virol 65:3919–3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bruss V, Ganem D. 1991. Mutational analysis of hepatitis B surface antigen particle assembly and secretion. J Virol 65:3813–3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pagratis NC. 1996. Rapid preparation of single stranded DNA from PCR products by streptavidin induced electrophoretic mobility shift. Nucleic Acids Res 24:3645–3646. doi: 10.1093/nar/24.18.3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tuerk C, Gold L. 1990. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 22.Wynne SA, Crowther RA, Leslie AG. 1999. The crystal structure of the human hepatitis B virus capsid. Mol Cell 3:771–780. doi: 10.1016/S1097-2765(01)80009-5. [DOI] [PubMed] [Google Scholar]
- 23.Zlotnick A, Cheng N, Conway JF, Booy FP, Steven AC, Stahl SJ, Wingfield PT. 1996. Dimorphism of hepatitis B virus capsids is strongly influenced by the C-terminus of the capsid protein. Biochemistry 35:7412–7421. doi: 10.1021/bi9604800. [DOI] [PubMed] [Google Scholar]
- 24.Summers J, Mason WS. 1982. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell 29:403–415. doi: 10.1016/0092-8674(82)90157-X. [DOI] [PubMed] [Google Scholar]
- 25.Dhason MS, Wang JCY, Hagan MF, Zlotnick A. 2012. Differential assembly of hepatitis B virus core protein on single- and double-stranded nucleic acid suggest the dsDNA-filled core is spring-loaded. Virology 430:20–29. doi: 10.1016/j.virol.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ning X, Nguyen D, Mentzer L, Adams C, Lee H, Ashley R, Hafenstein S, Hu J. 2011. Secretion of genome-free hepatitis B virus—single strand blocking model for virion morphogenesis of para-retrovirus. PLoS Pathog 7:e1002255. doi: 10.1371/journal.ppat.1002255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan WS, Dyson MR, Murray K. 1999. Two distinct segments of the hepatitis B virus surface antigen contribute synergistically to its association with the viral core particles. J Mol Biol 286:797–808. doi: 10.1006/jmbi.1998.2525. [DOI] [PubMed] [Google Scholar]
- 28.Seitz S, Urban S, Antoni C, Bottcher B. 2007. Cryo-electron microscopy of hepatitis B virions reveals variability in envelope capsid interactions. EMBO J 26:4160–4167. doi: 10.1038/sj.emboj.7601841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bottcher B, Tsuji N, Takahashi H, Dyson MR, Zhao S, Crowther RA, Murray K. 1998. Peptides that block hepatitis B virus assembly: analysis by cryomicroscopy, mutagenesis and transfection. EMBO J 17:6839–6845. doi: 10.1093/emboj/17.23.6839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kluge B, Schlager M, Pairan A, Bruss V. 2005. Determination of the minimal distance between the matrix and transmembrane domains of the large hepatitis B virus envelope protein. J Virol 79:7918–7921. doi: 10.1128/JVI.79.12.7918-7921.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feng H, Beck J, Nassal M, Hu KH. 2011. A SELEX-screened aptamer of human hepatitis B virus RNA encapsidation signal suppresses viral replication. PLoS One 6:e27862. doi: 10.1371/journal.pone.0027862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bellecave P, Cazenave C, Rumi J, Staedel C, Cosnefroy O, Andreola ML, Ventura M, Tarrago-Litvak L, Astier-Gin T. 2008. Inhibition of hepatitis C virus (HCV) RNA polymerase by DNA aptamers: mechanism of inhibition of in vitro RNA synthesis and effect on HCV-infected cells. Antimicrob Agents Chemother 52:2097–2110. doi: 10.1128/AAC.01227-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luckenbaugh L, Kitrinos KM, Delaney WE IV, Hu J. 14 November 2014. Genome-free hepatitis B virion levels in patient sera as a potential marker to monitor response to antiviral therapy. J Viral Hepat doi: 10.1111/jvh.12361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pan XB, Wei L, Han JC, Ma H, Deng K, Cong X. 2011. Artificial recombinant cell-penetrating peptides interfere with envelopment of hepatitis B virus nucleocapsid and viral production. Antiviral Res 89:109–114. doi: 10.1016/j.antiviral.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 35.Asif-Ullah M, Choi KJ, Choi KI, Jeong YJ, Yu YG. 2006. Identification of compounds that inhibit the interaction between core and surface protein of hepatitis B virus. Antiviral Res 70:85–90. doi: 10.1016/j.antiviral.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 36.Bruno JG. 2015. Predicting the uncertain future of aptamer-based diagnostics and therapeutics. Molecules 20:6866–6887. doi: 10.3390/molecules20046866. [DOI] [PMC free article] [PubMed] [Google Scholar]