

Understanding how different antiretrovirals reduce immune activation in human immunodeficiency virus type 1 infection is research priority. In this study, we found no consistent differential effects of raltegravir vs protease inhibitors on inflammation and immune activation among treatment-naive individuals after 96 weeks of therapy.
Keywords: protease inhibitors, integrase inhibitors, human immunodeficiency virus, inflammation, immune activation
Abstract
Background. It is unclear whether the integrase inhibitor raltegravir (RAL) reduces inflammation and immune activation compared with ritonavir-boosted protease inhibitors (PIs).
Methods. In a prospective, randomized, multicenter clinical trial that included 328 human immunodeficiency type 1 (HIV-1)–infected, treatment-naive participants were randomized to receive tenofovir disoproxil fumarate-emtricitabine (TDF/FTC) plus atazanavir/ritonavir (ATV/r), darunavir/ritonavir (DRV/r), or RAL. A total of 234 participants (71%) with HIV-1 RNA levels <50 copies/mL by week 24 were included. Plasma biomarkers of inflammation and coagulation that were analysed included high-sensitivity C-reactive protein, interleukin-6 (IL-6), GlycA, D-dimer, soluble CD14 (sCD14), sCD163, and sIL-2r; blood cellular markers included %CD38+DR+ of T-cell subsets and %CD14+CD16+ and%CD14(dim)CD16+ monocyte subsets. Changes from baseline were examined at earlier (24 or 48 weeks) and later (96 weeks) time points, with 95% confidence intervals on fold-change. Pairwise treatment groups were compared using Wilcoxon rank sum tests, with P values adjusted for false discovery rate control.
Results. Changes in biomarkers varied by regimen during the 96 weeks of follow-up as follows: hsCRP declined with ATV/r and RAL, IL-6 declined only with RAL, and GLycA decreased in all groups. D-dimer declined with ATV/r and DRV/r and was unchanged with RAL. Markers of T-cell activation and sCD163 (but not sCD14 and CD14-+CD16+) declined in all groups.
Conclusions. Despite some differences in specific markers of inflammation and immune activation between the antiretroviral therapy (ART) regimens, we found no consistent evidence that the reduction of inflammation and immune activation with ART initiation was different between RAL and PI-based regimens.
Clinical Trials Registration. NCT00811954 and NCT00851799.
Human immunodeficiency virus type 1 (HIV-1) infection is characterized by a state of inflammation and immune activation that persists despite suppressive antiretroviral therapy (ART) and may contribute to the development of end-organ disease [1, 2]. Persistent immune activation in HIV-infected persons on ART may be the result of 1 or more drivers of increased inflammation, such as microbial translocation through a damaged gut [3], coinfections [4], persistence of HIV replication in sanctuary sites, and/or abnormal production of bioactive lipids [5] and homeostatic cytokines [6].
Recently, biomarkers of inflammation (including interleukin-6 [IL-6], high-sensitivity C-reactive protein [hsCRP], and soluble CD14 [sCD14]) and coagulation (D-dimer) have been shown to correlate with all-cause mortality in HIV-1 infection [7–9]. Thus, there is increased interest to examine these biomarkers as predictors of clinical outcomes in this population [7–9]. Macrophage activation markers of sCD14 and soluble CD163 (sCD163) and the proinflammatory monocyte subsets, including nonclassical (or patrolling, CD14(dim)CD16+) and intermediate (CD14+CD16+) monocytes, have also been linked to cardiovascular disease (CVD) risk in HIV-1–infected persons [10–13]. T-cell activation (particularly CD38 expression on the CD8+ subset) is the best-defined correlate of mortality and disease progression in untreated HIV-1–infected persons [14]. Emerging evidence suggests that protein glycans such as GlycA, a novel biomarker of protein glycan N-acetyl groups, play a possible role in CVD and inflammation; however, their role in HIV-1 infection remains unknown [15]. Thus, further study of changes in biomarkers of systemic inflammation and immune activation during effective ART may enhance our understanding of the pathogenesis of end-organ disease in HIV-1–infected persons.
Understanding how different initial ART regimens reduce chronic immune activation in treated HIV-1 infection is an ongoing research priority. Several studies have shown a significant reduction in markers of microbial translocation in people randomized to raltegravir (RAL) compared with people randomized to nonnucleoside reverse transcriptase inhibitors (NNRTIs) [16–18]. However, prior switch and intensification studies with RAL have not consistently demonstrated reduced low-level viral replication or changes in systemic inflammation and immune activation [19, 20].
Our objective in this study was to characterize and evaluate longitudinally the changes in biomarkers of inflammation and immune activation among treatment-naive individuals undergoing randomized ART initiation with an integrase-based regimen containing RAL or a protease inhibitor–based regimen containing either atazanavir/ritonavir (ATV/r) or darunavir/ritonavir (DRV/r). Our study was part of the AIDS Clinical Trials Group (ACTG) A5260s, a prospective, metabolic substudy of the clinical trial ACTG A5257 [21].
