

Abstract
The fruit fly Drosophila melanogaster is one of the premier model organisms for studying the function and evolution of immune defense. Many aspects of innate immunity are conserved between insects and mammals, and since Drosophila can readily be genetically and experimentally manipulated, they are powerful for studying immune system function and the physiological consequences of disease. The procedure demonstrated here allows infection of flies by introduction of bacteria directly into the body cavity, bypassing epithelial barriers and more passive forms of defense and allowing focus on systemic infection. The procedure includes protocols for the measuring rates of host mortality, systemic pathogen load, and degree of induction of the host immune system. This infection procedure is inexpensive, robust and quantitatively repeatable, and can be used in studies of functional genetics, evolutionary life history, and physiology.
Keywords: Immunology, Issue 99, Drosophila, immunity, infection, resistance, tolerance, bacteria, Providencia, antimicrobial peptides, immune defense
Introduction
The fruit fly Drosophila melanogaster is one of the premier model organisms for studying the function and evolution of immune defense. Drosophila are inexpensive and easy to rear, are highly amenable to experimental manipulation, and are backed by an extensive scientific community that has developed a broad array of research tools. Many aspects of innate immunity are conserved between insects and mammals, including signal transduction mediated by Toll-like receptors and NF-kB family transcription factors, JAK/STAT signaling, and JNK pathway responses.1,2 The function of these genes and pathways can be queried in D. melanogaster using mutations or RNAi knockdowns that increase or decrease pathway activities.3–6 Additionally, Drosophila can be used to study the physiological consequences of infection and disease, including in the context of evolutionary life history theory.7–9 All such studies, however, depend on the ability to reliably infect experimental flies under defined treatment conditions. The procedure described here presents a methodological framework for delivering robust and repeatable bacterial infections to Drosophila melanogaster and subsequently measuring infection severity and quantifying the host immune response.
Drosophila can be naturally and experimentally infected by a wide variety of parasites and pathogens, including bacteria, fungi, viruses, nematodes and parasitoid wasps. The current protocol is focused on delivering systemic bacterial infection. Many different bacteria can be used to infect flies, and the experimenter’s choice should be based on the precise scientific questions being asked. For example, human clinical isolates may be employed to study bacterial virulence mechanisms10, or ecologically relevant isolates may be preferred for evolutionary study.11 Some bacteria are competent pathogens of D. melanogaster, proliferating upon infection and causing host sickness or death. Other bacteria are effectively managed by the host immune system and cleared within a few days. In this demonstration, Providencia rettgeri will be used as a proliferative pathogen that can cause host mortality and persists in surviving hosts. Escherichia coli will be used as a non-pathogen that is cleared by the host immune system.
Infection will be established by introduction of bacteria directly into the body cavity of the fly. This approach bypasses epithelial barriers and protective behaviors, allowing investigation of systemic infection irrespective of the natural mode of transmission. There are two primary methods for experimentally establishing systemic infection. In the first, a nanoinjector and pulled glass capillary needles are used to inject a precise number of bacteria into the fly. This method has the advantages of allowing a large dynamic range of infection doses and of being quantitatively highly repeatable. The second approach is to deliver infection with a septic pinprick. This approach has the advantages of being rapid and requiring no special equipment. Once the infections are established, it becomes possible to measure systemic pathogen load, host mortality, and inducible immune system activity. Of course, any number of additional phenotypes could conceivably be measured in infected D. melanogaster, including post-infection fecundity12, learning ability13, metabolic status14, or virtually any other trait that can be imagined.
Protocol
1. Collect and Prepare Flies
Rear D. melanogaster under the desired experimental conditions. Take care not to overcrowd the flies during rearing and make sure that larval densities are consistent across treatments, since environmental conditions during the larval stage can profoundly affect immune defense phenotypes during adult stage.15
Collect experimental flies 0 - 3 days after eclosion from the pupal case and transfer them onto fresh medium.
House the collected flies at a desired temperature (temperatures between 22 °C and 28 °C are generally suitable) until they are aged 5 - 7 days post-eclosion. NOTE: This allows sufficient time for the flies to complete metamorphosis and become mature adults, but is well before senescence begins.
Sort the desired number of flies into a separate vial prior to infection, again taking care to avoid overcrowding. NOTE: Only males are infected in this demonstration but is equally possible to infect females using the procedure described.
2. Culture and Prepare Bacteria
Prepare a master plate of the chosen bacteria at least 2 days prior to infection. Streak the bacteria from a 15% glycerol stock stored indefinitely at -80 °C. Store the master plate at 4 °C for up to 2 weeks. Always streak the master plates directly from the frozen glycerol stock. Avoid serial passage of bacteria from plate to plate, as repeated passage in culture can cause bacteria to evolve attenuated virulence. NOTE: This example makes use of Providencia rettgeri and Escherichia coli.
- Use the following procedure to prepare a bacterial suspension for injection.
