

ABSTRACT
The coxsackievirus and adenovirus receptor (CAR) is a cell adhesion molecule used as a docking molecule by some adenoviruses (AdVs) and group B coxsackieviruses. We previously proposed that the preferential transduction of neurons by canine adenovirus type 2 (CAV-2) is due to CAR-mediated internalization. Our proposed pathway of CAV-2 entry is in contrast to that of human AdV type 5 (HAdV-C5) in nonneuronal cells, where internalization is mediated by auxiliary receptors such as integrins. We therefore asked if in fibroblast-like cells the intracellular domain (ICD) of CAR plays a role in the internalization of the CAV-2 fiber knob (FKCAV), CAV-2, or HAdV-C5 when the capsid cannot engage integrins. Here, we show that in fibroblast-like cells, the CAR ICD is needed for FKCAV entry and efficient CAV-2 transduction but dispensable for HAdV-C5 and an HAdV-C5 capsid lacking the RGD sequence (an integrin-interacting motif) in the penton. Moreover, the deletion of the CAR ICD further impacts CAV-2 intracellular trafficking, highlighting the crucial role of CAR in CAV-2 intracellular dynamics. These data demonstrate that the CAR ICD contains sequences important for the recruitment of the endocytic machinery that differentially influences AdV cell entry.
IMPORTANCE Understanding how viruses interact with the host cell surface and reach the intracellular space is of crucial importance for applied and fundamental virology. Here, we compare the role of a cell adhesion molecule (CAR) in the internalization of adenoviruses that naturally infect humans and Canidae. We show that the intracellular domain of CAR differentially regulates AdV entry and trafficking. Our study highlights the mechanistic differences that a receptor can have for two viruses from the same family.
INTRODUCTION
Adenoviruses (AdVs) infect mammals, reptiles, amphibians, and birds (1). In humans, AdVs cause diseases ranging from mild respiratory and ocular infections to severe or lethal pathologies in immunocompromised hosts (2). There are over 200 AdVs identified, which include more than 56 human AdV (HAdV) types that have been partially characterized. Most of the studies addressing receptor usage and intracellular trafficking have used HAdV type 5 (HAdV-C5) or type 2 (HAdV-C2) and paved the way toward the characterization of how AdVs interact with surface molecules and use the endocytic machinery to access the cytoplasm (2–4).
AdVs engage cell surface molecules via their fiber, penton, and hexon proteins (2). The knob region of the fiber (FK) of some of HAdV species A, C, D, E, and F interacts with the coxsackievirus and adenovirus receptor (CAR) (2, 5). CAR is a single-pass transmembrane protein containing an extracellular domain (ECD) composed of two Ig-like domains (D1 and D2), a transmembrane domain (TM), and an intracellular domain (ICD) (6). Several motifs present in the ICD, such as the PDZ domain and the clathrin adaptor protein (AP) binding site, mediate protein-protein interactions (6, 7). Moreover, posttranslational modifications, including glycosylation of the ECD, palmitoylation, and phosphorylation in the ICD, have been reported for CAR (8, 9). These sequences and posttranslational modifications could influence CAR's role during AdV engagement/internalization.
In epithelial cells, CAR is thought to be mainly a docking factor for HAdV-C5 because CAR lacking the ICD is not notably different from full-length CAR during HAdV-C5 vector transduction assays (10). These data led to the conclusion that HAdV-C5 internalization is mediated by integrins via the engagement of the conserved RGD motif in the AdV penton (4, 11, 12). Interestingly, CAR is coendocytosed upon engagement of the canine adenovirus type 2 (CAdV-2, commonly referred to as CAV-2) and HAdV-C5 ex vivo in neurons and neuronal cell lines, raising the possibility that CAR actively participates in the endocytosis of some viruses (13, 14). CAV-2 vectors are powerful tools for gene transfer to the brain because they preferentially transduce neurons and undergo efficient axonal retrograde transport (15–18). CAV-2 engages CAR on the neuronal membrane, which leads to internalization and access to the axonal transport machinery (13, 14). Moreover, using the CAV-2 FK (FKCAV), which also triggers CAR endocytosis, we delineated the endocytic mechanisms during CAR internalization and showed that FKCAV entry requires lipid raft integrity, dynamin, actin dynamics, and the first 16 amino acids (aa) of the ICD (14). Because CAR appears to be the exclusive attachment molecule for CAV-2 (19), understanding CAR's role and membrane dynamics is crucial for applied and fundamental studies.
In this study, we characterized the role of the CAR ICD for FKCAV, CAV-2, and HAdV-C5 internalization in fibroblast-like cells. We show that CAR is endocytosed in NIH 3T3 cells and that the deletion of the ICD impacts FKCAV and CAV-2 intracellular trafficking and/or transduction efficacy but does not impact the transduction of HAdV-C5 or HAdV-C5 lacking the penton RGD motif (HAdV-C5ΔRGD), which partially recapitulates CAV-2 capsid. We report a role for the CAR ICD in AdV internalization and identify a differential use of CAR by AdV types.
MATERIALS AND METHODS
Cell culture.
NIH 3T3 cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Life Technologies) with UltraGlutamine (Lonza) supplemented with 10% fetal calf serum (FCS) (Sigma), nonessential amino acids (NEAA) (Gibco), and penicillin-streptomycin. Cells were incubated at 37°C in saturated humidity with 5% CO2.
DNA constructs and reagents.