METHODS
Study Design and Participants
ACTG A5260s was a substudy of ACTG 5257, a prospective, 14-week longitudinal evaluation of 328 HIV-infected, ART-naive persons aged ≥18 years without known CVD or diabetes mellitus, uncontrolled thyroid disease, or use of lipid-lowering medications. The design of this substudy has been previously reported [22, 23]. Participants were randomized equally to 1 of 3 regimens of tenofovir disoproxil fumarate-emtricitabine (TDF/FTC) plus ATV/r, DRV/r, or RAL. Randomization was stratified by screening HIV-1 RNA level (>100 000 or ≤100 000 copies/mL) and Framingham 10-year coronary heart disease risk score (<6% or ≥6% risk). The virologic, tolerability, and metabolic outcomes of these regimens were previously reported in A5257 [21]. The institutional review boards at all participating institutions approved the parent study and substudy (clinicalTrials.gov identifier: NCT00811954 and NCT00851799), and all participants provided written informed consent.
In this preplanned biomarker study, we were interested in understanding how markers of inflammation and immune activation change in response to successful ART. The A5260s population was restricted to the subset of virologically suppressed individuals. This cohort included participants who remained on randomized treatment throughout the substudy follow-up with no ART interruptions longer than 7 days and who achieved HIV-1 RNA suppression of <50 copies/mL by study week 24 and thereafter.
Biomarker and Laboratory Assessment
Blood samples were drawn at study entry prior to ART initiation and after 24, 48, 96, and 144 weeks on treatment. All blood samples were sent to core laboratories for processing. Plasma biomarkers were measured at the University of Vermont Laboratory for Clinical Biochemistry Research (Burlington) on batched plasma samples that were stored at −70°C. The samples had not been previously thawed and included hsCRP by nephelometry (interassay coefficient of variation (CV) range, 2.96%–6.24%), D-dimer with immunoturbidometric methods, and soluble IL-2 receptor (sIL-2R), sCD14, sCD163, and IL-6 by enzyme-linked immunosorbent assay (interassay CV for all range, 4.14%–14.07%). GlycA was quantified by nuclear magnetic resonance (NMR) spectroscopy at LipoScience (Raleigh, North Carolina). Intra- and interassay CVs were 1.9% and 2.6%, respectively.
Peripheral blood mononuclear cells (PBMCs) were isolated and cryopreserved for later analysis according to standard ACTG protocol [21]. Immunophenotyping was performed on these PBMCs using multicolor flow cytometry. The fluorochrome-conjugated antibodies (all from BD Biosciences, San Jose, California) were anti-CD3 phycoerythrin (PE)-Cy7 (clone SK7), anti-CD4 V450 (clone RPA-T4), anti-CD8 allophycocyanin (APC) (clone RPA-T8), anti-CD8 APC-Cy7 (clone SK1), anti-HLA-DR fluorescein isothiocyanate (clone L243), anti-CD38 PE (clone HB7), anti-CD14 APC (clone M5E2), and anti-CD16 PE-Cy7 (clone 3G8). Monocytes were identified as dump-gate (CD2, CD3, CD19, CD20, and CD56) negative and HLA-DR positive. Monocyte subpopulations were characterized as nonclassical (patrolling, CD14(dim)/CD16+) and intermediate (CD14+CD16+). CD4+ and CD8+ T-cell activation was determined as HLA-DR+CD38+. All samples had >75% viability; only live cells (as defined by 7-amino-actinomycin D negative staining) were included. An LSR-II flow cytometer (BD Biosciences) was used for all measurements, and data were analyzed using FlowJo software, version 9.3.3 (Treestar, Ashland, Oregon).
Statistical Analyses
Our primary objective was to characterize and compare changes in biomarkers of systemic inflammation and immune activation. Biomarkers were analyzed using all available measures at study entry (baseline) and further examined at weeks 24 and 96 (cellular markers), weeks 48 and 96 (plasma markers), and week 48 (GlycA). Measures outside the assay quantification limits were imputed at the respective limit. Biomarker changes over time were calculated as the mean difference of the on-treatment level compared with baseline on the log10 scale and back-transformed to represent mean fold-change from baseline. Evidence for change over time was assessed according to the bounds of the 95% confidence interval (CI), with 1 indicating no change. Shifts in the distribution of changes from baseline for all pairwise treatment group comparisons were evaluated using Wilcoxon rank sum tests and described as relative fold-change. While nominal P values are presented, P values adjusted using Benjamini–Hochberg methods to control the false discovery rate (FDR) are also provided [24], given the multiple comparisons across the multiple biomarkers for each study week. To provide additional error-rate control for the 3 pairwise comparisons, effect sizes are presented with 97.5% CIs and inference was guided by an FDR of 2.5%. Of note, since the study was not powered to detect effect sizes with such adjustment for multiple comparisons, consistency, direction, and magnitude of the effect sizes over time in conjunction with the nominal P values were considered in order to help distinguish true and false-positive findings. All analyses were performed with SAS, version 9.2 (SAS Institute, Cary, North Carolina).
RESULTS
Baseline Characteristics
Baseline demographic characteristics of the 328 participants from the A5260s study population were previously described [22, 23]. The 234 participants (71%) included in the virologically suppressed population for this analysis had similar baseline demographic characteristics across treatment groups (Figure 1, Table 1). A smaller sample size for the ATV/r group was driven by a higher rate of tolerability failure that was observed in this group [21]. Median age was 36 years; 90% were men; and 48% were white, 29% were black, and 19% were Hispanic. Median CD4+ cell count was 338 cells/mm3 and median HIV-1 RNA was 4.6 log10 copies/mL. Baseline levels of all biomarker parameters are presented in Table 1; no notable differences are apparent when the distributions and proportions are examined by treatment group.
Figure 1.

Study flow diagram. Abbreviations: ATV/r, atazanavir/ritonavir; DRV/r, darunavir/ritonavir; RAL, raltegravir.