- Grow a 2 ml culture of bacteria by inoculating sterile medium with a single colony isolated from the master plate. Grow the bacteria to stationary phase (e.g. O/N growth at 37 °C). NOTE: Both P. rettgeri and E. coli grow well in Luria Broth at temperatures from 20 - 37 °C with gentle shaking.
- Once the culture has reached stationary phase, gently pellet cells from approx. 600 µl of the culture in a tabletop centrifuge (3 min at 5,000 x g), discard the supernatant, and resuspend the bacteria in approx. 1,000 µl of sterile phosphate buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2PO4, 1.8 mM KH2PO4, pH = 7.4).
- Dilute the resuspended cells in sterile PBS to achieve an optical density (OD) appropriate for the bacterium being used, measured with a spectrophotometer as the absorbance at 600 nm. Use this suspension to infect the flies. NOTE: A600 = 0.1 or 1.0 corresponds respectively to approximately 108 and 109 Providencia rettgeri or E. coli per ml. Because both live and dead bacteria contribute to optical density, it is important to harvest cultures early in stationary phase before bacterial corpses accumulate and distort the relationship between optical density and the number of viable bacteria introduced during infection.
3. Infect the Flies
NOTE: As Drosophila immunity is influenced by circadian rhythm, it is important to perform infections at a similar time of day across experimental replicates.16
3.1) Using a Nanoinjector
- Prepare the glass needles for infection.
- Prepare a glass needle by pulling a borosilicate glass capillary using a micropipette puller.
- Using forceps, break off the tip of the needle to create an opening of approximately 50 µm diameter to allow ejection of liquid.
- Assemble the injector.
- Place the sealing O-ring and then the white spacer (large dimple facing outwards) onto the metal plunger.
- Fill the glass needle with mineral oil using a syringe with a 30 G needle.
- Put the filled glass needle through the collet and then place the larger O-ring around its base, about 1mm from the blunt end of the needle.
- Slide the needle onto the metal plunger and gently screw on the collet until secure.
- Eject most of the mineral oil from the injector, but make sure that there is still a small volume of oil in the needle to act as a barrier between the injector and the bacterial suspension. Make sure that there are no air bubbles within the mineral oil or bacterial suspension, or between the two liquids.
- Set the injector to the desired volume for injection (between 9 nl and 50 nl).
- Generate wounding controls.
- Fill the injector needle with sterile PBS by carefully inserting the tip of the capillary needle in a tube of the media and pressing the “fill” button on the injector. NOTE: Sterile bacterial growth media can also be used a wounding control if the experiment requires injection of bacteria suspended in their growth medium. However, because bacterial growth media contains components that may stimulate the immune system or have other effects on the host, an inert carrier such as PBS is preferable.
- Anaesthetize the desired number of flies under a light flow of CO2.
- Inject the flies in the anterior abdomen on the ventrolateral surface with the sterile PBS. NOTE: It is also possible to inject flies at other sites, such as the sternopleural plate of the thorax, but it is important to keep the injection site consistent within each experiment.17
- Place injected flies into fresh vials with new medium, laying the vials on their side until all of the flies have recovered from the anesthesia to prevent the flies from becoming stuck to the food. NOTE: It is recommended to inject the PBS control flies before injecting bacteria to experimental flies so the same needle can be used for both treatments. It will not always be possible to use the same needle for an entire experiment. In that case, it may be desirable to record which needle was used with which flies and to include needle identity as an experimental factor in statistical analysis.
- Inject the bacterial pathogen.
- Eject the remaining sterile media from the injector and refill the same needle with the bacterial suspension.
- Repeat the above procedure (3.1.3), now injecting the flies with the bacterial suspension prepared in procedure 2.2.
3.2) With Septic Pinprick
- Prepare the needle for pricking.
- Melt the end of a 200 µl micropipette tip and insert a 0.15 mm insect minutien pin into the molten plastic. Allow the plastic to solidify such that the pin is held in place with approximately 0.5 cm of pin extending from the plastic.
- Generate wounding controls.
- Anaesthetize the desired number of flies under a light flow of CO2.
- Prick the flies in the sternopleural plate of the thorax with the needle, avoiding the attachment sites of the wings and the legs. If necessary, gently remove flies from the minutien pin using soft forceps. NOTE: It is also possible to prick the flies at other sites, such as the anterior abdomen on the ventrolateral surface, but it is important to keep the injection site consistent within each experiment.17 Pricking through the cuticle of the abdomen tends to be more difficult than pricking of the thorax and is therefore less common.
- Place the pricked flies into a fresh vial with new fly medium, laying the vial on its side until all of the flies have recovered from the anesthesia to prevent the flies from becoming stuck to the food.
- Introduce the bacterial infection.
- Anaesthetize the desired number of flies under a light flow of CO2.
- Prick each fly in the same location as the media controls, dipping the tip of the pin into the bacterial suspension prepared in procedure 2.2 before pricking each fly.
- Place the pricked flies into a fresh vial with new fly medium, laying the vial on its side until all of the flies have recovered from the anesthesia to prevent the flies from becoming stuck to the food.