The plasmids pCARFL-RFP (encoding human CAR; RFP is red fluorescent protein) and pCARΔICD-RFP were a gift from Maddy Parsons (Kings College London) and have been previously described (20). All point mutations or deletions were performed by directed-mutagenesis PCR. Two mutants of CAR (isoform 1) were generated using the plasmid pCARFL-RFP (where FL is full-length) as the template. The pCARΔCC-RFP plasmid was obtained using the mutagenic primer 5′-CTCATTGGTCTTATCATCTTTGCCGCTCGTAAAAAGCGCAGAGAA-3′ (substituted residues are underlined), which replaces cysteine residues at positions 259 and 260 with two alanines (C259A and C260A). The plasmid pCAR274 was obtained by adding a stop codon after the histidine residue at position 274 with the following primer 5′-AAATATGAAAAGGAAGTTCATTAAGATATCAGGGAAGATGTGCCA-3′ (underlining indicates mutations/substitutions). All DNA constructs were verified by sequencing. The Rab7-green fluorescent protein (GFP) construct was previously described (21). Several anti-CAR antibodies were used: a mouse monoclonal RmcB (Millipore), a mouse monoclonal E1.1, and a goat polyclonal anti-CAR (R&D Systems). All CAR antibodies recognize the ECD. Anti-CAV-2 rabbit polyclonal (against the entire capsid) was purified in-house. FKCAV staining was performed with a rabbit polyclonal anti-FKCAV previously described (14). Lysosomes were labeled with a rat monoclonal anti-LAMP-1 (Abcam). Mouse monoclonal anti-β-tubulin (Sigma) was used for immunofluorescence and as a loading control for immunoblot analyses. Anti-mouse, anti-rabbit, or anti-goat horseradish peroxidase (HRP)-conjugated secondary antibodies (Sigma) were used according to the species of the primary antibody used. Fluorescent secondary antibodies coupled to Alexa 488, 555, and 647 (Life Technology) were used depending on the combination of the primary antibody (e.g., goat anti-CAR labeled with an anti-goat coupled to Alexa 488 [Fig. 1] or with an anti-goat coupled to Alexa 555 [Fig. 2, 4, 5]).
FIG 1.

Cell surface expression of CAR and its mutants in NIH 3T3 cells. (A) Schematic representation of constructs used in the study. The numbers indicate the amino acid residues, and the two Ig-like domains D1 and D2 are indicated. (B) Immunoblot analysis of CARFL-RFP, CARΔCC-RFP, CAR274, and CARΔICD-RFP expression. CAR was detected using an antibody against the ECD. Expected bands are indicated by an asterisk, and other bands correspond to CAR without the RFP tag. β-Tubulin was used as a loading control. Immunoblot analysis shows equal expression levels of the different DNA constructs in NIH 3T3 cells. NT, nontransfected. (C) Flow cytometry analysis of CAR cell surface levels. NIH 3T3 cells were transfected with plasmids expressing CAR constructs, and CAR cell surface expression was assayed by flow cytometry using an antibody against the CAR ECD on nonpermeabilized cells. (D) Confocal microscopy of CAR plasma membrane location using FKCAV. NIH 3T3 cells were transfected with the different constructs, and at 24 h posttransfection 1 ng/ml of FKCAV was added for 20 min on ice. Cells were fixed and stained for CAR (green) and for FKCAV (cyan). Scale bar, 15 μm.
FIG 4.

The role of the CAR ICD in CAV-2 internalization. (A) Flow cytometry analyses of CAV-GFP at the cell surface in NIH 3T3 cells expressing CARFL-RFP or CARΔICD-RFP. At 24 h posttransfection, 2,000 pp/cell of CAV-GFP was added to the cells for 30 min on ice. Cells were washed and collected at the indicated times. CAV-GFP labeling was performed prefixation by incubating cells with an anti-CAV-2 antibody. Results of three independent experiments are expressed as means ± SEM.*, P < 0.05; **, P < 0.01 (for values in transfected cells versus the control values; Student's t test). (B and C) Confocal microscopy of NIH 3T3 cells expressing either CARFL-RFP or CARΔICD-RFP and infected with 2,000 pp/cell of CAV-GFP for 30 min on ice (B) or washed and incubated for 30 min at 37°C before fixation (C). CAV-GFP was labeled with an anti-CAV-2. In panel B, virions are bound at the cell surface and colocalized with CAR (insets show ∼3× higher magnifications), and arrows indicate colocalization between CAR (magenta) and CAV-2 (green). Panel C shows colocalization at 30 min postinternalization. Insets show higher magnifications (3×) of internalized CAV-2 (green) colocalizing with CARFL-RFP (magenta; filled arrows) but not with CARΔICD-RFP (magenta, inset 1, empty arrows). In cells expressing CARΔICD-RFP, surface-bound virions are still found with CAR (inset 2, filled arrows). The asterisks indicate accumulation of CAV-2. (D) Colocalization quantification between CAR and CAV-GFP after 30 min of internalization. Results of three independent experiments are expressed as means ± SEM (≥504 structures counted per sample). ***, P < 0.001 (versus control values; Student's t test). (E) Confocal microscopy of NIH 3T3 cells expressing either CARFL-RFP or CARΔICD-RFP incubated with 2,000 pp/cell of CAV-GFP for 30 min on ice with transferrin coupled to Alexa 488 (Tf-488), washed, and placed at 37°C for 20 min before fixation. CARFL-RFP is found in Tf-positive (Tf+) structures, whereas CARΔICD-RFP is mostly absent from endocytic structures. Insets show higher magnifications (∼3×) of internalized structures (white) containing Tf (green), CAR (magenta), and CAV-2 (cyan), identified by arrows. (F) Quantification of colocalization between Tf-positive structures and CAV-GFP. Results of three independent experiments are expressed as means ± SEM (≥186 structures counted per sample). **, P < 0.01 (versus control values; Student's t test). Scale bar, 10 μm.
FIG 5.

The lack of CAR ICD triggers a delay in CAV-2 trafficking. (A) Confocal microscopy of NIH 3T3 cells expressing CARFL-RFP or CARΔICD-RFP and transfected with Rab7-GFP. Cells were incubated with 2,000 pp/cell of CAV-GFP for 30 min on ice, washed, and placed at 37°C for 1 h to allow internalization. CAV-2 in Rab7+ structures were visualized in cells expressing CARFL-RFP or CARΔICD-RFP. Insets show higher magnifications (∼3×) of internalized structures (white) containing Rab7 (green), CAR (magenta), and CAV-2 (cyan), identified by arrows. (B) Quantification of colocalization between Rab7+ structures and CAV-2. Results of three independent experiments are expressed as means ± SEM (≥406 structures counted per sample). **, P < 0.01 (versus the control values; Student's t test). (C and D) Confocal microscopy of NIH 3T3 cells expressing CARFL-RFP or CARΔICD-RFP. Cells were incubated with 2,000 pp/cell of CAV-GFP for 30 min on ice, washed, and placed at 37°C and visualized at 30 min (C) and 2 h (D) postinternalization. CAV-2 localized at the microtubule organizing center (MTOC; identified by arrows) in cells expressing CARFL-RFP or CARΔICD-RFP. (E) Quantification of MTOC accumulation. Results of three independent experiments are expressed as means ± SEM (≥132 cells per sample). * P, <0.05; **, P < 0.01 (versus control values; Student's t test). Scale bars, 15 μm (A), 20 μm (C), and 5 μm (D).