Table 1.
Baseline Characteristics by Randomized Treatment Group
| Characteristic | Total (N = 234) | Atazanavir/Ritonavir (N = 68) | Raltegravir (N = 82) | Darunavir/Ritonavir (N = 84) |
|---|---|---|---|---|
| Demographics | ||||
| Sex, male | 210 (90%) | 63 (93%) | 73 (89%) | 74 (88%) |
| Age, y | 36 (28–45) | 38 (31–44) | 36 (27–45) | 36 (28–47) |
| Race/ethnicity | ||||
| White non-Hispanic | 112 (48%) | 35 (51%) | 36 (44%) | 41 (49%) |
| Black non-Hispanic | 68 (29%) | 21 (31%) | 23 (28%) | 24 (29%) |
| Hispanic, regardless of race | 45 (19%) | 11 (16%) | 16 (20%) | 18 (21%) |
| Medium/High 10-year risk of hard coronary heart disease, % (≥6%)a | 29 (12%) | 7 (10%) | 7 (9%) | 15 (18%) |
| Human immunodeficiency virus type 1 RNA, log10 copies/mL | 4.6 (4.0–5.0) | 4.8 (4.0–5.2) | 4.5 (4.0–4.9) | 4.6 (4.0–5.0) |
| CD4+ cell count, cells/mm³ | 338 (191–448) | 294 (180–461) | 347 (246–450) | 337 (172–424) |
| CD8+ cell count, cells/mm³ | 823 (578–1089) | 823 (577–1055) | 884 (607–1086) | 764 (545–1118) |
| Body mass index, kg/m² | 25 (22, 28) | 26 (23, 29) | 25 (22, 28) | 24 (22, 27) |
| Current smoker | 77 (33%) | 22 (32%) | 28 (34%) | 27 (32%) |
| Biomarkers (available measures) | ||||
| High-sensitivity C-reactive protein, μg/mL (n = 231) | 1.5 (0.8–3.2) | 1.8 (1.0–3.2) | 1.3 (0.7–3.4) | 1.3 (0.7–3.2) |
| IL-6, pg/mL (n = 231) | 1.7 (1.2–2.8) | 1.9 (1.2–2.9) | 1.5 (1.0–2.8) | 1.7 (1.3–2.8) |
| D-dimer, μg/mL (n = 232) | 0.3 (0.1–0.6) | 0.3 (0.1–0.5) | 0.3 (0.1–0.5) | 0.3 (0.2–0.6) |
| GlycA, μmol/L (n = 230) | 411 (366–451) | 420 (388–456) | 399 (363–446) | 408 (357–443) |
| %CD4+: CD38+HLADR+ (n = 212) | 18 (12–30) | 18 (12–28) | 19 (12–32) | 17 (10–30) |
| %CD8+: CD38+HLADR+ (n = 212) | 43 (34–54) | 40 (33–57) | 45 (37–52) | 43 (32–54) |
| sIL-2r, pg/mL (n = 232) | 1790 (1178–2310) | 1898 (1340–2291) | 1664 (1065–2435) | 1651 (1179–2242) |
| soluble CD14, ng/mL (n = 229) | 1688 (1454–2023) | 1799 (1486–2157) | 1663 (1438–1924) | 1646 (1451–2024) |
| soluble CD163, ng/mL (n = 217) | 1090 (773–1560) | 1144 (799–1550) | 1249 (898–1564) | 1012 (708–1548) |
| %MNC: CD14(dim)CD16+ (n = 212) | 62 (48–71) | 58 (46–65) | 62 (47–73) | 63 (54–72) |
| %MNC: CD14+CD16+ (n = 212) | 8 (6–13) | 10 (6–16) | 7 (4–13) | 8 (6–12) |
Median (first and third quartiles) or number (%).
Abbreviations: HLADR, human leukocyte antigen DR; IL, interleukin; MNC, monocyte.
a Hard indicates severe cardiovascular event.
Changes in Markers of Systemic Inflammation and Coagulation
Sustained decreases from baseline in hsCRP levels were evident with ATV/r and RAL. Specifically, levels at 96 weeks were on average approximately 35% lower than baseline levels. A persistent decline in IL-6 from baseline was apparent only in the RAL group. While decreases in IL-6 were observed over 48 weeks with ATV/r, these decreases were not apparent over 96 weeks. Changes from baseline in hsCRP and IL-6 were not observed with DRV/r. In contrast, while consistent decreases in D-dimer were seen with ATV/r and DRV/r, week 96 levels were on average 50% and 35% lower than baseline levels, respectively. A reduction in D-dimer was not apparent with RAL. On average, a 10% decline in levels of GlycA was apparent over 48 weeks with all treatment combinations (Table 2, Figure 2). There was a high degree of variability across participants in the observed magnitude of changes in all biomarkers.
Table 2.
