

ABSTRACT
Myd88 signaling is critical to the control of numerous central nervous system (CNS) infections by promoting both innate and adaptive immune responses. Nevertheless, the extent to which Myd88 regulates type I interferon (IFN) versus proinflammatory factors and T cell function, as well as the anatomical site of action, varies extensively with the pathogen. CNS infection by neurotropic coronavirus with replication confined to the brain and spinal cord induces protective IFN-α/β via Myd88-independent activation of melanoma differentiation-associated gene 5 (MDA5). However, a contribution of Myd88-dependent signals to CNS pathogenesis has not been assessed. Infected Myd88−/− mice failed to control virus, exhibited enhanced clinical disease coincident with increased demyelination, and succumbed to infection within 3 weeks. The induction of IFN-α/β, as well as of proinflammatory cytokines and chemokines, was impaired early during infection. However, defects in both IFN-α/β and select proinflammatory factors were rapidly overcome prior to T cell recruitment. Myd88 deficiency also specifically blunted myeloid and CD4 T cell recruitment into the CNS without affecting CD8 T cells. Moreover, CD4 T cells but not CD8 T cells were impaired in IFN-γ production. Ineffective virus control indeed correlated most prominently with reduced antiviral IFN-γ in the CNS of Myd88−/− mice. The results demonstrate a crucial role for Myd88 both in early induction of innate immune responses during coronavirus-induced encephalomyelitis and in specifically promoting protective CD4 T cell activation. In the absence of these responses, functional CD8 T cells are insufficient to control viral spread within the CNS, resulting in severe demyelination.
IMPORTANCE During central nervous system (CNS) infections, signaling through the adaptor protein Myd88 promotes both innate and adaptive immune responses. The extent to which Myd88 regulates antiviral type I IFN, proinflammatory factors, adaptive immunity, and pathology is pathogen dependent. These results reveal that Myd88 protects from lethal neurotropic coronavirus-induced encephalomyelitis by accelerating but not enhancing the induction of IFN-α/β, as well as by promoting peripheral activation and CNS accumulation of virus-specific CD4 T cells secreting IFN-γ. By controlling both early innate immune responses and CD4 T cell-mediated antiviral IFN-γ, Myd88 signaling limits the initial viral dissemination and is vital for T cell-mediated control of viral loads. Uncontrolled viral replication in the absence of Myd88 leads to severe demyelination and pathology despite overall reduced inflammatory responses. These data support a vital role of Myd88 signaling in protective antimicrobial functions in the CNS by promoting proinflammatory mediators and T cell-mediated IFN-γ production.
INTRODUCTION
Rapid antiviral responses are initiated by a diverse array of pattern recognition receptors (PRRs) that respond to pathogen-associated molecular patterns. These include membrane-bound Toll-like receptors (TLRs) at the cell surface and endocytic compartments, as well as the cytoplasmic RNA helicases RIG-I and melanoma differentiation-associated gene 5 (MDA5) (1, 2). Both the structural components and the replication cycle of the virus, as well as the respective identities of the activated PRRs, dictate the magnitude and selectivity of the response. The viral structures and specific PRRs triggering innate immune responses, especially type I interferon (IFN), have been identified for numerous viruses (2, 3). However, analyses of distinct cell types in combination with in vivo studies are revealing a more complex picture in which the innate host response is coordinated by several pathways involving multiple PRRs (2–5). Efficient regulation of these pathways is especially crucial within the central nervous system (CNS), where innate immune activation is vital not only to limit viral spread via type I IFN but also to facilitate the recruitment of leukocytes and the expression of their effector functions via the induction of proinflammatory mediators. Nevertheless, the highly restricted and cell type-specific magnitude and diversity of PRR expression (3, 6–8) suggests tight regulation to initiate inflammation while avoiding irrelevant or excessive activation that may lead to bystander tissue damage.
The unifying factor required to transmit signals from most TLRs, excluding TLR-3, is the adaptor protein Myd88, which also transmits signals through the interleukin 1 (IL-1) and IL-18 receptors (1, 2). Myd88 is critical for the upregulation of proinflammatory genes and recruitment of leukocytes during numerous CNS infections and plays a protective role during vesicular stomatitis virus (VSV) (9), West Nile virus (WNV) (5), herpes simplex virus 1 (HSV-1) (10), HSV-2 (11), and Toxoplasma gondii (12) infection. However, the underlying mechanisms are only partially defined and differ among distinct infections and even for different virus strains. For example, both HSV-1 and HSV-2 activate TLR-2 and TLR-9 in vitro via a virus surface component and virus DNA, respectively. However, while infected Myd88−/− mice succumb to HSV-1, TLR-2−/− mice survive at rates similar to the survival of wild-type (wt) mice (10), suggesting that TLR-2 is redundant in vivo. In contrast, the results of studies with a distinct HSV-1 strain indicated that TLR-2 signaling mediates enhanced encephalitis and mortality (13). Peripheral infection with HSV-2 also demonstrated increased viral loads in the brains but not the livers of mice deficient in Myd88 or dually deficient in both TLR-2 and TLR-9; however, deficiency in either TLR alone did not alter the viral loads, despite affecting cytokine and chemokine profiles (11). Unlike HSV, WNV primarily activates innate immune responses within the CNS through MDA5 and RIG-I via the MAVS adapter (14, 15). Despite this Myd88-independent pathway, WNV infection of TLR-7−/− mice, as well as of Myd88−/− mice, revealed impaired viral control and leukocyte migration into the CNS that were associated with peripheral defects in IL-23 but not in IFN-α/β, IL-6, or tumor necrosis factor (TNF) (16). In a separate study, WNV-infected Myd88−/− mice also exhibited increased mortality coincident with enhanced viral spread specifically within the CNS, but not in peripheral organs. In this case, uncontrolled virus was associated with reduced proinflammatory responses and leukocyte recruitment into the CNS, yet no defects in peripheral T cell activation, CNS IFN-α/β, or CNS virus-specific T cell responses were detected (5).
Similar to WNV infection, PRR-dependent IFN-α/β production is vital to prevent peripheral dissemination of mouse hepatitis virus (MHV) strain A59, as well as the spread of the gliatropic MHV strain JHM (JHMV) within the CNS (17–19). While peripheral MHV infection is sensed via Myd88-dependent TLR-7 in plasmacytoid dendritic cells (pDCs) (18), MDA5 is the primary sensor inducing IFN-α/β in microglia/macrophages (20). A contribution of Myd88 signaling during encephalomyelitis mediated by infection with the gliatropic JHMV, which is associated with minimal if any productive replication in draining lymph node dendritic cells (21), has not been assessed. Following JHMV infection, IFN-α/β prevents neuronal infection and restrains viral spread within the CNS prior to the emergence of adaptive immunity (19). T cells subsequently control CNS viral replication within 2 weeks via IFN-γ- and perforin-mediated mechanisms but are insufficient to provide sterile immunity, resulting in viral RNA persistence (22–24). Given the crucial roles of both innate and adaptive components to antiviral protection within the CNS, the current studies assessed the role of Myd88 in regulating inflammation and antiviral activity during JHMV-induced encephalomyelitis.
Infection of Myd88−/− mice revealed that early innate immune responses were transiently impaired but that the impairment was rapidly overcome by Myd88-independent signals. Uncontrolled viral replication correlated with significantly diminished IFN-γ and IFN-γ-dependent major histocompatibility complex (MHC) upregulation, supporting the idea that antiviral T cell effector function was impaired in vivo. Moreover, myeloid and CD4 T cell but not CD8 T cell recruitment to the CNS was significantly blunted. The results demonstrate a crucial biphasic role for Myd88 in supporting a rapidly induced innate immune response early during coronavirus encephalomyelitis and subsequently promoting protective CD4 T cell functions during the adaptive phase.
MATERIALS AND METHODS
Mice, viruses, and infections.
C57BL/6 mice were purchased from the National Cancer Institute (Frederick, MD). Homozygous Myd88−/− mice [B6.129P2(SJL)-Myd88tm1Defr/J, stock number 008888; Jackson Laboratories, Bar Harbor, ME] on the C57BL/6 background were kindly provided by Robert Fairchild (Cleveland Clinic, Cleveland, OH) and bred locally. Mice were housed under pathogen-free conditions in an accredited facility at the Cleveland Clinic Lerner Research Institute. All animal procedures were performed in compliance with protocols approved by the Cleveland Clinic Institutional Animal Care and Use Committee (PHS assurance number A3047-01). Mice at 6 to 7 weeks of age were infected intracranially in the left hemisphere with a sublethal, gliatropic, monoclonal antibody (MAb)-derived variant of JHMV designated 2.2v-1 (25) at a dose of 1,000 PFU in 30 μl of endotoxin-free Dulbecco's phosphate-buffered saline (PBS). The clinical disease severity was graded daily using the following scale: 0, healthy; 1, ruffled fur/hunched back; 2, inability to turn upright/partial hind-limb paralysis; 3, complete hind-limb paralysis; 4, moribund or dead (26–28). Infectious virus in cell-free supernatants was determined by plaque assay on DBT astrocytoma monolayers as described previously (25). Briefly, individual brains were homogenized in 4 ml Dulbecco's PBS using chilled Tenbroeck glass homogenizers. Homogenates were clarified by centrifugation at 400 × g for 7 min at 4°C, and supernatants stored at −70°C until used for plaque assay.