Transferrin-Alexa 488 (Life Technology) was used at 1:100 in culture medium. Transfections were performed using Lipofectamine 2000 (Life Technology) according to the manufacturer's recommendations.
Vectors and recombinant viral ligand.
HAdV-C5 vectors expressing β-galactosidase (here called AdV-βGal and AdVΔRGD-βGal (a gift from A. Baker, University of Glasgow), CAV-GFP, and CAV-βGal were produced and purified as described previously (22, 23). The CAV-2 vectors used in the study had a ratio of 1 infectious particle/∼3 physical particles (pp). The HAdV-C5 vectors used in the study had a ratio of 1 infectious particle/∼10 pp. The recombinant CAV-2 fiber knob (FKCAV) was produced and purified as previously described (24).
Immunofluorescence staining and microscopy imaging.
NIH 3T3 cells were washed with phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde (PFA) in PBS for 15 min at room temperature (RT). After fixation, the coverslips were washed with PBS at RT, and cells were permeabilized with PBS containing 0.1% (vol/vol) Triton X-100 for 5 min. Cells were then blocked with a solution containing 2% (wt/vol) bovine serum albumin (BSA) and 10% (vol/vol) horse serum in PBS for 30 min to 1 h at RT. Primary and secondary antibodies were diluted in blocking solution and incubated overnight at 4°C and for 1 h at RT, respectively. Coverslips were mounted on slides with fluorescent mounting medium supplemented with 4′,6′-diamidino-2-phenylindole (DAPI; Sigma). Image acquisitions were performed using a Leica SP5 confocal microscope equipped with multiwavelength argon and helium-neon lasers and a 63× (1.4 numerical aperture [NA]) Plan Apochromat oil immersion objective. Immunofluorescence quantification was performed by imaging three independent experiments, with 5 to 10 fields/experiment and 5 to 20 cells/field. Details for each experiment are found in the figure legends.
Flow cytometry.
NIH 3T3 cells were washed and then scraped in ice-cold PBS and pelleted at 200 × g. Cells were either fixed with 2% PFA in PBS for 15 min at RT or incubated with primary and secondary antibodies before fixation. Staining was performed in cold PBS supplemented with 10% FCS for 1 h at 4°C for primary antibodies and for 30 min at 4°C for secondary antibodies. Washes were performed using cold PBS. Sample collections were analyzed using a FACSCalibur analyzer (BD Bioscience), and data analysis was performed with FlowJo software. The CAV-2-specific signal (see Fig. 4A) was quantified by measuring the total fluorescence compared to that of nontransfected and noninfected cells and setting the fluorescence level at time zero to 100%.
β-Galactosidase activity assay.
NIH 3T3 cells were incubated with 100 pp/cell of AdV-βGal, AdVΔRGD-βGal, or CAV-βGal for 24 h (AdV) or 48 h (CAV) in 12-well plates, washed in PBS, and then lysed with 100 μl/well of lysis buffer; cells were collected and centrifuged at 1,000 × g for 5 min. Protein concentration was determined using Bradford reagent (Sigma). A β-galactosidase activity assay was performed using a Galacto-Star system (Applied Biosystems) according to the manufacturer's recommendation. Ten microliters of sample (∼1 mg/ml) was added to 200 μl of reaction buffer and incubated for 15 min at RT. β-Galactosidase activity was measured with a luminometer during 1 s. Data analysis was performed by normalizing the β-galactosidase activity to the protein concentration for three independent experiments performed in duplicates.
Statistical analyses.
Data were analyzed using Student's t test for unpaired data or a Mann-Whitney test. Results are expressed as means ± standard errors of means (SEM).
RESULTS
The ICD is needed to target CAR and FKCAV to the endolysosomal pathway in fibroblast-like cells.
In neurons, CAR is coendocytosed following engagement of FKCAV, CAV-2, and HAdV-C5 (13, 14). However, it is unclear whether CAR is taken up by the endocytic pathway in nonneuronal cells or whether its ICD influences CAV-2 transduction. We therefore examined CAR membrane dynamics in NIH 3T3 cells, a murine fibroblast-like cell line that does not express CAR and is resistant to CAV-2 and HAdV-C5 vector transduction (19, 25). First, using CARFL-RFP (full-length), we generated a series of constructs. In one construct we replaced the palmitoylation sites (cysteines 259 and 260 [8]) with two alanines to create CARΔCC-RFP. We then deleted either the residues after the first 16 aa of the ICD (CAR274) or the intracellular tail except for the two first cysteines, C259C260 (CARΔICD) (Fig. 1A) (14). NIH 3T3 cells were then transfected with plasmids encoding each mutant, and global expression and membrane levels were examined. Immunoblot analyses showed CAR expression and confirmed the lack of CAR in mock-transfected cells (Fig. 1B). Flow cytometry analyses of transfected cells incubated with an anti-CAR antibody before fixation showed similar CAR levels at the plasma membrane between constructs (Fig. 1C). Incubation of transfected cells with FKCAV on ice for 20 min and detection using an anti-FKCAV antibody also showed binding on CAR-positive (CAR+) cells, confirming that the CAR constructs were efficiently targeted to the plasma membrane (Fig. 1D).
To determine whether FKCAV engagement of CAR induces its internalization and degradation by lysosomes in NIH 3T3 cells, we incubated transfected cells with FKCAV for 20 min on ice and shifted them to 37°C for 30 min to allow internalization. FKCAV binding to CARFL-RFP triggered efficient targeting to LAMP-1-positive lysosomes as seen by the colocalization of FKCAV, CAR, and LAMP-1 (Fig. 2). To determine the role of the ICD in FKCAV internalization, we performed the same assay in cells transfected with CARΔCC-RFP, CAR274, and CARΔICD-RFP. Only the cells transfected with CARΔICD-RFP were unable to internalize FKCAV, suggesting that the role of the CAR ICD in FKCAV endocytosis in fibroblast-like cells is similar to that in neurons (Fig. 2).
FIG 2.

CAR endocytosis triggered by FKCAV in NIH 3T3 cells. Confocal microscopy analyses were performed of FKCAV-mediated CAR endocytosis. NIH 3T3 cells were transfected with the different CAR constructs, and at 24 h posttransfection 1 ng/ml of FKCAV was added for 20 min on ice; samples were washed and incubated for 1 h at 37°C. Cells were labeled with anti-CAR against the ECD (magenta), anti-FKCAV (cyan), and anti-LAMP-1 (green) to visualize lysosomes. All constructs except CARΔICD-RFP allowed targeting of FKCAV to lysosomes. CAR lacking its ICD is not internalized upon FKCAV engagement. Insets show higher magnifications (∼3×) of internalized structures (white) containing CAR, FKCAV, and LAMP-1, identified by arrows. Scale bar, 10 μm.
