

Abstract
Protein hyperacetylation is associated with glucose intolerance and insulin resistance, suggesting that the enzymes regulating the acetylome play a role in this pathological process. Sirtuin 3 (SIRT3), the primary mitochondrial deacetylase, has been linked to energy homeostasis. Thus, it is hypothesized that the dysregulation of the mitochondrial acetylation state, via genetic deletion of SIRT3, will amplify the deleterious effects of a high-fat diet (HFD). Hyperinsulinemic-euglycemic clamp experiments show, for the first time, that mice lacking SIRT3 exhibit increased insulin resistance due to defects in skeletal muscle glucose uptake. Permeabilized muscle fibers from HFD-fed SIRT3 knockout (KO) mice showed that tricarboxylic acid cycle substrate–based respiration is decreased while fatty acid–based respiration is increased, reflecting a fuel switch from glucose to fatty acids. Consistent with reduced muscle glucose uptake, hexokinase II (HKII) binding to the mitochondria is decreased in muscle from HFD-fed SIRT3 KO mice, suggesting decreased HKII activity. These results show that the absence of SIRT3 in HFD-fed mice causes profound impairments in insulin-stimulated muscle glucose uptake, creating an increased reliance on fatty acids. Insulin action was not impaired in the lean SIRT3 KO mice. This suggests that SIRT3 protects against dietary insulin resistance by facilitating glucose disposal and mitochondrial function.
Introduction
Lysine acetylation has recently emerged as an important post-translational modification. Consisting of the reversible transfer of the acetyl group from acetyl-CoA to a lysine residue, this highly regulated event modulates diverse cellular processes, including protein-protein interactions, protein subcellular localization, protein stability, and enzymatic activity (1–3). Lysine acetylation is also increasingly linked to metabolism regulation and energy balance (4), and it has been shown that high-fat (HF) feeding induces a shift in the acetylation balance, causing protein hyperacetylation in liver (5). This dysregulation of the acetylation balance could be due to diet-induced increases in acetyl-CoA levels, as observed in liver and muscle (6,7). In a setting of overnutrition, deacetylases such as sirtuins (SIRTs) should play a key protective role by counterbalancing increased protein acetylation.
SIRTs are NAD+-dependent deacetylases and as such are metabolic sensors (8). The SIRT family is comprised of seven members that differ by their subcellular distribution, substrate specificity, and cellular functions (SIRT1–7) (9). Recent studies have highlighted the importance of mitochondrial SIRTs in the regulation of metabolism (10). Indeed, numerous mitochondrial proteins involved in metabolism are regulated by acetylation (11). It now appears that lysine acetylation could be the most important regulatory protein modification event within the mitochondria. Recent evidence suggests that mitochondrial protein acetylation is solely regulated by deacetylases, with acetylation being the consequence of unregulated nonenzymatic reactions, driven by acetyl-CoA levels (12). SIRT3 in particular has been identified as the primary mitochondrial deacetylase (13) and has been linked to the regulation of lipid metabolism, reactive oxygen species levels, and energy production (14,15).
SIRT3 has specifically been shown to direct mitochondrial respiration, and its targets include subunits of the respiratory chain complex (16,17). Importantly, SIRT3 has been implicated in the development of metabolic disease in both humans and rodents (5). SIRT3 activity is increased by nutrient distress such as fasting, caloric restriction, and forced exercise, while SIRT3 activity is decreased in skeletal muscle of models of type 1 and type 2 diabetes (18). Given that SIRT3 is implicated in mitochondrial function, understanding how SIRT3 is involved in the regulation of metabolism is critical in understanding the link between mitochondrial function and metabolic disease. Mice lacking SIRT3 exhibit a decrease in oxygen consumption in isolated mitochondria from liver (16) and reduced glucose tolerance when placed on a high-fat diet (HFD) (5). However, the in vivo contribution of distinct peripheral tissues to this phenotype remains to be elucidated, as the results of efforts to decipher the distinct roles of SIRT3 in tissue-specific models have remained inconclusive. Indeed, while various global SIRT3 knockout (KO) models consistently exhibit metabolic impairments (5,16,18), liver and muscle-specific KO models have no overt metabolic phenotype (19) as assessed by primary screening tools (20).
Here, we report, for the first time, the assessment of insulin sensitivity in chow and HF-fed SIRT3 KO mice using the hyperinsulinemic-euglycemic clamp. In this study, we show that HF-fed SIRT3 KO mice have exacerbated insulin resistance compared with their wild-type (WT) littermates and that this phenotype is due to defects in skeletal muscle metabolism. Furthermore, we show that SIRT3-deficient muscle exhibits profound mitochondrial dysfunction, with decreased reliance on glycolytic substrates and increased reliance on fatty acid substrates, as assessed by high-resolution respirometry on permeabilized muscle fibers.
Research Design and Methods
Mouse Models
Mice lacking the SIRT3 protein (SIRT3 KO mice) and WT littermates on a C57BL/6J strain (16) were fed chow (13.5% calories from fat; catalog #5001, LabDiet) or an HFD (60% calories from fat; catalog #F3282, BioServ) for 12 weeks from 3 weeks of age. Mice were housed in a temperature- and humidity-controlled environment (light cycle 6:00 a.m. to 6:00 p.m.). Studies were performed on 15-week-old 5-h–fasted male mice. Vanderbilt Institutional Animal Care and Use Committee approved the procedures. Body composition was determined by nuclear magnetic resonance.
Hyperinsulinemic-Euglycemic Clamp
One week before hyperinsulinemic-euglycemic clamps, carotid artery and jugular vein catheters were surgically placed for sampling and infusions. Mice were fasted for 5 h and then clamped as previously described (21,22). [3-3H]glucose was primed and continuously infused (t = −90 to 0 min, 0.04 µCi/min). The clamp was initiated at t = 0 min with a continuous insulin infusion (4 mU/kg/min). Arterial glucose levels were monitored every 10 min to provide feedback for adjustment of the glucose infusion rate (GIR) (50% dextrose + [3-3H]glucose). [3-3H]glucose kinetics were determined at −10 min and from 80 to 120 min. 2[14C]deoxyglucose (13 μCi) was administered at 120 min to determine the tissue-specific Rg. Plasma insulin was determined by ELISA. Plasma nonesterified fatty acids (NEFAs) and tissue triglycerides were measured spectrophotometrically. See Supplementary Data for details.