3.3) Assess the Infectious Dose Delivered
Infect a set of flies as described above in either procedure 3.1 or 3.2, only instead of returning the flies to food vial to recover from anesthesia, place each fly into a microcentrifuge tube on ice.
Add 250 µl PBS to each tube and homogenize the flies either using a pestle or a bead beater.
- Plate the homogenate on an LB agar plate, either using a spiral plater or a serial dilution.
- To plate using serial dilution, transfer each fly homogenate to the first row of a 96 well plate.
- Fill each well of the remaining rows with 90 µl of PBS.
- Using a multichannel pipette, take 10 µl from the first row containing fly homogenate and dispense into the second row.
- Pipette up and down at least 10 times to thoroughly mix, and then take 10 µl and transfer to the third row. Repeat this procedure using the remaining rows.
- Starting from the bottom row (most dilute bacteria suspension), use the multichannel pipette to take 10 µl from each well and deposit on an LB plate, taking care that the samples are dispensed as discrete spots that do not touch each other. Repeat until all wells have been sampled from each row, dispensing them in descending order of dilution on the LB plate.
- Leave the plate at RT until the spots have completely soaked into the LB plate. NOTE: Drying the LB plate for a few days at RT prior to use is recommended to ensure that the liquid is readily absorbed, minimizing the chances of accidental contact between sample spots.
Incubate the plates O/N, taking care not to overgrow the plates so colonies remain small and discrete. NOTE: Depending on the bacterial growth medium and incubation temperature used, colonies derived from the fly’s endogenous gut microbiota may eventually appear on the plate. However, most bacteria used for pathogenic infection grow much faster than the gut microbiota on LB agar at 37 °C.
Remove plates from the incubator when the experimental bacteria have grown visible colonies (typically 8 - 12 hr), but before Drosophila gut microbiota colonies appear (approximately 36 hr). For slow growing experimental bacteria, use a selective antibiotic in the LB agar to remove any colonies from the gut microbiota.
- Count the number of colonies for each homogenate.
- For spiral plates, count the colonies that grow using an automated colony counter that can estimate the bacterial load per ml of homogenate based on the number and position of the colonies on the plate. NOTE: Spiral counts are most accurate when the bacterial concentration is in the range of 5 x 102 to 5 x 104 bacteria per ml.
- For spot plates, manually count the colonies for each fly from whichever dilution contains 30 - 300 colonies and calculate the number of bacteria per ml of original homogenate.
4. Characterize Survivorship of Infection
- Assay mortality post-injection.
- Place infected flies and wounded controls in fresh vials and maintain in an incubator at a desired temperature (25 °C with a 12:12 light:dark cycle is recommended). Do not put more than 15 flies into a single vial as it becomes difficult to count more than 15 living flies.
- Count the number of living and dead flies each day, or more frequently if appropriate.
- In order to maintain food quality, flip the flies to new food on a regular schedule.
- If the vial contains only males, transfer them to fresh vials every three days. If the vial contains females, transfer to fresh vials every two days because larval progeny will liquefy the food, increasing the risk that adult flies may get stuck and resulting in overestimated rates of mortality due to infection.
- Statistically analyze the data.
- Plot survival as a Kaplan-Meier curve, which indicates the probability that an individual will survive to a measured time-point post injection.18 For survival curves that do not cross, perform pair-wise comparisons using a Log-Rank Test (also called Mantel Cox Test) or perform more complex analyses using a Cox Proportional Hazards Model, which can incorporate several factors and their interactions. For survival curves that cross, perform a stratified Cox analysis.17
5. Assay Bacterial Load Post-infection
- Measure bacterial load.
- At a prescribed time post-injection, repeat the procedure used to assess infectious dose to determine bacterial load over the course of the infection (see protocol 3.3). NOTE: If using the spiral plater, dilute homogenates so that the bacterial suspension falls within a range of 1,000 - 100,000 bacterial per ml.
- Analyze the data.
- If bacterial load is non-normal, either transform the data to better approximate a normal distribution or analyze the data using non-parametric statistical analysis (e.g. Mann-Whitney U test). Use a Box-Cox analysis to find the optimal transformation; natural log or base -10 log transformations are often effective.19
- If the data are sufficiently normally distributed and there are only two treatments being contrasted, perform a t-test to determine whether the two treatments result in different mean bacterial loads. If there are multiple experimental variables or other potentially predictive factors, use Analysis of Variance (ANOVA) to determine which factors are significant predictors of bacterial load.19
6. Assay Transcriptional Activation of Immune System Genes
To coarsely visualize immune activation after infection, infect transgenic flies that express green fluorescent protein (GFP) under the control of a promoter from an antimicrobial peptide gene as previously described.18
View the infected flies under a fluorescence-capable microscope; GFP induction should become visible in the fat body within the first 6 - 10 hr after infection.