Together, these data demonstrate that in fibroblast-like cells, the first 16 aa (CAR274) are needed for FKCAV entry and targeting to lysosomes and that palmitoylation of the ICD is dispensable for ligand-induced CAR internalization.
CAR ICD influences CAV-βGal but not AdVΔRGD-βGal transduction.
The CAV-2 capsid lacks a readily identifiable integrin-interacting motif (15, 19, 26). We therefore hypothesized that CAR, and not integrins, regulates CAV-2 entry (19). Because CARΔICD cannot internalize FKCAV, we asked whether the ICD regulates CAV-2 vector transduction. We therefore tested NIH 3T3 cells expressing CARFL-RFP, CARΔCC-RFP, CAR274, and CARΔICD-RFP for their ability to be transduced by CAV-βGal (an E1-deleted CAV-2 vector harboring a β-galactosidase expression cassette). At 24 h posttransfection with the CAR plasmids, we incubated cells with 100 pp/cell of CAV-βGal for 48 h and then quantified β-Gal activity. Nontransfected cells incubated with CAV-βGal and mock (nontransfected noninfected) cells had no significant β-Gal activity (Fig. 3A). To compare the constructs, β-Gal activity in cells expressing CARFL-RFP and incubated with CAV-βGal was normalized to 1. CARΔCC-RFP-expressing cells had a ∼10% decrease in transduction compared to the level in CARFL-RFP-expressing cells, whereas CAR274-expressing cells did not show a significant decrease (Fig. 3A.). Although CARΔICD-RFP did not allow FKCAV internalization, CARΔICD-RFP-expressing cells were transduced by CAV-βGal but had significantly lower β-Gal activity than CARFL-RFP-expressing cells (Fig. 3A). These data suggest that the first 16 aa of the ICD and possibly the palmitoylation of the CAR ICD are necessary for efficient CAV-2 vector transduction.
FIG 3.
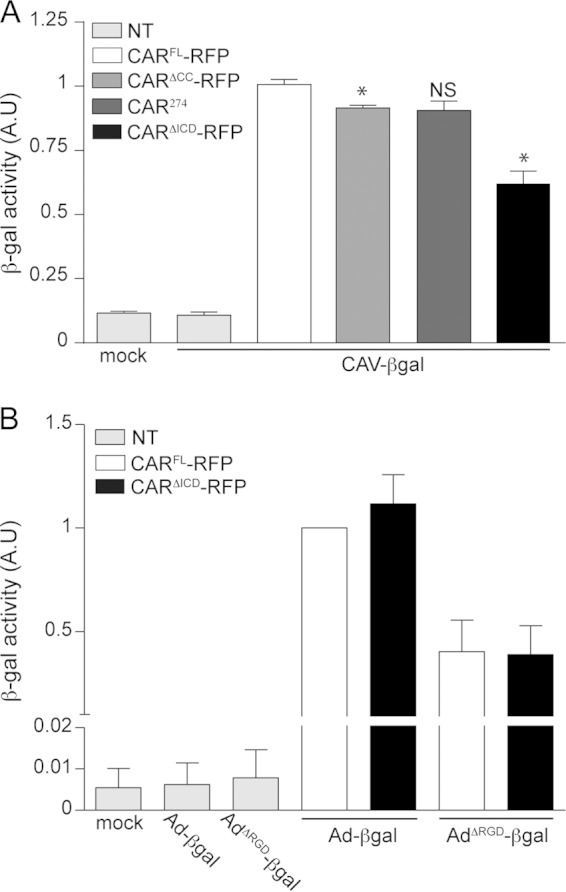
CAV-2 and HAdV-C5 vectors rely differently on the CAR ICD. (A) Differential efficacy of CAV-βGal cell transduction by CARFL-RFP and CARΔICD-RFP. Efficacy of cell transduction was measured by quantifying β-galactosidase (β-Gal) activity. Cells expressing either CARFL-RFP, CARΔCC-RFP, CAR274, or CARΔICD-RFP for 24 h were infected with 100 pp/cell of CAV-βGal for 48 h. Mock cells are nontransfected (NT) and mock transduced. Nontransfected cells were incubated with 100 pp/cell of CAV-βGal. Results of three independent experiments in duplicate are expressed as means ± SEM. *, P < 0.05 (for values in transfected cells versus the controls; Mann-Whitney test). (B) Quantification of β-Gal activity in cells expressing either CARFL-RFP or CARΔICD-RFP and incubated with 100 pp/cell of AdV-βGal or AdVΔRGD-βGal for 24 h. Mock cells are nontransfected and mock transduced. Nontransfected cells were incubated with 100 pp/cell of AdV-βGal or AdVΔRGD-βGal. Results of three independent experiments in duplicate are expressed as means ± SEM. AU, arbitrary units.
In contrast to CAV-2, the list of cell surface molecules that HAdV-C5 can engage is long (2). HAdV-C5 can dock to CAR, and, in most cell types, integrins act as auxiliary receptors and facilitate virus internalization through an RGD motif in the penton base (4, 11, 27). In contrast, the CAV-2 capsid lacks identifiable integrin-interacting motifs, is smoother, is neutral in charge, and has fibers with two potential hinges (26). In this context, an HAdV-C5 capsid lacking the RGD motif (HAdV-C5ΔRGD) may mirror the possible integrin-independent CAV-2 entry mechanism. Previous studies in CAR-expressing cells showed that HAdV-C5ΔRGD has a delay in internalization (23, 28). In neurons, CAR is cointernalized with HAdV-C5, suggesting a potential CAR-mediated internalization of HAdV-C5. Therefore, we asked if HAdV-C5ΔRGD transduction was impacted by the absence of the CAR ICD. To address this, NIH 3T3 cells were transfected with CARFL-RFP or CARΔICD-RFP and incubated with AdV-βGal or AdVΔRGD-βGal. In mock-treated and nontransfected cells incubated with vectors, β-Gal activity was undetectable (Fig. 3B). In cells transfected with CARFL-RFP or CARΔICD-RFP and incubated with AdV-βGal, no significant difference in the levels of β-Gal activity was observed (Fig. 3B). Interestingly, AdVΔRGD-βGal efficiently transduced cells expressing CARFL-RFP or CARΔICD-RFP (Fig. 3B), demonstrating that the ICD of CAR was not crucial for transduction of an HAdV-C5 vector that is unlikely to engage integrins.