High-Resolution Respirometry on Permeabilized Muscle Fibers
Fibers from gastrocnemius of 5-h–fasted mice were prepared in BIOPS (23). Respiration was measured in Buffer Z at 37°C (Oxygraph O2k, Oroboros Instruments) with 2 mmol/L malate + 10 mmol/L glutamate or 2 mmol/L malate + 75 μmol/L palmitoyl-carnitine (24). With 2 mmol/L ADP (malate-glutamate [MG] + ADP, malate-palmitoyl-carnitine [MPC] + ADP), coupled respiration is maximally stimulated. Citrate synthase activity was measured spectrophotometrically (25).
Immunoprecipitations and Western Blots
Whole-tissue lysates were obtained by homogenization in lysis buffer. Mitochondrial fractions were obtained by homogenization in Buffer M. The mitochondrial pellet was washed twice before resuspension in lysis buffer. For immunoprecipitation, lysates were incubated for 1 h with anti–insulin receptor substrate 1 (IRS1) or anti–voltage-dependent anion channel (VDAC) and then overnight with A/G PLUS Agarose. Beads were washed four times in PBS then resuspended in loading buffer. Samples were applied to 4–12% SDS-PAGE. Quantifications were performed using the Odyssey system.
Immunostaining
CD31 staining was assessed by immunohistochemistry in paraffin-embedded gastrocnemius sections. Gastrocnemius cryosections were stained with antibodies anti-GLUT4 and anti–caveolin-3 followed by visualization with fluorescent secondary antibodies. Images were acquired at ×40 magnification (LSM 510 confocal microscope; Zeiss). Image analysis was performed using ImageJ (for details, see Supplementary Data).
Statistics
Data are expressed as the mean ± SE. The Student t test or two-way ANOVA followed by Tukey post hoc tests were performed. P < 0.05 was considered significant.
Additional details on methods can be found in the Supplementary Data.
Results
Mice Lacking SIRT3 Exhibit Increased Skeletal Muscle Mitochondrial Protein Acetylation
SIRT3 protein in vastus lateralis was measured in muscle from WT and SIRT3 KO mice that were fed a chow diet or an HFD for 12 weeks. As expected, SIRT3 protein is absent in SIRT3 KO muscle (Fig. 1A). HF feeding does not affect SIRT3 protein expression in WT skeletal muscle. We were interested in determining how SIRT3 deletion or an HFD would impact protein acetylation in the mitochondrial and cytosolic compartments. SIRT3 deletion increased acetylation exclusively in skeletal muscle mitochondria (Fig. 1B and C). HF feeding alone induced an important increase in mitochondrial protein acetylation in muscle from both WT and SIRT3 KO mice but did not affect protein acetylation in the cytosol. Interestingly, the genetic and diet effects were additive, as SIRT3 KO mice that were fed an HFD exhibited the highest mitochondrial protein acetylation.
Figure 1.

Skeletal muscle mitochondrial protein acetylation is increased in chow-fed and HF-fed SIRT3 KO mice. A: Protein homogenates extracted from the vastus lateralis muscle were applied to 4–12% SDS-PAGE. Western blotting was performed using the anti-SIRT3 and β-actin antibodies. Quantifications below the blot were obtained by normalizing SIRT3 band intensity to β-actin band intensity for each lane (n = 4). B and C: Mitochondrial and cytosolic proteins were extracted from vastus lateralis muscle isolated from 5-h–fasted WT or SIRT3 KO mice that were fed a chow diet or an HFD for 12 weeks. Mitochondrial (B) and cytosolic (C) protein lysates were subjected to a 4–12% SDS-PAGE, and Western blotting was performed using an anti–acetyl-lysine antibody. Integrated intensities of acetyl-lysine bands from mitochondrial (B) or cytosolic (C) proteins were obtained by the Odyssey software and normalized to VDAC or GAPDH, respectively (n = 5). *P < 0.05; ***P < 0.001.
HF-Fed SIRT3 KO Mice Have Increased Skeletal Muscle Insulin Resistance
To study the role of SIRT3 in the pathogenesis of insulin resistance, we fed WT and SIRT3 KO mice a chow diet or an HFD for 12 weeks. We observed no difference in weight gain within their respective diets (Table 1). The WT and SIRT3 KO chow-fed mice were not different with respect to basal fasting blood glucose, plasma insulin, plasma NEFAs, and liver triglycerides (Table 1). HF feeding raised fasting blood glucose comparably in both genotypes. Basal plasma insulin levels was higher in HF-fed SIRT3 KO mice compared with their WT littermates, which is indicative of exaggerated insulin resistance (Table 1). We observed no difference in plasma NEFAs and liver triglycerides between HF-fed WT and SIRT3 KO mice.
Table 1.
Characteristics of the SIRT3 KO mice
| Chow-fed mice |
HF-fed mice |
|||
|---|---|---|---|---|
| WT (N = 9) | SIRT3 KO (N = 8) | WT (N = 6) | SIRT3 KO (N = 7) | |
| Weight (g) | 26 ± 1 | 25 ± 1 | 35 ± 2 | 39 ± 2 |
| Adiposity (%) | 7.1 ± 0.4 | 8.7 ± 0.4** | 33.3 ± 2.2 | 34.3 ± 1.3 |
| Age at study (weeks) | 15 | 15 | 15 | 15 |
| Duration of HF feeding (weeks) | 12 | 12 | ||
| Blood glucose (mg/dL) | ||||
| Basal | 123 ± 6 | 128 ± 9 | 134 ± 4 | 143 ± 5 |
| Clamp | 129 ± 3 | 133 ± 4 | 133 ± 3 | 138 ± 2 |
| Plasma insulin (ng/mL) | ||||
| Basal | 0.9 ± 0.1 | 1.1 ± 0.2 | 3.0 ± 0.9 | 7.4 ± 2.3 |
| Clamp | 4.2 ± 0.3 | 4.8 ± 0.4 | 9.1 ± 2.3 | 11.4 ± 2.9 |
| Plasma NEFAs (mmol/L) | ||||
| Basal | 0.64 ± 0.03 | 0.87 ± 0.14 | 0.62 ± 0.09 | 0.67 ± 0.06 |
| Clamp | 0.16 ± 0.01 | 0.21 ± 0.06 | 0.20 ± 0.04 | 0.18 ± 0.04 |
| Liver triglycerides (mg/g) | 25 ± 1 | 25 ± 1 | 79 ± 24 | 74 ± 10 |
Data are expressed as the mean ± SE, unless otherwise indicated.