- For more precise quantification of immune activation, use quantitative PCR on cDNA template (qRT-PCR) to measure the abundance of antimicrobial peptide gene transcripts. NOTE: The expression of immune genes can be measured at any time after (or before) infection. Generally speaking, induction of immune gene expression begins approximately 4 hr after injection and increases over the next 24 hr.
- Anesthetize flies using CO2.
- Place 15 flies into a microfuge tube for each RNA extraction. NOTE: It is recommended to collect at least three replicate samples for each treatment condition.
- Extract RNA from the flies and generate first-strand cDNA using any of a number of standard procedures or commercial kits. Store RNA at -80 °C. Store cDNA at -20 °C for periods of a few weeks and at -80 °C for long term storage. Prior to RNA extraction, store flies at -80 °C.
- Perform quantitative PCR (qPCR) on target genes of interest using the cDNA as a template. Refer to Table 1 for primer sequences to amplify the antibacterial peptide genes Diptericin A, Drosomycin, Defensin, Attacin A, and Metchnikowin as well as the housekeeping control gene rp49 (also known as rpL32). NOTE: The genes encoding the antimicrobial peptides Diptericin A and Drosomycin provide very good readouts of D. melanogaster immune activity induced mainly through the Imd and Toll pathways, respectively. In order to control for sample-to-sample variation in RNA yield and the efficiency of reverse transcription, standardize the recorded expression of target genes against a reference housekeeping gene whose expression is not expected to change with infection such as rp49 (also known as rpL32).
- Analyze the data.
- For rough approximation of expression differences, calculate the ratio of the starting quantity (SQ) value obtained from the test gene (e.g., Diptericin A) relative to the SQ value obtained from the reference gene (e.g., rp49) for each sample (calculated as SQ-DptA/SQ-rp49). Scale these ratios to a baseline control treatment such as an uninfected handling control. Arbitrarily set the control expression level equal to 1.0 and visualize experimental treatments as relative fold changes. Estimate the SQ value for each gene against a standard curve generated from a dilution series of known-quantity cDNA. NOTE: Statistical analysis of ratios can be complicated, so this approach is better for quick approximation than for rigorous analysis. If PCR efficiency is low, design new primers to improve PCR kinetics if possible. For qPCR reactions with near-perfect efficiency (a doubling of PCR product every cycle), the threshold cycle (CT) can be substituted for the SQ value.
- For simple experiments with optimally efficient amplification, data may be analyzed using the ΔΔCT method.19 For each sample, subtract the CT value of the reference gene (e.g., rp49) from the CT of the test gene (e.g., Diptericin A) to yield a ΔCT. Then subtract the ΔCT of the baseline control from the ΔCT for each experimental sample to yield a ΔΔCT value for each sample; this ΔΔCT is indicative of relative differences in gene expression levels between treatments, where 2(-ΔΔCT) approximates the fold change in expression. NOTE: This analysis can badly misestimate fold-change in expression of the target gene if the PCR of either the test gene or the reference gene has low efficiency.
- For the most rigorous analysis, conduct analysis of variance (ANOVA) using the estimated expression of the test gene (e.g. Diptericin A) as the response variable and estimated expression of the control gene (e.g. rp49) and other experimental factors as explanatory variables. NOTE: This approach is relatively robust to inefficiency in PCR amplification and allows analysis of more complicated experimental designs.
Representative Results
This section illustrates results that can be obtained after bacterial infection of Drosophila melanogaster. Figure 1 shows that infection dose varies with optical density of the bacterial suspension used for injection, and that the dose delivered can be reliably estimated by homogenizing and plating flies immediately after injection. As illustrated in Figure 2, different pathogens can cause different levels of host mortality (Figure 2A) and host mortality can be dose-dependent (Figure 2B). Importantly, this protocol allows for different types of infections to be achieved: Providencia rettgeri can cause a chronic, sub-lethal infection that persists for 20 days or longer (Figure 3A). However, other bacteria like Escerichia coli will mostly be cleared by the host fly within six hours post-infection (Figure 3B). Induction of the immune system can be estimated through RNA isolation and subsequent qRT-PCR of antibacterial peptide transcripts (Figure 4A and B). Analogously but less quantitatively, flies expressing GFP under the control of antimicrobial peptide gene promoters can be used to visualize induction of the immune system (Figure 4C).
