

Abstract
Hypoxic tumors are associated with poor clinical outcome for multiple types of human cancer. This may be due, in part, to hypoxic cancer cells being resistant to anticancer therapy, including radiation therapy, chemotherapy, and targeted therapy. Hypoxia inducible factor 1, a major regulator of cellular response to hypoxia, regulates the expression of genes that are involved in multiple aspects of cancer biology, including cell survival, proliferation, metabolism, invasion, and angiogenesis. Here, we review multiple pathways regulated by hypoxia/hypoxia inducible factor 1 in cancer cells and discuss the latest advancements in overcoming hypoxia-mediated tumor resistance.
Keywords: HIF-1, cancer, chemoresistance, signaling, apoptosis
Video abstract
Introduction
Maintaining oxygen concentration is critical for cellular function and survival. In normal tissues, oxygen concentration is maintained at 3%–7%. In contrast, in solid tumors, uncontrolled cell proliferation and the inability to form normal blood vessels results in impaired blood supply and low oxygen tension.1 Tumors with an oxygen concentration at or below 1% are considered to be hypoxic. Insufficient oxygen supply produces stressful environments for normal cells and can promote cell death in hypoxic regions by either apoptosis or necrosis. However, the effect of hypoxia on cancer cells is multifaceted, as cancer cells are able to survive hypoxic environments and hypoxia itself can activate adaptive cellular responses that contribute to tumor progression. It is well established that hypoxic cancers are associated with poor prognosis in different types of cancer.2 Moreover, hypoxic tumors are also associated with resistance to different therapies, including radiotherapy,3 chemotherapy,4 and targeted therapy,5 potentially contributing to poor patient survival.
Our understanding of how oxygen tension regulates cellular functions has been significantly advanced by the discovery of hypoxia inducible factors.6 Hypoxia inducible factor 1 (HIF-1) is a major player in cellular responses to low oxygen levels. HIF-1 is a transcription factor that functions as a heterodimer and contains two subunits: hypoxia inducible factor 1 alpha (HIF-1α) and hypoxia inducible factor 1 beta (HIF-1β). Under normal oxygen concentration, HIF-1α is rapidly degraded.7 HIF-1α is hydroxylated by prolyl hydroxylases (PHDs), which requires oxygen as a substrate. Hydroxylated HIF-1α is recognized by an E3-ubiquitin ligase Von Hippel–Lindau (VHL) and undergoes rapid proteasomal degradation.8 Under hypoxic conditions, PHDs cannot hydroxylate HIF-1α, leading to HIF-1α stabilization and translocation to the nucleus. Within the nucleus, HIF-1α interacts with its partner HIF-1β and, as heterodimers, binds to hypoxia response elements on promoters to induce expression of hundreds of different genes.9 Many HIF-1α-mediated biological effects are beneficial for tumor progression, including shifting metabolism toward glycolysis,10 induction of angiogenesis,11 regulation of apoptosis, and induction of migration12 to escape hostile hypoxic environments. Although a lot of progress has been made in understanding the effect of hypoxia on cancer cells, we are just beginning to uncover the mechanisms by which hypoxia contributes to resistance to anticancer therapy (Figure 1). In this review we discuss different mechanisms of hypoxia-mediated resistance to cancer therapy and potential approaches to overcome this resistance.
Figure 1.
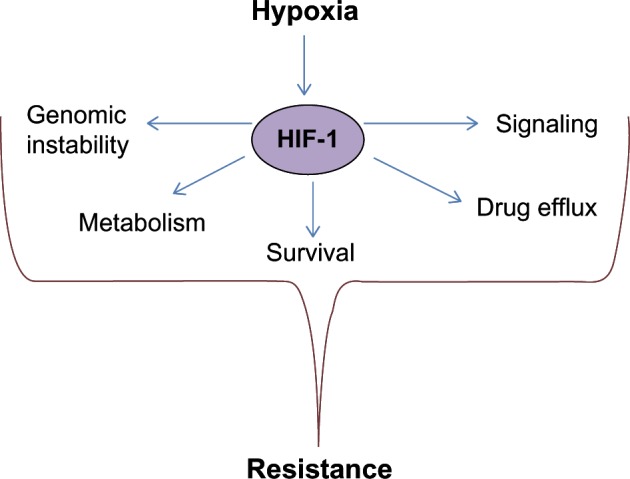
Hypoxia/Hypoxia inducible factor 1 (HIF-1)-mediated mechanisms leading to anticancer therapy resistance.
Notes: Under hypoxic conditions HIF-1 is stabilized, translocates to the nucleus, and activates expression of its target genes. Activity of many of these genes promotes resistance to anticancer therapy through regulation of metabolism, survival, drug efflux, signaling, and DNA repair.
Tumor hypoxia pathophysiology
Hypoxia can alter multiple aspects of cancer cell biology, including proliferation, survival, invasion, and metastasis, affecting both early and late stages of carcinogenesis. In breast cancer, hypoxic markers can be observed in premalignant ductal carcinoma in situ lesions as cancer cells begin to form multilayers and move away from stroma-containing blood vessels.13 At later stages of tumorigenesis, cancer cells experience hypoxia due to both limited vascularization in rapidly proliferating tumors and poorly organized blood vessels due to angiogenesis.14 Hypoxia also plays an important role in reducing reactive oxygen species (ROS) during metastatic colonization.15
Metabolic reprogramming is one of the hallmarks of hypoxia.16 HIF-1 mediates the expression of genes encoding glucose transporters (GLUT1, GLUT3) and many glycolytic enzymes such as aldolase A (ALDOA), hexokinase 1 (HK1), pyruvate kinase isozyme M2 (PKM2), and lactate dehydrogenase A (LDHA).17 HIF-1 can also suppress mitochondrial oxidative metabolism by increasing the expression of pyruvate dehydrogenase kinase (PDK) 1.18 PDK1 regulates pyruvate dehydrogenase (PDH), the enzyme that converts pyruvate to acetyl-coenzyme A for entry into the tricarboxylic acid cycle. HIF-1 also induces the expression of pentose phosphate-pathway enzymes transketolase and transketolase-like 2 that promote the production of ribose that is required for nucleic acid synthesis to support cell division.19 Hypoxia also modulates lipid metabolism via uptake and lipid droplet accumulation through the upregulation of lipin-1.20 Activation of different metabolic pathways is critical for cell survival and proliferation under low oxygen tension. Thus, the targeting of hypoxia-activated tumor metabolism is a promising strategy in anticancer therapy.
Hypoxia is a hostile environment and has been shown to either promote or inhibit cell death depending on cancer type and experimental setting. For example, energy deprivation and ROS production induce apoptosis in human neuroblastoma cells under hypoxic conditions.21 On the other hand, hypoxia was shown to inhibit apoptosis in breast cancer cell lines treated with paclitaxel through activation of the c-JUN N-terminal kinase pathway.22 Cancer cells also respond differently depending on the severity of the hypoxia. Prolonged hypoxia can lead to cell death. However, short exposure to low oxygen concentration may allow cells to adapt to hypoxia. One adaptive mechanism is the induction of autophagy. Autophagy has been demonstrated to play an important role in hypoxia-mediated cell survival and chemoresistance.23 For example, HIF-1 promotes selective mitochondrial autophagy by upregulating the expression of BNIP3 and BNIP3L. These act as potent inducers of autophagy by reducing oxidative metabolism and inhibiting ROS production.24 Although the role that hypoxia/HIF-1 plays in apoptosis may be context and cancer cell dependent, it is generally accepted that, in many cancers, hypoxia inhibits cell death, resulting in resistance to different anticancer agents.25 Reversing hypoxic-mediated adaptation to cell death signals may also be a critical in targeting hypoxic tumors.