RNA extraction, reverse transcription, and gene expression analysis.
RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions and subjected to real-time PCR analysis as described previously (17, 26, 27). In brief, snap-frozen tissues were dissociated with TRIzol in a TissueLyser II (Qiagen, Valencia, CA) and treated with chloroform, and RNA was precipitated with isopropyl alcohol. Following washing with 75% ethanol, RNA was resuspended in RNase-free water (Gibco/Invitrogen, Grand Island, NY) and treated for 30 min at 37°C with DNase I using a DNA-free kit (Ambion, Austin, TX) according to the manufacturer's instructions. Two micrograms RNA was converted to cDNA using Moloney murine leukemia virus reverse transcriptase (Invitrogen) in buffer containing 10 mM deoxynucleoside triphosphate mix, 250 ng random hexamer primers, and oligo(dT) (1:1 ratio) (Invitrogen). cDNA samples were diluted 10-fold in RNase-free water before analysis by quantitative real-time PCR using either SYBR green master mix (Applied Biosystems, Foster City, CA) or TaqMan technology as described previously (17, 26). The primer sequences for SYBR green PCR analysis were as follows: (F, forward; R, reverse): Gapdh, F, 5′-CATGGCCTTCCGTGTTCCTA-3′, and R, 5′-ATGCCTGCTTCACCACCTTCT-3′; Il6, F, 5′-ACACATGTTCTCTGGGAAATCGT-3′, and R, 5′-AAGTGCATCATCGTTGTTCATACA-3′; Il10, F, 5′-TTTGAATTCCCTGGGTGAGAA-3′, and R, 5′-GCTCCACTGCCTTGCTCTTATT-3′; Il21, F, 5′-GGACAGTATAGACGCTCACGAATG-3′, and R, 5′-CGTATCGTACTTCTCCACTTGCA-3′; Tnf, F, 5′-GCCACCACGCTCTTCTGTCT-3′, and R, 5′-GGTCTGGGCCATAGAACTGATG-3′; Nos2, F, 5′-CCTGGTACGGGCATTGCT-3′, and R, 5′-CATGCGGCCTCCTTTGAG-3′; Ccl3, F, 5′-CCAAGTCTTCTCAGCGCCAT-3′, and 5′-GAATCTTCCGGCTGTAGGAGAAG-3′; Ccl5, F, 5′-GCAAGTGCTCCAATCTTGCA-3′, and R, 5′-CTTCTCTGGGTTGGCACACA-3′; Cxcl10, F, 5′-GACGGTCCGCTGCAACTG-3′, and R, 5′-GCTTCCCTATGGCCCTCATT-3′; Cxcl13, F, 5′-CATAGATCGGATTCAAGTTACGCC-3′, and 5′-TCTTGGTCCAGATCACAACTTCA-3′; and viral nucleocapsid (N) protein-encoding RNA, F, 5′-GCCAAATAATCGCGCTAGAA-3′, and R, 5′-CCGAGCTTAGCCAAAACAAG-3′. All samples were run in duplicate on a 96-well plate using a 7500 fast real-time PCR system (Applied Biosystems) with an automatic set baseline and a manually set critical threshold cycle (CT) at which the fluorescent signal becomes higher than the signals of all of the PCR primer pairs. Dissociation curves were used to confirm the amplification of a single product for each primer pair per sample. The expression levels of Gapdh, Cxcl1, Ccl2, Ifnα4, Ifnα5, Ifnβ1, Ifnγ, Ifit1, Ifit2, Isg15, and Il1β were determined using TaqMan primer and probe sets and 2× universal TaqMan fast master mix (Applied Biosystems). Data were calculated relative to the results for the housekeeping gene Gapdh using the following formula: 2[CT(Gapdh) − CT(target gene)] × 1,000.
Cell isolation, flow cytometry, and intracellular cytokine staining.
Cells for flow cytometric analysis were isolated from brains as described previously (17, 26, 28). Briefly, mice were perfused with PBS, and their brains removed and homogenized in 4 ml of Dulbecco's PBS (pH 7.4) using Tenbroeck tissue homogenizers. Following centrifugation at 400 × g for 7 min, cell pellets were resuspended in RPMI containing 25 mM HEPES (pH 7.2) that was adjusted to 30% Percoll (Pharmacia, Uppsala, Sweden) and underlain with 1 ml of 70% Percoll. Following centrifugation at 800 × g for 30 min at 4°C, cells were recovered from the 30%–70% interface, washed with RPMI, and suspended in fluorescence-activated cell sorter (FACS) buffer (0.5% bovine serum albumin in Dulbecco's PBS). Fcγ receptors were blocked with 1% mouse serum and 1% rat anti-mouse CD16/CD32 MAb (clone 2.4G2; BD Biosciences, San Jose, CA) for 20 min. Specific cell types were identified by staining with fluorescein isothiocyanate (FITC)-, phycoerythrin (PE)-, peridinin chlorophyll protein (PerCP)-, or allophycocyanin (APC)-conjugated MAb for 30 min on ice in FACS buffer. The expression of surface markers was characterized with MAb (from BD Biosciences except when indicated) specific for CD45 (clone Ly-5), CD4 (clone GK1.5), CD8 (clone 53-6.7), F4/80 (Serotec, Raleigh, NC), Ly-6G (clone 1A8), and MHC class II (clone 2G9). Virus-specific CD8 T cells were identified using H-2Db/S510 MHC class I tetramers as described previously (19, 26, 27, 29). Samples were analyzed using a FACSCalibur flow cytometer (BD Biosciences) and Flow-Jo 7 software (Treestar, Inc., Ashland, OR).
IFN-γ production by T cells was measured by intracellular flow cytometry following stimulation of 5 × 105 CNS-derived cells with 3 × 105 EL4 or CHB3 feeder cells with or without virus-specific MHC class I- or class II-restricted peptides for 5 h as described previously (26, 29). Briefly, CD8 and CD4 T cells were stimulated with 1 μM S510 or 10 μM M133, respectively, in a total volume of 200 μl of RPMI supplemented with 10% fetal calf serum for 5 h at 37°C with Golgi Stop (BD Biosciences). Following surface staining for CD8, CD4, and CD45, cells were fixed and permeabilized using the Cytofix/Cytoperm kit (BD Biosciences) according to the manufacturer's protocol. Intracellular cytokines were detected using FITC-conjugated anti-IFN-γ MAb. Cells were analyzed by flow cytometry as described above.
Immunohistochemistry.
Brains from PBS-perfused mice were divided along the mid-sagittal plane, fixed with 10% Zn+ formalin, and embedded in paraffin. The spinal cords were divided into 6 equivalent-length segments from the cervical to the lumbar end and embedded in paraffin together, allowing cross sections from individual mice to be examined at each of the 6 levels. Sections were stained with either hematoxylin and eosin (H&E) or Luxol fast blue (LFB) as described previously (30–32). Cells containing the viral N protein were identified by immunoperoxidase staining using MAb J.3.3 as the primary antibody, biotinylated horse anti-mouse antibody as the secondary antibody, and streptavidin-conjugated horseradish peroxidase and 3,3′-diaminobenzidine substrate (Vectastain-ABC kit; Vector Laboratory, Burlingame, CA) (33). High-resolution whole-slide scanning was performed using an Aperio ScanScope digital slide scanner (Carlsbad, CA) with the 40× objective as described previously (17). Sections from each experimental group were evaluated in a blinded fashion, and representative fields identified.
Cytokine and chemokine ELISA.
Virus-infected-brain supernatants were assessed for IL-1β and IL-6 levels using the mouse IL-1β enzyme-linked immunosorbent assay (ELISA) Ready-SET-Go (catalog number 88-7013; eBioscience) and IL-6 ELISA Ready-SET-Go (catalog number 88-7064; eBioscience) kits, while CCL5 and CXCL10 levels were measured using mouse CCL5 ELISA (catalog number MMR00; R&D systems; Minneapolis, MN) and CXCL10 ELISA (catalog number MCX100; R&D systems, Minneapolis, MN) kits according to the respective manufacturer's instructions. The IFN-γ levels in brain supernatants were measured by ELISA as described previously (26, 31). Briefly, 96-well plates were coated overnight at 4°C with 1 μg/ml rat anti-mouse IFN-γ MAb (R4-6A2; BD Bioscience) in 100 μl 0.1 M disodium hydrogen phosphate, pH 9.5, followed by blocking of nonspecific binding with 10% fetal calf serum (FCS) in PBS for 1 h. Samples and recombinant IFN-γ standard (BD Biosciences) were added overnight at 4°C. Bound IFN-γ was detected using biotinylated rat anti-mouse IFN-γ MAb (XMG1.2; BD Biosciences) and avidin peroxidase, followed by 3,3′,5,5′-tetramethylbenzidine (TMB reagent set; BD Biosciences) 30 min later. Optical densities were read at 450 nm with a Bio-Rad model 680 microplate reader and analyzed using Microplate Manager 5.2 software (Bio-Rad Laboratories, Hercules, CA).
Statistical analysis.
Survival curves and real-time PCR data were analyzed by using the unpaired, two-tailed Student t test. A P value of <0.05 was considered significant. Data were analyzed using Microsoft Excel.
RESULTS
Myd88 deficiency results in lethal viral encephalomyelitis.