Mean Fold-Change (95% Confidence Interval) From Baseline Over Time by Treatment Group
| Biomarker | Atazanavir/Ritonavir |
Raltegravir |
Darunavir/Ritonavir |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | Week 24 or 48 | N | Week 96 | N | Week 24 or 48 | N | Week 96 | N | Week 24 or 48 | N | Week 96 | |
| High-sensitivity C-reactive protein | 64 | 0.57 (0.40, 0.82) | 66 | 0.64 (0.46, 0.90) | 80 | 0.78 (0.59, 1.04) | 77 | 0.66 (0.51, 0.87) | 76 | 0.90 (0.69, 1.16) | 75 | 1.21 (0.91, 1.62) |
| IL-6 | 64 | 0.66 (0.52, 0.83) | 66 | 0.87 (0.69, 1.09) | 80 | 0.85 (0.67, 1.07) | 77 | 0.76 (0.65, 0.88) | 76 | 0.83 (0.67, 1.02) | 75 | 0.97 (0.78, 1.22) |
| D-dimer | 64 | 0.58 (0.42, 0.80) | 64 | 0.48 (0.35, 0.66) | 77 | 0.93 (0.72, 1.19) | 76 | 0.82 (0.65, 1.03) | 78 | 0.60 (0.44, 0.82) | 75 | 0.65 (0.48, 0.87) |
| GlycA | 67 | 0.89 (0.85, 0.93) | … | 79 | 0.90 (0.86, 0.94) | … | 78 | 0.91 (0.88, 0.94) | … | |||
| soluble CD14 | 63 | 1.00 (0.94, 1.06) | 65 | 0.96 (0.89, 1.04) | 80 | 0.91 (0.86, 0.96) | 77 | 0.89 (0.84, 0.94) | 75 | 1.00 (0.95, 1.05) | 74 | 0.96 (0.90, 1.03) |
| soluble CD163 | 61 | 0.56 (0.51, 0.61) | 62 | 0.50 (0.45, 0.56) | 76 | 0.59 (0.54, 0.64) | 73 | 0.55 (0.50, 0.61) | 71 | 0.61 (0.56, 0.67) | 70 | 0.58 (0.52, 0.65) |
| sIL-2r | 64 | 0.70 (0.61, 0.80) | 65 | 0.65 (0.58, 0.72) | 78 | 0.65 (0.58, 0.73) | 76 | 0.61 (0.54, 0.70) | 78 | 0.70 (0.62, 0.80) | 75 | 0.65 (0.57, 0.74) |
| %CD4+: CD38+HLADR+ | 58 | 0.47 (0.42, 0.53) | 61 | 0.33 (0.28, 0.39) | 65 | 0.53 (0.46, 0.62) | 70 | 0.38 (0.33, 0.45) | 73 | 0.53 (0.47, 0.61) | 72 | 0.36 (0.31, 0.42) |
| %CD8+: CD38+HLADR+ | 58 | 0.49 (0.44, 0.56) | 61 | 0.33 (0.29, 0.37) | 65 | 0.55 (0.50, 0.61) | 70 | 0.36 (0.30, 0.42) | 73 | 0.52 (0.47, 0.58) | 72 | 0.34 (0.29, 0.39) |
| %MNC: CD14(dim)CD16+ | 58 | 1.06 (0.99, 1.14) | 61 | 1.08 (1.00, 1.18) | 65 | 1.15 (1.05, 1.25) | 70 | 1.09 (1.01, 1.17) | 73 | 1.05 (0.99, 1.12) | 71 | 1.08 (1.00, 1.17) |
| %MNC: CD14+CD16+ | 58 | 0.58 (0.46, 0.72) | 61 | 0.58 (0.47, 0.70) | 65 | 0.93 (0.71, 1.23) | 70 | 0.85 (0.66, 1.10) | 73 | 0.78 (0.61, 1.02) | 71 | 0.71 (0.54, 0.93) |
GlycA was not assayed at week 96. N gives the number of participants with baseline and follow-up markers available for calculation of change.
Values in italics indicate statistically significant observations.
Abbreviations: HLADR, human leukocyte antigen DR; IL, interleukin; MNC, monocyte.
Figure 2.
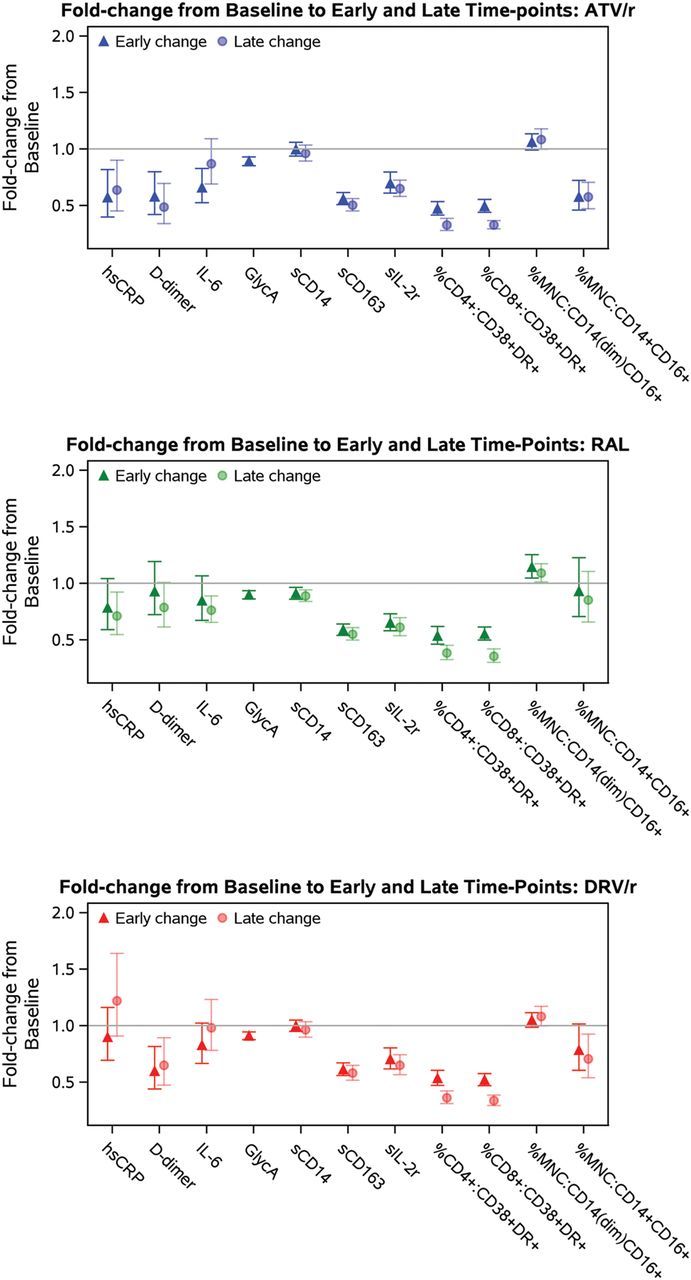
Differential effects of antiretroviral therapy on markers of inflammation and coagulation and markers of immune activation. Point estimates and error bars reflect mean and 95% confidence intervals, respectively. GlycA was not assayed at week 96. Early change represents change from baseline to week 24 (cellular markers) or week 48 (plasma markers); late change represents change from baseline to week 96 (soluble and cellular markers). Abbreviations: ATV/r, atazanavir/ritonavir; DRV/r, darunavir/ritonavir; hsCRP, high-sensitivity C-reactive protein; IL, interleukin; MNC, monocytes; RAL, raltegravir; sCD14, soluble CD14; sCD163, soluble CD163.