Genomic instability is a hallmark of cancer that has been shown to be associated with hypoxia.26 Genetic stability depends on DNA repair mechanisms that occur during the gap 1 (G1) or synthesis (S) checkpoints. Tumor hypoxia has been shown to inhibit G1 and S checkpoints, resulting in the accumulation of DNA replication errors or DNA breaks.27 The altered checkpoint controls indicate that hypoxic cells could acquire increased rates of mutation.28 Human DNA double-strand breaks (DSBs) are usually repaired through two main pathways: homologous recombination and non-homologous end joining. Homologous recombination is an error-free DNA repair pathway and its inhibition has been linked to increased genetic instability and carcinogenesis.29 Interestingly, hypoxia has been shown to inhibit homologous recombination, resulting in increased genetic instability.30 In addition hypoxia has been implicated in decreasing the activity of the mismatch repair pathway. Hypoxia downregulates of two mismatch repair genes (MLH1 and MSH2) promoting mutagenesis.31 Although genomic instability and defects in DNA repair machinery are beneficial for tumorigenesis, it may provide a target for anticancer therapy. Indeed, hypoxia sensitizes cancer cell lines to DNA damaging agents such as mitomycin C and the poly(ADP-ribose) polymerase inhibitor.32
Another notable feature of hypoxia is its ability to promote epithelial-to-mesenchymal transition (EMT).12 EMT is characterized by loss of cell polarity, cell–cell adhesions in epithelial cells, and increased motility. EMT is a physiological process that is important during embryonic development and wound healing.33 Interestingly, EMT is also a feature of many cancers, in which it facilitates cell motility and thus contributes to cancer cell invasion and metastasis.34 Many studies have demonstrated that hypoxia activates different signaling pathways that promote an EMT phenotype. Transforming growth factor beta (TGFβ) is one of the signaling pathways regulated by hypoxia.35 For example, TGFβ is a HIF-1 target gene and at the later stages of tumorigenesis TGFβ expression is associated with increased tumor cell growth, proliferation, and motility.36 In addition TGFβ signaling is known to activate multiple EMT-promoting transcription factors such as TWIST and SNAIL.37 The Notch signaling pathway controls stem-cell renewal, multipotency, and EMT during tissue development and is also often activated in different types of cancer.38 Interestingly, HIF-1α interacts with the Notch intracellular domain and increases its transcriptional activity contributing to hypoxia-mediated EMT.39
Cancer cell invasion has also been linked to hypoxia. The ability of cancer cells to modify and degrade the extracellular matrix (ECM) is an essential stage of tumorigenesis.40 The ECM contains many proteins, including collagen, that provide the structural framework for most tissues. Increased or abnormal collagen deposition is one of the most well-described ECM alterations that is observed in different types of cancer, including lung and breast tumors.41,42 Recent studies have shown that hypoxia participates in collagen biogenesis. Hypoxia and HIF-1 have been implicated in abnormal collagen I deposition that results in increased cross-linking and stiffness. Specifically, hypoxia increases the expression of intracellular collagen-modifying enzymes prolyl 4-hydroxylases (P4HAs) in cancer-associated fibroblasts and endothelial cells. P4HAs are required for collagen cross-linking and the expression of P4HA1 and P4HA2 is directly regulated by HIF-1.43 Depletion of HIF-1α or P4HA led to decreased tissue stiffness, fibrosis, and invasion in a breast cancer xenograft model.44 HIFs can also regulate the expression of extracellular collagen-modifying enzymes. Collagen cross-linking is initiated by a family of lysyl oxidase (LOX) enzymes. Three members of this family, LOX, lysyl oxidase-like protein 2 (LOXL2), and LOXL4, are regulated by HIF-1 and all three have been implicated in collagen cross-linking and tumor stiffness.45 Simtuzumab, an anti-LOXL2 antibody used to treat liver fibrosis, has emerged as an anticancer therapy and is currently in clinical trials for the treatment of pancreatic cancer. Finally, HIF-1 and HIF-2 can regulate the expression of genes involved in ECM degradation, such as matrix metalloproteinases (MMPs).46 MMP2 and MMP9 expression is increased in hypoxic breast and colon cancers.47,48 Thus, the hypoxic regulation of ECM dynamics may contribute to its role in metastatic dissemination and ECM-modifying enzymes could potentially be targeted by anticancer agents.
Mechanisms of hypoxia resistance to cancer therapy
Clinically, the most significant feature of hypoxia is its ability to promote resistance to cancer therapy including radiotherapy, chemotherapy, and targeted therapy. Most of the protective effects of hypoxia require HIF-1 and HIF-2. However, hypoxia can promote resistance independently of HIFs. Ionizing radiation produces intracellular ROS that damage DNA. Since oxygen is required for ROS generation, hypoxic tumors are resistant to the cytotoxic effects of radiotherapy.49 In addition to the direct effect of decreased oxygen tension on chemo- and radiosensitivity, hypoxia also promotes resistance through the HIF-1-mediated upregulation of different genes and signaling pathways. As mentioned earlier, hypoxia/HIF regulates the expression of hundreds of genes to control cell proliferation, survival, metabolism, angiogenesis, and many other cellular functions that contribute to drug resistance. In the following sections, we discuss hypoxia/HIF-1 regulated pathways that may contribute to resistance to cancer therapy.
Hypoxia-induced drug resistance through increased drug efflux
Chemoresistance often involves the induction of proteins that increase drug efflux. Multidrug resistance protein (MDR1) and multidrug resistance-associated protein (MRP1) encode membrane glycoproteins that pump foreign molecules out of the cells. MDR1 and MRP2 promote resistance by decreasing the intracellular concentrations of chemotherapeutic drugs.50 A few multidrug resistance genes including MRP2 and MDR1 are upregulated by hypoxia. An induction of MDR1 by HIF-1α has been reported in breast, colon, and stomach tumors.51,52 Upregulation of MRP2 was reported to be involved in HIF-1α-mediated doxorubicin efflux and resistance to doxorubicin in breast cancer cell lines.53 Thus, hypoxia/HIF can contribute to chemoresistance by increasing the drug efflux ability of cancer cells.
Hypoxia-induced drug resistance through inhibition of apoptotic pathways
Damage to DNA is sensed by several DNA damage-response pathways that prevent the transmission of DNA alterations and maintain genomic stability in a cell. Normally, DNA damage is recognized by ataxia telangiectasia mutated/ataxia telangiectasia and Rad3-related protein kinases that promote cell-cycle arrest and apoptosis when too much damage has occurred. Hypoxia can attenuate DNA damage-induced cell-cycle arrest and apoptosis. For example, hypoxia inhibited the expression of pro-apoptotic proteins, such as Bax and Bid, that contribute to drug resistance in a colon xenograft model.54 In normal breast epithelial cells, hypoxia suppresses anoikis and luminal clearing during mammary morphogenesis through the downregulation of the pro-apoptotic proteins Bim and Bmf, resulting in a ductal carcinoma in situ-like phenotype.55 Expression of survivin, an anti-apoptotic protein, is also regulated by HIF-1α, and its expression has been shown to correlate with HIF-1α levels in non-small lung carcinomas in vivo.56 High levels of survivin expression also correlated with doxorubicin resistance in a panel of organotypic human breast tumors.57 Through the inhibition of pro-apoptotic and induction of anti-apoptotic genes, HIF-1 can inhibit apoptosis and promote tumor cell survival in chemotherapy-treated cancer cells.
Overcoming tumor hypoxia
Targeting metabolism in hypoxic tumors
Currently, a large effort is being made to target metabolic pathways activated in cancer cells. Since hypoxia is known to enhance glycolysis and increase the expression of many metabolic genes, it is expected that targeting metabolic pathways may kill hypoxic cells or increase sensitivity to standard chemotherapy or targeted therapy.58
The first rate-limiting step of glucose metabolism is the transport of glucose across the plasma membrane. WZB117 is a small molecule inhibitor of glucose transporter GLUT1 and treatment with WZB117 and cisplatin or paclitaxel can synergistically kill hypoxic lung cancer cells in vitro and in vivo as demonstrated with a xenograft model.59 Another GLUT1 inhibitor, phloretin, significantly enhances daunorubicin effects by increasing daunorubicin-induced apoptosis under hypoxia and overcoming hypoxia-conferred drug resistance in colon, non-small cell lung carcinoma, and renal carcinoma cancer cell lines.60
LDHA catalyzes the final step in the glycolytic pathway, the conversion of pyruvate and reduced nicotinamide adenine dinucleotide to lactate and oxidized nicotinamide adenine dinucleotide, and has a critical role in tumor maintenance.61 Knockdown of LDHA in tumor cells produced increased mitochondrial respiration, decreased cellular ability to proliferate, and reduced survival under hypoxic conditions in breast cancer cell lines and in an in vivo breast cancer model.62 Inhibition of LDHA by small molecule FX11 reduces adenosine triphosphate (ATP) levels, induces oxidative stress and inhibits tumor progression in human lymphoma xenografts.63 Importantly, combining FX11 with paclitaxel promotes tumor regression in paclitaxel-resistant triple negative breast cancer xenografts.64 These results indicate that LDHA and lactate metabolism have an important role in resistance to paclitaxel. Since LDHA is a well-known HIF target, the combination of FX11 and paclitaxel may prove efficient against hypoxic tumors.