To assess how Myd88 signaling affects pathogenesis associated with a demyelinating neurotropic virus, wt and Myd88−/− mice were infected with the sublethal gliatropic coronavirus JHMV. Both groups of mice began to exhibit clinical signs of encephalitis at day 7 postinfection (p.i.), which progressed to partial or complete hind-limb paralysis by day 10 p.i. (Fig. 1). Disease symptoms remained relatively stable in wt mice, with recovery starting at day 14 p.i. In contrast, Myd88−/− mice suffered from more severe paralytic disease, which did not regress. In contrast to the 90% survival of infected wt mice, Myd88−/− mice gradually succumbed to infection, with a survival rate of only ∼10% by day 21 (Fig. 1). Although Myd88 signaling did not alter the initial acute phase of viral encephalomyelitis, it affected clinical disease at the time coinciding with T cell-mediated control of virus replication, during days 7 to 14 p.i., in wt mice (26, 28, 31).
FIG 1.
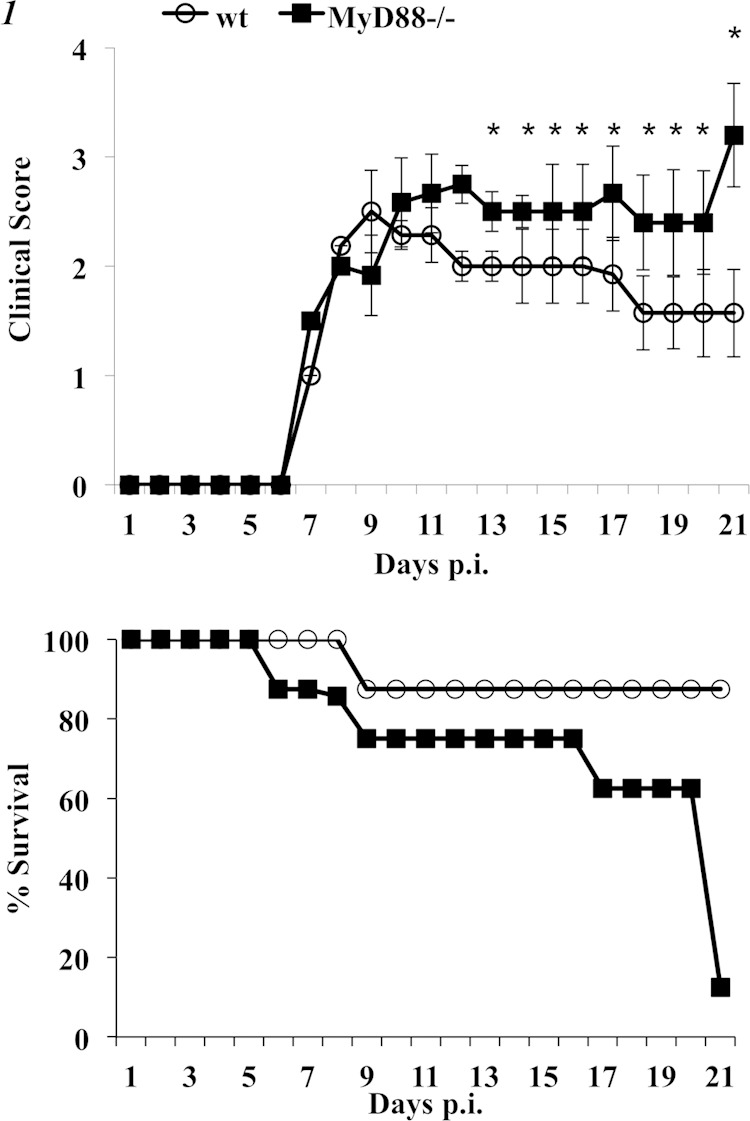
Myd88−/− mice exhibit severe clinical disease and succumb to JHMV infection. Wild-type (open symbols) and Myd88−/− (closed symbols) mice infected intracranially with JHMV were monitored for clinical disease (top) and for survival (bottom). Clinical scores, determined as described in Materials and Methods, represent the average results of two independent experiments (n ≥ 20) ± standard errors of the means (SEM). *, P < 0.05. Survival rates are representative of two independent experiments (n = 18 per group).
Analysis of the viral loads within the CNS demonstrated that the sustained disease severity in Myd88−/− mice correlated with ineffective virus control (Fig. 2). Whereas wt mice displayed progressively reduced viral loads between days 5 and 10 p.i., Myd88−/− mice already harbored higher viral loads at day 5 p.i. and showed no evidence of viral control until day 10 p.i., when the virus titers exceeded those in wt mice by 3 log10 (Fig. 2A). Infectious virus was modestly further reduced in Myd88−/− mice surviving to day 14 p.i., at which time virus was already below the detection limit in wt mice. The viral N mRNA levels in brains confirmed increased early virus replication and modest control by days 10 and 14 p.i. in Myd88−/− mice compared to the virus replication and control in wt mice (Fig. 2A). Similarly, the viral mRNA levels in spinal cords were vastly increased by day 7 in infected Myd88−/− relative to the levels in wt mice, despite being similar at day 5 p.i. The differences remained prominent at days 10 and 14 p.i., when the levels had already progressively declined in spinal cords of wt mice. Poorly controlled viral replication in spinal cords was confirmed by ∼2-log10 higher viral loads in Myd88−/− mice relative to the loads in wt mice through days 10 to 14 p.i. (data not shown).
FIG 2.
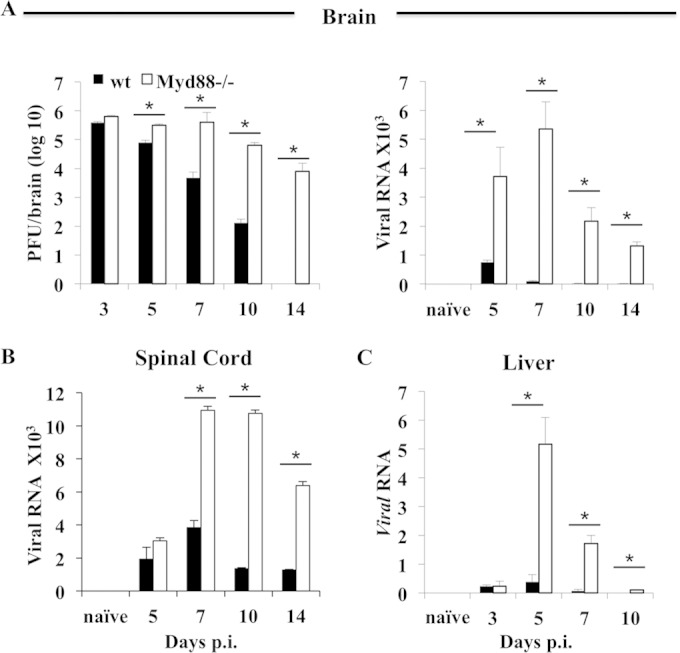
Myd88 deficiency impairs CNS viral control. The levels of virus replication in the brains, spinal cords, and livers of infected wt and Myd88−/− mice were determined by plaque assay or quantitative real-time PCR analysis of viral N protein-encoding transcripts, as indicated. The levels of viral mRNA were determined relative to the level of Gapdh. Data represent the average results of two or three independent experiments (n = 6 to 9) ± SEM. *, P < 0.05.
The gliatropic JHMV variant used herein is highly neurotropic, with barely detectable mRNA in the periphery of wt mice; however, it can disseminate to the liver in immunocompromised mice (30). Viral mRNA was measured in the liver to determine whether Myd88 deficiency affected the immune system sufficiently to allow virus dissemination to the periphery. Viral mRNA levels in the liver were similarly low at day 3 p.i. in both groups (Fig. 2C). However, while viral mRNA in wt mice remained sparse in the liver compared to the levels in the CNS at days 5 and 7 p.i., it increased by ∼10-fold by day 5 p.i. in the absence of Myd88. Nevertheless, a significant drop in viral mRNA by day 10 p.i. suggested control by adaptive immunity distinct from the limited control of virus in the CNS. Furthermore, the absence of hepatitis as assessed by lesion formation (data not shown) indicated that liver infection did not contribute to mortality.
JHMV predominantly infects glia, with very sparse neuronal tropism, and rapidly transitions from brain to spinal cord, where virus is predominantly found within white matter tracts (24, 31, 34, 35). To determine whether Myd88 deficiency alters viral tropism, brains and spinal cords were analyzed for the distribution of infected cells at days 7 and 10 p.i. (Fig. 3). In contrast to the focal nature of infection in the brains of wt mice at day 7 p.i., the numbers of viral antigen-positive cells were markedly increased in infected Myd88−/− mice, and the foci were much more widely distributed throughout the brain (Fig. 3A). The dominant cell type infected in wt mice exhibited a morphology consistent with glia/macrophages, although rare infected neurons were also identified (Fig. 3A). Despite the increase in the numbers of infected cells in the brains of Myd88−/− mice, the relative proportions of infected glia to infected neurons remained similar. Striking differences in the distribution of virus antigen-positive cells were also apparent in spinal cords (Fig. 3B). Whereas sections from wt mice harbored only a few if any infected cells in white matter by day 10 p.i., sections from Myd88−/− mice exhibited prominently increased numbers of infected cells in white matter, as well as viral dissemination to gray matter.
FIG 3.