These data suggest that the ICD of CAR is necessary for efficient CAV-2 transduction but is dispensable for HAdV-C5 and HAdV-C5 lacking the highly conserved integrin binding motif, thereby identifying differential use of CAR by AdV types.
CARΔICD poorly enters the endosomal pathway upon CAV-2 engagement and triggers a delayed uptake.
The above results regarding the role of the CAR ICD suggest that CAV-2 and FKCAV are internalized via different mechanisms. Indeed, although CAV-2 transduction is less efficient in the absence of CAR ICD, it still occurs. We therefore used flow cytometry to quantify CAV-2 endocytosis kinetics. NIH 3T3 cells transfected with CARFL-RFP or CARΔICD-RFP were incubated with CAV-GFP for 30 min on ice. Cells were then shifted to 37°C for the indicated amount of time (Fig. 4A), put back on ice, incubated with an anti-CAV-2 antibody, washed, incubated with an Alexa 488-secondary antibody, fixed, and analyzed by flow cytometry. Using this assay, we detected a time-dependent decrease in fluorescence that reflects the internalization of CAV-2 and its inaccessibility to the antibody in cells transfected with CARFL-RFP or CARΔICD-RFP (Fig. 4A). In CARΔICD-RFP-expressing cells, the kinetics of internalization of CAV-2 was slower since the decrease in fluorescence was less pronounced than in CARFL-RFP-expressing cells (Fig. 4A). We then asked if CARΔICD-RFP was internalized following CAV-2 engagement. NIH 3T3 cells expressing either CARFL-RFP or CARΔICD-RFP were incubated with CAV-GFP for 30 min on ice and assayed for CAV-2/CAR colocalization at various times of internalization. When the cells were fixed at 0 min, CAV-2 colocalized with CARFL-RFP and CARΔICD-RFP (Fig. 4B). At 30 min, CAV-2/CARFL-RFP colocalized inside the cells (Fig. 4C). In contrast, at 30 min postengagement, CAV-2 poorly colocalized with CARΔICD-RFP (Fig. 4C). Quantitative analyses showed that CARΔICD-RFP/CAV-2 colocalization was significantly lower than that of CARFL-RFP/CAV-2 (Fig. 4D), demonstrating that CARΔICD was less efficiently cointernalized with CAV-2.
We then used Alexa-labeled transferrin (Tf-Alexa 488) to visualize endocytic structures containing internalized virions and CAR. NIH 3T3 cells transfected with CARFL-RFP or CARΔICD-RFP were incubated with Tf-Alexa 488 and CAV-GFP for 30 min on ice. Cells were then shifted to 37°C for 20 min to allow endocytosis. CAV-2/CARFL-RFP colocalized with Tf-Alexa 488-positive (Tf-Alexa 488+) structures, demonstrating that, similar to results in neurons, cointernalization of virion and receptor in endosomes occurred (Fig. 4E). However, CARΔICD-RFP and CAV-2 were seldom seen in Tf-Alexa 488+ structures (Fig. 4E). Quantification of CAR in Tf-Alexa 488+ endocytic structures showed little CARΔICD-RFP internalized, consistent with a defect of entry (Fig. 4F).
Together, these data demonstrate that the ICD of CAR plays a role during CAV-2 endocytosis. This is consistent with the decrease in transduction efficiency in cells expressing CARΔICD-RFP. These observations suggest a requirement that the CAR ICD interact with other proteins to allow efficient internalization.
The ICD of CAR affects CAV-2 intracellular trafficking.
In axons, CAV-2 vectors and CAR are transported in Rab7+/pH-neutral endosomes until they reach the cell body (13, 29). We therefore monitored whether CAV-2 vectors could reach Rab7+ organelles in NIH 3T3 cells. To this end, we cotransfected NIH 3T3 cells with plasmids encoding the CAR-RFP constructs and Rab7-GFP, incubated them with CAV-GFP, and assayed for colocalization. We detected CAV-2 in Rab7+ structures together with CARFL-RFP, suggesting that CAV-2 reaches the late stage of endocytosis in nonpolarized cells, where Rab7 is associated with acidic endosomes (Fig. 5A and B). In CARΔICD-RFP-expressing cells, CAV-2 was found in Rab7+ structures at a lower rate, showing impaired or delayed CAV-2 endosomal trafficking (Fig. 5A and B).
Postendocytosis, HAdVs are thought to escape endosomal structures via a protein VI-dependent lysis of the vesicular membrane (3, 30), and once in the cytoplasm, they undergo dynein-dependent transport (4, 31). In cells incubated with high doses of AdVs, the capsids accumulate at the microtubule organizing center (MTOC) (4). Here, we found that in NIH 3T3 cells incubated with 2,000 pp/cell of CAV-GFP, capsids accumulated close to the nucleus. CAR staining suggested that CAV-2 was not associated with CAR, suggesting that virions had escaped endosomes and trafficked to the MTOC (Fig. 4C, asterisks). To determine whether this structure was the MTOC, we fixed cells at 30 min, 60 min, 2 h, and 6 h postincubation and labeled the microtubule network with an anti-β-tubulin antibody. At 30 min postinternalization in cells expressing CARFL-RFP, CAV-2 colocalized with the gathering of microtubules (Fig. 5C and E). In contrast, in cells expressing CARΔICD-RFP, CAV-2 was significantly less concentrated at the MTOC at 30 min and was more dispersed inside the cytoplasm at 2 h (Fig. 5C, D, and E). Quantitative analyses showed that this was also observed at late time points postentry (6 h), suggesting a defect in intracellular trafficking in the absence of the CAR ICD (Fig. 5E).
Together, these data suggest that the lack of the CAR ICD impacts downstream steps of CAV-2 trafficking in fibroblast-like cells. Ultimately, transgene expression will be altered and have a quantitative consequence for gene transfer by CAV-2 vectors (Fig. 6).
FIG 6.

