

ABSTRACT
Since it was first recognized in 2004 that human parechoviruses (HPeV) are a significant cause of central nervous system and neonatal sepsis, their clinical importance, primarily in children, has started to emerge. Intravenous immunoglobulin treatment is the only treatment available in such life-threatening cases and has given moderate success. Direct inhibition of parechovirus infection using monoclonal antibodies is a potential treatment. We have developed two neutralizing monoclonal antibodies against HPeV1 and HPeV2, namely, AM18 and AM28, which also cross-neutralize other viruses. Here, we present the mapping of their epitopes using peptide scanning, surface plasmon resonance, fluorescence-based thermal shift assays, electron cryomicroscopy, and image reconstruction. We determined by peptide scanning and surface plasmon resonance that AM18 recognizes a linear epitope motif including the arginine-glycine-aspartic acid on the C terminus of capsid protein VP1. This epitope is normally used by the virus to attach to host cell surface integrins during entry and is found in 3 other viruses that AM18 neutralizes. Therefore, AM18 is likely to cause virus neutralization by aggregation and by blocking integrin binding to the capsid. Further, we show by electron cryomicroscopy, three-dimensional reconstruction, and pseudoatomic model fitting that ordered RNA interacts with HPeV1 VP1 and VP3. AM28 recognizes quaternary epitopes on the capsid composed of VP0 and VP3 loops from neighboring pentamers, thereby increasing the RNA accessibility temperature for the virus-AM28 complex compared to the virus alone. Thus, inhibition of RNA uncoating probably contributes to neutralization by AM28.
IMPORTANCE Human parechoviruses can cause mild infections to severe diseases in young children, such as neonatal sepsis, encephalitis, and cardiomyopathy. Intravenous immunoglobulin treatment is the only treatment available in such life-threatening cases. In order to develop more targeted treatment, we have searched for human monoclonal antibodies that would neutralize human parechoviruses 1 and 2, associated with mild infections such as gastroenteritis and severe infections of the central nervous system, and thus allow safe treatment. In the current study, we show how two such promising antibodies interact with the virus, modeling the atomic interactions between the virus and the antibody to propose how neutralization occurs. Both antibodies can cause aggregation; in addition, one antibody interferes with the virus recognizing its target cell, while the other, recognizing only the whole virus, inhibits the genome uncoating and replication in the cell.
INTRODUCTION
Human parechoviruses (HPeV) are single-stranded, positive-sense RNA viruses in the Parechovirus genus within the Picornaviridae family (1). HPeV infections mainly cause mild gastrointestinal symptoms, although HPeV are also associated with more severe central nervous system symptoms, such as meningitis and neonatal sepsis (2–5). The HPeV genome is about 7,300 bases in length, enclosed in an icosahedrally symmetric capsid of 60 copies of each of the three capsid proteins VP0, VP3, and VP1 (1, 6). HPeV lack the maturation cleavage of the capsid protein VP0 into VP4 and VP2, which is present in most picornaviruses (7). They have a 30-amino-acid-long extension to the N terminus of VP3 and a unique nonstructural protein 2A, lacking proteolytic activity (8). HPeV1 contains an arginine-glycine-aspartic acid (RGD) motif close to the C terminus of VP1 (1). The RGD motif is found in a number of viral capsid proteins which recognize integrin receptors to gain entry into host cells, e.g., coxsackievirus A9 (CVA9), echovirus (EV) 9 (Echo9), and foot-and-mouth disease virus (9–11). Such a role for the RGD motif for HPeV1 has been shown through blocking experiments with RGD-containing peptides, antibodies (Abs), and mutations of the sequence, where deletion of the RGD motif is lethal (12–16). Studies of the HPeV1 virion in complex with both αVβ3 and αVβ6 integrins confirmed that they have overlapping binding sites on the predicted site of the RGD motif on the capsid surface (6). There are several potential neutralizing mechanisms for antibodies that bind specifically to viral capsid surfaces, e.g., antibodies may neutralize by obstructing a receptor-binding site, cause viral aggregation as a result of interlinking particles, or bind bivalently, preventing uncoating (17–19). The RGD motif has also been shown to be an important antigenic site. Diluted antiserum raised against a peptide containing the RGD motif neutralized 51% of HPeV1 infections in a plaque assay compared to a background of 1% in the preimmune serum (16). When the virion has been used as an antigen in rabbits, the immune sera recognize linear epitopes from VP0 and VP3. One hundred percent neutralization has been shown with rabbit sera raised against virions and VP1 (16, 20).
We have isolated two different human HPeV1 monoclonal antibodies (MAbs), of which MAb AM18 was shown to be a broadly cross-neutralizing MAb against HPeV1, -2, -4, -5, and -6 and MAb AM28 neutralized HPeV1 and HPeV2 (21). These results indicated two different neutralizing epitopes for AM18 and AM28. Here, we present the epitopes identified for AM18 and AM28 and also their probable neutralization mechanisms. The location of the virus neutralization epitopes on the capsid surface was revealed by peptide scanning and surface plasmon resonance (SPR) for AM18. For AM28, we generated homology models of the AM28 antibody and of the HPeV1 capsid constrained by an 8.5-Å-resolution reconstruction of HPeV1 (6), as there are no atomic models available, and used these to interpret data from electron cryomicroscopy (cryo-EM) and three-dimensional (3D) image reconstruction of AM28 complexed with HPeV1.
MATERIALS AND METHODS
Virus culture and purification.
HPeV1-Harris was provided by the Dutch National Institute for Public Health and the Environment (RIVM), Bilthoven, the Netherlands, and grown on a human colon carcinoma cell line (HT29). The HT29 cells were maintained in Eagle's minimum essential medium (EMEM) with l-glutamic acid (0.2×), nonessential amino acids (1×), streptomycin (0.1 μg/ml), and ampicillin (0.1 μg/ml), supplemented with 8% heat-inactivated fetal calf serum (FCS). The virus concentration was determined by the median tissue culture infective dose (TCID50) and calculated by the Reed and Muench method (22). For large-scale virus purification, 90% confluent cell layers in T-175 flasks were infected with HPeV1 at a multiplicity of infection of 0.1. After 75 to 100% infection of the cell monolayer, evident by the cytopathic effect, the cells and spent medium were freeze-thawed twice at −80°C/+37°C and centrifuged at 4,000 rpm for 15 min in an Eppendorf A-4-62 swing bucket rotor at 4°C, and the supernatant was filtered using a 0.22-μm filter. The virus was pelleted by ultracentrifugation at 32,000 rpm for 2 h at 4°C in a Beckman SW32 Ti rotor. The pellet was dissolved in 10 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM MgCl2 (1× TNM buffer), and loaded onto a cesium chloride step gradient with a 5-ml 40% (wt/vol) bottom layer and a 5-ml 15% (wt/vol) top layer and centrifuged at 32,000 rpm for 16 h at 4°C in a Beckman SW41 Ti rotor. The fraction containing the virus was concentrated with a 100-kDa-cutoff filter (Millipore) in 1× TNM.
Antibody preparation and virus labeling.
Human memory IgG+ B cells were cultured using the AIMSelect method (23), and antibody-containing culture supernatants were used to directly screen for HPeV1 neutralization (21). Two antibodies (AM18 and AM28) were selected, and the corresponding nucleotide sequences were cloned for expression in 293T cells and purified. The initial production and characterization of these MAbs have been recently described in detail (21). Initially, we tested binding of each MAb to HPeV1 virions at molar ratios of 60:1 and 300:1 at 37°C for 1 h in 1× TNM buffer followed by cryo-EM (see below). The MAbs both caused aggregation and were thus not suitable for structure determination by image reconstruction. We attempted to purify Fab fragments from both MAb preparations but were successful only with purified AM28, using a Pierce Fab micropreparation kit according to the manufacturer's protocol. The products were checked by reducing and nonreducing SDS-PAGE and shown to contain only pure Fab preparations. The resulting Fab was mixed with HPeV1 capsids at a molar ratio of 300:1 in 1× TNM buffer for 30 min at room temperature.
Peptide-scanning ELISA.