Myd88 deficiency enhances virus dissemination within the CNS. Viral antigen in the brains (A) and spinal cords (B) of infected wt and Myd88−/− mice at days 7 and 14 p.i., respectively, was detected using MAb specific for viral N protein. Note the increased numbers of foci of viral-antigen-positive cells in tissue samples from Myd88−/− mice. Enlargements of boxed areas show infected cells at higher magnification.
Early IFN-α/β expression in the CNS is Myd88 dependent.
IFN-α/β is essential in preventing early JHMV dissemination within the CNS prior to the emergence of adaptive immune responses (19). Thus, elevated levels of virus in the brains of Myd88−/− mice as early as day 5 p.i. implied impaired induction of IFN-α/β and/or IFN-α/β-mediated antiviral factors. Ifnα/β mRNA expression in the brains was analyzed as early as day 3 p.i., when virus replication was similar in both groups (Fig. 2). In wt mice, Ifnβ, Ifnα4, and Ifnα5 transcripts all peaked at day 3 p.i., declined by day 5 p.i., and subsequently subsided to background levels (Fig. 4A), confirming previous results (19, 26). Although all three mRNA species were also increased over naive levels in Myd88−/− mice, the levels were significantly reduced relative to those in wt mice at day 3 p.i. All transcripts, particularly Ifnβ mRNA, subsequently increased by day 5 p.i., exceeding wt levels, and remained elevated until day 10 p.i. in the absence of Myd88. Consistent with the distinct Ifnα/β mRNA expression pattern, analysis of the IFN-stimulated genes (ISG) Ifit1, Ifit2, and Isg15 also revealed impaired early induction in the brain but higher transcript levels subsequently in the absence of Myd88 (Fig. 4B). The distinct kinetics and magnitude of Ifnβ mRNA induction in Myd88−/− mice relative to the pattern in wt mice suggested that Myd88 signaling contributes to very early IFN-α/β induction but that a Myd88-independent pathway is triggered as virus continues to replicate. Overall, these results suggest that Myd88-dependent IFN-α/β activity stems early viral spread within the CNS. However, the subsequently impaired viral control in the absence of Myd88 is likely to reside in a defect in the adaptive immune response.
FIG 4.

Delayed type I interferon induction and IFN-stimulated gene expression in the CNS of Myd88−/− mice. The expression levels of Ifnβ, Ifna4, and Ifna5 mRNA (A) and of IFN-α/β-inducible genes Ifit1, Ifit2, and Isg15 (B) in the brains of naive and JHMV-infected wt and Myd88−/− mice relative to Gapdh expression were determined by real-time PCR. Data represent the average results ± SEM (n = 6 mice/group/time point) from two separate experiments. Values for naive wt mice are shown (n = 3). *, P < 0.05.
Reduced proinflammatory factors in Myd88−/− mice correlate with impaired accumulation of neutrophils, macrophages, and CD4 T cells.
Infections and sterile injury in Myd88−/− mice are both associated with reduced induction of proinflammatory cytokines and chemokines (11, 12, 36), even when IFN-α/β induction is mainly driven by RIG-I and MDA5 (5). To assess whether Myd88 deficiency affected proinflammatory factors during JHMV infection, the brains were analyzed for cytokine and chemokine mRNA and protein expression throughout the acute phase of encephalomyelitis. IL-1β, IL-6, and TNF, commonly upregulated during acute neuroinflammation, promote the disruption of the blood-brain barrier (BBB) and facilitate CNS entry of leukocytes. In wt mice, Il1β and Il6 mRNA levels were both maximal at day 3 p.i. (Fig. 5A). Subsequently, Il1β mRNA was sustained at high levels through day 7 p.i., while Il6 mRNA progressively declined. Similar to the induction of Ifnα/β mRNA, the induction of Il1β and Il6 mRNA was impaired early in infection in the absence of Myd88, but the mRNA levels of both increased by day 5 p.i., suggesting that the early deficit was overcome by Myd88-independent signaling. Furthermore, while Il1β mRNA continued to increase, Il6 mRNA levels dropped by day 7 p.i., despite uncontrolled viral replication. Tnf mRNA levels were also significantly reduced at day 3 p.i. in the absence of Myd88 but were upregulated by day 5 p.i. and remained stable through day 10 p.i. CNS supernatants revealed reduced levels of IL-1β in Myd88−/− mice as early as day 3 p.i. and even more distinctly at day 7 p.i., when expression peaked in wt mice (Fig. 5B). Similarly, IL-6 was reduced by >50% in the absence of Myd88 at days 3 and 5 p.i. Moreover, while IL-6 protein levels reflected the mRNA levels in wt mice, IL-6 appeared to be posttranscriptionally regulated in Myd88−/− mice. The discrepancy between mRNA and protein levels may reside in distinct translational regulation, e.g., impaired translation due to elevated levels of activated PKR (protein kinase RNA dependent) coincident with elevated viral replication (26).
FIG 5.

Reduced proinflammatory cytokine response in the absence of Myd88. (A) Levels of Il1β, Il6, and Tnf mRNA in the brains of naive and JHMV-infected wt and Myd88−/− mice relative to Gapdh expression were determined by real-time PCR. Data represent the average results ± SEM (n = 6 mice/group/time point) from two separate experiments. Values for naïve wt mice are shown (n = 3). (B) Levels of IL-1β and IL-6 proteins in brain supernatants of infected wt and Myd88−/− mice were determined by ELISA. Data are the average results ± SEM from two independent experiments (n = 6/group). *, P < 0.05.
Chemokine transcripts associated with neutrophil, monocyte, and lymphocyte recruitment into the CNS were also examined (37). Transcripts encoding CXCL1, a neutrophil chemoattractant, as well as CCL2 and CCL3, both associated with monocyte recruitment, were all significantly impaired at day 3 p.i. in the absence of Myd88. However, while these transcripts declined by days 5 and 7 p.i. in wt mice, their delayed induction reached similar or even elevated levels in Myd88−/− mouse brains (Fig. 6A). In wt mice, transcripts encoding CCL5, CXCL10, and CXCL13, chemoattractants for T and B cells, were all induced by day 3 p.i., and Ccl5 and Cxcl13 mRNAs continued to increase until day 7 p.i. In contrast, Ccl5, Cxcl10, and Cxcl13 mRNAs were barely detectable in Myd88−/− mice at day 3 p.i. While Cxcl13 mRNA remained near baseline levels through day 10 p.i., the induction of Ccl5 and Cxcl10 mRNAs was delayed in the absence of Myd88, but these mRNAs reached levels similar to those in wt mice at days 10 and 7 p.i., respectively. Although the levels of Cxcl10 mRNA dropped in both groups after day 7 p.i., they remained higher at days 10 and 14 p.i. in the absence of Myd88, consistent with the elevated virus and IFN-α/β levels. The levels of CCL5 and CXCL10 in CNS homogenates reflected the patterns of the mRNAs (Fig. 6B). Both were significantly reduced at days 5 and 7 p.i. in the absence of Myd88. By day 10 p.i., the level of CCL5 was equal to and the level of CXCL10 even higher than the levels in wt mice. The equilibration or even increased mRNA levels of chemokines after day 7 p.i. in the absence of Myd88, despite early deficits, presumably reflect sustained virus loads in the CNS.
FIG 6.

Impaired chemokine expression in the absence of Myd88. (A) Expression levels of Cxcl1, Ccl2, Ccl3, Ccl5, Cxcl10, and Cxcl13 mRNAs in the brains of infected wt and Myd88−/− mice relative to Gapdh expression were determined by real-time PCR. Data from naive wt mice are shown (n = 3). (B) Protein levels of T cell chemoattractants CCL5 and CXCL10 in brain supernatants from infected wt and Myd88−/− mice were determined by ELISA. Data represent the average results ± SEM from two independent experiments (n = 6/group). *, P < 0.05.
Consistent with impaired expression of proinflammatory chemokines, Myd88 deficiency is associated with reduced leukocyte recruitment into the CNS in various neuroinflammation models (5, 9, 12, 36). During JHMV infection, neutrophils and monocytes are early innate responders that aid in subsequent parenchymal entry of T cells, although they do not directly affect viral control or demyelination (28, 38). CD8 T cells are essential antiviral effectors via IFN-γ- and perforin-mediated mechanisms (23, 24). CD4 T cells contribute to antiviral IFN-γ production, promote local CD8 T cell effector function (39, 40), and are the major producers of anti-inflammatory IL-10 (41). We therefore assessed how the early impairment in chemokine expression affected CNS leukocyte recruitment during JHMV infection. Infiltrating CD45hi leukocytes were reduced by ∼50% in Myd88−/− mice relative to their levels in wt mice at days 3 and 5 p.i., but the differences were less prominent by day 7 p.i. and thereafter (Fig. 7A). The deficit in early inflammatory cells in Myd88−/− mouse brains reflected reductions in neutrophils (CD45hi Ly6G+) by >60% and in monocyte/macrophages (CD45hi F480+) by >70% at days 3 and 5 p.i. (Fig. 7A), supporting the idea that myeloid cells are early CNS infiltrates (28). Nevertheless, monocytes increased subsequently (Fig. 7A), consistent with delayed expression of the chemoattractants CCL2 and CCL3 (Fig. 6A). The total numbers of CD4 and CD8 T cells in the brains peaked between days 7 and 10 p.i. in both groups (Fig. 7A). However, CD4 T cell recruitment was significantly impaired in the absence of Myd88, reaching only 50% or less of the wt levels throughout the infection. In contrast, Myd88 deficiency did not alter the overall numbers of CD8 T cells or tetramer+ CD8 T cells in the CNS between days 7 and 14 p.i. (Fig. 7A). Myd88 signaling thus promoted early CNS accumulation of myeloid cells and CD4 T cells but did not affect CD8 T cell recruitment. Analysis of overall inflammation by immunohistochemistry confirmed diminished recruitment of inflammatory cells. Whereas wt mice showed numerous perivascular cuffs in the brain at day 7 p.i., areas of perivascular inflammation were sparse and less pronounced in Myd88−/− mice (Fig. 7B).