**P < 0.01, WT vs. SIRT3 KO.
To assess whole-body insulin sensitivity, we performed hyperinsulinemic-euglycemic clamps. During the clamp, blood glucose was clamped at 130 mg/dL in all groups (Fig. 2A and D), and insulin was raised to comparable levels between groups (Table 1). Circulating NEFAs were suppressed in all groups, suggesting equivalent insulin-induced suppression of lipolysis (Table 1). The GIR necessary to maintain euglycemia in the chow-fed animals was similar between genotypes (Fig. 2B). However, on an HFD, the GIR was significantly lower in the SIRT3 KO mice, indicating increased insulin resistance (Fig. 2E). Endogenous glucose production (EndoRa) was completely suppressed in both chow groups during the clamp (Fig. 2C and 2G). On an HFD, SIRT3 KO mice have slightly lower basal EndoRa and lower clamp EndoRa (Fig. 2F), which is likely due to the slightly higher insulin levels in these mice (Table 1). However, the insulin-induced suppression of EndoRa and the percent EndoRa suppression were similar between genotypes (Fig. 2F and G). In addition, the phosphorylated (P)-Akt/Akt ratios in WT and SIRT3 KO mouse livers were not different (Fig. 2H).
Figure 2.
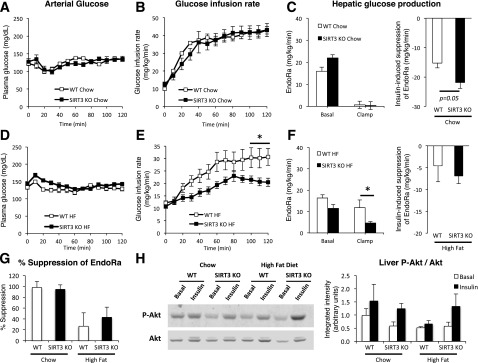
HF-fed SIRT3 KO mice exhibit increased insulin resistance during the hyperinsulinemic-euglycemic clamp. A and D: Blood glucose was monitored throughout the clamp at 10-min intervals by sampling from the arterial catheter. Blood glucose was maintained at euglycemia (130∼140 mg/dL) in both chow-fed (A) and HF-fed (D) mice. The GIR in the venous catheter needed to maintain euglycemia in chow-fed (B) or HF-fed (E) mice. EndoRa, a marker of hepatic glucose production, in chow-fed (C) or HF-fed (F) mice, determined by administration of [3-3H]glucose. The insulin-induced suppression of EndoRa is the difference between basal and clamp EndoRa values. G: Insulin-induced suppression of EndoRa expressed as the percent of basal levels. H: Liver protein homogenates obtained from 5-h–fasted (Basal) or clamped (Insulin) mice were applied to a 4–12% SDS-PAGE (n = 4). Western blotting was performed for Akt and P-Akt (Ser473). Integrated intensities were obtained by the Odyssey software. *P < 0.05.
Basal and clamp glucose disappearance rates (Rds), and consequently the insulin-stimulated increase in Rd, were not different between genotypes of mice on a chow diet (Fig. 3A). In addition, the tissue-specific Rg was not different between chow-fed WT and SIRT3 KO mice in gastrocnemius, vastus lateralis, soleus, diaphragm, and heart (Fig. 3B). These data are consistent with the similar GIR observed in lean WT and SIRT3 KO mice, suggesting that SIRT3 is not a critical determinant of insulin action in chow-fed mice. However, when fed an HFD, both basal and clamp Rd values were significantly decreased in SIRT3 KO mice (Fig. 3C). In addition, the insulin-stimulated increase in Rd was significantly lower in SIRT3 KO mice, indicating that the ability of insulin to stimulate peripheral glucose uptake is greatly diminished in HF-fed SIRT3 KO mice (Fig. 3C). Gastrocnemius, vastus lateralis, soleus, and diaphragm Rg values were also dramatically reduced in HF-fed SIRT3 KO mice (Fig. 3D). These data are consistent with the reduced GIR, as they reflect higher insulin resistance in SIRT3 KO mice. Moreover, these results clearly indicate that skeletal muscle insulin resistance to muscle glucose uptake is responsible for the observed whole-body phenotype.
Figure 3.
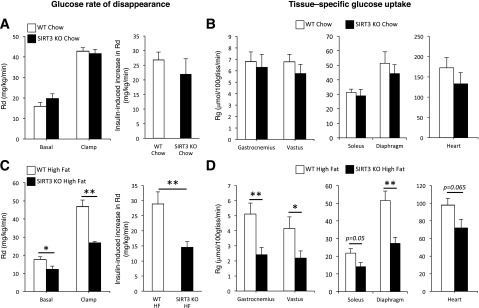
HF-fed SIRT3 KO mice have increased skeletal muscle insulin resistance. Rd in chow-fed (A) or HF-fed (C) WT and SIRT3 KO mice. The insulin-induced increase in Rd is the difference between clamp and basal Rd and represents the ability of insulin to stimulate peripheral tissue glucose uptake. Rg in gastrocnemius, vastus lateralis, soleus, diaphragm, and cardiac muscles is determined by the administration of nonmetabolizable glucose 2[14C]deoxyglucose in chow-fed (B) or HF-fed (D) WT and SIRT3 KO mice. 100gtiss, 100 g tissue. *P < 0.05; **P < 0.01.