Streptavidin-coated enzyme-linked immunosorbent assay (ELISA) plates (Greiner Bio-One) were blocked with 2% bovine serum albumin (BSA) in phosphate-buffered saline (PBS) for 2 h at room temperature. N-terminally biotin-labeled HPeV1 overlapping peptides (12 amino acids [aa] in length with 6-aa overlap and no flexible linker) (Antoni van Leeuwenhoek-Netherlands Cancer Institute, Peptide Synthesis) were diluted (1:500) in 1% BSA in PBS and bound to the plate for 1 h at room temperature. The plate was incubated with 2 μg/ml of AM18 or AM28 for 1 h at room temperature and washed 3 times with PBS-0.1% Tween. Horseradish peroxidase (HRP)-labeled anti-human IgG (0.3 μg/ml) was used as the secondary Ab, incubated for 1 h at room temperature, and washed three times with PBS-0.1% Tween. The substrate solution containing 3,3′,5,5′-tetramethylbenzidine was added and incubated for 10 min at room temperature in the dark. The reaction was stopped by the addition of 0.8 M H2SO4. The absorbance at 450 nm and 620 nm was measured with a microplate reader.
SPR.
The binding specificities of MAb AM18 to VP1 peptides spanning the LRGD region of HPeV1 containing the wild-type sequence or mutations in the LRGD region were determined using SPR on an Ibis Mx96 SPR instrument (Ibis Technologies). For this purpose, 12-mer peptides, with a single biotin attached to the N terminus of the peptides via a double aminohexanoic spacer, were synthesized. Antibody binding to VP1 peptides was measured in an epitope-mapping assay, where the biotinylated peptides were first immobilized on a streptavidin-coated gold SPR chip (G-strep; Ssens Technologies). After capture of the peptides, a concentration series (0.5 to 5.0 μg/ml) of MAb AM18 or AM28 (negative control) was injected over the chip. SPR data were processed with SprintX software (Ibis).
Neutralization assay.
A neutralization assay for AM28 MAb or Fab was performed as described earlier (21). Briefly, HPeV1 at a titer of 2,000 TCID50/ml was mixed with 25 μg/ml, 2.5 μg/ml, 0.25 μg/ml, or 0.025 μg/ml of AM28 MAb or Fab at 37°C for 1 h. The complex was added onto HT29 cells and scored for cytopathic effect for 7 days postinfection. Virus alone was used as the positive control.
Electron cryomicroscopy.
Aliquots of the MAb or Fab-labeled virus mixture were vitrified on Quantifoil R2/2 holey carbon nickel grids in a home-built guillotine by plunging into liquid ethane maintained in a liquid nitrogen bath. After vitrification, the grids were stored in liquid nitrogen until use. The grids were examined in an FEI F20 transmission electron microscope at 200 keV using a Gatan 626 cryostage. The images were recorded on a Gatan Ultrascan 4000 charge-coupled device (CCD) under low-dose conditions at a nominal magnification of ×69,000 with a sampling size of 2.17 Å per pixel.
The contrast transfer function of each micrograph was estimated using CTFFIND3, and images containing drift or astigmatism were discarded (24). Particles were picked using the program ETHAN (25) with a box size of 401 pixels and inspected by eye in the program suite EMAN (26). A previous reconstruction of HPeV1 from the work of Seitsonen et al. (6) (EMD-1690) was used as a starting model to initiate full orientation and origin determinations of the Fab-labeled set of images using AUTO3DEM ver4.03.1 (27). The final reconstruction calculated to the Nyquist frequency was used to estimate the B-factors with EM-Bfactor, and then the reconstruction was truncated to the resolution indicated by the Fourier shell correlation analysis with a threshold criterion of 0.5 (28–30). The statistics for the reconstruction are given in Table 1.
TABLE 1.
Statistics of the reconstruction
| Parameter | HPeV1-AM28 Fab reconstruction |
|---|---|
| No. of micrographs | 65 |
| No. of particles used in the reconstruction | 270 |
| Underfocus range (μm) | 1.65–4.06 |
| Resolution (Å) | 20 |
Homology modeling and fitting of models into cryo-EM maps.
The structures of the three HPeV1 capsid proteins were generated by multiple-template comparative modeling using the I-TASSER server (31). The template structures used by I-TASSER for VP0 were foot-and-mouth disease virus (PDB identifiers [IDs] 1QQP, 1FMD, and 1BBT) (32–34), poliovirus 1 (PDB ID 1POV) (35), bovine enterovirus (PDB ID 1BEV) (36), and Seneca Valley virus 001 (PDB ID 3CJI) (37). For VP1, they were Triatoma virus (PDB ID 3NAP) (38), human rhinovirus 14 (PDB ID 1D3I) (39), cricket paralysis virus (PDB ID 1B35) (40), rabbit hemorrhagic disease virus (PDB ID 4EJR) (41), echovirus 7 (PDB ID 1M11) (42), and bovine enterovirus (PDB ID 1BEV) (36). For VP3, they were human enterovirus 71 (PDB ID 3VBF) (43), Seneca Valley virus 001 (PDB ID 3CJI) (37), human rhinovirus 16 (PDB ID 1AYM) (44), and poliovirus Mahoney strain (PDB ID 1HXS) (45).
An atomic model of the echovirus 1 capsid (PDB ID 1EV1) (46) was placed into an 8.5-Å-resolution HPeV1 map (EMD-1690) (6). The homology models from I-TASSER were aligned with the echovirus 1 capsid to give the approximate relative positions of all three proteins in the context of the capsid. Then, these homology models were refined, using a protocol described earlier (47). Briefly, the three I-TASSER models were rigidly fitted into the HPeV1 map (EMD-1690) (6) using the “fit in map” feature in UCSF-Chimera (48). The N termini of VP0, VP3, and VP1 were truncated to avoid intersubunit clashes. Using the “zoning” feature in UCSF-Chimera (48), the HPeV1 capsid map was zoned to an asymmetric unit with a radius of 6 Å using the truncated VP0-VP3-VP1 rigidly fitted model. RIBFIND-based rigid bodies were identified for the truncated VP0-VP3-VP1 model (49), and the model was flexibly fitted into the asymmetric unit using one iteration in FlexEM (50) followed by iMODfit-based flexible fitting using the default settings (51). The resulting homology model of the complete HPeV1 capsid was then placed directly into the Fab-labeled reconstruction to identify the probable binding sites. The variable regions of AM28 Fab were modeled using the WAM webserver (52). We manually fitted the atomic models into the corresponding Fab density in the HPeV1-AM28 Fab reconstruction, and the fit was optimized with the “fit in map” feature in UCSF-Chimera, which is a rigid body method. Flexible fitting of the Fab could not be used as it relied only on the 20-Å-resolution reconstruction of the complex. The orientation of the Fab was decided based on the highest cross-correlation output by UCSF-Chimera. Once the Fab and capsid protein density had been assigned, the finger-like densities observed previously by Seitsonen et al. in contact with VP1 and VP3 on the inside of the capsid were assigned to RNA (6). We rigidly fitted an appropriately sized RNA density (PDB ID 3P22) (53) into the density using the “fit in map” feature in UCSF-Chimera. All the visualization was carried out in UCSF-Chimera (48).
Sequence alignment.
The P1 amino acid sequences of HPeV1 (GenBank IDs L02971, GQ183023, GQ183022, GQ183021, GQ183020, GQ183019, GQ183018, GQ183025, and GQ183024), HPeV2 (GenBank ID NC_001897), HPeV3 (GenBank ID GQ183026), HPeV4 (GenBank ID DQ315670), and HPeV5 (GenBank ID AF055846) used for AM18 and AM28 neutralization assays (21) were aligned using Clustal Omega (54) with additional HPeV strains for which the complete genome sequences were available in GenBank (GenBank IDs for HPeV1 are JX441355, JX575746, S45208, EF051629, FJ840477, GQ183035, GQ183034, HQ696574, HQ696572, HQ696570, HQ696573, HQ696571, and FM178558; those for HPeV3 are GQ183027, GQ183028, and GQ183029; those for HPeV4 are AB433629 and AM235750; that for HPeV5 is AM235749; those for HPeV6 are EU077518 and AB252583). The alignment was visualized with Jalview (55).