FIG 7.

Myd88 deficiency impairs recruitment of myeloid and CD4 T cells in the CNS. (A) The numbers of total infiltrating leukocytes (CD45hi), neutrophils (Ly6G+), macrophages (F480+), CD4 T cells, CD8 T cells, and virus-specific Db/S510 tetramer-reactive CD8 T cells in the CNS of infected wt and Myd88−/− mice were determined by flow cytometry at the indicated times p.i. Data represent the mean results ± SEM for mice (n = 6/group/time point and experiment) from three separate experiments. *, P < 0.05. (B) Representative results for inflammation in brains at day 7 p.i. as determined by hematoxylin-and-eosin staining. Images are representative of the results for 3 to 4 individuals from 2 separate experiments. Inset shows perivascular area at higher magnification.
Myd88 deficiency impairs IFN-γ production in the CNS.
Impaired CD4 T cell CNS infiltration yet similar CD8 T cell CNS infiltration in Myd88−/− mice compared to wt mice suggested that uncontrolled viral replication is due to inefficient T cell effector function (40). IFN-γ, the most critical T cell-derived antiviral mediator during JHMV infection (22, 24, 34), is expressed more abundantly by CD4 than by CD8 T cells in vivo (27). In addition to its antiviral effect, IFN-γ increases MHC expression on CNS target cells (42, 43), thus promoting both CD8 and CD4 T cell effector functions in vivo. In wt mice, Ifnγ mRNA starts to accumulate at day 5 and peaks at day 7 p.i. (Fig. 8A), coincident with T cell accumulation and antiviral effector function within the CNS. Despite similar kinetics of Ifnγ mRNA expression in infected Myd88−/− and wt mice, the levels were significantly reduced throughout acute infection in Myd88−/− mice. The pattern of IFN-γ protein expression in the CNS matched that of mRNA in both groups (Fig. 8A). However, IFN-γ was significantly lower in the CNS of Myd88−/− mice, specifically at day 7 p.i., when virus control by T cells is initially evident in wt mice. IFN-γ production remained low by day 10 p.i., despite the vastly increased viral load and similar levels of virus-specific CD8 T cells in Myd88−/− mice (Fig. 7). Reduced IFN-γ activity was supported by very sparse induction of IFN-γ-dependent Nos2 mRNA in the CNS (Fig. 8). Low IFN-γ levels also predicted impaired MHC upregulation and, consequently, inefficient T cell engagement and effector activity. MHC class II expression was undetectable on microglia in uninfected controls (data not shown), as well as in infected IFN-γ-deficient mice (42). Indeed, analysis of IFN-γ-dependent MHC class II expression on resident CD45lo microglia confirmed impaired MHC class II upregulation in Myd88−/− mice (Fig. 8B). Consistent with the IFN-γ kinetics, MHC class II expression was low on microglia at day 5 p.i. but was increased on ∼75% of microglia by day 7 p.i. Expression was sustained at day 10 p.i. in wt mice (Fig. 8B), despite the decline in IFN-γ. In contrast, in Myd88−/− mice, only 22% and 38% of microglia expressed MHC class II by day 7 p.i. and 10 p.i., respectively (Fig. 8B), reflecting reduced IFN-γ. Infiltrating macrophages (CD45hi F480+) exhibited similarly impaired and delayed MHC class II expression in Myd88−/− mice and wt mice (data not shown). Decreased IFN-γ in the CNS of Myd88−/− mice was thus functionally evident from reduced Nos2 mRNA and MHC class II expression, consistent with the diminished accumulation and effector function of CD4 T cells (40).
FIG 8.

Defective IFN-γ and MHC class II upregulation in the CNS of Myd88−/− mice. (A) Expression levels of Ifnγ mRNA, IFN-γ protein, and IFN-γ-inducible Nos2 mRNA in the brains of naive and infected wt and Myd88−/− mice at the indicated times. Transcript levels relative to Gapdh expression were measured by real-time PCR, and protein levels by ELISA. Data represent the mean results ± SEM from two independent experiments (n = 6). Data from naive wt mice are shown (n = 3). *, P < 0.05. (B) Upregulation of MHC class II on brain microglia in wt and Myd88−/− mice following JHMV infection was determined by flow cytometry. CNS-derived cells pooled from infected wt and Myd88−/− mice (n = 4 or 5) at days 5, 7, and 10 p.i. were stained with anti-CD45 and anti-MHC class II I-A/E antibodies. Density plots were gated on total CD45+ cells; quadrants were set to separate MHC class II-positive microglia (CD45lo) from infiltrating (CD45hi) cells as indicated. Numbers in parentheses represent the percentages of MHC class II-expressing cells within microglia. Data are representative of three independent experiments ± SEM.
Myd88 signaling specifically promotes CD4 T cell function within the CNS.
The extent to which reduced IFN-γ in the JHMV-infected CNS was associated with CD4 or CD8 T cells was evaluated by ex vivo stimulation with the immunodominant I-Ab-restricted M133 or Db-restricted S510 peptide, respectively. Whereas ∼25% of CNS-derived CD4 T cells from wt mice produced IFN-γ at days 7 and 10 p.i., only ∼7% of the already lower Myd88−/− CD4 T cell population was capable of IFN-γ production, representing an ∼65 to 70% reduction in frequency despite the sustained high viral loads (Fig. 9A). Importantly, stimulation of CNS-derived cells was carried out in the presence of MHC class II-expressing feeder cells, suggesting that CD4 T cell function was intrinsically impaired and not attributable to reduced MHC class II expression in vivo. In contrast to virus-specific CD4 T cells, ∼35% of CNS-derived CD8 T cells produced IFN-γ in both groups of mice at days 7 (Fig. 9A) and 10 p.i. (data not shown), revealing no defects in Myd88−/− CD8 T cells for IFN-γ production. This represented ∼76% of Db/S510 tetramer+ CD8 T cells and reflected similar percentages of tetramer-reactive cells within the CD8 populations.
FIG 9.

Myd88 signaling is critical for regulating virus-specific CD4 but not CD8 T cell activity. Cells from the brains (A) or CLN (B) of infected wt and Myd88−/− mice harvested at the indicated times were analyzed for frequencies of IFN-γ-producing CD8 or CD4 T cells in response to S510 or M133 peptide stimulation, respectively. Representative density plots of brain-derived cells were gated on CD4 or CD8 T cells in the absence (−) or presence (+) of peptide. IFN-γ-producing cells are depicted in ellipses or rectangles, and numbers show relative percentages of IFN-γ+ cells. Bar graphs of the frequencies of IFN-γ-producing cells within CD4 or CD8 T cells show the average results ± SEM from 3 individual mice. *, P < 0.05.
To assess whether defects in CD4 T cells were limited to the CNS or were imprinted during activation in the periphery, T cells from the cervical lymph nodes (CLN), the anatomical site of T cell priming following JHMV infection (33), were examined at days 5 and 7 p.i. The overall frequencies of M133-specific CD4 T cells were below 2% in CLN of both groups; however, the percentages of IFN-γ-producing cells were reduced by ∼50% in Myd88−/− mice relative to the percentages in wt populations (Fig. 9B). In contrast, CLN CD8 T cells showed no differences in the percentages of IFN-γ-producing cells at either day 5 or 7 p.i. (Fig. 9B). Unimpaired virus-specific CD8 T cell expansion in CLN was confirmed by similar percentages of S510 tetramer+ CD8 T cells (∼1% in both wt and Myd88−/− CD8 T cells at day 7 p.i.) (data not shown). These results imply that reduced IFN-γ in the CNS of JHMV-infected Myd88−/− mice is associated not only with reduced recruitment/survival of CD4 T cells but, also, severely impaired IFN-γ production by CD4 T cells. Moreover, the defect in CD4 T cell responsiveness is already imprinted during the priming/expansion phase in the CLN and is further exacerbated in the CNS.
Uncontrolled virus replication in the absence of Myd88 coincides with increased demyelination despite limited inflammation.
A hallmark of JHMV infection in wt mice is T cell-driven, immune-mediated demyelination (22, 35, 44). In contrast to immunocompetent mice, mice deficient in T and B cells do not develop demyelinating disease, despite the inability to control infectious virus (44). To assess how reduced T cell effector function in Myd88−/− mice influences JHMV-mediated pathology, demyelination was assessed in the brains and spinal cords. Interestingly, JHMV-infected Myd88−/− mice exhibited pronounced demyelination in the brain stem by day 10 p.i. (Fig. 10). In contrast, demyelination was very limited in the brains of wt mice. Although demyelination was clearly evident in the spinal cords of wt mice at day 14 p.i., the extent of demyelination in the spinal cord was prominently increased in the absence of Myd88 signaling (Fig. 10).
FIG 10.