Mice Lacking SIRT3 Exhibit Normal Skeletal Muscle Insulin Signaling and GLUT4 Translocation
To investigate whether skeletal muscle insulin resistance in HF-fed SIRT3 KO mice was associated with decreased insulin signaling, we performed Western blots for key proteins of the insulin signaling pathway. The P-Akt/Akt and P-AS160/AS160 ratios were increased by insulin during the clamp in muscle from both WT and SIRT3 KO HF-fed mice, with no difference between genotypes (Fig. 4A). We also investigated the phosphorylation of glycogen synthase kinase-3β (GSK-3β) as a surrogate marker for Akt activity and did not observe differences in the P-GSK-3β/GSK-3β ratio (Supplementary Fig. 1). By immunoprecipitating IRS1 in insulin-stimulated muscle homogenates, we showed that p85 binding to IRS1, while decreased by HF feeding, was not different between genotypes (Fig. 4B). Furthermore, the P-IRS1/IRS1 ratio was not different between groups (Fig. 4B). Western blotting for GLUT4 and GLUT1 in insulin-stimulated muscle lysates revealed that GLUT4 and GLUT1 levels were not different between groups (Fig. 4C). In addition, GLUT4 intensity at the muscle plasma membrane, as detected by immunofluorescence, was not different, suggesting no defects in the insulin-induced GLUT4 translocation event (Fig. 4D). Taken together, these results suggest that the insulin signaling pathway was not impaired in SIRT3 KO mouse muscle and do not explain the insulin resistance observed in mice lacking SIRT3.
Figure 4.

Insulin signaling and GLUT4 translocation are not affected in skeletal muscle of SIRT3 KO mice. A: Gastrocnemius protein homogenates obtained from 5-h–fasted (Basal) or clamped (Insulin) HF-fed mice were applied to a 4–12% SDS-PAGE (n = 4). Western blotting was performed for Akt, P-Akt (Ser473), AS160, and P-AS160 (Thr588). Integrated intensities were obtained by the Odyssey software. B: Immunoprecipitation (IP) from gastrocnemius muscle extracts obtained from clamped (Insulin) mice were performed with total IRS1 antibody; then, immunoblots were completed for IRS1, P-IRS1 (Ser612), and the p85 subunit of PI3K (n = 6). Integrated intensities were obtained by the ImageJ software. C: Western blotting was performed for GLUT1 and GLUT4 on gastrocnemius protein homogenates obtained from 5-h–fasted (Basal) or clamped (Insulin) mice (n = 4). Integrated intensities were obtained by the Odyssey software and normalized to GAPDH intensities. D: Confocal imaging of GLUT4 and plasma membrane marker caveolin-3 (CAV3) was performed on clamped (insulin) gastrocnemius cryosections (n = 5–8). *P < 0.05.
Decreased muscle vascularization is sufficient to induce insulin resistance in mice (26). We therefore investigated whether this decreased vascularization factors into the muscle insulin resistance observed in HF-fed SIRT3 KO mice. We found that staining for endothelial cell marker CD31 in gastrocnemius muscle was not different between genotypes, regardless of diet (Supplementary Fig. 2).
Mitochondrial Respiration Is Decreased in Skeletal Muscle of SIRT3 KO Mice
We hypothesized that the loss of SIRT3 would impair mitochondrial respiration, as SIRT3 tightly regulates respiration in isolated mitochondria (16,18). We performed high-resolution respirometry on permeabilized fibers prepared from red gastrocnemius obtained from chow-fed and HF-fed WT and SIRT3 KO mice. We observed that respiration was decreased in fibers from chow-fed SIRT3 KO mice in the presence of MG, both in basal (MG) and ADP-stimulated (MG + ADP) states (Fig. 5A). This indicates decreased respiration through complex I in SIRT3 KO muscle. We observed no difference in respiration between genotypes in the presence of fatty acid substrates (Fig. 5C).
Figure 5.
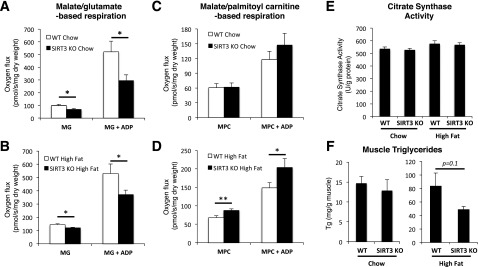
Oxygen flux is reduced in permeabilized fibers from gastrocnemius muscle of SIRT3 KO mice that were fed a chow diet or an HFD. High-resolution respirometry is performed on permeabilized red gastrocnemius fiber bundles isolated from 5-h–fasted WT or SIRT3 KO mice that were fed a chow diet (A) or an HFD (B). Oxygen flux is first measured in the presence of malate (2 mmol/L) and glutamate (10 mmol/L) (MG). Saturating ADP (2 mmol/L) is then added to the chamber (MG + ADP). Oxygen flux is normalized to fiber dry weight (n = 8–10). High-resolution respirometry is performed on permeabilized red gastrocnemius fiber bundles isolated from 5-h–fasted WT or SIRT3 KO mice that were fed a chow diet (C) or an HFD (D) in the presence of malate (2 mmol/L) and palmitoyl-carnitine (75 μmol/L) (MPC), followed by the addition of saturating ADP (2 mmol/L) (MPC + ADP). Oxygen flux is normalized to fiber dry weight (n = 8–10). E: Citrate synthase activity was measured in red gastrocnemius muscle from 5-h–fasted WT or SIRT3 KO mice that were fed a chow diet or an HFD (n = 8). F: Triglyceride content was determined in the vastus lateralis muscle from 5-h–fasted WT or SIRT3 KO mice that were fed a chow diet or an HFD (n = 6–9). *P < 0.05; **P < 0.01.
As in chow-fed mice, respiration was decreased in MG and MG + ADP conditions in fibers of HFD-fed SIRT3 KO mice (Fig. 5B). Interestingly, when supplied with MPC, oxygen consumption was significantly higher in both basal and ADP-stimulated conditions in fibers of SIRT3 KO mice (Fig. 5D). Citrate synthase activity and oxidative phosphorylation proteins were not different between groups, indicating similar mitochondrial abundance in these muscles (Fig. 5E and Supplementary Fig. 3) (27). Therefore, the differences in respiration observed between WT and SIRT3 KO mice cannot be attributed to differences in mitochondrial content. In addition, HF-fed SIRT3 KO mice have decreased muscle triglyceride content compared with their WT littermates (Fig. 5F). Taken together, these results show that muscle fibers of SIRT3 KO mice have defective complex I–dependent respiration and, when subjected to an HFD, have increased reliance on fatty acids to provide substrates for the respiratory chain.