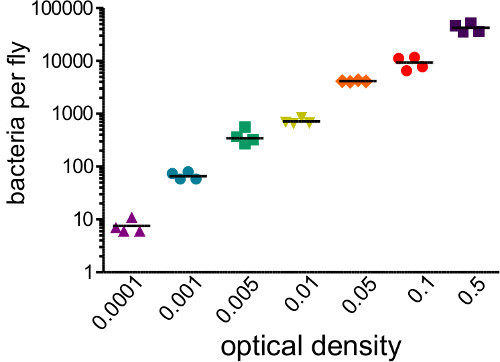 Figure 1: Determining Infectious Dose. Flies were injected with 50 nl of bacterial suspensions covering a range of optical densities (0.0001 - 0.05). The flies were immediately homogenized and plated to determine the number of bacterial introduced by the injection. Initial bacterial load strongly correlates with initial OD injected (r2 = 0.96)
Figure 1: Determining Infectious Dose. Flies were injected with 50 nl of bacterial suspensions covering a range of optical densities (0.0001 - 0.05). The flies were immediately homogenized and plated to determine the number of bacterial introduced by the injection. Initial bacterial load strongly correlates with initial OD injected (r2 = 0.96)
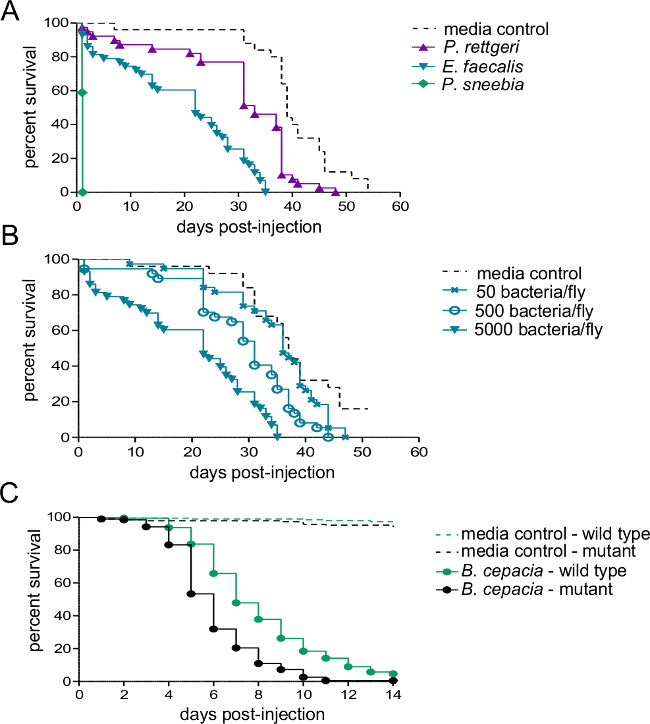 Figure 2: Survival after Injection with Bacterial Pathogens. Five to seven day old males were injected with 50 nl of either bacterial suspension or sterile media and monitored for survival. (A) Wild type flies were injected with approximately 5,000 bacteria from of one of three different species. The rate of host mortality depends heavily on the bacterial species used for infection. (B) Wild type flies were injected with three different doses of Enterococcous faecalis - 50, 500, and 5,000 bacteria per fly. Flies die more quickly when infected with higher infectious doses (C) An immune system mutant and its wild-type isogenic control line were injected with approximately 500 Burkholderia cepacia. A pairwise Log-Rank comparison shows that the wild-type fly survives the infection significantly better than the mutant (χ2 = 59.02, df = 1, p <0.0001).
Figure 2: Survival after Injection with Bacterial Pathogens. Five to seven day old males were injected with 50 nl of either bacterial suspension or sterile media and monitored for survival. (A) Wild type flies were injected with approximately 5,000 bacteria from of one of three different species. The rate of host mortality depends heavily on the bacterial species used for infection. (B) Wild type flies were injected with three different doses of Enterococcous faecalis - 50, 500, and 5,000 bacteria per fly. Flies die more quickly when infected with higher infectious doses (C) An immune system mutant and its wild-type isogenic control line were injected with approximately 500 Burkholderia cepacia. A pairwise Log-Rank comparison shows that the wild-type fly survives the infection significantly better than the mutant (χ2 = 59.02, df = 1, p <0.0001).
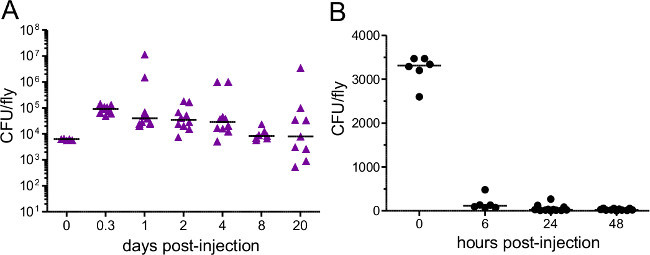 Figure 3: Bacterial Load after Injection. Flies were injected with approximately (A) 5,000 P. rettgeri (B) 3,400 E. coli. Bacterial load was determined immediately after injection and at various subsequent time points. Each data point represents the bacterial load of a single fly. E. coli is quickly cleared from challenged hosts while P. rettgeri persists for the remainder of the host’s life,.
Figure 3: Bacterial Load after Injection. Flies were injected with approximately (A) 5,000 P. rettgeri (B) 3,400 E. coli. Bacterial load was determined immediately after injection and at various subsequent time points. Each data point represents the bacterial load of a single fly. E. coli is quickly cleared from challenged hosts while P. rettgeri persists for the remainder of the host’s life,.