Proposed mechanism of CAR-mediated CAV-2 entry in fibroblast-like cells. This scheme recapitulates CAR-mediated endocytosis upon FKCAV and CAV-2 engagement (14). CAR is located in lipid rafts (lipid bilayer in brown).The trimeric FKCAV binds with high affinity to D1 and triggers CAR endocytosis, which requires lipid raft integrity, dynamin, and actin, leading to targeting to endolysosomes for degradation. CARΔICD is not internalized upon FKCAV binding (black cross). CARFL is coendocytosed with CAV-2 and reaches transferrin-positive endosomal structures within 30 min and Rab7+ structures in the late stage of endocytosis prior to endosomal escape. Virions further traffic via microtubules to reach the MTOC. CARΔICD is poorly cointernalized with CAV-2 and triggers delay in CAV-2 trafficking in each step, which ultimately impacts vector transduction. The absence of the CAR ICD does not impact HAdV-C5 and HAdV-C5ΔRGD cell transduction (far right), highlighting different uses of CAR among AdV types.
DISCUSSION
CAR is likely the unique receptor for CAV-2 (19) and is responsible for the preferential transduction of neurons and efficient retrograde transport of CAV-2 vectors (13, 15). We therefore asked whether the CAR ICD influences CAV-2 uptake. In this study, we show that CAR is taken up to the endocytic pathway in nonpolarized cells upon interaction with FKCAV and that the first 16 aa of the CAR ICD are needed to promote efficient endocytosis. We concluded that palmitoylation does not play a significant role in CAR entry since CAR was targeted equally to lysosomes with or without the palmitoylation sites. Moreover, in accordance with CAR's inability to be endocytosed after FKCAV engagement, a tailless CAR (CARΔICD) reduces CAV-2 transduction efficacy. In contrast, an HAdV-C5 capsid lacking the RGD domain—which presumably precludes its interaction with some integrins—is not significantly different in cell transduction efficacy with or without the CAR ICD. We show that CAV-2 poorly induces CARΔICD targeting to endocytic structures, suggesting that mechanisms not directly involving the CAR ICD potentiate CAV-2 entry. Finally, we show that CAV-2 trafficking from the plasma membrane to the MTOC is perturbed by the absence of the CAR ICD (Fig. 6).
We previously proposed that the disruption of the homodimeric interaction between CAR ECDs, triggered by viral ligands that bind with more efficiency to the site responsible for homodimeric interaction, is necessary for endocytosis (14). This disruption, in addition to sequences in the ICD, lipid raft integrity, actin, and the small GTPase dynamin, targets CAR to the endolysosomal pathway upon interaction with FKCAV (13, 14) (Fig. 6). Interestingly, not all ligands trigger CAR endocytosis (e.g., polyclonal antibodies failed to induce CAR internalization) (14). Here, our data demonstrate significant differences in the roles of the CAR ICD in internalization, depending on whether the ligand is FKCAV, CAV-2, HAdV-C5, or HAdV-C5ΔRGD (Fig. 6). This highlights the use of different endocytic mechanisms for these CAR ligands that can be totally or partially dependent on, or independent of, the ICD. What could be the explanation for the differential use of the CAR ICD following FKCAV versus CAV-2 engagement? It is certain that the trimeric FKCAV engages fewer CAR molecules than CAV-2 (three CAR-binding sites/trimeric fiber, with 12 fibers) (Fig. 6) and in a different spacing. In addition, this clustering of CAR molecules at the cell surface by FKCAV or CAV-2 could differentially trigger signaling involved in optimal internalization. Finally, one could envisage scenarios in which the CAR ICD influences extracellular interacting partners necessary for endocytosis. Notably, the ICD of CAR interacts with actin (32) and microtubules (33), which could influence the internalization and the intracellular trafficking of its ligands, such as FKCAV.
Another explanation for impaired CAV-2 vector transduction in the context of CARΔICD could be related to plasma membrane dynamics/targeting of CAR. In epithelial cells (34) and in neurons (14), CAR is found in lipid rafts, and lipid raft integrity is necessary for FKCAV-induced CAR endocytosis. During internalization of FKCAV, CAV-2, and HAdV-C5, CAR also remains in lipid rafts (14). Previous studies showed that the replacement of the transmembrane domain and the ICD of CAR with those of a non-lipid raft protein (low-density lipoprotein receptor [LDLR]) does not affect its location in lipid rafts (34), suggesting that impaired targeting to rafts is not responsible for the effect of CARΔICD on CAV-2 vector transduction. Furthermore, it was proposed that CAR drifting at the plasma membrane is necessary for AdV internalization (35). CAR can mediate HAdV-C2 motion at the cell surface and influence capsid engagement of integrins in an actin-dependent manner (35). Whether CAR-mediated drifting also occurs for CAV-2 vector transduction is not yet characterized, but one could envisage that CAR lacking the ICD could impact such a process.
Because we detected CAR/HAdV-C5 cointernalization in neurons (14), we reevaluated CAR's role in HAdV-C5 entry in the absence of the penton-mediated integrin binding. In contrast to CAV-2 and as reported before (10, 36), the absence of the CAR ICD does not perturb HAdV-C5 vector transduction. While HAdV-C5ΔRGD capsids partially mimic the RGD-less CAV-2 capsid, the CAV-2 fiber shaft is more flexible than the shaft of HAdV-C5 and probably increases CAV-2 flexibility (26). However, in contrast to CAV-2 and similar to wild-type HAdV-C5 (HAdV-C5WT), the absence of the CAR ICD does not perturb HAdV-C5ΔRGD-mediated transduction. This observation highlights the difference in terms of receptor usage, in particular CAR, between AdVs. That CAV-2 FK interacts with CAR with a greater affinity than HAdV-C5 FK (1.1 nM versus 7.9 nM, respectively) (24, 26) and that the lack of CAR ICD affected its transduction efficiency are consistent with a role for CAR in CAV-2 internalization because CAV-2 engagement would be more efficient than that of HAdV-C5. Moreover, because HAdV-C5 can interact with other secondary receptors besides integrins (2), internalization could still be mediated post-CAR engagement, or integrin interaction could take place in an RGD-independent manner (37) and influence HAdV-C5 entry.