Thermofluor assay.
In order to test the capsid stability in the presence of antibody, AM28 MAb was mixed with HPeV1 virions at a molar ratio of 66:1 and incubated at room temperature for 30 min. Dye accessibility to the RNA as a function of temperature was detected with a fluorescent dye. The reaction volumes were set up per well in a 96-well PCR plate in triplicate. Each reaction mixture contained 2.5 μl of 200× Sybr Safe DNA gel stain (Invitrogen; also binds RNA) and the protein sample, which was one of the following: HPeV1 (0.4 μg/μl), AM28 (0.8 μg/μl), or HPeV1-AM28 complex (0.4 μg/μl HPeV1, 0.8 μg/μl AM28). The total volume was made up to 25 μl for each reaction volume using 1× TNM buffer. The assay was run from 25°C to 95°C with readout every 0.33 s in an Mx3005P quantitative PCR (qPCR) instrument (Agilent Technologies). The ramp rate was 1°C/30 s. The Sybr Safe DNA gel stain dye was excited at 492 nm, and emission was read at 516 nm (56). The assay was also run at a constant temperature of 37°C for 12 h.
Protein structure accession numbers.
The HPeV1-AM28 Fab density map was deposited in the Protein Data Bank in Europe (accession number EMD-2761). The fitted models of the capsid proteins and Fab are deposited in the Protein Data Bank in Europe with the PDB ID 4UDF, with the accompanying sequence data.
RESULTS
Peptide scanning.
To determine the specific binding region of the MAbs AM18 and AM28, overlapping 12-mer peptides designed to cover the P1 (VP0, VP3, and VP1 coding region) sequence of HPeV1 were bound directly to biotin for use in peptide ELISA. The AM18 antibody showed strong binding to one peptide containing the RGD motif (peptide 85, VTSSRALRGDMA), binding to a lesser extent to the second peptide containing the RGD motif (peptide 86, ALRGDMANLTNQ), no binding to the preceding peptide (peptide 84, FFFPLPAPKVTS), and low binding to a peptide in the VP0 region (peptide 18, YGQSRYFAAVRC) (Fig. 1A). This difference in binding between the two RGD-containing peptides and the nonbinding peptide 84 indicates that the residues VTSSR N-terminal to the RGD increase the binding specificity, most probably by increasing the accessibility of the RGD motif due to the position of the RGD in the peptide. If it were due to recognition of the residues VTS, then peptide 84 should also show binding. To further confirm RGD as the epitope for AM18, we tested binding efficiency of AM18 against a peptide (peptide 1, VTSSRALRGDMANL, a combination of peptides 85 and 86) and its variants (peptide 2, VTSSRALAGDMANL; peptide 3, VTSSRALRADMANL; peptide 4, VTSSRALRGAMANL; peptide 5, VTSSRAAAAAMANL) (Fig. 2). From the SPR assay, it was evident that peptide 1 had the strongest binding compared to its variants, confirming RGD as the epitope for AM18 (Fig. 2). Weaker binding for the variant peptides 2 and 3 than for peptide 4 indicated that the residues RG are the major contributors to the epitope (Fig. 2). Peptide 5, in which the residues LRGD were replaced by AAAA, behaved like the negative control (AM28) (Fig. 2); this result, moreover, demonstrates that the LRGD sequence is essential for AM18 binding. Moreover, it explains the broad antibody reactivity with other RGD-containing viruses (21).
FIG 1.

Peptide-scanning ELISA of AM18 and AM28. Plot of peptide number versus absorbance. The x axis shows the peptides used to test binding with AM18 (A) or with AM28 (B), and the y axis shows the absorbance at 540 nm after background subtraction from absorbance at 620 nm. Peptide 85 (VTSSRALRGDMA) showed significant binding to AM18 compared to peptide 86 (ALRGDMANLTNQ) even though both peptides contain the RGD motif.
FIG 2.
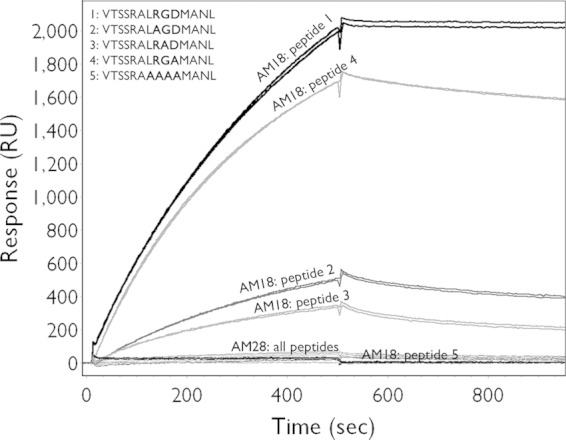
Mapping of AM18 epitope. Binding to variants of the peptide VTSSRALRGDMANL was tested by SPR in order to verify whether the RGD motif was the epitope recognized by AM18. Shown are the binding curves of injected antibody on the immobilized peptides.
The AM28 antibody showed no binding to the linear overlapping peptides in the ELISA (Fig. 1B). This supported Western blot data where AM28 did not bind to any of the viral structural proteins (21), strongly suggesting that the epitope recognized by it is a nonlinear, conformation-dependent epitope. Hence, we progressed with three-dimensional epitope mapping on the intact virions for AM28 Fabs.
Virus-antibody complex.
In order to shed light on the possible neutralization mechanisms and the exact epitopes of the MAbs AM18 and AM28, we imaged HPeV1 virions complexed with either MAb in a transmission electron microscope under cryogenic conditions. The micrographs did not show any noticeable disruption of HPeV1 virions in the presence of the MAb AM18 or AM28, but they did induce heavy aggregation of the HPeV1 virions compared to the virus alone (Fig. 3A, B, and D).
FIG 3.

AM18 and AM28 antibodies recognize virus capsids leading to aggregation. (A and B) Raw micrographs showing the aggregation (arrow) of HPeV1 when mixed with AM18 Ab (A) or AM28 (B) at a molar ratio of 1 (asymmetric unit of the capsid):5 (antibody). (C) Raw micrograph of HPeV1 in complex with AM28 Fab. (D) Raw micrograph of HPeV1. Bar, 50 nm. (E) Endpoint neutralization assay of HPeV1 using either AM28 MAb or AM28 Fab.
Since no linear epitopes were determined for AM28 using peptide scanning (Fig. 1B), we used cryo-EM, 3D reconstruction, modeling, and fitting to determine the binding site of AM28 on HPeV1. Due to aggregation of the virus in the presence of the MAb, we prepared AM28 Fab-labeled virus (Fig. 3C). The Fab neutralized the HPeV1 Harris strain with a 50% inhibitory concentration (IC50) of 90 ng compared to 40 ng for the MAb (Fig. 3E). The reconstruction statistics of HPeV1-AM28 Fab are summarized in Table 1, and the reconstruction is shown in Fig. 4 and 5. In comparison with the virus reconstruction alone (6), the HPeV1-AM28 Fab reconstruction showed clear additional density attributable to the Fab density either side of the 2-fold axes, bridging neighboring pentamers (Fig. 4B and C). This Fab footprint is distinct from the integrin footprint encompassing the VP1 RGD epitope that we have shown previously (6) (Fig. 4D).
FIG 4.