PDH is responsible for the conversion of pyruvate to acetyl-coenzyme A, which enters the tricarboxylic acid cycle to produce ATP. PDK phosphorylates PDH and inhibits its enzymatic activity. Hypoxia induces PDK expression in an HIF-1α-dependent manner, resulting in a switch from mitochondrial respiration to glycolysis.18 Hypoxia-mediated PDK3 upregulation significantly inhibited cell apoptosis and increased resistance to cisplatin or paclitaxel in a cervical cancer cell line.65 Knockdown of PDK3 inhibits hypoxia-induced glycolysis and increases the sensitivity of colon cancer cell lines to chemotherapeutic agents such as cisplatin, paclitaxel, or oxaliplatin.66 Dichloroacetate, a potent PDK inhibitor, has shown effectiveness in a variety of preclinical models via induction of apoptosis.67 Early stages of clinical trials demonstrated dichloroacetate safety in adults with recurrent malignant brain tumors (NCT00540176) and it is currently being studied in clinical trial for its efficacy against hypoxic head and neck tumors (NCT01386632).
The fatty acid biosynthesis pathway is important for the survival of hypoxic tumors. Fatty acid synthase (FAS), a key enzyme in lipid biogenesis, has been found to be upregulated in breast cancer cells under hypoxia.68 Several FAS inhibitors have shown antitumor activity, including cerulenin, C75, orlistat, C93, and GSK837149A.69 Lipid synthesis and lipid droplet accumulation is essential for cancer cell survival under hypoxic conditions.70 Thus, targeting FAS may be a viable option for hypoxic tumor treatment although this has not been directly tested.20
PKM2 is the last rate-limiting enzyme in the glycolytic pathway. PKM2 is expressed predominantly in tumor cells and is important for cancer metabolism and tumor growth.71 PKM2 is a direct HIF-1 target and hypoxic tumors show increased levels of PKM2.72 Thus, the chemical inhibition of PKM2 could be used to sensitize hypoxic tumors to chemotherapy.
Drugs targeting HIF-1
Since the discovery of HIF-1 as a major player in cellular responses to hypoxia, there have been multiple attempts to develop specific HIF-1 inhibitors.73 Despite these efforts, a specific and selective inhibitor of HIF-1 has not yet been developed. Many agents have been shown to affect HIF-1 activity through different mechanisms and some compounds are currently in clinical trials. Chemical inhibitors of HIF-1α could be divided into two major groups: agents that modulate HIF-1α transcriptional activity and agents that modulate HIF-1α expression. HIF-1 requires the recruitment of a transcription co-activator p300 in order to initiate transcription of its target genes. A few agents currently approved by the US Food and Drug Administration have demonstrated disruption of the HIF-1/p300 interaction complex. Chetomin disrupts the structure of the CH1 domain of p300, which is required for its interaction with HIF-1α.74 Chetomin exhibited in vivo antitumor activity in a preclinical model of prostate cancer.74 Bortezomib, a proteasome inhibitor, has been shown to paradoxically increase the levels of HIF-1α protein by blocking its degradation while simultaneously blocking HIF-1α interaction with p300.75 Bortezomib has been approved against hematological malignancies; at the same, it has failed in multiple clinical trials (NCT00512798, NCT00061932) against solid tumors as a single agent and in combination with different chemotherapeutic compounds. However, bortezomib’s effect might be more pronounced under low oxygen concentration and the effect of bortezomib specifically on hypoxic non-small cell lung tumors is currently being studied in a clinical trial.76 HIF-1 binding to hypoxia response elements is required for its transcriptional activity. Echinomycin, a cyclic peptide with antimicrobial properties, has been shown to bind to the core of the HIF-1 recognition sequence 5′-CGTG-3′ and inhibit HIF-1 transcriptional activity, promoting apoptosis in glioma and breast cancer cell lines.77 However, despite its efficacy in vivo and in vitro, echinomycin failed clinical trials due to severe side effects.78
Another group of agents that target the phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt)/mammalian target of rapamycin (mTOR) signaling pathway demonstrated effects on HIF-1 expression. The PI3K/Akt/mTOR signaling pathway is known to upregulate HIF-1 levels primarily by increasing the rate of HIF-1α protein translation.79 Thus, many cancer cells can stabilize HIF-1α under normal oxygen tension and require HIF-1 for oncogenesis.80 Therapeutic inhibition of PI3K by the dual PI3K/mTOR inhibitor BEZ235 decreases HIF-1α protein synthesis in head and neck squamous cell carcinoma and glioblastoma cell lines.81 EZN-2968, an oligonucleotide, specifically binds to HIF-1 mRNA and promotes its degradation in vitro and in vivo.82 EZN-2968 has been shown to decrease HIF-1 levels in both animal models and patients with refractory advanced solid tumors including breast and colon carcinomas.83 Inhibition of type I topoisomerase activity by topotecan was also shown to downregulate HIF-1 expression in glioma cells in vitro and in vivo.84 More importantly, topotecan blocked HIF-1 synthesis in patients with advanced solid tumors of different origin.85 Currently, topotecan chemotherapy is used in the clinic for treatment of small cell lung cancer and ovarian cancer. An inhibitor of tubulin polymerization, 2-methoxyestradiol (2ME2), reduces HIF-1α translation and its nuclear translocation.86 It is a well-tolerated compound and has been shown to be efficient in a Phase II clinical trial as a single agent for platinum-resistant epithelial ovarian cancer.87 The compound is currently being tested in clinical trials in patients with multiple myeloma, breast, prostate and ovarian cancer (NCT00028821).
A few agents have been show to affect HIF-1 protein stability and degradation. The molecular chaperone Hsp90 is required for HIF-1 protein stability.88 Geldanamycin is a small molecular Hsp90 ATP binding site inhibitor that promotes the ubiquitination and proteasomal degradation of HIF-1α in a VHL-independent manner in renal cell carcinoma and prostate cancer cell lines.89 An analog of geldanamycin, 17-AAG, has been shown to be efficient in Phase II clinical trials in combination with sorafenib, a Raf-kinase inhibitor, against VHL mutant renal tumors (NCT00088374),90 and in combination with trastuzumab, an anti-ERBB2 antibody, against metastatic ERBB2-positive breast cancers (NCT00773344).91 Multiple Phase I studies are currently testing the efficiency of 17-AAG in combination with other chemotherapeutic agents in solid cancers of different origin (NCT00121264).
Developing drugs activated by hypoxia
Hypoxia-activated prodrugs are chemotherapeutic agents that undergo activation under reductive metabolism by cellular oxidoreductases. This process is usually inhibited by molecular oxygen, making these drugs specific for hypoxic tumors. Five classes of bioreductive agents have been described: nitro(hetero)cyclic compounds, aromatic N-oxides, aliphatic N-oxides, quinones, and metal complexes.92 Several hypoxia-activated prodrugs are at various stages of clinical trial.
The N-oxide tirapazamine (TPZ) is the most studied hypoxia-activated prodrug. TPZ toxicity is 300-fold higher under hypoxic conditions than under normoxia in vitro. Preclinical in vivo studies, in which TPZ was combined with radiotherapy or cisplatin, showed great promise.93 However, poor tissue penetration and toxicity unrelated to its activation by hypoxia has limited the usage of TPZ in clinic.94 PR-104 and TH-302 (evofosfamide) are nitroaromatic compounds that exhibit cytotoxicity only under hypoxic conditions.95,96 Both drugs promote apoptosis through DNA interstrand cross-links.97 PR-104 has shown efficacy in Phase II clinical trials against non-small cell lung carcinoma and is currently moving to Phase III (NCT00544674). AQ4N (banoxantrone), an aliphatic N-oxide, is metabolized under low oxygen concentration into to a high-affinity DNA intercalator that inhibited topoisomerase II activity in vitro and in a bladder cell carcinoma xenograft model.98 AQ4N is currently undergoing tests in clinical trials for glioma treatment (NCT00394628). Another bioreductive prodrug, E09 (apaziquone), belonging to the quinine family, has shown effectiveness in a variety of cancers, including breast, bladder, and head and neck cancers.99 EO9 administration to patients with low-grade non-muscle invasive bladder tumors resulted in a statistically significant difference in the rate of tumor recurrence in a Phase III clinical trial (NCT01469221). These results are especially encouraging since no drugs have been approved in the USA for more than 20 years for low-grade non-muscle invasive bladder cancers.