Myd88 deficiency enhances demyelination. Brain stems were removed at day 10 p.i. (top) and spinal cords at day 14 p.i. (bottom) from infected wt and Myd88−/− mice and stained with LFB. Images are representative of 2 separate experiments with 3 to 4 individuals per experiment and 6 cross sections per cord.
DISCUSSION
Myd88 signaling is involved in the induction of type I IFN, proinflammatory factors, and leukocyte recruitment to the CNS in various neuroinflammation models, including microbial infection and stab wound injury (3–5, 12, 36). However, the effects exerted by Myd88 depend on the insult and can be disassociated from IFN-α/β induction if viruses are predominantly sensed by Myd88-independent PRRs (5). Coronaviruses, including MHV, are very poor inducers of IFN-α/β in most cell types, which has been attributed to the similarity of the capped viral mRNA to host mRNA (45). Nevertheless IFN-α/β is critical to block viral dissemination in both the CNS and the periphery (18, 19). The results of this study demonstrate that Myd88 is essential for control of CNS infection by the sublethal gliatropic coronavirus JHMV. Moreover, protection from lethal encephalomyelitis via Myd88 signaling was mediated at both the innate and adaptive immune levels.
Early during JHMV infection, Myd88 deficiency significantly impaired the initial induction of IFN-α/β and downstream transcription of ISGs. As virus continued to replicate, impaired IFN-α/β induction was overcome by Myd88-independent signals. Nevertheless, the failure to rapidly establish a protective antiviral state provided a window for viral dissemination within the CNS prior to the recruitment of adaptive immune cells. Myd88-independent IFN-α/β production, although delayed, is consistent with the notion that MHV infection of glia is mainly sensed by cytosolic PRRs. MDA5 is the main sensor required to induce IFN-α/β in microglia/macrophages following infection with the MHV strain A59 (20), while both MDA5 and RIG-I sense MHV A59 in an oligodendroglial cell line (46). However, although oligodendrocytes are major targets of JHMV, they do not induce IFN-α/β in response to JHMV in vivo (7). The delayed yet increased IFN-α/β induction in the Myd88−/− CNS is thus likely attributable to MDA5 activation in infected microglia/macrophages. The failure to limit viral spread despite enhanced Myd88-independent IFN-α/β responses presumably resides in the virus's ability to outpace IFN-α/β-mediated protection, similar to uncontrolled infection in SCID or Rag-deficient mice (47, 48).
The contributions of Myd88 to promoting innate and adaptive immunity associated with the cytosolic sensors RIG-I and MDA5 during CNS infections are only sparsely explored. Despite the overall paucity of IFN-α/β induction due to evasion of MDA5 recognition, plasmacytoid dendritic cells (pDCs) make a critical contribution to TLR-7-mediated IFN-α/β induction following peripheral infection with MHV A59 (18). While we cannot exclude a contribution of pDCs to early IFN-α/β production in the CNS, the minimal peripheral replication of the gliatropic JHMV, relative to that of MHV A59, is less likely to activate pronounced pDC signaling. Moreover, although recruitment of pDCs to the CNS has been demonstrated (49–51), it has not been described during MHV infection. We thus favor the notion that nonviral factors, such as cellular stress responses, may contribute to triggering Myd88-dependent innate immune responses (52). Moreover, other resident cells that are infected early, such as ependymal cells (53), may contribute to the early innate immune response.
Myd88 was also essential for early induction of numerous proinflammatory factors, including IL-1β, IL-6, TNF, and various chemokines. Similar to delayed IFN-α/β responses, the diminished proinflammatory responses were, for the most part, overcome by day 7 p.i. and correlated with uncontrolled virus replication. It remains to be elucidated whether IFN-α/β and innate proinflammatory factors are activated via similar temporally distinct Myd88 and Myd88-independent pathways. However, in this context, it is interesting to note that Myd88 deficiency was only associated with a defect in chemokines and not IFN-α/β production in the CNS following WNV infection (5). Neither the expression of systemic IFN-α/β nor that of Ifnα/β mRNA was impaired in the cerebral cortex of WNV-infected Myd88−/− mice relative to their expression in wt mice at day 6 p.i. However, the WNV load was elevated in the absence of Myd88 in several CNS anatomical regions as early as day 4 p.i. As the corresponding Ifnα/β mRNA levels at this early time were not provided, it remains unclear whether enhanced WNV replication was solely due to the absence of a Myd88-mediated antiviral activity independent of IFN-α/β (5).
Consistent with the delayed and blunted induction of proinflammatory mediators and the chemokines CXCL1, CCL2, and CCL5, the recruitment of neutrophils and monocytes into the CNS during the innate immune response was significantly dampened in JHMV-infected Myd88−/− mice. Neutrophils and monocytes play subtle roles during JHMV infection by accelerating leukocyte entry into the CNS parenchyma. Although abrogation of either myeloid subset alone does not significantly impair viral control or pathology (28), we cannot exclude that reduced recruitment of these myeloid populations may contribute to enhanced pathogenesis in Myd88−/− mice. Furthermore, impaired CXCL10, coincident with a defect in CD4 T cell accumulation, is consistent with a prominent role of CXCL10 in promoting CD4 but not CD8 T cell recruitment into the CNS (54, 55). Importantly, the impaired IFN-γ production by virus-specific CD4 T cells was directly correlated with uncontrolled viral replication after day 5 p.i. Early CD4 T cell-mediated IFN-γ production enhances both class I and class II MHC upregulation (31, 42, 43). Although neither total nor virus-specific CD8 T cell accumulation was affected by Myd88 deficiency, reduced antigen presentation by CNS-resident cells and CD4 T cell help may limit CD8 T cell function in vivo. Indeed, CD4 T cells are critical to promote CD8 function within the CNS during JHMV infection (40), partially by prolonging CD8 T cell activity via IL-21 production (39). Il21 mRNA, expressed mainly by CD4 T cells, was reduced by ∼40% in the CNS at day 7 p.i. in the absence of Myd88. Although CD8 T cell function was not affected ex vivo, a contribution of impaired CD4 T cell help to the limited CD8 T cell function in vivo cannot be excluded. Finally, NK cells are redundant for JHMV pathogenesis (29), supporting insufficient CD4 T cell activation as a critical Myd88-dependent component of viral control within the CNS.
Myeloid and total T cell but not virus-specific CD8 T cell accumulation were also blunted in the CNS of WNV-infected Myd88−/− mice, although peripheral T cell responses were not impaired (5). Similar to JHMV infection, virus control was primarily affected in the CNS and not the periphery. Myd88−/− mice are also highly susceptible to cerebral infection by Toxoplasma gondii (12). Increased parasite burdens and severe pathology in the CNS are also directly correlated with reduced expression of proinflammatory molecules and local production of IFN-γ, a mediator of protective immunity. In this case, impaired IFN-γ coincided with significantly decreased recruitment of CD8 T cells. However, in contrast with JHMV infection, the accumulation of myeloid cells in the CNS was increased relative to that in wt mice (12), suggesting that Myd88 signaling can also inhibit CNS inflammation in some circumstances.
The mechanisms underlying impaired T cell function in the absence of Myd88 have recently been explored during WNV-induced encephalitis. Consistent with unimpaired peripheral responses in Myd88−/− mice, IL-1R deficiency did not impair peripheral T cell activation or T cell recruitment into the CNS (56). Rather, IL-1R signaling was specifically required to reactivate T cells by promoting maturation of antigen-presenting cells (APC) within the CNS. Defective T cell antiviral activity within the CNS was thus attributed to impaired local reactivation, in contrast to a defect at the priming stage in the periphery (57). A similar mechanism involving IL-1R-dependent infiltrating APC may underlie poor CD4 T cell responses following JHMV infection. However, defective CD4 T cell responses were already evident in CLN during priming, consistent with the ability of IL-1β to induce both phagocytosis and antigen-presenting function during monocyte differentiation (58). The APC-reactivating JHMV-specific CD4 T cells in the CNS have not been identified; however, JHMV infects dendritic cells only poorly (21). Moreover, while microglia and macrophages are both infected, they require IFN-γ for class II upregulation. As oligodendrocytes, the most prominent cell type infected in wt mice, do not express class II MHC, CD4 T cells are most likely activated by cross-presentation (59).
An unanticipated result was the increased extent of demyelination in both the brain and spinal cord of JHMV-infected Myd88−/− mice, despite more limited induction of proinflammatory factors and reduced inflammatory infiltrates. Although it can be argued that enhanced demyelination results from uncontrolled viral load and direct virus-induced oligodendrocyte damage, demyelination is very sparse in mice devoid of adaptive immune responses (53, 60). Moreover, enhanced oligodendrocyte infection in mice incapable of responding to IFN-γ specifically in oligodendrocytes does not lead to increased demyelination despite effective T cell function (34). This suggests a protective function of Myd88 in demyelination independent of the viral load in oligodendrocytes. While Myd88 is also redundant for spontaneous autoimmune-mediated demyelination (61), it is essential for the mediation of experimental allergic encephalomyelitis following autoantigen immunization (62). These diverse results implicate distinct contributions of Myd88 to demyelination depending on the local environment and responder cell types.