In conclusion, unlike for HAdV-C5, CAR is more than a docking factor for CAV-2. How CAR is linked to the endocytic machinery and whether this plays other biological roles than pathogen binding remain to be characterized. The ICD of CAR contains numerous sequences that could be involved in protein-protein interaction and influence endocytosis (32, 33). CAR could also regulate the trafficking of other proteins potentially important for CAV-2 entry, as described for E-cadherin (38) or acid sensing ion channel 3 (39). The ICD of CAR could also influence the ECD of CAR. In this light, interaction with intracellular partners may modulate CAR adhesive function, suggestive of an ICD-dependent effect on the anchoring and/or membrane location of CAR (40). Although not relevant for unpolarized cells, differences in intracellular sequences also have a direct impact on the localization of different CAR isoforms (41). In the same line, CAR engagement can lead to activation of signaling pathways (20, 42) and could therefore modulate the trafficking of AdVs indirectly.
ACKNOWLEDGMENTS
We thank Julia Durand for experimental help, M. Parsons (Kings College London) and A. Baker (University of Glasgow) for sharing reagents, and Montpellier Rio Imaging for microscopy studies.
The Kremer lab has been supported by grants from the European Commission (grants 290002, 286071, and 222992), Fondation pour la Recherche Médicale, E-Rare Fondation Maladies Rare, Agence Nationale de la Recherche, Fondation de France, and the Region Languedoc Roussillon. F.L. is a recipient of a French Government-funded MNERT fellowship. E.J.K. and S.S. are INSERM fellows.
REFERENCES
- 1.Harrach B, Benko M, Both GW, Brown M, Davison AJ, Echavarria M, Hess M, Jones MS, Kajon A, Lehmkuhl HD, Mautner V, Mittal SK, Wadell G. 2011. Family Adenoviridae, p 125–141. In King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (ed), Virus taxonomy: classification and nomenclature of viruses. Ninth report of the International Committee on Taxonomy of Viruses. Academic Press, London, United Kingdom. [Google Scholar]
- 2.Arnberg N. 2012. Adenovirus receptors: implications for targeting of viral vectors. Trends Pharmacol Sci 33:442–448. doi: 10.1016/j.tips.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 3.Kremer EJ, Nemerow GR. 2015. Adenovirus tales: from the cell surface to the nuclear pore complex. PLoS Pathog 11:e1004821. doi: 10.1371/journal.ppat.1004821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Henaff D, Salinas S, Kremer EJ. 2011. An adenovirus traffic update: from receptor engagement to the nuclear pore. Future Microbiol 6:179–192. doi: 10.2217/fmb.10.162. [DOI] [PubMed] [Google Scholar]
- 5.Bergelson JM, Cunningham JA, Droguett G, Kurt-Jones EA, Krithivas A, Hong JS, Horwitz MS, Crowell RL, Finberg RW. 1997. Isolation of a common receptor for coxsackie B viruses and adenoviruses 2 and 5. Science 275:1320–1323. doi: 10.1126/science.275.5304.1320. [DOI] [PubMed] [Google Scholar]
- 6.Coyne CB, Bergelson JM. 2005. CAR: a virus receptor within the tight junction. Adv Drug Deliv Rev 57:869–882. doi: 10.1016/j.addr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 7.Diaz F, Gravotta D, Deora A, Schreiner R, Schoggins J, Falck-Pedersen E, Rodriguez-Boulan E. 2009. Clathrin adaptor AP1B controls adenovirus infectivity of epithelial cells. Proc Natl Acad Sci U S A 106:11143–11148. doi: 10.1073/pnas.0811227106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van't Hof W, Crystal RG. 2002. Fatty acid modification of the coxsackievirus and adenovirus receptor. J Virol 76:6382–6386. doi: 10.1128/JVI.76.12.6382-6386.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Excoffon KJ, Gansemer N, Traver G, Zabner J. 2007. Functional effects of coxsackievirus and adenovirus receptor glycosylation on homophilic adhesion and adenoviral infection. J Virol 81:5573–5578. doi: 10.1128/JVI.02562-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang X, Bergelson JM. 1999. Coxsackievirus and adenovirus receptor cytoplasmic and transmembrane domains are not essential for coxsackievirus and adenovirus infection. J Virol 73:2559–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mathias P, Wickham T, Moore M, Nemerow G. 1994. Multiple adenovirus serotypes use αv integrins for infection. J Virol 68:6811–6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nemerow G, Stewart P. 1999. Role of αv integrins in adenovirus cell entry and gene delivery. Microbiol Mol Biol Rev 63:725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salinas S, Bilsland LG, Henaff D, Weston AE, Keriel A, Schiavo G, Kremer EJ. 2009. CAR-associated vesicular transport of an adenovirus in motor neuron axons. PLoS Pathog 5:e1000442. doi: 10.1371/journal.ppat.1000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salinas S, Zussy C, Loustalot F, Henaff D, Menendez G, Morton PE, Parsons M, Schiavo G, Kremer EJ. 2014. Disruption of the coxsackievirus and adenovirus receptor-homodimeric interaction triggers lipid microdomain- and dynamin-dependent endocytosis and lysosomal targeting. J Biol Chem 289:680–695. doi: 10.1074/jbc.M113.518365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bru T, Salinas S, Kremer E. 2010. An update on canine adenovirus type 2 and its vectors. Viruses 2:2134–2153. doi: 10.3390/v2092134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ekstrand MI, Nectow AR, Knight ZA, Latcha KN, Pomeranz LE, Friedman JM. 2014. Molecular profiling of neurons based on connectivity. Cell 157:1230–1242. doi: 10.1016/j.cell.2014.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu Q, Clark MS, Palmiter RD. 2012. Deciphering a neuronal circuit that mediates appetite. Nature 483:594–597. doi: 10.1038/nature10899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwarz LA, Miyamachi K, Gao XJ, Beier TK, Weissbourd B, DeLoach KE, Ren J, Ibanes S, Malenka RC, Kremer EJ, Luo L. 1 July 2015 Viral-genetic tracing of the input-output organization of a central noradrenaline circuit. Nature doi: 10.1038/nature14600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soudais C, Boutin S, Hong SS, Chillon M, Danos O, Bergelson JM, Boulanger P, Kremer EJ. 2000. Canine adenovirus type 2 attachment and internalization: coxsackievirus-adenovirus receptor, alternative receptors, and an RGD-independent pathway. J Virol 74:10639–10649. doi: 10.1128/JVI.74.22.10639-10649.