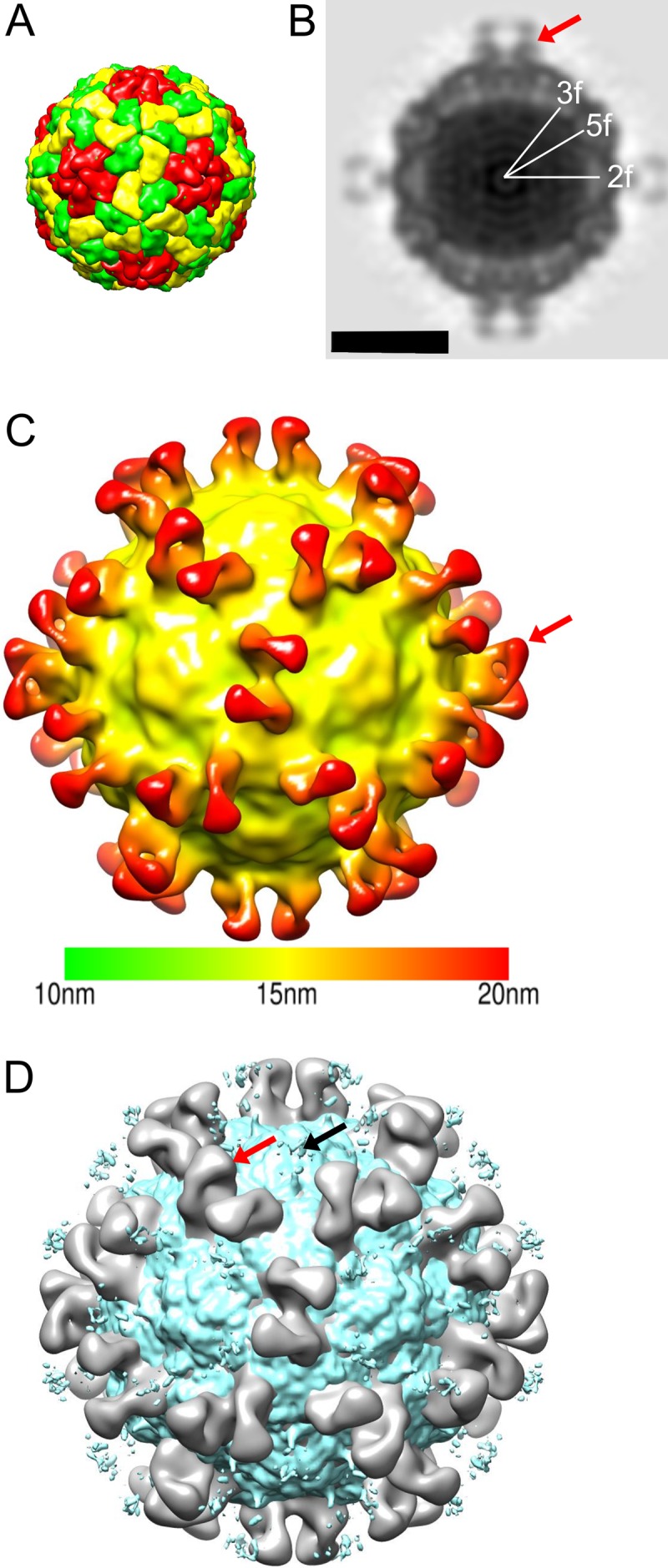
HPeV1-AM28 Fab reconstruction. (A) Schematic diagram of the HPeV1 capsid. VP1 is shown in red, VP0 in yellow, and VP3 in green. A pentamer would have 5 copies of VP1, VP0, and VP3. (B) Central cross-section of HPeV1-AM28 Fab complex with 2-fold (2f), 5-fold (5f), and 3-fold (3f) symmetry axes marked. Bar, 15 nm. (C) Three-dimensional radially depth-cued reconstruction of the HPeV1 capsid with 60 Fab molecules bound. The reconstruction is colored according to the distance from the center of the particle. The color key is shown below the reconstruction. (D) Overlay of the integrin-bound form of HPeV1 (EMD-1689; greenish blue) with the HPeV1-AM28 (gray) complex showing different binding sites for integrin and antibody. A red arrow indicates one of the Fab molecules on the capsid surface in panels C and D, and a black arrow in panel D indicates one of the positions where the integrin is bound.
FIG 5.

Epitopes on HPeV1 for AM28. (A) Homology models of VP1 (red), VP0 (yellow), and VP3 (green) built using I-TASSER. (B) Final fits of VP1, VP0, and VP3 homology models into an asymmetric unit of HPeV1 (EMD-1690). (C) One of the capsid protein interactions with RNA in HPeV1 is shown. An RNA stem-loop structure (PDB ID 3P22, chain A; orange) was rigidly fitted into the previously described (6) finger-like density in the HPeV1 EM density map (EMD-1690). The majority of the RNA interactions appear to be with VP1 and VP3. (D) Mapping the epitope region (black) on other picornaviruses by superimposing asymmetric units of echovirus 1 (PDB ID 1EV1; magenta), poliovirus 1 (PDB ID 1POV; gray), enterovirus 71 (PDB ID 3VBF; orange), and foot-and-mouth disease virus (PDB ID 1QQP; cyan) on final fits of HPeV1 VP0 (yellow) and VP3 (green). For ease of visualization, only the regions encompassing the epitopes in VP0 and VP3 are shown. (E) Mapping epitopes for AM28 on HPeV1 surface by fitting the AM28 Fab variable region homology model into the Fab density seen in HPeV1-AM28 Fab reconstruction and superimposing VP1 (red), VP0 (yellow), and VP3 (green) fits for HPeV1 (EMD-1690) into the HPeV1-AM28-Fab reconstruction (mesh). AM28 variable heavy chain is shown in magenta, and variable light chain is shown in blue. The Cα distance between the heavy-chain C-terminal residues of the symmetry-related Fabs is 58 Å and is marked by dashed lines. (F) Road map showing the density of AM28 Fab (red line contour; radius, 155 to 156 Å) and the epitopes HEWTPSWA (VP0; yellow), HQDKP (VP0; cyan), PLSIPTGSANQVD (VP0; magenta), MADSTTPSENHG (VP3; blue), ATTAPQSIVH (VP3; green), and FFPNATTDST (VP3; red). An asymmetric unit is marked by black lines.
In order to find the approximate location of the AM28 binding site, we needed a detailed model of the HPeV1 capsid. A homology model of the HPeV1 capsid was generated and constrained by a previous 8.5-Å-resolution reconstruction of HPeV1 (Fig. 5A) (6). The highest confidence model was obtained for VP3 with an I-TASSER-based confidence score (C-score) of −0.38 followed by VP0 with a C-score of −1.60. In contrast, the VP1 had a C-score of only −3.77. The typical C-score ranges from −5 to 2, where a higher score means better confidence in the quality of the modeling (31). In general, a C-score of −1.5 means that more than 90% of the quality predictions of the structures are correct; thus, the VP1 model was used only to constrain the fitting of VP0 and VP3 in the asymmetric unit (Fig. 5B and C). All the models had the characteristic eight-stranded β-barrels found in all picornavirus capsid proteins (Fig. 5C). Since the termini in picornaviruses are least conserved in the 3D conformation within the capsids and prediction was unreliable, we truncated the termini of the homology models. The placement of the individual capsid proteins within the capsid shell was improved using flexible fitting, resulting in improved fitting of the β-barrels and long helices of the models (47, 50, 51). This fit enabled us to identify VP1 and VP3 as the interaction partners for the previously described finger-like densities inside the capsid and to identify them as RNA into which we could easily place an arbitrary RNA stem-loop structure (Fig. 5C) (6, 53). The 8.5-Å-resolution HPeV1 reconstruction with the fitted homology model of the capsid was then aligned with the 20-Å-resolution HPeV1-AM28 Fab reconstruction. In addition, a model of the AM28 Fab variable region was also generated based on homology with known structures (52, 57, 58). The AM28 Fab model was fitted into the Fab density in the HPeV1-AM28 Fab reconstruction. This showed that the antibody recognizes a conformational epitope which has contributions from both VP0 and VP3 (Fig. 5D). Modeling and fitting of the HPeV1 VP0, VP1, and VP3 proteins indicated that amino acids in the following loops in HPeV1 are involved in the footprint: βB-βC (VP0), αA-βD (VP0), βI-βH (VP0), αZ-βB (VP3), βB-βC (VP3), and βE-αB (VP3) (Fig. 5D and E; also summarized in Table 2, residues in italics). These identified antigenic regions are distinct from linear epitopes of VP0 and VP3 that have been described previously by peptide scanning (Table 2, underlined amino acids) (16). To determine the orientation of the Fab, we compared the cross-correlation values of the Fab variable region fit in the density. The chosen orientation had a cross-correlation value of 0.9 compared to 0.8 if the molecule was rotated by 180° on the long axis of the Fab density, which we considered to be significant despite the relatively low resolution of the model. The Cα distance between the heavy-chain C-terminal residues of the two Fab molecules across the 2-fold symmetry axis was about 58 Å (Fig. 5F). This distance does not allow for bivalent binding of a MAb (59).