Targeting pathways activated by hypoxia
Receptor tyrosine kinase (RTK) signaling plays an important role in tumorigenesis.100 Activation of RTKs induces downstream signaling that is beneficial for tumor progression and often associated with anticancer therapy resistance.101 The expression of multiple RTKs and their ligands is increased under hypoxic conditions.102 Moreover, many of these signaling pathways can increase HIF-1 and HIF-2 expression and stabilization, establishing a positive feedback loop and enhancing the protective effect of hypoxia.103 Thus, targeting these pathways is a potent way to abrogate hypoxia-mediated resistance.
c-MET kinase is an oncogenic RTK that plays an important role in the regulation of embryogenesis by promoting invasion and survival.104 Interestingly, c-MET is upregulated in many cancers.105 Elevated expression of c-MET results in increased motility, invasion, and survival.106 Moreover, its expression is elevated under hypoxic conditions both in vitro and in vivo.107 A selective small molecule MET inhibitor, PHA665752, demonstrated radiosensitization in gastric cancer cells.108 In addition, this compound was able to abrogate hypoxia-induced invasiveness in non-small cell lung cancer cell lines.109
Another member of the MET kinase family, AXL kinase, was reported to participate in hypoxia-mediated resistance in prostate cancer.110 AXL expression and activation by its ligand, growth arrest-specific 6 (GAS6), is increased under hypoxic conditions in renal cell cancer cell lines.111 Moreover, overexpression of AXL kinase interacts with and activate c-SRC and MET kinases, resulting in increased invasion in renal cancer cell xenografts.111 Additionally, AXL inhibition decreased tumorigenesis in breast cancer cells in vitro and in vivo.112 BGB324, an AXL inhibitor, has shown low toxicity in Phase I clinical trials and is being currently studied in Phase II clinical trials against non-small cell lung carcinoma (NCT02424617).
Macrophage-stimulating protein receptor, known as RON kinase, can dimerize with MET kinases, and several human tumor tissues show increased RON expression.113 Interestingly, in a panel of human breast cancer cell lines, RON was reported to be transcriptionally activated through HIF-1.114 RON kinase was also shown to activate c-JUN signaling, resulting in cell proliferation, survival, and invasion both in vitro and in vivo.115 These results suggest that targeting RON may provide a potential therapeutic strategy to block tumorigenesis under hypoxic conditions.
ERBB is another family of RTKs that important for tumorigenesis and chemoresistance. The ERBB family consists of epidermal growth factor receptor (EGFR), ERBB2, ERBB3, and ERBB4.116 Increased activation of ERBB receptors, due to gene amplification, overexpression, or mutations, is tightly associated with poor prognosis and resistance in many types of cancer.117 Both HIF-1 and HIF-2 have been shown to increase EGFR expression in renal cell carcinoma and breast cancers.118 Thus, targeting EGFR in combination with anticancer agents targeting hypoxia may be beneficial to abrogate drug resistance in hypoxic tumors.
ERBB2 is another member of the ERBB family often overexpressed in breast cancers.119 Breast cancer patients with ERBB2 amplifications are currently treated with ERBB2 and EGFR inhibitors; however in many cases patients develop resistance.120 Hypoxia can maintain activation of signaling pathways downstream from ERBB2, including extracellular signal-regulated kinase (ERK), in the presence of lapatinib, a dual ERBB2–EGFR inhibitor. Hypoxia and HIF-1α were shown to circumvent RTK inhibition by inhibiting expression of the mitogen-activated protein (MAP) kinase phosphatase DUSP2 and maintaining ERK activation in ERBB2-positive breast cancers. Activation of ERK signaling is required for lapatinib resistance, as treatment with the MAPK/ERK kinase (MEK) inhibitor trametinib reverses hypoxia-mediated lapatinib resistance.121 Therefore, treatment of hypoxic ERBB2-expressing breast tumors with a combination of lapatinib and MEK inhibitors may serve as a viable therapeutic strategy.
The PI3K/Akt pathway is activated in multiple cancers of different origin.122 This pathway regulates multiple mechanisms of survival under hypoxic conditions including metabolism, proliferation, and inhibition of apoptosis.122 As mentioned previously, PI3K/Akt has been implicated in the regulation of HIF-1α expression. Regulation is not unidirectional, as hypoxia/HIF-1 induces PI3K/Akt activation through upregulation of RTKs in many different cancers.123 While BEZ235, a mTOR/PI3K inhibitor, is cytotoxic against normoxic cells, it has a greater effect against hypoxic head and neck squamous carcinoma cells.81 Whether the effect of BEZ235 in decreasing survival under hypoxia is actually due to HIF-1α inhibition is not clear. An alternate possibility is that the effect of BEZ235 on survival under hypoxia is due to its ability to inhibit Akt-mediated anti-apoptotic effects. Thus, targeting PI3K/Akt may be very promising since it inhibits multiple aspects of hypoxia-mediated resistance including metabolism, HIF-1 expression, metabolism and survival.
Hypoxia is also associated with the elevated expression of several RTK ligands. Vascular endothelial growth factor (VEGF)-A is upregulated by hypoxia in many cancer cells, both in vitro and in vivo.124 Under low oxygen concentration, tumor cells secrete enhanced levels of VEGF family members to stimulate proliferation and migration of endothelial cells, thereby promoting tumor-associated angiogenesis. Discovery of VEGF-A as the primary tumor angiogenesis factor prompted the development of a number of drugs that target VEGF or its receptor. Bevacizumab and ranibizumab are two antibodies, targeting VEGF-A, used in antiangiogenic therapy.125 These drugs were successful in inhibiting tumor angiogenesis and decreasing tumor progression in animal models. Unfortunately, results in clinic were less impressive. Although some of the patients initially responded well and had regression of primary tumors, over time tumors became resistant and in many cases the tumors become more aggressive and contain increased metastasis.126 One possibility is that antiangiogenic therapy promotes hypoxia that activates survival, invasion and proliferation by the mechanisms described above. New strategies may be needed to combine anti-VEGF therapy with anti-HIF-1 or anti-hypoxic therapies. A summary of drugs targeting multiple aspects of hypoxia and HIF-1 pathways is presented in Table 1.
Table 1.
Examples of drugs targeting multiple pathways associated with hypoxia and hypoxia inducible factor 1 (HIF-1)
| Targeted mechanism | Target | Agent | Study/studies |
|---|---|---|---|
| Metabolism | Glut1 | WZB117 | Liu et al59 |
| Glut1 | Phloretin | Cao et al60 | |
| LDH1 | FX11 | Le et al,63 Zhou et al64 | |
| PDK3 | Dichloroacetate | Michelakis et al67 | |
| FASN | GSK837149A | Flavin et al,69 Baenke et al70 | |
| Signaling | MET kinase | PHA665752 | Funakoshi et al,108 Puri et al109 |
| AXL kinase | BGB324 | Holland et al112 | |
| MEK | Trametinib | Karakashev et al121 | |
| PI3K/mTOR | BEZ235 | Karar et al81 | |
| HIF-1 activity | DNA binding | Echinomycin | Kong et al77 |
| Stability | 17-AAG, geldanamycin | Mabjeesh et al,89 Vaishampayan et al,90 Mode et al91 | |
| Synthesis | BEZ235, 2ME2, topotecan | Karar et al,81 Rapisarda et al,84 Kummar et al,85 Mabjeesh et al86 | |
| mRNA | EZN-2968 | Greenberger et al,82 Jeong et al83 | |
| p300 interaction | Chetomin, bortezomib | Kung et al,74 Shin et al,75 Falchook et al76 | |
| Hypoxia-activated drugs | DNA | Tirapazamine | Marcu and Olver,93 Rischin et al94 |
| DNA | TH-302 | Guise et al95 | |
| DNA | PR-104 | Sun et al,96 McKeage et al97 | |
| DNA | AQ4NE09 | Williams et al98 | |
| Angiogenesis | VEGF-A | Bevacizumab, ranibizumab | Ellis and Hicklin125 |
Abbreviations: 2ME2, 2-methoxyestradiol; mTOR, mammalian target of rapamycin; PI3K, phosphoinositide 3-kinase; VEGF-A, vascular endothelial growth factor-A.
Future directions and conclusion
Immunotherapy is one of the most promising anticancer treatments. Very recent studies have demonstrated the importance of hypoxia and HIF-1 in antitumor immune response regulation.127 Hypoxia has been shown to promote the accumulation of immunosuppressive cells that inhibit T-cell activation and T-cell cytotoxicity.128 HIF-1 stabilization in T-cells under hypoxia has been also shown to inhibit T-cell function and HIF-1 depletion significantly improves the anticancer immune response.129 Hypoxic tumors can be also targeted by engineered T-cells recognizing hypoxia-associated proteins such as carbonic anhydrase 9.130 This approach has been shown to be effective in VHL-mutated renal carcinoma in vitro and in vivo and can also be used against solid hypoxic malignancies. Thus targeting hypoxia/HIF-1 in immune cells or targeting T-cells against hypoxic cancer cells may provide novel ways to treat therapy-resistant hypoxic tumors.
Cancer is the leading cause of death worldwide, especially in developed countries. Despite recent advances in anticancer radio-, chemo-, and targeted therapy, the mortality rate remains very high. Unfortunately, in many cases, patients develop resistance to therapy. Although the mechanisms of resistance to anticancer therapy vary from cancer to cancer, some are common among different types of solid tumors. Hypoxia and stabilization of HIF-1 have been shown to promote resistance in different types of cancer and are associated with poor prognosis. However, a better understanding of mechanisms of resistance by hypoxia/HIF-1 is needed to develop more specific therapies to treat hypoxia-mediated drug resistance. Nevertheless, blocking any step of the HIF-1 pathway remains an important therapeutic area for targeting solid cancers (Figure 2).