Hexokinase II Association With Mitochondria Is Decreased in Skeletal Muscle of SIRT3 KO Mice
To further examine the defects in muscle glucose uptake in SIRT3 KO mice, we investigated hexokinase (HK)II cellular compartmentation. HKII binds to the outer mitochondrial membrane, and this directs its activity, with active HKII being bound to VDAC, an outer mitochondrial transmembrane protein (28). Immunoblots for HKII performed on muscle mitochondrial and whole-tissue fractions showed that mitochondria-bound HKII was significantly decreased in the muscles of HF-fed SIRT3 KO mice, compared with WT controls (Fig. 6A). As no difference in total HKII was observed between groups, the ratio of mitochondria-bound to total HKII, which is indicative of overall HKII activity, was significantly reduced in the muscles of HF-fed SIRT3 KO mice (Fig. 6A). Consistent with this finding, we observed decreased HK activity in gastrocnemius and vastus lateralis muscles in HF-fed SIRT3 KO mice compared with their WT littermates (Fig. 6B). As a control, HK activity was not affected in the brain. In addition, the ability of insulin to stimulate glycogen synthesis during the clamp was severely blunted in muscles of HF-fed SIRT3 KO mice (Fig. 6C). In accordance with this result, the net glycogen synthetic rate was reduced in these tissues (Fig. 6D).
Figure 6.
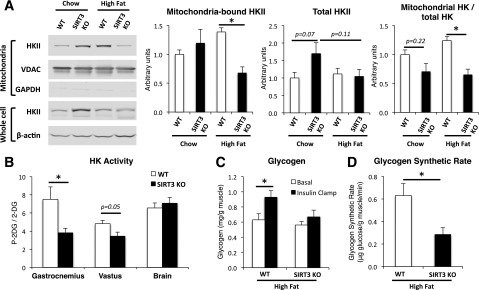
SIRT3 deletion decreases HKII binding to the mitochondria. A: Mitochondrial or whole muscle proteins were extracted from the vastus lateralis muscle isolated from 5-h–fasted WT or SIRT3 KO mice, and lysates were subjected to a 4–12% SDS-PAGE (n = 4–6). Western blotting was performed using anti-HKII, anti-VDAC, anti-GAPDH, or anti–β-actin antibodies. HKII band intensities from the mitochondrial fraction were quantified using the Odyssey software and normalized to VDAC. HKII band intensities from the whole muscle fraction were quantified using the Odyssey software and normalized to β-actin. The ratio of mitochondria-bound HKII to total HKII intensities was then calculated. B: HK activity was determined by dividing the amount of 2[14C]deoxyglucose-6-phosphate by the amount of 2[14C]deoxyglucose (2-DG) found in gastrocnemius, vastus lateralis, and brain in 5-h–fasted WT or SIRT3 KO HF-fed mice at the end of the insulin clamp (n = 6–9). C: Glycogen content was determined in vastus lateralis muscle samples obtained from 5-h–fasted WT or SIRT3 KO mice that were fed an HFD (n = 6–9). D: The glycogen synthetic rate was determined in vastus lateralis muscle in 5-h–fasted WT or SIRT3 KO HF-fed mice by measuring the rate of incorporation of [3-3H]glucose into glycogen during the insulin clamp (n = 6–9). *P < 0.05.
HKII docking to the mitochondria is dependent on the binding of VDAC to the adenine nucleotide transporter (ANT), which is located in the inner mitochondrial membrane. We therefore hypothesized that VDAC binding to ANT would be decreased, causing dissociation of the HKII-VDAC-ANT complex in muscles of SIRT3 KO mice. VDAC was immunoprecipitated from mitochondrial protein fractions to assess the association of HKII, VDAC, and ANT. In accordance with our previous results, HKII binding to VDAC was decreased in SIRT3 KO mice (Fig. 7A and B). We also showed that the VDAC-ANT association was decreased in skeletal muscle lacking SIRT3 (Fig. 7A and C). Taken together, these results show that HF-fed SIRT3 KO mice exhibit decreased HKII binding to the mitochondria associated with decreased VDAC-ANT complex formation.
Figure 7.
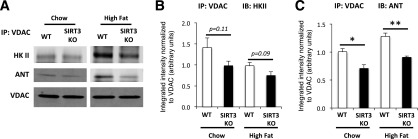
SIRT3 deletion affects mPTP formation in skeletal muscle of HF-fed mice. A: Mitochondrial proteins were extracted from vastus lateralis muscle isolated from 5-h–fasted WT or SIRT3 KO mice, and immunoprecipitation (IP) for VDAC was performed on 400 μg of proteins. Western blotting was performed on immunoprecipitated proteins using anti-HKII, anti-ANT, and anti-VDAC antibodies. Quantifications of bands for HKII and ANT obtained by Odyssey software are presented in panels B and C, respectively (n = 4–6). *P < 0.05; **P < 0.01.
Discussion
HF feeding in mice is a well-described model of overnutrition and has been shown to induce liver mitochondrial protein hyperacetylation (5). Nutrient overload is associated with increased levels of acetyl-CoA, a substrate for protein acetylation, in liver (7) and muscle (6). This is consistent with the idea that mitochondrial protein acetylation is driven by acetyl-CoA abundance (12). In the current study, we challenged WT and SIRT3 KO mice with nutrient overload to amplify protein acetylation. Our results show that SIRT3 is a crucial regulator of glucose fluxes under conditions of overnutrition, as whole-body insulin sensitivity and insulin-stimulated muscle glucose uptake are markedly impaired. We further report the novel finding of the disassociation of the mitochondrial permeability transition pore (mPTP) complex and the decrease in mitochondrial HKII binding. Importantly, HKII activity is a key site of control of muscle glucose uptake (29–31). In addition, SIRT3 KO resulted in impaired mitochondrial carbohydrate–based substrate oxidation.