In summary, the present study reveals that Myd88 protects from lethal JHMV encephalomyelitis by at least two pathways. During the innate immune phase, Myd88 promotes the initial induction of IFN-α/β, thereby stemming virus dissemination within the CNS and its spread to the liver prior to the expansion of virus-specific T cells. Although the impaired early innate immune responses are overcome, Myd88 is essential in promoting the activation of virus-specific CD4 T cells in CLN, as well as enhancing their accumulation and IFN-γ secretion within the CNS. Analogous to the knowledge of WNV- and Toxoplasma gondii-induced encephalitis, these data support a vital role of Myd88 signaling in protective antimicrobial functions in the CNS by promoting the early induction of proinflammatory mediators, as well as supporting T cell-mediated IFN-γ production.
ACKNOWLEDGMENTS
This study was supported by National Institutes of Health grant P01 NS064932.
We sincerely thank Wenqiang Wei, Eric Barron, and Ernesto Barron for exceptional technical assistance with histopathology.
This paper is dedicated to Niranjan Butchi and his family.
REFERENCES
- 1.Takeuchi O, Akira S. 2010. Pattern recognition receptors and inflammation. Cell 140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 2.Thompson MR, Kaminski JJ, Kurt-Jones EA, Fitzgerald KA. 2011. Pattern recognition receptors and the innate immune response to viral infection. Viruses 3:920–940. doi: 10.3390/v3060920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carty M, Reinert L, Paludan SR, Bowie AG. 2014. Innate antiviral signalling in the central nervous system. Trends Immunol 35:79–87. doi: 10.1016/j.it.2013.10.012. [DOI] [PubMed] [Google Scholar]
- 4.Furr SR, Marriott I. 2012. Viral CNS infections: role of glial pattern recognition receptors in neuroinflammation. Front Microbiol 3:201. doi: 10.3389/fmicb.2012.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Szretter KJ, Daffis S, Patel J, Suthar MS, Klein RS, Gale M Jr., Diamond MS. 2010. The innate immune adaptor molecule MyD88 restricts West Nile virus replication and spread in neurons of the central nervous system. J Virol 84:12125–12138. doi: 10.1128/JVI.01026-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cho H, Proll SC, Szretter KJ, Katze MG, Gale M Jr., Diamond MS. 2013. Differential innate immune response programs in neuronal subtypes determine susceptibility to infection in the brain by positive-stranded RNA viruses. Nat Med 19:458–464. doi: 10.1038/nm.3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kapil P, Butchi NB, Stohlman SA, Bergmann CC. 2012. Oligodendroglia are limited in type I interferon induction and responsiveness in vivo. Glia 60:1555–1566. doi: 10.1002/glia.22375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paul S, Ricour C, Sommereyns C, Sorgeloos F, Michiels T. 2007. Type I interferon response in the central nervous system. Biochimie 89:770–778. doi: 10.1016/j.biochi.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 9.Lang KS, Navarini AA, Recher M, Lang PA, Heikenwalder M, Stecher B, Bergthaler A, Odermatt B, Akira S, Honda K, Hengartner H, Zinkernagel RM. 2007. MyD88 protects from lethal encephalitis during infection with vesicular stomatitis virus. Eur J Immunol 37:2434–2440. doi: 10.1002/eji.200737310. [DOI] [PubMed] [Google Scholar]
- 10.Mansur DS, Kroon EG, Nogueira ML, Arantes RM, Rodrigues SC, Akira S, Gazzinelli RT, Campos MA. 2005. Lethal encephalitis in myeloid differentiation factor 88-deficient mice infected with herpes simplex virus 1. Am J Pathol 166:1419–1426. doi: 10.1016/S0002-9440(10)62359-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sorensen LN, Reinert LS, Malmgaard L, Bartholdy C, Thomsen AR, Paludan SR. 2008. TLR2 and TLR9 synergistically control herpes simplex virus infection in the brain. J Immunol 181:8604–8612. doi: 10.4049/jimmunol.181.12.8604. [DOI] [PubMed] [Google Scholar]
- 12.Torres M, Guiton R, Lacroix-Lamande S, Ryffel B, Leman S, Dimier-Poisson I. 2013. MyD88 is crucial for the development of a protective CNS immune response to Toxoplasma gondii infection. J Neuroinflammation 10:19. doi: 10.1186/1742-2094-10-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kurt-Jones EA, Chan M, Zhou S, Wang J, Reed G, Bronson R, Arnold MM, Knipe DM, Finberg RW. 2004. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci U S A 101:1315–1320. doi: 10.1073/pnas.0308057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fredericksen BL, Keller BC, Fornek J, Katze MG, Gale M Jr. 2008. Establishment and maintenance of the innate antiviral response to West Nile virus involves both RIG-I and MDA5 signaling through IPS-1. J Virol 82:609–616. doi: 10.1128/JVI.01305-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suthar MS, Ma DY, Thomas S, Lund JM, Zhang N, Daffis S, Rudensky AY, Bevan MJ, Clark EA, Kaja MK, Diamond MS, Gale M Jr. 2010. IPS-1 is essential for the control of West Nile virus infection and immunity. PLoS Pathog 6:e1000757. doi: 10.1371/journal.ppat.1000757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Town T, Bai F, Wang T, Kaplan AT, Qian F, Montgomery RR, Anderson JF, Flavell RA, Fikrig E. 2009. Toll-like receptor 7 mitigates lethal West Nile encephalitis via interleukin 23-dependent immune cell infiltration and homing. Immunity 30:242–253. doi: 10.1016/j.immuni.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Butchi NB, Hinton DR, Stohlman SA, Kapil P, Fensterl V, Sen GC, Bergmann CC. 2014. Ifit2 deficiency results in uncontrolled neurotropic coronavirus replication and enhanced encephalitis via impaired alpha/beta interferon induction in macrophages. J Virol 88:1051–1064. doi: 10.1128/JVI.02272-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cervantes-Barragan L, Zust R, Weber F, Spiegel M, Lang KS, Akira S, Thiel V, Ludewig B. 2007. Control of coronavirus infection through plasmacytoid dendritic-cell-derived type I interferon. Blood 109:1131–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ireland DD, Stohlman SA, Hinton DR, Atkinson R, Bergmann CC. 2008. Type I interferons are essential in controlling neurotropic coronavirus infection irrespective of functional CD8 T cells. J Virol 82:300–310. doi: 10.1128/JVI.01794-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roth-Cross JK, Bender SJ, Weiss SR. 2008. Murine coronavirus mouse hepatitis virus is recognized by MDA5 and induces type I interferon in brain macrophages/microglia. J Virol 82:9829–9838. doi: 10.1128/JVI.01199-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou H, Perlman S. 2006. Preferential infection of mature dendritic cells by mouse hepatitis virus strain JHM. J Virol 80:2506–2514. doi: 10.1128/JVI.80.5.2506-2514.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bergmann CC, Lane TE, Stohlman SA. 2006. Coronavirus infection of the central nervous system: host-virus stand-off. Nat Rev Microbiol 4:121–132. doi: 10.1038/nrmicro1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin MT, Stohlman SA, Hinton DR. 1997. Mouse hepatitis virus is cleared from the central nervous systems of mice lacking perforin-mediated cytolysis. J Virol 71:383–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parra B, Hinton DR, Marten NW, Bergmann CC, Lin MT, Yang CS, Stohlman SA. 1999. IFN-gamma is required for viral clearance from central nervous system oligodendroglia. J Immunol 162:1641–1647. [PubMed] [Google Scholar]
- 25.Fleming JO, Trousdale MD, el-Zaatari FA, Stohlman SA, Weiner LP. 1986. Pathogenicity of antigenic variants of murine coronavirus JHM selected with monoclonal antibodies. J Virol 58:869–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kapil P, Stohlman SA, Hinton DR, Bergmann CC. 2014. PKR mediated regulation of inflammation and IL-10 during viral encephalomyelitis. J Neuroimmunol 270:1–12. doi: 10.1016/j.jneuroim.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Phares TW, Stohlman SA, Hinton DR, Atkinson R, Bergmann CC. 2010. Enhanced antiviral T cell function in the absence of B7-H1 is insufficient to prevent persistence but exacerbates axonal bystander damage during viral encephalomyelitis. J Immunol 185:5607–5618. doi: 10.4049/jimmunol.1001984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Savarin C, Stohlman SA, Atkinson R, Ransohoff RM, Bergmann CC. 2010. Monocytes regulate T cell migration through the glia limitans during acute viral encephalitis. J Virol 84:4878–4888. doi: 10.1128/JVI.00051-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zuo J, Stohlman SA, Hoskin JB, Hinton DR, Atkinson R, Bergmann CC. 2006. Mouse hepatitis virus pathogenesis in the central nervous system is independent of IL-15 and natural killer cells. Virology 350:206–215. doi: 10.1016/j.virol.2006.