Figure 2.

Potential strategies to target hypoxic cancer cells in solid tumors.
Notes: Multiple pathways may be targeted to reverse hypoxia-mediated anticancer therapy resistance. (1) Hypoxia inducible factor 1 (HIF-1) plays a critical role in cellular adaptation to hypoxic environment. Several agents can impair HIF-1 function by inhibiting its expression, translation, or transcriptional activity, or by promoting HIF-1 degradation. (2) Hypoxia-mediated angiogenesis plays an important role in tumorigenesis and could be targeted by anti-vascular endothelial growth factor (VEGF) therapy. (3) Multiple metabolic pathways are activated under hypoxia and the inhibition of these pathways targets hypoxic cells. (4) Activation of cancer signaling under hypoxia promotes proliferation, survival, and invasion. Targeting hypoxia/HIF-1-mediated signaling pathways has been shown to be efficient at decreasing tumorigenesis in vitro and in vivo. (5) Hypoxia and HIF-1 can remodel the extracellular matrix (ECM) in cancer cells that can also contribute to altered signaling by adhesion receptors such as integrins. The targeting ECM-regulating enzymes in hypoxic tumors may be used to reverse hypoxia-mediated resistance to cancer therapy.
Abbreviations: Akt, protein kinase B; DCA, dichloroacetate; EGFR, epidermal growth factor receptor; FAS, fatty acid synthase; HIF-1α/β, hypoxia inducible factor 1 alpha/beta; HRE, hypoxia response element; LDHA, lactate dehydrogenase A; LOXL2, lysyl oxidase-like protein 2; PDK3, pyruvate dehydrogenase kinase 3; PI3K, phosphoinositide 3-kinase; RTK, receptor tyrosine kinase; VEGF-A, vascular endothelial growth factor-A.
As we move into the era of personalized medicine, each tumor will have to be monitored for markers of hypoxia and HIF-1. Patients that have tumors with hypoxic markers will be treated with a combination of targeted therapy but also with hypoxia-specific therapy. Clinically, some HIF-1-targeting agents, in combination with radio-, chemo-, and targeted therapy, have the potential to reduce tumorigenesis and in some cases extend patient survival. However, the efficiency of hypoxia/HIF-1 targeting therapy remains low. Continued research focusing of understanding how hypoxia/HIF pathways regulate oncogenesis and identifying more agents that target molecules in the HIF pathway will be of the utmost priority in the future.
Acknowledgments
We thank Valerie L Sodi for her helpful comments on this review. MJR is supported by grants from the National Institutes of Health (RO1CA155413).
Footnotes
Disclosure
The authors declare no conflicts of interest in this work.
References
- 1.Brahimi-Horn MC, Chiche J, Pouysségur J. Hypoxia and cancer. J Mol Med (Berl) 2007;85(12):1301–1307. doi: 10.1007/s00109-007-0281-3. [DOI] [PubMed] [Google Scholar]
- 2.Vaupel P, Mayer A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev. 2007;26(2):225–239. doi: 10.1007/s10555-007-9055-1. [DOI] [PubMed] [Google Scholar]
- 3.Moeller BJ, Richardson RA, Dewhirst MW. Hypoxia and radiotherapy: opportunities for improved outcomes in cancer treatment. Cancer Metastasis Rev. 2007;26(2):241–248. doi: 10.1007/s10555-007-9056-0. [DOI] [PubMed] [Google Scholar]
- 4.Shannon AM, Bouchier-Hayes DJ, Condron CM, Toomey D. Tumour hypoxia, chemotherapeutic resistance and hypoxia-related therapies. Cancer Treat Rev. 2003;29(4):297–307. doi: 10.1016/s0305-7372(03)00003-3. [DOI] [PubMed] [Google Scholar]
- 5.Ellis LM, Hicklin DJ. Resistance to Targeted Therapies: Refining Anticancer Therapy in the Era of Molecular Oncology. Clin Cancer Res. 2009;15(24):7471–7478. doi: 10.1158/1078-0432.CCR-09-1070. [DOI] [PubMed] [Google Scholar]
- 6.Wang GL, Semenza GL. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc Natl Acad Sci U S A. 1993;90(9):4304–4308. doi: 10.1073/pnas.90.9.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Semenza GL. HIF-1: mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol. 2000;88(4):1474–1480. doi: 10.1152/jappl.2000.88.4.1474. [DOI] [PubMed] [Google Scholar]
- 8.Tyers M, Rottapel R. VHL: a very hip ligase. Proc Natl Acad Sci U S A. 1999;96(22):12230–12232. doi: 10.1073/pnas.96.22.12230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene. 2010;29(5):625–634. doi: 10.1038/onc.2009.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Masson N, Ratcliffe PJ. Hypoxia signaling pathways in cancer metabolism: the importance of co-selecting interconnected physiological pathways. Cancer Metab. 2014;2(1):3. doi: 10.1186/2049-3002-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liao D, Johnson RS. Hypoxia: a key regulator of angiogenesis in cancer. Cancer Metastasis Rev. 2007;26(2):281–290. doi: 10.1007/s10555-007-9066-y. [DOI] [PubMed] [Google Scholar]
- 12.Jiang J, Tang YL, Liang XH. EMT: a new vision of hypoxia promoting cancer progression. Cancer Biol Ther. 2011;11(8):714–723. doi: 10.4161/cbt.11.8.15274. [DOI] [PubMed] [Google Scholar]
- 13.Wykoff CC, Beasley N, Watson PH, et al. Expression of the hypoxia-inducible and tumor-associated carbonic anhydrases in ductal carcinoma in situ of the breast. Am J Pathol. 2001;158(3):1011–1019. doi: 10.1016/S0002-9440(10)64048-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chung AS, Lee J, Ferrara N. Targeting the tumour vasculature: insights from physiological angiogenesis. Nat Rev Cancer. 2010;10(7):505–514. doi: 10.1038/nrc2868. [DOI] [PubMed] [Google Scholar]
- 15.Zhao T, Zhu Y, Morinibu A, et al. HIF-1-mediated metabolic reprogramming reduces ROS levels and facilitates the metastatic colonization of cancers in lungs. Sci Rep. 2014;4:3793. doi: 10.1038/srep03793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Semenza GL. Regulation of cancer cell metabolism by hypoxia-inducible factor 1. Semin Cancer Biol. 2009;19(1):12–16. doi: 10.1016/j.semcancer.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 17.Semenza GL. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev. 2010;20(1):51–56. doi: 10.1016/j.gde.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim J, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3(3):177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 19.Gao P, Zhang H, Dinavahi R, et al. HIF-dependent anti-tumorigenic effect of anti-oxidants in vivo. Cancer Cell. 2007;12(3):230–238. doi: 10.1016/j.ccr.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Santos CR, Schulze A. Lipid metabolism in cancer. FEBS J. 2012;279(15):2610–2623. doi: 10.1111/j.1742-4658.2012.08644.x. [DOI] [PubMed] [Google Scholar]
- 21.Greijer AE, van der Wall E. The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J Clin Pathol. 2004;57(10):1009–1014. doi: 10.1136/jcp.2003.015032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Notte A, Ninane N, Arnould T, Michiels C. Hypoxia counteracts taxol-induced apoptosis in MDA-MB-231 breast cancer cells: role of autophagy and JNK activation. Cell Death Dis. 2013;4(5):e638. doi: 10.1038/cddis.2013.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mazure NM, Pouysségur J. Hypoxia-induced autophagy: cell death or cell survival? Curr Opin Cell Biol. 2010;22(2):177–180. doi: 10.1016/j.ceb.2009.11.015. [DOI] [PubMed] [Google Scholar]
- 24.Bellot G, Garcia-Medina R, Gounon P, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29(10):2570–2581. doi: 10.1128/MCB.00166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sullivan R, Paré GC, Frederiksen LJ, Semenza GL, Graham CH. Hypoxia-induced resistance to anticancer drugs is associated with decreased senescence and requires hypoxia-inducible factor-1 activity. Mol Cancer Ther. 2008;7(7):1961–1973. doi: 10.1158/1535-7163.MCT-08-0198. [DOI] [PubMed] [Google Scholar]