Consistent with the role of SIRT3 as the major mitochondrial deacetylase, we find that SIRT3 deletion increases skeletal muscle protein acetylation, specifically in the mitochondrial compartment. HFD increases acetyl-CoA in skeletal muscle, expanding the precursor pool for acetylation (6). This resulted in greater acetylation of mitochondrial proteins. This imbalance is relatively small in the presence of SIRT3 but is increased synergistically in the absence of SIRT3. This is in line with the hypothesis that SIRT3 is a master regulator of the mitochondrial acetylome. Its presence effectively counteracts hyperacetylation. Accordingly, its absence combined with nutrient overload causes a marked increase in mitochondrial protein acetylation with functional consequences.
For the first time, insulin sensitivity was assessed in conscious chow-fed SIRT3 KO mice and HF-fed mice. Consistent with previous studies (5,13), we showed that lean SIRT3 KO mice have no overt metabolic dysfunction, despite the protein hyperacetylation present in the muscle mitochondria. In contrast, HF feeding dramatically increased insulin resistance in SIRT3 KO mice. Thus, while SIRT3 may not be critical to muscle insulin action in lean mice, it becomes crucial in the face of nutrient overload, where it is needed to offset the increased mitochondrial protein acetylation. Interestingly, clamp studies revealed that this insulin-resistant phenotype was due solely to the effects on skeletal muscle. Indeed, the Rd was strongly reduced in HF-fed SIRT3 KO mice at basal and during the clamp, and the insulin-induced increase in Rd was also markedly decreased. In accordance with this result, muscle glucose uptake was strongly impaired in skeletal muscle. Indeed, the insulin-induced and percent suppression of EndoRa are equal between genotypes, suggesting that insulin action on the liver is similar between genotypes. In addition, liver triglycerides were similar between genotypes. Others have concluded (5,15) that the liver is a driver of the metabolic phenotype in SIRT3-deficient mice. The techniques used in those studies, such as glucose tolerance tests, do not delineate whether the mouse is more insulin sensitive or identify the tissues responsible for phenotypic differences. More importantly, those studies used 24-h–fasted mice, as opposed to our 5-h–fasted mice. These differences in fast duration are critical in mice, particularly as liver fatty acid and glucose metabolism are extremely different in those two states (32,33). By combining the insulin clamp technique with isotopic methods, we show that SIRT3 deletion in HF-fed mice strongly impairs skeletal muscle metabolism and that this induces the severe insulin-resistant phenotype observed in our mice.
Fernandez-Marcos et al. (19) concluded that muscle-specific SIRT3 KO mice were not affected by HF feeding, as assessed by glucose and insulin tolerance tests. The techniques used were designed to assess not insulin sensitivity but, rather, glucose tolerance and the hypoglycemic responsiveness of the hypothalamic-pituitary-adrenal axis (34,35). Furthermore, the effects on muscle were not assessed in that study. One cannot rule out that SIRT3 deficiency in multiple tissues may be necessary to exacerbate insulin resistance in HF-fed mice. We have shown (26) that a reduction in capillary density is sufficient to cause muscle insulin resistance. We therefore hypothesized that the absence of SIRT3 in endothelial cells could affect capillary density and aggravate the muscle insulin resistance in our model. However, our results show that the endothelial cell marker CD31 is not different in WT and SIRT3 KO mice.
Studies (15,17) have shown that SIRT3 deletion impairs ATP generation and that this could be due to decreased mitochondrial respiration. Indeed, SIRT3 activity has been shown to positively correlate with oxygen consumption in isolated muscle mitochondria (16) and cell lines (18,36). The assessment of muscle respiratory capacity by high-resolution respirometry on permeabilized fibers, as performed here, enables the analysis of mitochondrial respiration in situ, with intact intermitochondrial morphology, cytoskeleton, and myofilaments (23,37). We showed that respiration is markedly decreased in the presence of complex I substrates in the muscle fibers of SIRT3 KO mice. This result is consistent with studies (16) showing that complex I is a direct SIRT3 target and that SIRT3 deletion decreases complex I activity. Moreover, we showed that SIRT3 KO muscle undergoes a fuel switch, whereby the mitochondria rely more on fatty acids as substrates for oxidative phosphorylation. While this trend is observed in chow-fed mice, it is dramatic in HF-fed mice. Interestingly, fatty acids feed substrates to complexes II and III, suggesting that mitochondria of SIRT3 KO muscle may compensate for complex I deficiency by increasing electron flux through complexes II and III. Given that these mice have markedly reduced muscle glucose uptake, we hypothesize that muscle mitochondria in SIRT3 KO mice increase their reliance on fatty acid substrates to compensate for reduced glycolytic substrates. In support of this hypothesis, we show that muscle triglycerides are reduced in HF-fed SIRT3 KO mice. However, despite this compensatory mechanism, the overall oxygen consumption remains reduced in these tissues. This likely contributes to the decreased ATP seen in the skeletal muscle of this model (17).
Finally, regulatory events that couple mitochondrial function to muscle glucose uptake were investigated. HK activity is a key determinant of muscle glucose uptake (38). HK binding to the mitochondria forms the most direct link from mitochondria function to muscle glucose uptake. The mPTP is upstream of the respiration chain and links oxidative phosphorylation to glucose uptake by forming a complex with HKII on the outer mitochondrial membrane. HKII is a crucial step in determining glucose uptake, as it is responsible for creating the downhill concentration gradient that favors glucose transport into the cell. It irreversibly commits glucose to glucose-utilizing pathways such as glycolysis in skeletal muscle (39). HKII activity is modulated by its binding to VDAC on the outer mitochondrial membrane (39). VDAC itself binds ANT in the inner mitochondrial membrane to form the mPTP, a pore that allows the exchange of ADP and ATP across the mitochondrial membrane (28,40). The binding of HKII to mPTP increases HKII activity and gives it “privileged access” to its substrate ATP (41,42), while allowing rechanneling of its product ADP directly back into the mitochondria (39). Therefore, the highly efficient HKII-mPTP complex promotes high rates of glycolysis. In the muscle of HF-fed SIRT3 KO mice, we found that not only was HKII dissociated from the mPTP and its activity reduced, but VDAC and ANT were disassociated, suggesting a closed mPTP and reduced rates of glycolysis. In line with this hypothesis, we found reduced insulin-stimulated glycogen content and glycogen synthetic rate in the muscle of HF-fed SIRT3 KO mice.