TABLE 2.
Mapping conformational epitopes for AM28 and linear epitopes from peptide scanning of sera on the capsid protein amino acid sequence
| Capsid protein | Amino acid sequencea |
|---|---|
| VP0 (GenBank ID L02971, amino acids 1–289) | METIKSIADMATGVVSSVDSTINAVNEKVESVGNEIGGNLLTKVADDASNILGPNCFATTAEPENKNVVQATTTVNTTNLTQHPSAPTMPFSPDFSNVDNFHSMAYDITTGDKNPSKLVRLETHEWTPSWARGYQITHVELPKVFWDHQDKPAYGQSRYFAAVRCGFHFQVQVNVNQGTAGSALVVYEPKPVVTYDSKLEFGAFTNLPHVLMNLAETTQADLCIPYVADTNYVKTDSSDLGQLKVYVWTPLSIPTGSANQVDVTILGSLLQLDFQNPRVFAQDVNIYDN |
| VP3 (GenBank ID L02971, amino acids 290–542) | APNGKKKNWKKIMTMSTKYKWTRTKIDIAEGPGSMNMANVLCTTGAQSVALVGERAFYDPRTAGSKSRFDDLVKIAQLFSVMADSTTPSENHGVDAKGYFKWSATTAPQSIVHRNIVYLRLFPNLNVFVNSYSYFRGSLVLRLSVYASTFNRGRLRMGFFPNATTDSTSTLDNAIYTICDIGSDNSFEITIPYSFSTWMRKTNGHPIGLFQIEVLNRLTYNSSSPSEVYCIVQGKMGQDARFFCPTGSVVTFQ |
Underlined residues indicate antigenic sites identified by peptide scanning in reference 13. Italicized residues indicate antigenic sites for AM28 identified by fitting pseudoatomic model in EM density of HPeV1-Fab AM28.The loops in consecutive order are βB-βC (VP0), αA-βD (VP0), βI-βH (VP0), αZ-βB (VP3), βB-βC (VP3), and βE-αB (VP3).
Conservation of the conformational epitope recognized by AM28.
We compared amino acid sequence alignments of the newly identified VP0 and VP3 antigenic regions from different HPeV1 isolates with those of HPeV2 to HPeV6. They were well conserved in HPeV1, moderately conserved in HPeV2, and poorly conserved in HPeV3 to HPeV6 (Fig. 6), which explains why MAb AM28 cross-binds and cross-neutralizes HPeV2 but no reactivity could be detected against HPeV3 to HPeV6 (21).
FIG 6.

Conservation of VP0 and VP3 epitopes recognized by AM28. Amino acid sequences of HPeV1 to -5 used for neutralization in the work of Westerhuis et al. (21) were aligned against complete genome sequences for HPeV1 to -6. The sequence annotation on the left side is virus genotype/GenBank ID. The epitopes are marked in black on the HPeV1 Harris strain (GenBank ID L02971) that was used as the basis for the HPeV1 homology modeling. The alignment is colored according to percent sequence identity, on a scale of white (no identity) to dark blue (full identity). The conservation panel in yellow below the alignment is based on the BLOSUM 62 score of the physicochemical conservation of the amino acids (70). The blue arrowheads indicate irrelevant regions of the sequence that have been hidden in the final representation for simplicity (1 to 120, 160 to 246, 270 to 372, and 413 to 453). The figure was made with Jalview (55).
Capsid stabilization by AM28 MAb.
To further consolidate the observation from the HPeV1-AM28 structure that the binding of AM28 MAb may stabilize the capsid, we performed a capsid thermal stability assay using an RNA binding dye, which has previously been used, for example, to explain the mode of action of echovirus 71 (EV71) neutralizing antibodies and anti-poliovirus 1 single-domain antibody fragments and to show pleconaril stabilizing the EV-D68 capsid (18, 60, 61). This assay measures the dye accessibility to the RNA that correlates with the capsid becoming porous to the dye. The temperature with the sharpest transition in the fluorescence intensity was taken as the temperature at which the dye interacts with the RNA. These temperatures for HPeV1 alone and HPeV1-AM28 were 53.2°C and 56.2°C, respectively (Fig. 7). The 3°C shift in RNA accessibility temperature for HPeV1-AM28 compared to HPeV1 alone was statistically significant (P < 0.001) and indicates that the binding of one arm of AM28 across neighboring pentamers probably stabilizes the capsid, inhibiting access to the RNA within the capsid. No effect was seen if the experiments were carried out at 37°C for 12 h.
FIG 7.

Thermofluor assay. Plot of temperature (x axis) versus first derivative of fluorescence (y axis) showing the change in fluorescence per degree rise in temperature when HPeV1 is bound to AM28 MAb compared to HPeV1 alone. Arrows indicate the RNA accessibility temperature for each sample. AM28 was used as the negative control for the RNA binding dye.
DISCUSSION
HPeV infections can be severe and even life-threatening, especially for neonates. Severe infections in neonates can be due to a lack of protective maternal Abs (62). No treatment is currently available against these severe HPeV infections, making it an unmet medical need (62). For HPeV, Ab-based therapies are a feasible option as HPeV are a relatively small group of highly similar viruses for which cross-reactive, neutralizing polyclonal Abs have already been described (16, 20). Although HPeV1 is less frequently associated with severe diseases than HPeV3 (4, 62), the work presented here shows that potential therapeutic MAbs against HPeV can successfully be generated.
For a successful antiviral approach, knowledge of specific and overlapping viral antigenic sites is important, and MAbs are preferred. We developed two different MAbs, AM18 and AM28, which were able to neutralize HPeV1-Harris efficiently (21). These MAbs were cross-reactive with HPeV2 strain 751312, and AM18 also showed cross-reactivity against HPeV4 strain 251176, HPeV5 strain 552322, and HPeV6 strain 550389 (21). Peptide-scanning ELISA confirmed that the site of the AM18 binding was within the sequence ALRGDMA, as this was the common sequence between two positive overlapping peptides from the VP1 C terminus. Of this, RGDMA is also common to HPeV4 strain 251176 VP1 and RGD is common to HPeV2 strain 751312 VP1; it is absent in HPeV3. The binding site of AM18 within the sequence ALRGDMA was further narrowed down to RGD by systematic replacement of the sequence with alanine and testing by SPR and consideration of the viruses neutralized. Hence, we hypothesize that the minimally recognized epitope is RGD and AM18 most likely neutralizes by aggregation and by directly competing with the cellular receptor (integrins) for this binding site, diminishing the ability to infect cells through this route. AM18 recognizes and neutralizes other RGD-containing viruses like coxsackievirus A9 (21), but it did not neutralize echovirus 9 strains, presumably because they can use alternative receptors to enter the host cells. For HPeV1, it has been shown that RGD-less mutants with reduced binding to integrin are less viable (12). Therefore, it may be difficult for the virus to generate escape mutants resistant to AM18 neutralization. Hence, AM18 shows potential as a useful therapeutic molecule for HPeV1 to -6 infections.