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Farmer C, Morton PE, Snippe M, Santis G, Parsons M. 2009. Coxsackie adenovirus receptor (CAR) regulates integrin function through activation of p44/42 MAPK. Exp Cell Res 315:2637–2647. doi: 10.1016/j.yexcr.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 21.Deinhardt K, Salinas S, Verastegui C, Watson R, Worth D, Hanrahan S, Bucci C, Schiavo G. 2006. Rab5 and Rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron 52:293–305. doi: 10.1016/j.neuron.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 22.Kremer EJ, Boutin S, Chillon M, Danos O. 2000. Canine adenovirus vectors: an alternative for adenovirus-mediated gene transfer. J Virol 74:505–512. doi: 10.1128/JVI.74.1.505-512.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bradshaw AC, Parker AL, Duffy MR, Coughlan L, van Rooijen N, Kahari VM, Nicklin SA, Baker AH. 2010. Requirements for receptor engagement during infection by adenovirus complexed with blood coagulation factor X. PLoS Pathog 6:e1001142. doi: 10.1371/journal.ppat.1001142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seiradake E, Lortat-Jacob H, Billet O, Kremer EJ, Cusack S. 2006. Structural and mutational analysis of human Ad37 and canine adenovirus 2 fiber heads in complex with the D1 domain of coxsackie and adenovirus receptor. J Biol Chem 281:33704–33716. doi: 10.1074/jbc.M605316200. [DOI] [PubMed] [Google Scholar]
- 25.Tomko R, Xu R, Philipson L. 1997. HCAR and MCAR: the human and mouse cellular receptors for subgroup C adenoviruses and group B coxsakieviruses. Proc Natl Acad Sci U S A 94:3352–3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schoehn G, El Bakkouri M, Fabry CM, Billet O, Estrozi LF, Le L, Curiel DT, Kajava AV, Ruigrok RW, Kremer EJ. 2008. Three-dimensional structure of canine adenovirus serotype 2 capsid. J Virol 82:3192–3203. doi: 10.1128/JVI.02393-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nemerow G, Cheresh D, Wickham T. 1994. Adenovirus entry into host cells: a role for αv integrins. Trends Cell Biol 4:52–55. [DOI] [PubMed] [Google Scholar]
- 28.Shayakhmetov DM, Eberly AM, Li ZY, Lieber A. 2005. Deletion of penton RGD motifs affects the efficiency of both the internalization and the endosome escape of viral particles containing adenovirus serotype 5 or 35 fiber knobs. J Virol 79:1053–1061. doi: 10.1128/JVI.79.2.1053-1061.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salinas S, Schiavo G, Kremer EJ. 2010. A hitchhiker's guide to the nervous system: the complex journey of viruses and toxins. Nat Rev Microbiol 8:645–655. doi: 10.1038/nrmicro2395. [DOI] [PubMed] [Google Scholar]
- 30.Wiethoff CM, Wodrich H, Gerace L, Nemerow GR. 2005. Adenovirus protein VI mediates membrane disruption following capsid disassembly. J Virol 79:1992–2000. doi: 10.1128/JVI.79.4.1992-2000.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bremner KH, Scherer J, Yi J, Vershinin M, Gross SP, Vallee RB. 2009. Adenovirus transport via direct interaction of cytoplasmic dynein with the viral capsid hexon subunit. Cell Host Microbe 6:523–535. doi: 10.1016/j.chom.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang KC, Yasruel Z, Guerin C, Holland PC, Nalbantoglu J. 2007. Interaction of the coxsackie and adenovirus receptor (CAR) with the cytoskeleton: binding to actin. FEBS Lett 581:2702–2708. doi: 10.1016/j.febslet.2007.05.019. [DOI] [PubMed] [Google Scholar]
- 33.Fok PT, Huang KC, Holland PC, Nalbantoglu J. 2007. The coxsackie and adenovirus receptor binds microtubules and plays a role in cell migration. J Biol Chem 282:7512–7521. [DOI] [PubMed] [Google Scholar]
- 34.Ashbourne Excoffon KJ, Moninger T, Zabner J. 2003. The coxsackie B virus and adenovirus receptor resides in a distinct membrane microdomain. J Virol 77:2559–2567. doi: 10.1128/JVI.77.4.2559-2567.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burckhardt CJ, Suomalainen M, Schoenenberger P, Boucke K, Hemmi S, Greber UF. 2011. Drifting motions of the adenovirus receptor CAR and immobile integrins initiate virus uncoating and membrane lytic protein exposure. Cell Host Microbe 10:105–117. doi: 10.1016/j.chom.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 36.Walters RW, van't Hof W, Yi SM, Schroth MK, Zabner J, Crystal RG, Welsh MJ. 2001. Apical localization of the coxsackie-adenovirus receptor by glycosyl-phosphatidylinositol modification is sufficient for adenovirus-mediated gene transfer through the apical surface of human airway epithelia. J Virol 75:7703–7711. doi: 10.1128/JVI.75.16.7703-7711.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Humphries JD, Byron A, Humphries MJ. 2006. Integrin ligands at a glance. J Cell Sci 119:3901–3903. doi: 10.1242/jcs.03098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morton PE, Hicks A, Nastos T, Santis G, Parsons M. 2013. CAR regulates epithelial cell junction stability through control of E-cadherin trafficking. Sci Rep 3:2889. doi: 10.1038/srep02889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Excoffon KJ, Kolawole AO, Kusama N, Gansemer ND, Sharma P, Hruska-Hageman AM, Petroff E, Benson CJ. 2012. Coxsackievirus and adenovirus receptor (CAR) mediates trafficking of acid sensing ion channel 3 (ASIC3) via PSD-95. Biochem Biophys Res Commun 425:13–18. doi: 10.1016/j.bbrc.2012.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Excoffon KJ, Hruska-Hageman A, Klotz M, Traver GL, Zabner J. 2004. A role for the PDZ-binding domain of the coxsackie B virus and adenovirus receptor (CAR) in cell adhesion and growth. J Cell Sci 117:4401–4409. doi: 10.1242/jcs.01300. [DOI] [PubMed] [Google Scholar]
- 41.Excoffon KJ, Gansemer ND, Mobily ME, Karp PH, Parekh KR, Zabner J. 2010. Isoform-specific regulation and localization of the coxsackie and adenovirus receptor in human airway epithelia. PLoS One 5:e9909. doi: 10.1371/journal.pone.0009909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tamanini A, Nicolis E, Bonizzato A, Bezzerri V, Melotti P, Assael BM, Cabrini G. 2006. Interaction of adenovirus type 5 fiber with the coxsackievirus and adenovirus receptor activates inflammatory response in human respiratory cells. J Virol 80:11241–11254. doi: 10.1128/JVI.00721-06. [DOI] [PMC free article] [PubMed] [Google Scholar]