- 26.Luoto KR, Kumareswaran R, Bristow RG. Tumor hypoxia as a driving force in genetic instability. Genome Integr. 2013;4(1):5. doi: 10.1186/2041-9414-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gardner LB, Li Q, Park MS, Flanagan WM, Semenza GL, Dang CV. Hypoxia inhibits G1/S transition through regulation of p27 expression. J Biol Chem. 2001;276(11):7919–7926. doi: 10.1074/jbc.M010189200. [DOI] [PubMed] [Google Scholar]
- 28.Kondo A, Safaei R, Mishima M, Niedner H, Lin X, Howell SB. Hypoxia-induced enrichment and mutagenesis of cells that have lost DNA mismatch repair. Cancer Res. 2001;61(20):7603–7607. [PubMed] [Google Scholar]
- 29.Shammas MA, Shmookler Reis RJ, Koley H, Batchu RB, Li C, Munshi NC. Dysfunctional homologous recombination mediates genomic instability and progression in myeloma. Blood. 2009;113(10):2290–2297. doi: 10.1182/blood-2007-05-089193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bindra RS, Schaffer PJ, Meng A, et al. Down-regulation of Rad51 and decreased homologous recombination in hypoxic cancer cells. Mol Cell Biol. 2004;24(19):8504–8518. doi: 10.1128/MCB.24.19.8504-8518.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakamura H, Tanimoto K, Hiyama K, et al. Human mismatch repair gene, MLH1, is transcriptionally repressed by the hypoxia-inducible transcription factors, DEC1 and DEC2. Oncogene. 2008;27(30):4200–4209. doi: 10.1038/onc.2008.58. [DOI] [PubMed] [Google Scholar]
- 32.Chan N, Koritzinsky M, Zhao H, et al. Chronic hypoxia decreases synthesis of homologous recombination proteins to offset chemoresistance and radioresistance. Cancer Res. 2008;68(2):605–614. doi: 10.1158/0008-5472.CAN-07-5472. [DOI] [PubMed] [Google Scholar]
- 33.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 34.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu X, Kang Y. Hypoxia and hypoxia-inducible factors: master regulators of metastasis. Clin Cancer Res. 2010;16(24):5928–5935. doi: 10.1158/1078-0432.CCR-10-1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heldin CH, Vanlandewijck M, Moustakas A. Regulation of EMT by TGFβ in cancer. FEBS Lett. 2012;586(14):1959–1970. doi: 10.1016/j.febslet.2012.02.037. [DOI] [PubMed] [Google Scholar]
- 37.Zavadil J, Böttinger EP. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene. 2005;24(37):5764–5774. doi: 10.1038/sj.onc.1208927. [DOI] [PubMed] [Google Scholar]
- 38.Wang Z, Li Y, Kong D, Sarkar FH. The role of Notch signaling pathway in epithelial-mesenchymal transition (EMT) during development and tumor aggressiveness. Curr Drug Targets. 2010;11(6):745–751. doi: 10.2174/138945010791170860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pear WS, Simon MC. Lasting longer without oxygen: The influence of hypoxia on Notch signaling. Cancer Cell. 2005;8(6):435–437. doi: 10.1016/j.ccr.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 40.Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol. 2012;196(4):395–406. doi: 10.1083/jcb.201102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kauppila S, Stenbäck F, Risteli J, Jukkola A, Risteli L. Aberrant type I and type III collagen gene expression in human breast cancer in vivo. J Pathol. 1998;186(3):262–268. doi: 10.1002/(SICI)1096-9896(1998110)186:3<262::AID-PATH191>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 42.Liu SS, Wang HY, Tang JM, Zhou XM. Hypoxia-induced collagen synthesis of human lung fibroblasts by activating the angiotensin system. Int J Mol Sci. 2013;14(12):24029–24045. doi: 10.3390/ijms141224029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gilkes DM, Bajpai S, Chaturvedi P, Wirtz D, Semenza GL. Hypoxia-inducible factor 1 (HIF-1) promotes extracellular matrix remodeling under hypoxic conditions by inducing P4HA1, P4HA2, and PLOD2 expression in fibroblasts. J Biol Chem. 2013;288(15):10819–10829. doi: 10.1074/jbc.M112.442939. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 44.Gilkes DM, Chaturvedi P, Bajpai S, et al. Collagen prolyl hydroxylases are essential for breast cancer metastasis. Cancer Res. 2013;73(11):3285–3296. doi: 10.1158/0008-5472.CAN-12-3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Erler JT, Giaccia AJ. Lysyl oxidase mediates hypoxic control of metastasis. Cancer Res. 2006;66(21):10238–10241. doi: 10.1158/0008-5472.CAN-06-3197. [DOI] [PubMed] [Google Scholar]
- 46.Muñoz-Nájar UM, Neurath KM, Vumbaca F, Claffey KP. Hypoxia stimulates breast carcinoma cell invasion through MT1-MMP and MMP-2 activation. Oncogene. 2006;25(16):2379–2392. doi: 10.1038/sj.onc.1209273. [DOI] [PubMed] [Google Scholar]
- 47.Choi JY, Jang YS, Min SY, Song JY. Overexpression of MMP-9 and HIF-1α in Breast Cancer Cells under Hypoxic Conditions. J Breast Cancer. 2011;14(2):88–95. doi: 10.4048/jbc.2011.14.2.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Krishnamachary B, Berg-Dixon S, Kelly B, et al. Regulation of colon carcinoma cell invasion by hypoxia-inducible factor 1. Cancer Res. 2003;63(5):1138–1143. [PubMed] [Google Scholar]
- 49.Ward JF. DNA damage produced by ionizing radiation in mammalian cells: identities, mechanisms of formation, and reparability. Prog Nucleic Acid Res Mol Biol. 1988;19(2):95–108. doi: 10.1016/s0079-6603(08)60611-x. [DOI] [PubMed] [Google Scholar]
- 50.Kruh GD, Belinsky MG. The MRP family of drug efflux pumps. Oncogene. 2003;22(47):7537–7552. doi: 10.1038/sj.onc.1206953. [DOI] [PubMed] [Google Scholar]
- 51.Ding Z, Yang L, Xie X, et al. Expression and significance of hypoxia-inducible factor-1 alpha and MDR1/P-glycoprotein in human colon carcinoma tissue and cells. J Cancer Res Clin Oncol. 2010;136(11):1697–1707. doi: 10.1007/s00432-010-0828-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu L, Ning X, Sun L, et al. Hypoxia-inducible factor-1 alpha contributes to hypoxia-induced chemoresistance in gastric cancer. Cancer Sci. 2008;99(1):121–128. doi: 10.1111/j.1349-7006.2007.00643.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krishnamurthy P, Schuetz JD. The ABC transporter Abcg2/Bcrp: role in hypoxia mediated survival. Biometals. 2005;18(4):349–358. doi: 10.1007/s10534-005-3709-7. [DOI] [PubMed] [Google Scholar]
- 54.Erler JT, Cawthorne CJ, Williams KJ, et al. Hypoxia-mediated down-regulation of Bid and Bax in tumors occurs via hypoxia-inducible factor 1-dependent and -independent mechanisms and contributes to drug resistance. Mol Cell Biol. 2004;24(7):2875–2889. doi: 10.1128/MCB.24.7.2875-2889.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Whelan KA, Caldwell SA, Shahriari KS, et al. Hypoxia suppression of Bim and Bmf blocks anoikis and luminal clearing during mammary morphogenesis. Mol Biol Cell. 2010;21(22):3829–3837. doi: 10.1091/mbc.E10-04-0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen YQ, Zhao CL, Li W. Effect of hypoxia-inducible factor-1alpha on transcription of survivin in non-small cell lung cancer. J Exp Clin Cancer Res. 2009;28:29. doi: 10.1186/1756-9966-28-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Faversani A, Vaira V, Moro GP, et al. Survivin family proteins as novel molecular determinants of doxorubicin resistance in organotypic human breast tumors. Breast Cancer Res. 2014;16(3):R55. doi: 10.1186/bcr3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bristow RG, Hill RP. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer. 2008;8(3):180–192. doi: 10.1038/nrc2344. [DOI] [PubMed] [Google Scholar]
- 59.Liu Y, Cao Y, Zhang W, et al. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol Cancer Ther. 2012;11(8):1672–1682. doi: 10.1158/1535-7163.MCT-12-0131. [DOI] [PubMed] [Google Scholar]
- 60.Cao X, Fang L, Gibbs S, et al. Glucose uptake inhibitor sensitizes cancer cells to daunorubicin and overcomes drug resistance in hypoxia. Cancer Chemother Pharmacol. 2007;59(4):495–505. doi: 10.1007/s00280-006-0291-9. [DOI] [PubMed] [Google Scholar]