AM28 did not recognize linear epitopes from denatured proteins for HPeV1, -2, -4, -5, and -6 strains in Western blot assays or overlapping peptides in Western blot assays or ELISA, indicating that the epitopes are conformational. We used homology models of VP0, VP1, VP3, and AM28 fitted into an HPeV1-AM28 Fab reconstruction to identify a conformational epitope, which has contributions from VP0 and VP3 of neighboring pentamers. Although we have few experimental data to show the validity of the homology models, as there are no atomic models for parechoviruses that we are aware of, the position of the RGD epitope in VP1 agrees to within 5 Å with the position of the integrin footprint found earlier (6) and the fit of the obvious elements of secondary structure, such as the β-barrels and the VP0 helices forming the interface at the 2-fold axes, is consistent with what is expected from the literature. Comparison of known picornavirus structures with that of our capsid model indicates that this conformational epitope is a region commonly occupied by the equivalent loops from VP2 and VP3, defined by the conserved position of the β-barrels. Hence, even though we could not trace the chain of the capsid proteins or identify amino acid side chains, we are confident in the prediction of the loops contributing to the footprint. The sequence conservation between HPeV1 and HPeV2 supports this prediction, as both are neutralized by AM28. From the reconstruction and the fitting, a single AM28 Fab arm appears to interact with epitopes from neighboring pentamers. The distance of ∼58 Å between the two Fabs around the 2-fold axis was too long for bivalent binding of the MAb, so we expected that the monovalent binding of the MAb across the neighboring pentamers would be enough to stabilize the capsid (57–59, 63, 64). This hypothesis was tested in two ways: first, we showed that the Fab alone can neutralize the virus but not cause the aggregation seen with the MAb (Fig. 3C); second, we investigated the protection of RNA inside the capsid. We measured the fluorescence of an RNA binding dye when the virus capsid was heated in a stepwise manner from 25°C to 95°C. The idea was that upon heating, the capsid would destabilize at a certain temperature, thus allowing the dye access to the RNA. AM28 had a stabilizing effect on the capsid. The RNA accessibility temperature was increased by 3°C compared to the virus alone. A similar degree of capsid stabilization has recently been shown for EV-D68, where addition of the small molecule pleconaril increased the RNA accessibility temperature by 4°C (61). Measuring the dye accessibility at physiological temperature in HPeV1-AM28 complex for 12 h did not affect the fluorescence, which is in contrast to E18 MAb binding to EV71 at physiological temperature, where a significant increase in fluorescence was observed as a result of capsid destabilization (18). Significantly, increased porosity of picornavirus capsids for RNA release has been shown to be dependent on domain movements of the major capsid proteins opening up the interfaces at the 2-fold axes (24, 43, 65–69). Our model of the HPeV1 capsid now includes identification of RNA stem-loops interacting directly with VP1 and VP3. Thus, conformational changes in the capsid proteins should have a direct effect on RNA accessibility. We hypothesize that the monovalent binding of one Fab arm of AM28 across the 2-fold axis could have a major neutralizing effect, preventing conformational changes in the capsid, thus stabilizing the capsid and preventing RNA release on cell entry (17–19). This conformational epitope did not overlap any of the linear immunogenic epitopes identified previously by peptide scanning (16), indicating that such single-dimensional epitope mapping techniques may miss some of the crucial epitopes on the capsid surface which are presented only in the tertiary form. The importance of this area of the capsid in capsid assembly and RNA delivery is shown in the conservation of these loops in multiple HPeV1 and HPeV2 isolates but not in the other serotypes. Hence, this MAb also shows therapeutic and diagnostic promise for HPeV1 and HPeV2 infections. More work is needed, for instance, in efficacy testing in animal models before these MAbs can be taken into clinical settings. The atomic model of HPeV1 will be of use in understanding mutations in the capsid that affect the tropism of the virus—an area of great interest in understanding the transmission from the respiratory and gastrointestinal tract to the central nervous system. The next challenge is to utilize these approaches and others to develop neutralizing MAbs to HPeV3 to treat neonatal sepsis.
ACKNOWLEDGMENTS
We thank Pasi Laurinmäki, Konstantin Kogan, Eveliina Karelehto, and Eevakaisa Vesanen for excellent technical assistance and Susan Hafenstein for helpful discussions. The Biocenter Finland National Cryo Electron Microscopy Unit; the Crystallization Facility in the Institute of Biotechnology, Helsinki University; and the CSC-IT Center for Science Ltd. are thanked for providing facilities.
This study was supported by the Academy of Finland (139178 and 275199 to S.J.B.), the Sigrid Juselius Foundation (S.J.B.), the Helsinki Graduate Program in Biotechnology and Molecular Biology (S.S.), grants from the Netherlands Organization for Health Research and Development's Clinical Fellowship (to K.C.W.) and the AMC Research Council (to K.C.W.), and short-term fellowships from the European Molecular Biology Organization and the European Society of Clinical Virology (to B.M.W.). This work was supported by the Seventh Framework Programme of the European Union AIPP under contract PIAPP-GA-2013-612308 to K.C.W. and S.J.B.
AIMM Therapeutics employed Y.C., T.B., and A.Q.B. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript, except AIMM Therapeutics, who discovered, patented, and furthered the development of AM18 and AM28 antibodies. They supported the research collaboration of Y.C., T.B., K.W., and A.Q.B. with the other authors and were consulted prior to the decision to publish. AIMM Therapeutics agreed to publish these results.
REFERENCES
- 1.Hyypiä T, Horsnell C, Maaronen M, Khan M, Kalkkinen N, Auvinen P, Kinnunen L, Stanway G. 1992. A distinct picornavirus group identified by sequence analysis. Proc Natl Acad Sci U S A 89:8847–8851. doi: 10.1073/pnas.89.18.8847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Verboon-Maciolek MA, Groenendaal F, Hahn CD, Hellmann J, van Loon AM, Boivin G, de Vries LS. 2008. Human parechovirus causes encephalitis with white matter injury in neonates. Ann Neurol 64:266–273. doi: 10.1002/ana.21445. [DOI] [PubMed] [Google Scholar]
- 3.Verboon-Maciolek MA, Krediet TG, Gerards LJ, de Vries LS, Groenendaal F, van Loon AM. 2008. Severe neonatal parechovirus infection and similarity with enterovirus infection. Pediatr Infect Dis J 27:241–245. doi: 10.1097/INF.0b013e31815c1b07. [DOI] [PubMed] [Google Scholar]
- 4.Benschop KS, Schinkel J, Minnaar RP, Pajkrt D, Spanjerberg L, Kraakman HC, Berkhout B, Zaaijer HL, Beld MG, Wolthers KC. 2006. Human parechovirus infections in Dutch children and the association between serotype and disease severity. Clin Infect Dis 42:204–210. doi: 10.1086/498905. [DOI] [PubMed] [Google Scholar]
- 5.Pajkrt D, Benschop KS, Westerhuis B, Molenkamp R, Spanjerberg L, Wolthers KC. 2009. Clinical characteristics of human parechoviruses 4-6 infections in young children. Pediatr Infect Dis J 28:1008–1010. doi: 10.1097/INF.0b013e3181a7ab5f. [DOI] [PubMed] [Google Scholar]