Although formal treatment group comparisons of changes in these markers of inflammation and coagulation did not hold after FDR adjustment at all time points, the consistency in the magnitude and direction of the effects is suggestive of treatment group differences for the comparisons of ATV/r vs DRV/r and DRV/r vs RAL for hsCRP, as well as for each protease inhibitor (PI)/r regimen vs RAL for D-dimer (Table 3). No other treatment group differences were apparent.
Table 3.
Pairwise Treatment Group Comparisons by Biomarker and Study Week
| Week 24 |
Week 48 |
Week 96 |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Biomarker | Mean (97.5% CI) | Pw Value | PFDR Value | Mean (97.5% CI) | Pw Value | PFDR Value | Mean (97.5% CI) | Pw Value | PFDR Value |
| ATV/r vs DRV/r | |||||||||
| hsCRP | 0.64 (.39, 1.04) | .039 | .58 | 0.53 (.32, .88) | .007 | .08 | |||
| IL-6 | 0.80 (.56, 1.14) | .15 | .58 | 0.89 (.62, 1.29) | .49 | .82 | |||
| D-dimer | 0.97 (.58, 1.61) | .78 | .99 | 0.74 (.45, 1.21) | .25 | .61 | |||
| GlycA | 0.98 (.92, 1.04) | .29 | .37 | ||||||
| sCD14 | 1.00 (.91, 1.09) | .96 | .99 | 1.00 (.89, 1.12) | .96 | .96 | |||
| sCD163 | 0.91 (.78, 1.06) | .12 | .58 | 0.87 (.73, 1.04) | .07 | .42 | |||
| sIL-2r | 0.99 (.80, 1.23) | .79 | .99 | 1.00 (.82, 1.22) | .72 | .96 | |||
| %CD4+: CD38+HLADR+ | 0.88 (.72, 1.08) | .18 | .32 | 0.90 (.70, 1.17) | .67 | .84 | |||
| %CD8+: CD38+HLADR+ | 0.95 (.80, 1.13) | .24 | .32 | 0.98 (.79, 1.20) | .55 | .84 | |||
| %MNC: CD14(dim)CD16+ | 1.01 (.91, 1.12) | .23 | .32 | 1.00 (.88, 1.14) | .32 | .76 | |||
| %MNC: CD14+CD16+ | 0.74 (.49, 1.10) | .08 | .31 | 0.82 (.55, 1.21) | .32 | .76 | |||
| ATV/r vs RAL | |||||||||
| hsCRP | 0.73 (.44, 1.22) | .60 | .77 | 0.97 (.59, 1.57) | .73 | .80 | |||
| IL-6 | 0.78 (.53, 1.13) | .16 | .43 | 1.14 (.85, 1.55) | .49 | .69 | |||
| D-dimer | 0.62 (.39, .99) | .015 | .13 | 0.59 (.38, .91) | .001 | .015 | |||
| GlycA | 0.99 (.93, 1.06) | .74 | .74 | ||||||
| sCD14 | 1.10 (1.00, 1.21) | .012 | .13 | 1.08 (.97, 1.20) | .06 | .13 | |||
| sCD163 | 0.95 (.82, 1.10) | .18 | .43 | 0.92 (.78, 1.08) | .09 | .19 | |||
| sIL-2r | 1.07 (.88, 1.31) | .48 | .71 | 1.06 (.87, 1.29) | .24 | .40 | |||
| %CD4+: CD38+HLADR+ | 0.88 (.71, 1.10) | .26 | .39 | 0.86 (.66, 1.11) | .42 | .77 | |||
| %CD8+: CD38+HLADR+ | 0.89 (.75, 1.06) | .11 | .23 | 0.93 (.73, 1.17) | .86 | .93 | |||
| %MNC: CD14(dim)CD16+ | 0.93 (.81, 1.06) | .68 | .68 | 0.99 (.88, 1.13) | .48 | .77 | |||
| %MNC: CD14+CD16+ | 0.62 (.41, .94) | .011 | .09 | 0.68 (.46, .99) | .007 | .06 | |||
| DRV/r vs RAL | |||||||||
| hsCRP | 1.14 (.74, 1.77) | .046 | .18 | 1.82 (1.16, 2.86) | <.001 | .012 | |||
| IL-6 | 0.98 (.68, 1.40) | .84 | .94 | 1.28 (.94, 1.74) | .12 | .28 | |||
| D-dimer | 0.64 (.41, 1.01) | .029 | .18 | 0.79 (.51, 1.21) | .046 | .15 | |||
| GlycA | 1.01 (.95, 1.08) | .52 | .55 | ||||||
| sCD14 | 1.10 (1.01, 1.20) | .008 | .07 | 1.08 (.98, 1.20) | .050 | .15 | |||
| sCD163 | 1.04 (.90, 1.20) | .99 | .99 | 1.06 (.89, 1.25) | .82 | .95 | |||
| sIL-2r | 1.08 (.89, 1.32) | .34 | .58 | 1.06 (.86, 1.31) | .41 | .55 | |||
| %CD4+: CD38+HLADR+ | 1.00 (.81, 1.24) | .98 | .98 | 0.95 (.73, 1.22) | .71 | .93 | |||
| %CD8+: CD38+HLADR+ | 0.94 (.80, 1.11) | .80 | .98 | 0.95 (.74, 1.21) | .81 | .93 | |||
| %MNC: CD14(dim)CD16+ | 0.92 (.81, 1.03) | .12 | .45 | 0.99 (.88, 1.12) | .68 | .93 | |||
| %MNC: CD14+CD16+ | 0.84 (.55, 1.30) | .25 | .50 | 0.83 (.54, 1.27) | .18 | .82 | |||