- 61.Doherty J, Cleveland J. Targeting lactate metabolism for cancer therapeutics. J Clin Invest. 2013;123(9):3685–3692. doi: 10.1172/JCI69741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9(6):425–434. doi: 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 63.Le A, Cooper CR, Gouw AM, et al. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A. 2010;107(5):2037–2042. doi: 10.1073/pnas.0914433107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhou M, Zhao Y, Ding Y, et al. Warburg effect in chemosensitivity: targeting lactate dehydrogenase-A re-sensitizes taxol-resistant cancer cells to taxol. Mol Cancer. 2010;9:33. doi: 10.1186/1476-4598-9-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lu CW, Lin SC, Chen KF, Lai YY, Tsai SJ. Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J Biol Chem. 2008;283(42):28106–28114. doi: 10.1074/jbc.M803508200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lu CW, Lin SC, Chien CW, et al. Overexpression of pyruvate dehydrogenase kinase 3 increases drug resistance and early recurrence in colon cancer. Am J Pathol. 2011;179(3):1405–1414. doi: 10.1016/j.ajpath.2011.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Michelakis ED, Webster L, Mackey JR. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer. 2008;99(7):989–994. doi: 10.1038/sj.bjc.6604554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Furuta E, Pai SK, Zhan R, et al. Fatty acid synthase gene is up-regulated by hypoxia via activation of Akt and sterol regulatory element binding protein-1. Cancer Res. 2008;68(4):1003–1011. doi: 10.1158/0008-5472.CAN-07-2489. [DOI] [PubMed] [Google Scholar]
- 69.Flavin R, Peluso S, Nguyen P, Loda M. Fatty acid synthase as a potential therapeutic target in cancer. Futur Oncol. 2010;6(4):551–562. doi: 10.2217/fon.10.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Baenke F, Peck B, Miess H, Schulze A. Hooked on fat: the role of lipid synthesis in cancer metabolism and tumour development. Dis Model Mech. 2013;6(6):1353–1363. doi: 10.1242/dmm.011338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Luo W, Semenza GL. Emerging roles of PKM2 in cell metabolism and cancer progression. Trends Endocrinol Metab. 2012;23(11):560–566. doi: 10.1016/j.tem.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Luo W, Hu H, Chang R, et al. Pyruvate kinase M2 is a PHD3- stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011;145(5):732–744. doi: 10.1016/j.cell.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Semenza GL. Evaluation of HIF-1 inhibitors as anticancer agents. Drug Discov Today. 2007;12(19–20):853–859. doi: 10.1016/j.drudis.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 74.Kung AL, Zabludoff SD, France DS, et al. Small molecule blockade of transcriptional coactivation of the hypoxia-inducible factor pathway. Cancer Cell. 2004;6(1):33–43. doi: 10.1016/j.ccr.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 75.Shin DH, Chun YS, Lee DS, Huang LE, Park JW. Bortezomib inhibits tumor adaptation to hypoxia by stimulating the FIH-mediated repression of hypoxia-inducible factor-1. Blood. 2008;111(6):3131–3136. doi: 10.1182/blood-2007-11-120576. [DOI] [PubMed] [Google Scholar]
- 76.Falchook GS, Wheler JJ, Naing A, et al. Targeting hypoxia-inducible factor-1α (HIF-1α) in combination with antiangiogenic therapy: a phase I trial of bortezomib plus bevacizumab. Oncotarget. 2014;5(21):10280–10292. doi: 10.18632/oncotarget.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kong D, Park EJ, Stephen AG, et al. Echinomycin, a small-molecule inhibitor of hypoxia-inducible factor-1 DNA-binding activity. Cancer Res. 2005;65(19):9047–9055. doi: 10.1158/0008-5472.CAN-05-1235. [DOI] [PubMed] [Google Scholar]
- 78.Gradishar WJ, Vogelzang NJ, Kilton LJ, et al. A phase II clinical trial of echinomycin in metastatic soft tissue sarcoma. An Illinois Cancer Center Study. Invest New Drugs. 1995;13(2):171–174. doi: 10.1007/BF00872868. [DOI] [PubMed] [Google Scholar]
- 79.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3(10):721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 80.Whelan Ka, Schwab LP, Karakashev SV, et al. The oncogene HER2/neu (ERBB2) requires the hypoxia-inducible factor HIF-1 for mammary tumor growth and anoikis resistance. J Biol Chem. 2013;288(22):15865–15877. doi: 10.1074/jbc.M112.426999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Karar J, Cerniglia GJ, Lindsten T, Koumenis C, Maity A. Dual PI3K/mTOR inhibitor NVP-BEZ235 suppresses hypoxia-inducible factor (HIF)-1α expression by blocking protein translation and increases cell death under hypoxia. Cancer Biol Ther. 2012;13(11):1102–1111. doi: 10.4161/cbt.21144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Greenberger LM, Horak ID, Filpula D, et al. A RNA antagonist of hypoxia-inducible factor-1alpha, EZN-2968, inhibits tumor cell growth. Mol Cancer Ther. 2008;7(11):3598–3608. doi: 10.1158/1535-7163.MCT-08-0510. [DOI] [PubMed] [Google Scholar]
- 83.Jeong W, Rapisarda A, Park SR, et al. Pilot trial of EZN-2968, an antisense oligonucleotide inhibitor of hypoxia-inducible factor-1 alpha (HIF-1α), in patients with refractory solid tumors. Cancer Chemother Pharmacol. 2014;73(2):343–348. doi: 10.1007/s00280-013-2362-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rapisarda A, Uranchimeg B, Sordet O, Pommier Y, Shoemaker RH, Melillo G. Topoisomerase I-mediated inhibition of hypoxia-inducible factor 1: mechanism and therapeutic implications. Cancer Res. 2004;64(4):1475–1482. doi: 10.1158/0008-5472.can-03-3139. [DOI] [PubMed] [Google Scholar]
- 85.Kummar S, Raffeld M, Juwara L, et al. Multihistology, target-driven pilot trial of oral topotecan as an inhibitor of hypoxia-inducible factor-1α in advanced solid tumors. Clin Cancer Res. 2011;17(15):5123–5131. doi: 10.1158/1078-0432.CCR-11-0682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mabjeesh NJ, Escuin D, LaVallee TM, et al. 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell. 2003;3(4):363–375. doi: 10.1016/s1535-6108(03)00077-1. [DOI] [PubMed] [Google Scholar]
- 87.Matei D, Schilder J, Sutton G, et al. Activity of 2 methoxyestradiol (Panzem NCD) in advanced, platinum-resistant ovarian cancer and primary peritoneal carcinomatosis: a Hoosier Oncology Group trial. Gynecol Oncol. 2009;115(1):90–96. doi: 10.1016/j.ygyno.2009.05.042. [DOI] [PubMed] [Google Scholar]
- 88.Minet E, Mottet D, Michel G, et al. Hypoxia-induced activation of HIF-1: role of HIF-1alpha-Hsp90 interaction. FEBS Lett. 1999;460(2):251–256. doi: 10.1016/s0014-5793(99)01359-9. [DOI] [PubMed] [Google Scholar]
- 89.Mabjeesh NJ1, Post DE, Willard MT, et al. Geldanamycin induces degradation of hypoxia-inducible factor 1alpha protein via the proteosome pathway in prostate cancer cells. Cancer Res. 2002;62(9):2478–2482. [PubMed] [Google Scholar]
- 90.Vaishampayan UN, Burger AM, Sausville EA, et al. Safety, efficacy, pharmacokinetics, and pharmacodynamics of the combination of sorafenib and tanespimycin. Clin Cancer Res. 2010;16(14):3795–3804. doi: 10.1158/1078-0432.CCR-10-0503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Modi S, Stopeck A, Linden H, et al. HSP90 inhibition is effective in breast cancer: a phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clin Cancer Res. 2011;17(15):5132–5139. doi: 10.1158/1078-0432.CCR-11-0072. [DOI] [PubMed] [Google Scholar]
- 92.Guise CP, Mowday AM, Ashoorzadeh A, et al. Bioreductive prodrugs as cancer therapeutics: targeting tumor hypoxia. Chin J Cancer. 2014;33(2):80–86. doi: 10.5732/cjc.012.10285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Marcu L, Olver I. Tirapazamine: from bench to clinical trials. Curr Clin Pharmacol. 2006;1(1):71–79. doi: 10.2174/157488406775268192. [DOI] [PubMed] [Google Scholar]
- 94.Rischin D, Peters LJ, O’Sullivan B, et al. Tirapazamine, cisplatin, and radiation versus cisplatin and radiation for advanced squamous cell carcinoma of the head and neck (TROG 02.02, HeadSTART): a phase III trial of the Trans-Tasman Radiation Oncology Group. J Clin Oncol. 2010;28(18):2989–2995. doi: 10.1200/JCO.2009.27.4449. [DOI] [PubMed] [Google Scholar]