- 6.Seitsonen J, Susi P, Heikkilä O, Sinkovits RS, Laurinmäki P, Hyypiä T, Butcher SJ. 2010. Interaction of alphaVbeta3 and alphaVbeta6 integrins with human parechovirus 1. J Virol 84:8509–8519. doi: 10.1128/JVI.02176-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stanway G, Kalkkinen N, Roivainen M, Ghazi F, Khan M, Smyth M, Meurman O, Hyypiä T. 1994. Molecular and biological characteristics of echovirus 22, a representative of a new picornavirus group. J Virol 68:8232–8238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schultheiss T, Emerson SU, Purcell RH, Gauss-Muller V. 1995. Polyprotein processing in echovirus 22: a first assessment. Biochem Biophys Res Commun 217:1120–1127. doi: 10.1006/bbrc.1995.2885. [DOI] [PubMed] [Google Scholar]
- 9.Roivainen M, Piirainen L, Hovi T, Virtanen I, Riikonen T, Heino J, Hyypiä T. 1994. Entry of coxsackievirus A9 into host cells: specific interactions with alpha v beta 3 integrin, the vitronectin receptor. Virology 203:357–365. doi: 10.1006/viro.1994.1494. [DOI] [PubMed] [Google Scholar]
- 10.Berinstein A, Roivainen M, Hovi T, Mason PW, Baxt B. 1995. Antibodies to the vitronectin receptor (integrin alpha V beta 3) inhibit binding and infection of foot-and-mouth disease virus to cultured cells. J Virol 69:2664–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nelsen-Salz B, Eggers HJ, Zimmermann H. 1999. Integrin alpha(v) beta3 (vitronectin receptor) is a candidate receptor for the virulent echovirus 9 strain Barty. J Gen Virol 80:2311–2313. [DOI] [PubMed] [Google Scholar]
- 12.Boonyakiat Y, Hughes PJ, Ghazi F, Stanway G. 2001. Arginine-glycine-aspartic acid motif is critical for human parechovirus 1 entry. J Virol 75:10000–10004. doi: 10.1128/JVI.75.20.10000-10004.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Joki-Korpela P, Marjomäki V, Krogerus C, Heino J, Hyypiä T. 2001. Entry of human parechovirus 1. J Virol 75:1958–1967. doi: 10.1128/JVI.75.4.1958-1967.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pulli T, Koivunen E, Hyypiä T. 1997. Cell-surface interactions of echovirus 22. J Biol Chem 272:21176–21180. doi: 10.1074/jbc.272.34.21176. [DOI] [PubMed] [Google Scholar]
- 15.Triantafilou K, Triantafilou M, Takada Y, Fernandez N. 2000. Human parechovirus 1 utilizes integrins alphavbeta3 and alphavbeta1 as receptors. J Virol 74:5856–5862. doi: 10.1128/JVI.74.13.5856-5862.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Joki-Korpela P, Roivainen M, Lankinen H, Poyry T, Hyypiä T. 2000. Antigenic properties of human parechovirus 1. J Gen Virol 81:1709–1718. [DOI] [PubMed] [Google Scholar]
- 17.Kandiah E, Watts NR, Cheng N, Cardone G, Stahl SJ, Heller T, Liang TJ, Wingfield PT, Steven AC. 2012. Cryo-EM study of hepatitis B virus core antigen capsids decorated with antibodies from a human patient. J Struct Biol 177:145–151. doi: 10.1016/j.jsb.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Plevka P, Lim PY, Perera R, Cardosa J, Suksatu A, Kuhn RJ, Rossmann MG. 2014. Neutralizing antibodies can initiate genome release from human enterovirus 71. Proc Natl Acad Sci U S A 111:2134–2139. doi: 10.1073/pnas.1320624111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith TJ, Olson NH, Cheng RH, Chase ES, Baker TS. 1993. Structure of a human rhinovirus-bivalently bound antibody complex: implications for viral neutralization and antibody flexibility. Proc Natl Acad Sci U S A 90:7015–7018. doi: 10.1073/pnas.90.15.7015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alho A, Marttila J, Ilonen J, Hyypiä T. 2003. Diagnostic potential of parechovirus capsid proteins. J Clin Microbiol 41:2294–2299. doi: 10.1128/JCM.41.6.2294-2299.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Westerhuis BM, Benschop KS, Koen G, Claassen Y, Wagner K, Bakker AQ, Wolthers KC, Beaumont T. 2015. Human memory B cells producing potent cross-neutralizing antibodies against human parechovirus; implications for prevalence, treatment and diagnosis. J Virol 89:7457–7464. doi: 10.1128/JVI.01079-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reed LJ, Muench H. 1938. A simple method of estimating fifty percent endpoints. Am J Hyg 27:493–497. [Google Scholar]
- 23.Kwakkenbos MJ, Diehl SA, Yasuda E, Bakker AQ, van Geelen CM, Lukens MV, van Bleek GM, Widjojoatmodjo MN, Bogers WM, Mei H, Radbruch A, Scheeren FA, Spits H, Beaumont T. 2010. Generation of stable monoclonal antibody-producing B cell receptor-positive human memory B cells by genetic programming. Nat Med 16:123–128. doi: 10.1038/nm.2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shakeel S, Seitsonen JJ, Kajander T, Laurinmäki P, Hyypiä T, Susi P, Butcher SJ. 2013. Structural and functional analysis of coxsackievirus A9 integrin alphavbeta6 binding and uncoating. J Virol 87:3943–3951. doi: 10.1128/JVI.02989-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kivioja T, Ravantti J, Verkhovsky A, Ukkonen E, Bamford D. 2000. Local average intensity-based method for identifying spherical particles in electron micrographs. J Struct Biol 131:126–134. doi: 10.1006/jsbi.2000.4279. [DOI] [PubMed] [Google Scholar]
- 26.Ludtke SJ, Baldwin PR, Chiu W. 1999. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol 128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 27.Yan X, Sinkovits RS, Baker TS. 2007. AUTO3DEM—an automated and high throughput program for image reconstruction of icosahedral particles. J Struct Biol 157:73–82. doi: 10.1016/j.jsb.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosenthal PB, Henderson R. 2003. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol 333:721–745. doi: 10.1016/j.jmb.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 29.van Heel M, Harauz G. 1986. Resolution criteria for three dimensional reconstruction. Optik 73:119–212. [Google Scholar]
- 30.Fernandez JJ, Luque D, Caston JR, Carrascosa JL. 2008. Sharpening high resolution information in single particle electron cryomicroscopy. J Struct Biol 164:170–175. doi: 10.1016/j.jsb.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 31.Roy A, Kucukural A, Zhang Y. 2010. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fry EE, Lea SM, Jackson T, Newman JW, Ellard FM, Blakemore WE, Abu-Ghazaleh R, Samuel A, King AM, Stuart DI. 1999. The structure and function of a foot-and-mouth disease virus-oligosaccharide receptor complex. EMBO J 18:543–554. doi: 10.1093/emboj/18.3.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fry E, Acharya R, Stuart D. 1993. Methods used in the structure determination of foot-and-mouth disease virus. Acta Crystallogr A 49:45–55. doi: 10.1107/S0108767392005737. [DOI] [PubMed] [Google Scholar]
- 34.Lea S, Hernandez J, Blakemore W, Brocchi E, Curry S, Domingo E, Fry E, Abu-Ghazaleh R, King A, Newman J, Stuart D, Mateu MG. 1994. The structure and antigenicity of a type C foot-and-mouth disease virus. Structure 2:123–139. doi: 10.1016/S0969-2126(00)00014-9. [DOI] [PubMed] [Google Scholar]
- 35.Basavappa R, Syed R, Flore O, Icenogle JP, Filman DJ, Hogle JM. 1994. Role and mechanism of the maturation cleavage of VP0 in poliovirus assembly: structure of the empty capsid assembly intermediate at 2.9 A resolution. Protein Sci 3:1651–1669. doi: 10.1002/pro.5560031005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smyth M, Tate J, Hoey E, Lyons C, Martin S, Stuart D. 1995. Implications for viral uncoating from the structure of bovine enterovirus. Nat Struct Biol 2:224–231. doi: 10.1038/nsb0395-224. [DOI] [PubMed] [Google Scholar]
- 37.Venkataraman S, Reddy SP, Loo J, Idamakanti N, Hallenbeck PL, Reddy VS. 2008. Structure of Seneca Valley virus-001: an oncolytic picornavirus representing a new genus. Structure 16:1555–1561. doi: 10.1016/j.str.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Squires G, Pous J, Agirre J, Rozas-Dennis GS, Costabel MD, Marti GA, Navaza J, Bressanelli S, Guerin DM, Rey FA. 2013. Structure of the Triatoma virus capsid. Acta Crystallogr D Biol Crystallogr 69:1026–1037. doi: 10.1107/S0907444913004617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kolatkar PR, Bella J, Olson NH, Bator CM, Baker TS, Rossmann MG. 1999. Structural studies of two rhinovirus serotypes complexed with fragments of their cellular receptor. EMBO J 18:6249–6259. doi: 10.1093/emboj/18.22.6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tate J, Liljas L, Scotti P, Christian P, Lin T, Johnson JE. 1999. The crystal structure of cricket paralysis virus: the first view of a new virus family. Nat Struct Biol 6:765–774. doi: 10.1038/11543. [DOI] [PubMed] [Google Scholar]