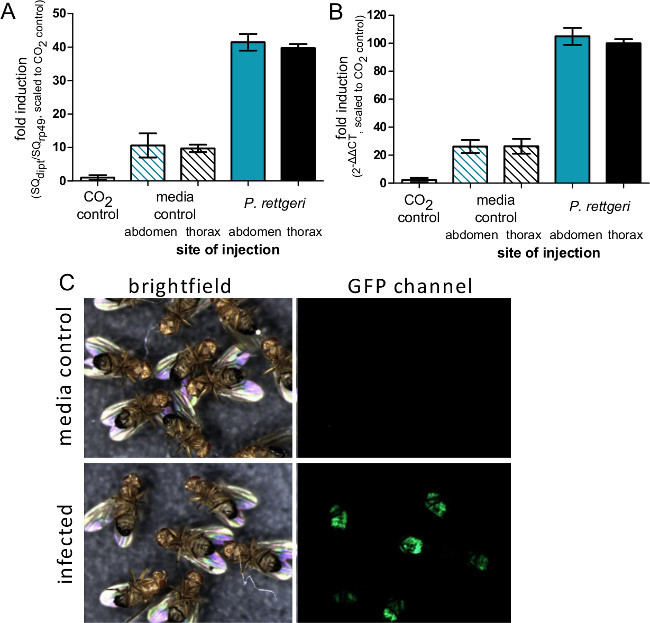 Figure 4: Induction of Immune Gene Expression after Injection. (A) Flies were injected with 50 nl of P. rettgeri or sterile PBS in either the abdomen or thorax,or were left unmanipulated aside from CO2 anesthesia. Six hours after infection, flies were collected for RNA isolation and expression of the Diptericin A gene was determined using qRT-PCR. (A) Expression levels are graphed as the ratio of Diptericin A transcript to rp49 transcript and scaled such that the CO2 control is defined as having an expression level of 1. The bars represent the mean and standard error of ratios from each condition (n = 4). (B) Expression levels are graphed as a 2ΔΔCT with the average CT value from CO2 aesthesia control flies used as the standard uninduced condition. The bars represent the mean and standard error of ΔΔCT from each condition (n = 4). While there is no difference in transcript induction due to site of injection, comparison of panels A and B shows how the ΔΔCT method can potentially overestimate induction levels. (C) Flies expressing GFP under control of the Diptericin A promoter were injected with 50 nl of P. rettgeri (OD = 0.1) and then imaged 7 days later. The GFP panel shows expression of GFP in the abdominal fat body of infected flies, indicating activation of the Diptericin A promoter in bacterially-infected flies but not media-injected controls.
Figure 4: Induction of Immune Gene Expression after Injection. (A) Flies were injected with 50 nl of P. rettgeri or sterile PBS in either the abdomen or thorax,or were left unmanipulated aside from CO2 anesthesia. Six hours after infection, flies were collected for RNA isolation and expression of the Diptericin A gene was determined using qRT-PCR. (A) Expression levels are graphed as the ratio of Diptericin A transcript to rp49 transcript and scaled such that the CO2 control is defined as having an expression level of 1. The bars represent the mean and standard error of ratios from each condition (n = 4). (B) Expression levels are graphed as a 2ΔΔCT with the average CT value from CO2 aesthesia control flies used as the standard uninduced condition. The bars represent the mean and standard error of ΔΔCT from each condition (n = 4). While there is no difference in transcript induction due to site of injection, comparison of panels A and B shows how the ΔΔCT method can potentially overestimate induction levels. (C) Flies expressing GFP under control of the Diptericin A promoter were injected with 50 nl of P. rettgeri (OD = 0.1) and then imaged 7 days later. The GFP panel shows expression of GFP in the abdominal fat body of infected flies, indicating activation of the Diptericin A promoter in bacterially-infected flies but not media-injected controls.
| Gene | Forward | Reverse |
| rp49 (also called rpL32) | 5’ AGGCCCAAGATCGTGAAGAA 3’ | 5’ GACGCACTCTGTTGTCGATACC 3’ |
| Diptericin A | 5’ GCGGCGATGGTTTTGG 3’ | 5’ CGCTGGTCCACACCTTCTG 3’ |
| Drosomycin | 5’ CTGCCTGTCCGGAAGATACAA 3’ | 5’ TCCCTCCTCCTTGCACACA 3’ |
| Defensin | 5’ GAGGATCATGTCCTGGTGCAT 3’ | 5’ TCGCTTCTGGCGGCTATG 3’ |
| Attacin A | 5’ CGTTTGGATCTGACCAAGG 3’ | 5’ AAAGTTCCGCCAGGTGTGAC 3’ |
| Metchnikowan | 5’ AACTTAATCTTGGAGCGATTTTTCTG 3’ | 5’ ACGGCCTCGTATCGAAAATG 3’ |
Table 1: Primers for qRT-PCR.
Discussion
The procedure described here yields rigorous and high quality infection of Drosophila melanogaster. The illustrated examples primarily focused on infection with Providencia rettgeri and E. coli, but the protocol is highly adaptable and can be applied to infections with diverse bacteria over a range of host rearing and maintenance conditions.