- 95.Guise CP, Abbattista MR, Singleton RS, et al. The bioreductive prodrug PR-104A is activated under aerobic conditions by human aldo-keto reductase 1C3. Cancer Res. 2010;70(4):1573–1584. doi: 10.1158/0008-5472.CAN-09-3237. [DOI] [PubMed] [Google Scholar]
- 96.Sun JD, Liu Q, Wang J, et al. Selective tumor hypoxia targeting by hypoxia-activated prodrug TH-302 inhibits tumor growth in preclinical models of cancer. Clin Cancer Res. 2012;18(3):758–770. doi: 10.1158/1078-0432.CCR-11-1980. [DOI] [PubMed] [Google Scholar]
- 97.McKeage MJ, Jameson MB, Ramanathan RK, et al. PR-104 a bioreductive pre-prodrug combined with gemcitabine or docetaxel in a phase Ib study of patients with advanced solid tumours. BMC Cancer. 2012;12(1):496. doi: 10.1186/1471-2407-12-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Williams KJ, Albertella MR, Fitzpatrick B, et al. In vivo activation of the hypoxia-targeted cytotoxin AQ4N in human tumor xenografts. Mol Cancer Ther. 2009;8(12):3266–3275. doi: 10.1158/1535-7163.MCT-09-0396. [DOI] [PubMed] [Google Scholar]
- 99.Phillips RM, Hendriks HR, Peters GJ, EORTC-Pharmacology and Molecular Mechanism Group EO9 (Apaziquone): from the clinic to the laboratory and back again. Br J Pharmacol. 2013;168(1):11–18. doi: 10.1111/j.1476-5381.2012.01996.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer. 2004;4(5):361–370. doi: 10.1038/nrc1360. [DOI] [PubMed] [Google Scholar]
- 101.Arora A, Scholar E. Role of tyrosine kinase inhibitors in cancer therapy. J Pharmacol Exp. 2005;315(3):971–979. doi: 10.1124/jpet.105.084145. [DOI] [PubMed] [Google Scholar]
- 102.Glück AA, Aebersold DM, Zimmer Y, Medová M. Interplay between receptor tyrosine kinases and hypoxia signaling in cancer. Int J Biochem Cell Biol. 2015;62:101–114. doi: 10.1016/j.biocel.2015.02.018. [DOI] [PubMed] [Google Scholar]
- 103.Nilsson MB, Zage PE, Zeng L, et al. Multiple receptor tyrosine kinases regulate HIF-1alpha and HIF-2alpha in normoxia and hypoxia in neuroblastoma: implications for antiangiogenic mechanisms of multikinase inhibitors. Oncogene. 2010;29(20):2938–2949. doi: 10.1038/onc.2010.60. [DOI] [PubMed] [Google Scholar]
- 104.Organ SL, Tsao MS. An overview of the c-MET signaling pathway. Ther Adv Med Oncol. 2011;3(1 Suppl):S7–S19. doi: 10.1177/1758834011422556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gentile A, Trusolino L, Comoglio PM. The Met tyrosine kinase receptor in development and cancer. Cancer Metastasis Rev. 2008;27(1):85–94. doi: 10.1007/s10555-007-9107-6. [DOI] [PubMed] [Google Scholar]
- 106.Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4(12):915–925. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 107.Pennacchietti S, Michieli P, Galluzzo M, Mazzone M, Giordano S, Comoglio PM. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell. 2003;3(4):347–361. doi: 10.1016/s1535-6108(03)00085-0. [DOI] [PubMed] [Google Scholar]
- 108.Funakoshi Y, Mukohara T, Tomioka H, et al. Excessive MET signaling causes acquired resistance and addiction to MET inhibitors in the MKN45 gastric cancer cell line. Invest New Drugs. 2013;31(5):1158–1168. doi: 10.1007/s10637-013-9959-2. [DOI] [PubMed] [Google Scholar]
- 109.Puri N, Khramtsov A, Ahmed S, et al. A selective small molecule inhibitor of c-Met, PHA665752, inhibits tumorigenicity and angiogenesis in mouse lung cancer xenografts. Cancer Res. 2007;67(8):3529–3534. doi: 10.1158/0008-5472.CAN-06-4416. [DOI] [PubMed] [Google Scholar]
- 110.Mishra A, Wang J, Shiozawa Y, et al. Hypoxia stabilizes GAS6/Axl signaling in metastatic prostate cancer. Mol Cancer Res. 2012;10(6):703–712. doi: 10.1158/1541-7786.MCR-11-0569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rankin EB, Fuh KC, Castellini L, et al. Direct regulation of GAS6/AXL signaling by HIF promotes renal metastasis through SRC and MET. Proc Natl Acad Sci. 2014;111(37):13373–13378. doi: 10.1073/pnas.1404848111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Holland SJ, Powell MJ, Franci C, et al. Multiple roles for the receptor tyrosine kinase Axl in tumor formation. Cancer Res. 2005;65(20):9294–9303. doi: 10.1158/0008-5472.CAN-05-0993. [DOI] [PubMed] [Google Scholar]
- 113.Wagh PK, Peace BE, Waltz SE. Met-related receptor tyrosine kinase Ron in tumor growth and metastasis. Adv Cancer Res. 2008;100:1–33. doi: 10.1016/S0065-230X(08)00001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Thangasamy A, Rogge J, Ammanamanchi S. Recepteur d’origine nantais tyrosine kinase is a direct target of hypoxia-inducible factor-1 alpha-mediated invasion of breast carcinoma cells. J Biol Chem. 2009;284(21):14001–14010. doi: 10.1074/jbc.M809320200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chang HY, Liu HS, Lai MD, et al. Hypoxia promotes nuclear translocation and transcriptional function in the oncogenic tyrosine kinase RON. Cancer Res. 2014;74(16):4549–4562. doi: 10.1158/0008-5472.CAN-13-3730. [DOI] [PubMed] [Google Scholar]
- 116.Yarden Y, Pines G. The ERBB network: at last, cancer therapy meets systems biology. Nat Rev Cancer. 2012;12(8):553–563. doi: 10.1038/nrc3309. [DOI] [PubMed] [Google Scholar]
- 117.Tebbutt N, Pedersen MW, Johns TG. Targeting the ERBB family in cancer: couples therapy. Nat Rev Cancer. 2013;13(9):663–673. doi: 10.1038/nrc3559. [DOI] [PubMed] [Google Scholar]
- 118.Franovic A, Gunaratnam L, Smith K, Robert I, Patten D, Lee S. Translational up-regulation of the EGFR by tumor hypoxia provides a nonmutational explanation for its overexpression in human cancer. Proc Natl Acad Sci U S A. 2007;104(32):13092–13097. doi: 10.1073/pnas.0702387104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Landgraf R. HER2 therapy. HER2 (ERBB2): functional diversity from structurally conserved building blocks. Breast Cancer Res. 2007;9(1):202. doi: 10.1186/bcr1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Révillion F, Bonneterre J, Peyrat JP. ERBB2 oncogene in human breast cancer and its clinical significance. Eur J Cancer. 1998;34(6):791–808. doi: 10.1016/s0959-8049(97)10157-5. [DOI] [PubMed] [Google Scholar]
- 121.Karakashev SV, Reginato MJ. Hypoxia/HIF1α induces lapatinib resistance in ERBB2-positive breast cancer cells via regulation of DUSP2. Oncotarget. 2015;6(4):1967–1980. doi: 10.18632/oncotarget.2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9(8):550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 123.Alvarez-Tejado M, Naranjo-Suarez S, Jiménez C, Carrera AC, Landázuri MO, del Peso L. Hypoxia induces the activation of the phosphati-dylinositol 3-kinase/Akt cell survival pathway in PC12 cells: protective role in apoptosis. J Biol Chem. 2001;276(25):22368–22374. doi: 10.1074/jbc.M011688200. [DOI] [PubMed] [Google Scholar]
- 124.Carmeliet P. VEGF as a key mediator of angiogenesis in cancer. Oncology. 2005;69(Suppl 3):4–10. doi: 10.1159/000088478. [DOI] [PubMed] [Google Scholar]
- 125.Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer. 2008;8(8):579–591. doi: 10.1038/nrc2403. [DOI] [PubMed] [Google Scholar]
- 126.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8(8):592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480(7378):480–489. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Corzo CA, Condamine T, Lu L, et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. 2010;207(11):2439–2453. doi: 10.1084/jem.20100587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.McNamee EN, Korns Johnson D, Homann D, Clambey ET. Hypoxia and hypoxia-inducible factors as regulators of T cell development, differentiation, and function. Immunol Res. 2013;55(1–3):58–70. doi: 10.1007/s12026-012-8349-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lamers CH, Sleijfer S, van Steenbergen S, et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther. 2013;21(4):904–912. doi: 10.1038/mt.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]