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ireland DD, Stohlman SA, Hinton DR, Kapil P, Silverman RH, Atkinson RA, Bergmann CC. 2009. RNase L mediated protection from virus induced demyelination. PLoS Pathog 5:e1000602. doi: 10.1371/journal.ppat.1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Phares TW, Ramakrishna C, Parra GI, Epstein A, Chen L, Atkinson R, Stohlman SA, Bergmann CC. 2009. Target-dependent B7-H1 regulation contributes to clearance of central nervous system infection and dampens morbidity. J Immunol 182:5430–5438. doi: 10.4049/jimmunol.0803557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Phares TW, Stohlman SA, Hinton DR, Bergmann CC. 2012. Enhanced CD8 T-cell anti-viral function and clinical disease in B7-H1-deficient mice requires CD4 T cells during encephalomyelitis. J Neuroinflammation 9:269. doi: 10.1186/1742-2094-9-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marten NW, Stohlman SA, Zhou J, Bergmann CC. 2003. Kinetics of virus-specific CD8+ T-cell expansion and trafficking following central nervous system infection. J Virol 77:2775–2778. doi: 10.1128/JVI.77.4.2775-2778.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gonzalez JM, Bergmann CC, Ramakrishna C, Hinton DR, Atkinson R, Hoskin J, Macklin WB, Stohlman SA. 2006. Inhibition of interferon-gamma signaling in oligodendroglia delays coronavirus clearance without altering demyelination. Am J Pathol 168:796–804. doi: 10.2353/ajpath.2006.050496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang FI, Stohlman SA, Fleming JO. 1990. Demyelination induced by murine hepatitis virus JHM strain (MHV-4) is immunologically mediated. J Neuroimmunol 30:31–41. doi: 10.1016/0165-5728(90)90050-W. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Babcock AA, Toft-Hansen H, Owens T. 2008. Signaling through MyD88 regulates leukocyte recruitment after brain injury. J Immunol 181:6481–6490. doi: 10.4049/jimmunol.181.9.6481. [DOI] [PubMed] [Google Scholar]
- 37.Hosking MP, Lane TE. 2010. The role of chemokines during viral infection of the CNS. PLoS Pathog 6:e1000937. doi: 10.1371/journal.ppat.1000937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Savarin C, Stohlman SA, Rietsch AM, Butchi N, Ransohoff RM, Bergmann CC. 2011. MMP9 deficiency does not decrease blood-brain barrier disruption, but increases astrocyte MMP3 expression during viral encephalomyelitis. Glia 59:1770–1781. doi: 10.1002/glia.21222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Phares TW, DiSano KD, Hinton DR, Hwang M, Zajac AJ, Stohlman SA, Bergmann CC. 2013. IL-21 optimizes T cell and humoral responses in the central nervous system during viral encephalitis. J Neuroimmunol 263:43–54. doi: 10.1016/j.jneuroim.2013.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Phares TW, Stohlman SA, Hwang M, Min B, Hinton DR, Bergmann CC. 2012. CD4 T cells promote CD8 T cell immunity at the priming and effector site during viral encephalitis. J Virol 86:2416–2427. doi: 10.1128/JVI.06797-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Puntambekar SS, Bergmann CC, Savarin C, Karp CL, Phares TW, Parra GI, Hinton DR, Stohlman SA. 2011. Shifting hierarchies of interleukin-10-producing T cell populations in the central nervous system during acute and persistent viral encephalomyelitis. J Virol 85:6702–6713. doi: 10.1128/JVI.00200-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bergmann CC, Parra B, Hinton DR, Chandran R, Morrison M, Stohlman SA. 2003. Perforin-mediated effector function within the central nervous system requires IFN-gamma-mediated MHC up-regulation. J Immunol 170:3204–3213. doi: 10.4049/jimmunol.170.6.3204. [DOI] [PubMed] [Google Scholar]
- 43.Malone KE, Stohlman SA, Ramakrishna C, Macklin W, Bergmann CC. 2008. Induction of class I antigen processing components in oligodendroglia and microglia during viral encephalomyelitis. Glia 56:426–435. doi: 10.1002/glia.20625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu GF, Dandekar AA, Pewe L, Perlman S. 2000. CD4 and CD8 T cells have redundant but not identical roles in virus-induced demyelination. J Immunol 165:2278–2286. doi: 10.4049/jimmunol.165.4.2278. [DOI] [PubMed] [Google Scholar]
- 45.Zust R, Cervantes-Barragan L, Habjan M, Maier R, Neuman BW, Ziebuhr J, Szretter KJ, Baker SC, Barchet W, Diamond MS, Siddell SG, Ludewig B, Thiel V. 2011. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat Immunol 12:137–143. doi: 10.1038/ni.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li J, Liu Y, Zhang X. 2010. Murine coronavirus induces type I interferon in oligodendrocytes through recognition by RIG-I and MDA5. J Virol 84:6472–6482. doi: 10.1128/JVI.00016-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bergmann CC, Parra B, Hinton DR, Ramakrishna C, Dowdell KC, Stohlman SA. 2004. Perforin and gamma interferon-mediated control of coronavirus central nervous system infection by CD8 T cells in the absence of CD4 T cells. J Virol 78:1739–1750. doi: 10.1128/JVI.78.4.1739-1750.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pewe L, Perlman S. 2002. Cutting edge: CD8 T cell-mediated demyelination is IFN-gamma dependent in mice infected with a neurotropic coronavirus. J Immunol 168:1547–1551. doi: 10.4049/jimmunol.168.4.1547. [DOI] [PubMed] [Google Scholar]
- 49.Brehin AC, Mouries J, Frenkiel MP, Dadaglio G, Despres P, Lafon M, Couderc T. 2008. Dynamics of immune cell recruitment during West Nile encephalitis and identification of a new CD19+B220-BST-2+ leukocyte population. J Immunol 180:6760–6767. doi: 10.4049/jimmunol.180.10.6760. [DOI] [PubMed] [Google Scholar]
- 50.Lande R, Gafa V, Serafini B, Giacomini E, Visconti A, Remoli ME, Severa M, Parmentier M, Ristori G, Salvetti M, Aloisi F, Coccia EM. 2008. Plasmacytoid dendritic cells in multiple sclerosis: intracerebral recruitment and impaired maturation in response to interferon-beta. J Neuropathol Exp Neurol 67:388–401. doi: 10.1097/NEN.0b013e31816fc975. [DOI] [PubMed] [Google Scholar]
- 51.Wuest TR, Carr DJ. 2008. Dysregulation of CXCR3 signaling due to CXCL10 deficiency impairs the antiviral response to herpes simplex virus 1 infection. J Immunol 181:7985–7993. doi: 10.4049/jimmunol.181.11.7985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smith JA. 2014. A new paradigm: innate immune sensing of viruses via the unfolded protein response. Front Microbiol 5:222. doi: 10.3389/fmicb.2014.00222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang FI, Hinton DR, Gilmore W, Trousdale MD, Fleming JO. 1992. Sequential infection of glial cells by the murine hepatitis virus JHM strain (MHV-4) leads to a characteristic distribution of demyelination. Lab Invest 66:744–754. [PubMed] [Google Scholar]
- 54.Liu MT, Keirstead HS, Lane TE. 2001. Neutralization of the chemokine CXCL10 reduces inflammatory cell invasion and demyelination and improves neurological function in a viral model of multiple sclerosis. J Immunol 167:4091–4097. doi: 10.4049/jimmunol.167.7.4091. [DOI] [PubMed] [Google Scholar]
- 55.Marques CP, Kapil P, Hinton DR, Hindinger C, Nutt SL, Ransohoff RM, Phares TW, Stohlman SA, Bergmann CC. 2011. CXCR3-dependent plasma blast migration to the central nervous system during viral encephalomyelitis. J Virol 85:6136–6147. doi: 10.1128/JVI.00202-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Durrant DM, Daniels BP, Klein RS. 2014. IL-1R1 signaling regulates CXCL12-mediated T cell localization and fate within the central nervous system during West Nile Virus encephalitis. J Immunol 193:4095–4106. doi: 10.4049/jimmunol.1401192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Durrant DM, Robinette ML, Klein RS. 2013. IL-1R1 is required for dendritic cell-mediated T cell reactivation within the CNS during West Nile virus encephalitis. J Exp Med 210:503–516. doi: 10.1084/jem.20121897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schenk M, Fabri M, Krutzik SR, Lee DJ, Vu DM, Sieling PA, Montoya D, Liu PT, Modlin RL. 2014. Interleukin-1beta triggers the differentiation of macrophages with enhanced capacity to present mycobacterial antigen to T cells. Immunology 141:174–180. doi: 10.1111/imm.12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aleyas AG, Han YW, Patil AM, Kim SB, Kim K, Eo SK. 2012. Impaired cross-presentation of CD8alpha+ CD11c+ dendritic cells by Japanese encephalitis virus in a TLR2/MyD88 signal pathway-dependent manner. Eur J Immunol 42:2655–2666. doi: 10.1002/eji.201142052. [DOI] [PubMed] [Google Scholar]
- 60.Fleming JO, Wang FI, Trousdale MD, Hinton DR, Stohlman SA. 1993. Interaction of immune and central nervous systems: contribution of anti-viral Thy-1+ cells to demyelination induced by coronavirus JHM. Reg Immunol 5:37–43. [PubMed] [Google Scholar]
- 61.Wexler AG, Frielle C, Berry G, Budgeon LR, Baccon J, Christensen ND, Waldner H. 2013. The innate immune adaptor MyD88 is dispensable for spontaneous autoimmune demyelination in a mouse model of multiple sclerosis. J Neuroimmunol 255:60–69. doi: 10.1016/j.jneuroim.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 62.Miranda-Hernandez S, Gerlach N, Fletcher JM, Biros E, Mack M, Korner H, Baxter AG. 2011. Role for MyD88, TLR2 and TLR9 but not TLR1, TLR4 or TLR6 in experimental autoimmune encephalomyelitis. J Immunol 187:791–804. doi: 10.4049/jimmunol.1001992. [DOI] [PubMed] [Google Scholar]