The manner in which SIRT3 modulates HKII binding to VDAC is currently unknown, and there are many different mechanisms by which this could occur. We know that acetylation status is changing within the mitochondria, and we believe acetylation events are modulating the formation of the mPTP. However, we do not know whether HKII binding is a consequence of a disrupted mPTP, or whether pore dissociation is subsequent to HKII dissociation. Given that little is known about the regulation of HKII binding to the mPTP in muscle, we cannot state whether it is a direct or indirect event, or whether it is mediated from within the mitochondria or from the cytosol. The latter is indeed a possibility since both HKII and VDAC have post-translational modifications that have been described to affect HKII binding (42–44). The pathways and proteins involved in these events could include SIRT3 targets and signaling events that are, as of yet, unidentified. Further studies are necessary to identify specific SIRT3 targets that mediate HKII binding to VDAC.
The reduced activity and binding of HKII to VDAC are critical components in our model, as it is established that glucose phosphorylation by HKII is a key step in muscle glucose uptake in insulin-stimulated conditions (29,30,45). We have shown that a 50% reduction in HKII in mice is sufficient to impair muscle glucose uptake (29). We therefore postulate that decreased HKII binding to the mitochondria contributes to the SIRT3 KO mouse muscle phenotype. In addition to decreased HKII activity in the muscle of HF-fed SIRT3 KO mice, we show a dissociated mPTP, as evidenced by decreased VDAC-ANT binding, likely leading to reduced trafficking of nucleotides in and out of the mitochondria. Taken together, these events may lead to decreased glycolytic rates and glucose-based respiration and a compensatory increase in fatty acid–based respiration. Our work is consistent with previous studies (36) showing reduced glycolytic rates and increased fatty acid use in SIRT3-deficient muscle. It has long been suggested that SIRT3 is involved in reprogramming metabolism during calorie restriction, allowing respiration to continue during times of low nutrient availability (46). We show here that SIRT3 is absolutely critical in times of overnutrition, when high acetyl-CoA levels lead to mitochondrial protein hyperacetylation. SIRT3, as the major mitochondrial deacetylase, becomes crucial for removal of these nonspecific acetylations and ultimately maintains efficient respiration, glycolysis, and ATP generation in the muscle.
Our findings demonstrate for the first time an insulin-resistant permissive phenotype in the SIRT3 KO mice, specifically due to skeletal muscle insulin resistance. Moreover, we show that SIRT3 promotes, from within the mitochondria, muscle glucose uptake and, subsequently, insulin sensitivity. SIRT3 is therefore a major mitochondrial protein that could, when activated, effectively protect against severe insulin resistance by promoting muscle glucose uptake and mitochondrial respiration. SIRT3 is in fact associated with metabolic disease in humans, as a single nucleotide polymorphism recently identified in the human Sirt3 gene causing decreased SIRT3 activity is associated with metabolic syndrome (5). Therefore, novel therapeutic approaches targeting SIRT3 activity may be key in providing new opportunities to treat insulin resistance and type 2 diabetes.
Supplementary Material
Article Information
Acknowledgments. The authors thank the Vanderbilt Mouse Metabolic Phenotyping Center for the 2[14C]deoxyglucose tissue processing and the Vanderbilt Translational Pathology Core for performing the immunohistochemistry. The authors also thank Dr. Jeffrey E. Pessin (Albert Einstein College of Medicine of Yeshiva University, Bronx, NY) for the gift of the GLUT4 antibody.
Funding. This work was supported by National Institutes of Health grants DK-054902, DK-050277, and DK-059637. D.G. was supported by National Cancer Institute grants 1-RO1-CA-152601-01, 1-RO1-CA-152799-01A1, and 1-RO1-CA-168292-01A1 and a Northwestern Avon Foundation Breast Care Center of Excellence grant.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. L.L. researched the data and wrote the article. A.S.W. researched the data, contributed to the discussion, and reviewed the article. I.M.W., K.K.Y., D.P.B., M.G., and F.D.J. researched the data. D.G. supplied material, contributed to the discussion, and reviewed the article. D.H.W. oversaw the project, supplied material, contributed to the discussion, and reviewed the article. D.H.W. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db14-1810/-/DC1.
See accompanying article, p. 3058.
References
- 1.Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene 2005;363:15–23 [DOI] [PubMed] [Google Scholar]
- 2.Kouzarides T. Chromatin modifications and their function. Cell 2007;128:693–705 [DOI] [PubMed] [Google Scholar]
- 3.Ozden O, Park SH, Kim HS, et al. Acetylation of MnSOD directs enzymatic activity responding to cellular nutrient status or oxidative stress. Aging (Albany, NY) 2011;3:102–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwer B, Verdin E. Conserved metabolic regulatory functions of sirtuins. Cell Metab 2008;7:104–112 [DOI] [PubMed] [Google Scholar]
- 5.Hirschey MD, Shimazu T, Jing E, et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol Cell 2011;44:177–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seiler SE, Martin OJ, Noland RC, et al. Obesity and lipid stress inhibit carnitine acetyltransferase activity. J Lipid Res 2014;55:635–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Satapati S, Sunny NE, Kucejova B, et al. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J Lipid Res 2012;53:1080–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol 2012;13:225–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu Rev Biochem 2004;73:417–435 [DOI] [PubMed] [Google Scholar]
- 10.Verdin E, Hirschey MD, Finley LW, Haigis MC. Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends Biochem Sci 2010;35:669–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim SC, Sprung R, Chen Y, et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell 2006;23:607–618 [DOI] [PubMed] [Google Scholar]
- 12.Wagner GR, Hirschey MD. Nonenzymatic protein acylation as a carbon stress regulated by sirtuin deacylases. Mol Cell 2014;54:5–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lombard DB, Alt FW, Cheng HL, et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol 2007;27:8807–8814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi T, Wang F, Stieren E, Tong Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J Biol Chem 2005;280:13560–13567 [DOI] [PubMed] [Google Scholar]