- 41.Wang X, Xu F, Liu J, Gao B, Liu Y, Zhai Y, Ma J, Zhang K, Baker TS, Schulten K, Zheng D, Pang H, Sun F. 2013. Atomic model of rabbit hemorrhagic disease virus by cryo-electron microscopy and crystallography. PLoS Pathog 9:e1003132. doi: 10.1371/journal.ppat.1003132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.He Y, Lin F, Chipman PR, Bator CM, Baker TS, Shoham M, Kuhn RJ, Medof ME, Rossmann MG. 2002. Structure of decay-accelerating factor bound to echovirus 7: a virus-receptor complex. Proc Natl Acad Sci U S A 99:10325–10329. doi: 10.1073/pnas.152161599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang X, Peng W, Ren J, Hu Z, Xu J, Lou Z, Li X, Yin W, Shen X, Porta C, Walter TS, Evans G, Axford D, Owen R, Rowlands DJ, Wang J, Stuart DI, Fry EE, Rao Z. 2012. A sensor-adaptor mechanism for enterovirus uncoating from structures of EV71. Nat Struct Mol Biol 19:424–429. doi: 10.1038/nsmb.2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hadfield AT, Lee W, Zhao R, Oliveira MA, Minor I, Rueckert RR, Rossmann MG. 1997. The refined structure of human rhinovirus 16 at 2.15 A resolution: implications for the viral life cycle. Structure 5:427–441. doi: 10.1016/S0969-2126(97)00199-8. [DOI] [PubMed] [Google Scholar]
- 45.Miller ST, Hogle JM, Filman DJ. 2001. Ab initio phasing of high-symmetry macromolecular complexes: successful phasing of authentic poliovirus data to 3.0 Å resolution. J Mol Biol 307:499–512. doi: 10.1006/jmbi.2001.4485. [DOI] [PubMed] [Google Scholar]
- 46.Filman DJ, Wien MW, Cunningham JA, Bergelson JM, Hogle JM. 1998. Structure determination of echovirus 1. Acta Crystallogr D Biol Crystallogr 54:1261–1272. doi: 10.1107/S0907444998002790. [DOI] [PubMed] [Google Scholar]
- 47.Pandurangan AP, Shakeel S, Butcher S, Topf M. 2014. Combined approaches to flexible fitting and assessment in virus capsids undergoing conformational change. J Struct Biol 185:427–439. doi: 10.1016/j.jsb.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 49.Pandurangan AP, Topf M. 2012. RIBFIND: a web server for identifying rigid bodies in protein structures and to aid flexible fitting into cryo EM maps. Bioinformatics 28:2391–2393. doi: 10.1093/bioinformatics/bts446. [DOI] [PubMed] [Google Scholar]
- 50.Topf M, Lasker K, Webb B, Wolfson H, Chiu W, Sali A. 2008. Protein structure fitting and refinement guided by cryo-EM density. Structure 16:295–307. doi: 10.1016/j.str.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lopez-Blanco JR, Chacon P. 2013. iMODFIT: efficient and robust flexible fitting based on vibrational analysis in internal coordinates. J Struct Biol 184:261–270. doi: 10.1016/j.jsb.2013.08.010. [DOI] [PubMed] [Google Scholar]
- 52.Whitelegg NR, Rees AR. 2000. WAM: an improved algorithm for modelling antibodies on the WEB. Protein Eng 13:819–824. doi: 10.1093/protein/13.12.819. [DOI] [PubMed] [Google Scholar]
- 53.Mitton-Fry RM, DeGregorio SJ, Wang J, Steitz TA, Steitz JA. 2010. Poly(A) tail recognition by a viral RNA element through assembly of a triple helix. Science 330:1244–1247. doi: 10.1126/science.1195858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. 2009. Jalview version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Walter TS, Ren J, Tuthill TJ, Rowlands DJ, Stuart DI, Fry EE. 2012. A plate-based high-throughput assay for virus stability and vaccine formulation. J Virol Methods 185:166–170. doi: 10.1016/j.jviromet.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hewat EA, Blaas D. 1996. Structure of a neutralizing antibody bound bivalently to human rhinovirus 2. EMBO J 15:1515–1523. [PMC free article] [PubMed] [Google Scholar]
- 58.Hewat EA, Marlovits TC, Blaas D. 1998. Structure of a neutralizing antibody bound monovalently to human rhinovirus 2. J Virol 72:4396–4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hafenstein S, Bowman VD, Sun T, Nelson CD, Palermo LM, Chipman PR, Battisti AJ, Parrish CR, Rossmann MG. 2009. Structural comparison of different antibodies interacting with parvovirus capsids. J Virol 83:5556–5566. doi: 10.1128/JVI.02532-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schotte L, Strauss M, Thys B, Halewyck H, Filman DJ, Bostina M, Hogle JM, Rombaut B. 2014. Mechanism of action and capsid-stabilizing properties of VHHs with an in vitro antipolioviral activity. J Virol 88:4403–4413. doi: 10.1128/JVI.03402-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu Y, Sheng J, Fokine A, Meng G, Shin WH, Long F, Kuhn RJ, Kihara D, Rossmann MG. 2015. Structure and inhibition of EV-D68, a virus that causes respiratory illness in children. Science 347:71–74. doi: 10.1126/science.1261962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wildenbeest JG, Harvala H, Pajkrt D, Wolthers KC. 2010. The need for treatment against human parechoviruses: how, why and when? Expert Rev Anti Infect Ther 8:1417–1429. doi: 10.1586/eri.10.130. [DOI] [PubMed] [Google Scholar]
- 63.Lee H, Cifuente JO, Ashley RE, Conway JF, Makhov AM, Tano Y, Shimizu H, Nishimura Y, Hafenstein S. 2013. A strain-specific epitope of enterovirus 71 identified by cryo-electron microscopy of the complex with Fab from neutralizing antibody. J Virol 87:11363–11370. doi: 10.1128/JVI.01926-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shingler KL, Cifuente JO, Ashley RE, Makhov AM, Conway JF, Hafenstein S. 2015. The enterovirus 71 procapsid binds neutralizing antibodies and rescues virus infection in vitro. J Virol 89:1900–1908. doi: 10.1128/JVI.03098-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Seitsonen JJ, Shakeel S, Susi P, Pandurangan AP, Sinkovits RS, Hyvonen H, Laurinmäki P, Ylä-Pelto J, Topf M, Hyypiä T, Butcher SJ. 2012. Structural analysis of coxsackievirus A7 reveals conformational changes associated with uncoating. J Virol 86:7207–7215. doi: 10.1128/JVI.06425-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ren J, Wang X, Hu Z, Gao Q, Sun Y, Li X, Porta C, Walter TS, Gilbert RJ, Zhao Y, Axford D, Williams M, McAuley K, Rowlands DJ, Yin W, Wang J, Stuart DI, Rao Z, Fry EE. 2013. Picornavirus uncoating intermediate captured in atomic detail. Nat Commun 4:1929. doi: 10.1038/ncomms2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Garriga D, Pickl-Herk A, Luque D, Wruss J, Caston JR, Blaas D, Verdaguer N. 2012. Insights into minor group rhinovirus uncoating: the X-ray structure of the HRV2 empty capsid. PLoS Pathog 8:e1002473. doi: 10.1371/journal.ppat.1002473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Belnap DM, Filman DJ, Trus BL, Cheng N, Booy FP, Conway JF, Curry S, Hiremath CN, Tsang SK, Steven AC, Hogle JM. 2000. Molecular tectonic model of virus structural transitions: the putative cell entry states of poliovirus. J Virol 74:1342–1354. doi: 10.1128/JVI.74.3.1342-1354.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Levy HC, Bostina M, Filman DJ, Hogle JM. 2010. Catching a virus in the act of RNA release: a novel poliovirus uncoating intermediate characterized by cryo-electron microscopy. J Virol 84:4426–4441. doi: 10.1128/JVI.02393-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Livingstone CD, Barton GJ. 1993. Protein sequence alignments: a strategy for the hierarchical analysis of residue conservation. Comput Appl Biosci 9:745–756. [DOI] [PubMed] [Google Scholar]