The details of an optimal experimental approach will depend on the bacterium used for infection, the genotype of the host, and the overall experimental conditions. It is strongly recommended to pilot test any new experimental conditions before initiating more ambitious projects. A good starting point is to test three infection doses over a 100-fold range. Highly virulent pathogens are often best introduced at very low infectious doses, on the order of 10 - 100 bacterial cells per fly. More moderate pathogens can be injected at higher doses of around 1,000 bacteria per fly, and non-pathogens may be injected at doses as high as 10,000 bacteria per fly. It is often instructive to define the kinetics of novel infections by tracking pathogen load, host mortality, and immune system activity over a longitudinal time series. Because measurement of pathogen load and host gene expression are destructive assays, it is necessary to infect distinct flies at the outset of the experiment for every time point that is to be measured.
When deciding whether to use pinprick or microcapillary-based injection, it is important to note there are advantages and limitations to each approach. Capillary injection introduces a volume of liquid into the fly, which both modestly increases turgor pressure and introduces salts or other molecules that are suspended or dissolved in the carrier. Capillary injection also requires access to an injection facility or purchase of the required equipment. Septic pinprick requires no special equipment and introduces negligible media into the fly, and is typically more efficient for infecting large numbers of flies. However, pinprick infections do not allow the precise control over infection dose that can be achieved with capillary injection. The present protocol focused on an injection apparatus that mechanically regulates injection volume, but there are also injection systems based on discrete pulses of pressurized air.20,21 These are typically more expensive than the apparatus featured here and require calibration of the air pulse to each needle in order to ensure consistent injection volumes.
There is considerable debate but very little data on how flies become systemically infected with bacteria in the wild. Some investigators posit that the majority of natural infections occur when Drosophila ingest pathogenic bacteria and the bacteria are subsequently able to escape the gut to establish a systemic infection. However, there are very few bacteria known to be able to cross the gut of D. melanogaster, and those that do have this ability are highly lethal to flies22,23. An alternative theory is that flies regularly sustain cuticular injuries through escape from failed predation attempts or attack by ectoparasitic mites. This hypothesis is supported by the frequent collection of wild D. melanogaster bearing melanization spots that are indicative of healed wounds (unpublished observations). Mites have been shown to transmit bacterial infections in Drosophila24 and wounds left by mites can be secondarily infected by bacteria in honey bees25. However, the frequency in nature of mite-driven or otherwise opportunistic infection of D. melanogaster through cuticle breaches is not known. The protocol described here allows introduction of bacteria directly into the hemolymph through quantitative injection which bypasses any epithelial barriers or behavioral immunity. Methods for feeding pathogenic bacteria to D. melanogaster have been described in Vodovar et al.22 and Nehme et al.23.
Many entomopathogenic bacteria pose little or no human health risk, allowing researchers to work with them comfortably. Furthermore, very few bacteria have the capacity to infect Drosophila on contact without experimental intervention, so the risk of “epidemic” spread of bacterial infection through a laboratory via contaminated surfaces or escaped flies is generally very low. Nonetheless, it is advisable to ensure that adequate containment measures are in place to prevent infected flies from escaping and for recapturing any escaped flies. The laboratory should be outfitted at a biological safety level commensurate with that of the pathogens being used, and standard best practices in microbiology should be employed.
The experimental infection method described here allows infections of Drosophila melanogaster with any dose of any arbitrary bacterium. Once infection has been established, it is straightforward to measure the kinetics of bacterial proliferation or clearance, to track host mortality, and to assay induction of the host immune system. Infected flies can easily be subjected to other phenotypic assays, including tests of physiological functions that may shape or be shaped by the infection. The procedures described are inexpensive, require relatively little specialized equipment, and are easily learned, making them amenable for use in diverse projects across a breadth of research and teaching labs.
Disclosures
None of the authors have competing interests or conflicting interests.
Acknowledgments
We would like to thank the entire Lazzaro lab, and especially Susan Rottschaefer, for their help in both reviewing and testing these protocols. This is a product of their cumulative expertise. Work in the Lazzaro lab is supported by grants R01 AI083932 and R01 AI064950 from the US National Institutes of Health.