Relative mean fold-change from baseline with 97.5% confidence interval. Pairwise treatment group comparisons were separately conducted for the group of plasma biomarkers and for the group of GlycA and cellular biomarkers.
Values in italics indicate statistically significant observations.
Abbreviations: ATV/r, atazanavir/ritonavir; CI, confidence interval; DRV/r, darunavir/ritonavir; HLADR, human leukocyte antigen DR; hsCRP, high-sensitivity C-reactive protein; IL, interleukin; MNC, monocyte; PFDR, adjusted P values using Benjamini–Hochberg methods to control false discovery rate; Pw, unadjusted P values using Wilcoxon rank-sum test; RAL, raltegravir; sCD14, soluble CD14; sCD163, soluble CD163.
Changes in Markers of Macrophage and T-Cell Activation
Declines of a consistent magnitude from baseline over time were evident for cellular (%CD38+Human Leukocyte Antigen DR [HLADR]+ on CD4+ and CD8+ T cells) and plasma (sIL-2r) biomarkers of T-cell activation across all treatment groups (Table 2, Figure 2). After 24 weeks, mean expression of %CD38+HLADR+ on CD4+ and CD8+ T cells was approximately 50% lower than baseline levels, while sIL-2r levels after 48 weeks were at least 25% lower than baseline levels. The magnitude of decline was even greater for these markers after 96 weeks in all treatment groups.
sCD14 decreased to levels 10% lower than baseline with RAL at weeks 48 and 96, while changes with the PI/r regimens were not apparent. In contrast, sCD163 decreased to at least 40% lower than baseline levels at week 96 across all treatment groups. A sustained decrease in frequency of intermediate CD14+CD16+ monocytes was evident in the ATV/r and DRV/r groups; after 96 weeks, mean expression was 40% and 30% lower than baseline, respectively. Increased expression of nonclassical monocytes CD14(dim)CD16+ was apparent across treatment groups over time, although this change was modest in magnitude (Table 2, Figure 2).
In general, differences in changes in these markers over time between treatment groups were not apparent (Table 3). One exception included a greater decline in sCD14 with RAL compared with ATV/r or DRV/r, although this difference was less than 10% in magnitude. Additionally, a marked decline in expression of inflammatory CD14+CD16+ with ATV/r compared with RAL was apparent at week 48 and sustained at week 96.
DISCUSSION
In this prospective study of ART-naive participants who initiated RAL, ATV/r, or DRV/r with TDF/FTC and successfully achieved virologic suppression, no consistent pattern in differences in biomarkers emerged that favored any of the ART regimens. To our knowledge, this is the most comprehensive study to describe changes in immune activation (both monocyte and lymphocyte) and inflammation after ART initiation and successful immune suppression with these agents. We focused our analyses on markers such as hsCRP, D-dimer, sCD14, and IL-6 that have been associated with serious clinical events in HIV-1–infected persons, including CVD and mortality [25, 26]. When comparing these 3 regimens, no clear pattern of differences in inflammation markers emerged and no consistent decreases in all measured markers were evident. In particular, our findings suggest that RAL did not have a more comprehensive impact on decreasing systemic inflammation and immune activation compared with PIs. Similarly, treatment group differences between the 2 PI-based regimens were inconsistent. In general, because some markers remained elevated despite successful ART therapy, our results also suggest incomplete reversal of inflammation and immune activation in the setting of effective treatment.