- 15.Hirschey MD, Shimazu T, Goetzman E, et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010;464:121–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ahn BH, Kim HS, Song S, et al. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci U S A 2008;105:14447–14452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vassilopoulos A, Pennington JD, Andresson T, et al. SIRT3 deacetylates ATP synthase F1 complex proteins in response to nutrient- and exercise-induced stress. Antioxid Redox Signal 2014;21:551–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jing E, Emanuelli B, Hirschey MD, et al. Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc Natl Acad Sci U S A 2011;108:14608–14613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fernandez-Marcos PJ, Jeninga EH, Canto C, et al. Muscle or liver-specific Sirt3 deficiency induces hyperacetylation of mitochondrial proteins without affecting global metabolic homeostasis. Sci Rep 2012;2:425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ayala JE, Samuel VT, Morton GJ, et al.; NIH Mouse Metabolic Phenotyping Center Consortium . Standard operating procedures for describing and performing metabolic tests of glucose homeostasis in mice. Dis Model Mech 2010;3:525–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ayala JE, Bracy DP, McGuinness OP, Wasserman DH. Considerations in the design of hyperinsulinemic-euglycemic clamps in the conscious mouse. Diabetes 2006;55:390–397 [DOI] [PubMed] [Google Scholar]
- 22.Berglund ED, Li CY, Poffenberger G, et al. Glucose metabolism in vivo in four commonly used inbred mouse strains. Diabetes 2008;57:1790–1799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuznetsov AV, Veksler V, Gellerich FN, Saks V, Margreiter R, Kunz WS. Analysis of mitochondrial function in situ in permeabilized muscle fibers, tissues and cells. Nat Protoc 2008;3:965–976 [DOI] [PubMed] [Google Scholar]
- 24.Perry CG, Kane DA, Lanza IR, Neufer PD. Methods for assessing mitochondrial function in diabetes. Diabetes 2013;62:1041–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hepple RT, Baker DJ, Kaczor JJ, Krause DJ. Long-term caloric restriction abrogates the age-related decline in skeletal muscle aerobic function. FASEB J 2005;19:1320–1322 [DOI] [PubMed] [Google Scholar]
- 26.Bonner JS, Lantier L, Hasenour CM, James FD, Bracy DP, Wasserman DH. Muscle-specific vascular endothelial growth factor deletion induces muscle capillary rarefaction creating muscle insulin resistance. Diabetes 2013;62:572–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Larsen S, Nielsen J, Hansen CN, et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J Physiol 2012;590:3349–3360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pastorino JG, Hoek JB. Regulation of hexokinase binding to VDAC. J Bioenerg Biomembr 2008;40:171–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fueger PT, Heikkinen S, Bracy DP, et al. Hexokinase II partial knockout impairs exercise-stimulated glucose uptake in oxidative muscles of mice. Am J Physiol Endocrinol Metab 2003;285:E958–E963 [DOI] [PubMed] [Google Scholar]
- 30.Fueger PT, Li CY, Ayala JE, et al. Glucose kinetics and exercise tolerance in mice lacking the GLUT4 glucose transporter. J Physiol 2007;582:801–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Halseth AE, Bracy DP, Wasserman DH. Overexpression of hexokinase II increases insulinand exercise-stimulated muscle glucose uptake in vivo. Am J Physiol 1999;276:E70–E77 [DOI] [PubMed] [Google Scholar]
- 32.Guan HP, Goldstein JL, Brown MS, Liang G. Accelerated fatty acid oxidation in muscle averts fasting-induced hepatic steatosis in SJL/J mice. J Biol Chem 2009;284:24644–24652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burgess SC, Jeffrey FM, Storey C, et al. Effect of murine strain on metabolic pathways of glucose production after brief or prolonged fasting. Am J Physiol Endocrinol Metab 2005;289:E53–E61 [DOI] [PubMed] [Google Scholar]
- 34.Hughey CC, Wasserman DH, Lee-Young RS, Lantier L. Approach to assessing determinants of glucose homeostasis in the conscious mouse. Mamm Genome 2014;25:522–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Erturk E, Jaffe CA, Barkan AL. Evaluation of the integrity of the hypothalamic-pituitary-adrenal axis by insulin hypoglycemia test. J Clin Endocrinol Metab 1998;83:2350–2354 [DOI] [PubMed] [Google Scholar]
- 36.Jing E, O’Neill BT, Rardin MJ, et al. Sirt3 regulates metabolic flexibility of skeletal muscle through reversible enzymatic deacetylation. Diabetes 2013;62:3404–3417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Picard M, Taivassalo T, Ritchie D, et al. Mitochondrial structure and function are disrupted by standard isolation methods. PLoS One 2011;6:e18317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wasserman DH. Four grams of glucose. Am J Physiol Endocrinol Metab 2009;296:E11–E21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Southworth R. Hexokinase-mitochondrial interaction in cardiac tissue: implications for cardiac glucose uptake, the 18FDG lumped constant and cardiac protection. J Bioenerg Biomembr 2009;41:187–193 [DOI] [PubMed] [Google Scholar]
- 40.Vyssokikh MY, Brdiczka D. The function of complexes between the outer mitochondrial membrane pore (VDAC) and the adenine nucleotide translocase in regulation of energy metabolism and apoptosis. Acta Biochim Pol 2003;50:389–404 [PubMed] [Google Scholar]
- 41.Golshani-Hebroni SG, Bessman SP. Hexokinase binding to mitochondria: a basis for proliferative energy metabolism. J Bioenerg Biomembr 1997;29:331–338 [DOI] [PubMed] [Google Scholar]
- 42.Robey RB, Hay N. Mitochondrial hexokinases, novel mediators of the antiapoptotic effects of growth factors and Akt. Oncogene 2006;25:4683–4696 [DOI] [PubMed] [Google Scholar]
- 43.Chiara F, Rasola A. GSK-3 and mitochondria in cancer cells. Front Oncol 2013;3:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roberts DJ, Tan-Sah VP, Smith JM, Miyamoto S. Akt phosphorylates HK-II at Thr-473 and increases mitochondrial HK-II association to protect cardiomyocytes. J Biol Chem 2013;288:23798–23806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morgan HE, Henderson MJ, Regen DM, Park CR. Regulation of glucose uptake in muscle. I. The effects of insulin and anoxia on glucose transport and phosphorylation in the isolated, perfused heart of normal rats. J Biol Chem 1961;236:253–261 [PubMed] [Google Scholar]
- 46.Schlicker C, Gertz M, Papatheodorou P, Kachholz B, Becker CF, Steegborn C. Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5. J Mol Biol 2008;382:790–801 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.