References
- Lemaitre B, Hoffmann J. The host defense of Drosophila melanogaster. Annual review of immunology. 2007;25:697–743. doi: 10.1146/annurev.immunol.25.022106.141615. [DOI] [PubMed] [Google Scholar]
- Dionne MS, Schneider DS. Models of infectious diseases in the fruit fly Drosophila melanogaster. Disease models & mechanisms. 2008;1(1):43–9. doi: 10.1242/dmm.000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutschmann S, Jung AC, Hetru C, Reichhart JM, Hoffmann JA, Ferrandon D. The Rel protein DIF mediates the antifungal but not the antibacterial host defense in Drosophila. Immunity. 2000;12(5):569–580. doi: 10.1016/s1074-7613(00)80208-3. [DOI] [PubMed] [Google Scholar]
- Ayres JS, Freitag N, Schneider DS. Identification of Drosophila mutants altering defense of and endurance to Listeria monocytogenes infection. Genetics. 2008;178(3):1807–1815. doi: 10.1534/genetics.107.083782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronin SJF, et al. Genome-wide RNAi screen identifies genes involved in intestinal pathogenic bacterial infection. Science. 2009;325(5938):340–343. doi: 10.1126/science.1173164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyen C, Bretscher AJ, Binggeli O, Lemaitre B. Methods to study Drosophila immunity. Methods. 2014. [DOI] [PubMed]
- Dionne MS, Pham LN, Shirasu-Hiza M, Schneider DS. Akt and FOXO dysregulation contribute to infection-induced wasting in Drosophila. Current biology CB. 2006;16(20):1977–1985. doi: 10.1016/j.cub.2006.08.052. [DOI] [PubMed] [Google Scholar]
- McKean Ka, Yourth CP, Lazzaro BP, Clark AG. The evolutionary costs of immunological maintenance and deployment. BMC evolutionary biology. 2008;8(1):76. doi: 10.1186/1471-2148-8-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo T-H, Pike DH, Beizaeipour Z, Williams JA. Sleep triggered by an immune response in Drosophila is regulated by the circadian clock and requires the NFkappaB Relish. BMC neuroscience. 2010;11:17. doi: 10.1186/1471-2202-11-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panayidou S, Ioannidou E, Apidianakis Y. Human pathogenic bacteria, fungi, and viruses in Drosophila: disease modeling, lessons, and shortcomings. Virulence. 2014;5(2):253–269. doi: 10.4161/viru.27524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keebaugh ES, Schlenke TA. Insights from natural host–parasite interactions: The Drosophila model. Developmental & Comparative Immunology. 2014;42(1):111–123. doi: 10.1016/j.dci.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howick VM, Lazzaro BP. Genotype and diet shape resistance and tolerance across distinct phases of bacterial infection. BMC evolutionary biology. 2014;14(1):56. doi: 10.1186/1471-2148-14-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babin A, Kolly S, Kawecki TJ. Virulent bacterial infection improves aversive learning performance in Drosophila. Brain, behavior, and immunity. 2014. [DOI] [PubMed]
- Chambers MC, Song KH, Schneider DS. Listeria monocytogenes infection causes metabolic shifts in Drosophila melanogaster. PLoS ONE. 2012;7(12):e50679. doi: 10.1371/journal.pone.0050679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellous S, Lazzaro BP. Larval food quality affects adult (but not larval) immune gene expression independent of effects on general condition. Molecular ecology. 2010;19(7):1462–1468. doi: 10.1111/j.1365-294X.2010.04567.x. [DOI] [PubMed] [Google Scholar]
- Stone EF, Fulton BO, Ayres JS, Pham LN, Ziauddin J, Shirasu-Hiza MM. The circadian clock protein timeless regulates phagocytosis of bacteria in Drosophila. PLoS pathogens. 2012;8(1):e1002445. doi: 10.1371/journal.ppat.1002445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers MC, Jacobson E, Khalil S, Lazzaro BP. Thorax injury lowers resistance to infection in Drosophila melanogaster. Infection and immunity. 2014. [DOI] [PMC free article] [PubMed]
- Rich JT, Neely JG, Paniello RC, Voelker CCJ, Nussenbaum B, Wang EW. A practical guide to understanding Kaplan-Meier curves. Otolaryngology--head and neck surgery official journal of American Academy of Otolaryngology-Head and Neck Surgery. 2010;143(3):331–336. doi: 10.1016/j.otohns.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokal RR, Rohlf FJ. Freeman WH. Biometry. 1995.
- Frydman H. Wolbachia bacterial infection in Drosophila. Journal of visualized experiments JoVE. 2007. p. 158. [DOI] [PMC free article] [PubMed]
- Kuo T-H, Handa A, Williams JA. Quantitative measurement of the immune response and sleep in Drosophila. Journal of visualized experiments JoVE. 2012. p. e4355. [DOI] [PMC free article] [PubMed]
- Vodovar N, et al. Drosophila host defense after oral infection by an entomopathogenic Pseudomonas species. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(32):11414–11419. doi: 10.1073/pnas.0502240102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehme NT, et al. A model of bacterial intestinal infections in Drosophila melanogaster. PLoS pathogens. 2007;3(11):e173. doi: 10.1371/journal.ppat.0030173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenike J, Polak M, Fiskin A, Helou M, Minhas M. Interspecific transmission of endosymbiotic Spiroplasma by mites. Biology. 2007;3(1):23–25. doi: 10.1098/rsbl.2006.0577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanbar G, Engels W. Ultrastructure and bacterial infection of wounds in honey bee ( Apis mellifera) pupae punctured by Varroa mites. Parasitology research. 2003;90(5):349–354. doi: 10.1007/s00436-003-0827-4. [DOI] [PubMed] [Google Scholar]