Other studies have suggested that integrase inhibitors may reduce inflammation more effectively than other antiretroviral agents [16, 27–29]. In the SPIRAL trial of 233 (119 RAL, 114 PI/r) otherwise healthy, virologically suppressed HIV-infected patients treated with PI/r who randomly switched from PI/r to RAL or continued with PI/r for 48 weeks, hsCRP, IL-6, tumor necrosis factor-alpha, and D-dimer decreased in the RAL group relative to the PI/r group [30]. Consistent findings show a rapid decrease of viral load in the first weeks of RAL treatment, with an almost absent second-phase decay [31, 32]. Although the reason for this effect remains unclear, the possibility of a differential effect of RAL on HIV-1 reservoirs, including monocytes and macrophages, has been suggested [33]. However, RAL intensification has demonstrated inconsistent improvements in T-cell activation [28, 29] and improved d-dimer [29] and lipopolysaccharide (LPS) levels [34] but not sCD14 levels [20, 34]. Despite the potential mechanisms mentioned above, our study does not corroborate any advantage of the integrase inhibitor RAL over PIs on reducing immune activation during the first 96 weeks of treatment. A strength of our study is the enrollment of ART-naive participants. Thus, the investigation of the effect of RAL vs PIs on immune activation is not confounded by prior ART experience; this is the case for ART switch or intensification-related studies. Unblinded RAL switch studies cannot rule out improvements in adherence that might be immunologically meaningful even with undetectable viremia. In addition, the higher background noise with regard to systemic inflammation during early ART may also explain the discrepancies in our findings compared with those from prior RAL switch studies. A limitation is also the relatively short follow-up period for patients who will remain on life-long therapy.
Inflammatory and thrombotic markers such as IL-6 and D-dimer are attracting increasing attention for their potential as predictive markers of HIV disease progression or death [9, 35, 36]. However, despite predicting mortality, we demonstrated no apparent treatment group differences with IL-6 and some differences with D-dimer for each PI/r regimen vs RAL. Thus, despite a theoretical advantage of integrase inhibitors to reduce macrophage products such as IL-6 [37], their effect on circulating plasma levels of markers such as IL-6 or D-dimer is inconsistent; these biomarkers may remain elevated despite suppressive ART [38].
We also found that levels of systemic immune activation as measured by cellular coexpression of CD38 and HLA-DR, sIL-2r, a plasma marker of T-cell activation, and sCD163 declined similarly across treatment groups. These data support the growing evidence that effective ART reduces immune activation. Prior studies have shown greater effects of RAL intensification on viral replication predominately in PI-treated patients [28, 29]. The decline of sCD14 with RAL is consistent with prior findings from smaller and shorter studies of treatment-naive patients that reported greater declines in sCD14 in RAL-based regimens compared with NNRTI-based regimens [16, 18] and with studies that reported a decrease in sCD14 (but not sCD163) after switching from an NNRTI or PI regimen to RAL [20, 39]. It is conceivable that the discordance seen between sCD163 and sCD14 results may be due to the fact that RAL has differential effects on reducing microbial translocation and not necessarily monocyte activation. Future studies should measure more specific indices of microbial translocation, such as LPS or measures of intestinal barrier integrity including intestinal fatty acid–binding protein.
Our study is also among the first to examine longitudinal changes in novel biomarkers of inflammation, such as GlycA in HIV-1 infection. The GlycA NMR signal originates from N-acetyl methyl groups of particular N-acetylglucosamine residues on the glycan branches of abundant acute-phase proteins (mainly α1-acid glycoprotein, haptoglobin, α1-antitrypsin, α1-antichymotrypsin, and transferrin), thereby serving as a composite biomarker of systemic inflammation. As described, GlycA levels decreased similarly across treatment groups. Recent data suggest that protein glycans such as GlycA may have a role in CVD and inflammation [15]. Future studies to determine the role of protein glycans in HIV-1 infection are warranted.
Interestingly, after initiation of ART, the pattern of changes in biomarkers of systemic inflammation over time appears similar to changes in markers of immune activation. For example, IL-6 and CRP reduced with ART over 48 weeks but then increased until week 96, so there was little residual effect of ART after 96 weeks. This pattern was different when compared with the effects of ART on markers of monocyte and T-cell activation, where there was marked durable suppression in levels over time. Thus, our study suggests that ART induces differential reductions on biomarkers of immune activation compared with biomarkers of systemic inflammation.
Our study had several limitations. As noted, this biomarker analysis was not the primary outcome of the A5260s study and thus was not powered to detect effect sizes with adjustment for multiple biomarker comparisons. However, in addition to inferences based on an FDR level of 2.5%, consistency, direction, and magnitude of the effect sizes over time in conjunction with the nominal P values were considered in order to help distinguish true and false-positive findings. Selection bias of A5260s participants when restricting to the cohort of virologically suppressed individuals is also possible. However, because the physiological variability when studying these biologic processes is high, we chose to examine the virologically suppressed cohort in order to minimize confounding due to uncontrolled viremia. Furthermore, our goal was to investigate changes in biomarkers of systemic inflammation and immune activation in the context of successful ART with these regimens. While longitudinal changes in plasma biomarkers were separately examined in the intention-to-treat population, the results did not demonstrate substantively different conclusions (data not included). We also did not control for concomitant medication use that may contribute to changes in these biomarkers (eg, statin, aspirin use), although it is noted that reported use of such agents was limited during study follow-up. Changes in systemic inflammation and immune activation that may occur during the initial phase of controlling HIV replication may not be present with longer follow-up. Thus, longer follow-up may be needed to determine the differential effects of ART on biomarkers of systemic inflammation and immune activation.
Also, our study included mostly of men, which may limit the generalizability of our findings. All study participants received TDF/FTC, but further research should investigate whether the effects of RAL would differ with alternative nucleoside reverse transcriptase inhibitor backbones. The levels of some biomarkers such as sCD14 were lower compared with those reported in previous studies that demonstrated end-organ events and similar to HIV-uninfected participants [40]. The lower levels of these plasma biomarkers may be related to the relatively younger age of our study population and differences in CD4 nadir count and ART exposure compared with those of the participants studied in previously reported cohorts [40]. Thus, it remains unknown whether any further decrease in certain biomarkers of inflammation and immune activation is clinically relevant. It is also recognized that the biomarkers reported in this analysis are not limited to discrete linear pathways but are part of a broader and interrelated group of biological systems that consist of multiple intermediary steps and feedback loops.
In conclusion, we found that differential changes in systemic inflammation and immune activation were not consistent after 96 weeks among treatment-naive individuals who initiated successful ART regimens of TDF/FTC with RAL, ATV/r, or DRV/r in this prospective study. Overall, these results suggest incomplete reversal of inflammation and immune activation in the setting of effective ART compared with ART-naive HIV-1–infected participants. These results also highlight the need to examine newer ART regimens with more potent antiinflammatory effects as a possible means to effectively prevent long-term comorbidities in the setting of HIV-1 infection.
Notes
Acknowledgments. The AIDS Clinical Trials Group (ACTG) 5260s team members included: H. Hodis, C. Godfrey, B. Jarocki, A. Benns, and K. Braun. We thank the staff and patients from the following hospitals who participate in ACTG (in alphabetical order): Beth Israel Deaconess Medical Center, Brigham and Womens Hospital, Case University Clinical Research Service (CRS), Duke University Medical Center, Harbor-University of California Los Angeles (UCLA) Medical Center, Houston AIDS Research Team CRS, John Hopkins Adult AIDS CRS, Metrohealth, New Jersey Medical School, New York University HIV/AIDS CRS, Northwestern University, Rush University Medical Center ACTG, The Ohio State University, The Ponce De Leon Center CRS, UCLA Care Center, University of California San Francisco AIDS CRS, University of Cincinnati, University of Colorado, University of North Carolina AIDS CRS, University of Pittsburgh CRS, University of Rochester ACTG AIDS Care, University of Southern California, University of Washington, Vanderbilt Therapeutics CRS, and Washington University.
Author contributions. J. S. C., J. H. S., G. A. M., T. T. B., and T. K. were responsible for the study concept and design. T. T. T. T., C. M., and H. J. R. carried out the statistical analyses. T. K., T. T. T. T., C. M., and G. A. M. drafted the manuscript. T. K., O. O. Y., M. P. D., R. M., J. H. S., J. S. C., G. A. M., and T. T. B. collected the data. All co-authors participated in discussions about the design of the study and interpretation of the findings and critically reviewed the manuscript.
Financial support. This research was supported by National Institutes of Health (NIH) grants HL095132, HL095126, AI 068636, AI068634, AI69471, and AI56933. The study received additional financial support from Gilead, Merck, Bristol Myers Squibb, and Janssen. The project described was supported by awards UM1 AI068634, UM1 AI068636, and UM1 AI106701 from the National Institute of Allergy and Infectious Diseases (NIAID) and supported by the National Institute of Mental Health and the National Institute of Dental and Craniofacial Research. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIAID or the NIH or any of the funders.
Potential conflicts of interest. T. T. B. has served as a consultant for Bristol-Myers Squibb (BMS), GlaxoSmithKline (GSK), Merck, Abbott, Gilead, and ViiV Healthcare and has received research funding from Merck and GSK. J. S. C. has served as a consultant for Gilead and has received research funding from Merck. R. M. has served as a consultant for Gilead and serves on a Data Safety Monitoring Board for Gilead. J. H. S. serves on a Data Safety Monitoring Board for Lilly. G. A. M. has served as consultant and speaker and has received research grants from BMS, Pfizer, Merck, Gilead, and GSK. H. J. R., C. M., and T. T. T. T. have no duality of interest disclosures. M. P. D. has served as a consultant for Gilead and Astra Zeneca and receives research funding from Gilead, ViiV Healthcare, and Merck. All other authors report no potential conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Brown TT, Tassiopoulos K, Bosch RJ, Shikuma C, McComsey GA. Association between systemic inflammation and incident diabetes in HIV-infected patients after initiation of antiretroviral therapy. Diabetes Care 2010; 33:2244–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McComsey GA, Kitch D, Daar ES, et al. Inflammation markers after randomization to abacavir/lamivudine or tenofovir/emtricitabine with efavirenz or atazanavir/ritonavir. AIDS 2012; 26:1371–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brenchley JM, Price DA, Schacker TW, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med 2006; 12:1365–71. [DOI] [PubMed] [Google Scholar]
- 4.Sacre K, Hunt PW, Hsue PY, et al. A role for cytomegalovirus-specific CD4+CX3CR1+ T cells and cytomegalovirus-induced T-cell immunopathology in HIV-associated atherosclerosis. AIDS 2012; 26:805–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rose H, Hoy J, Woolley I, et al. HIV infection and high density lipoprotein metabolism. Atherosclerosis 2008; 199:79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fry TJ, Connick E, Falloon J, et al. A potential role for interleukin-7 in T-cell homeostasis. Blood 2001; 97:2983–90. [DOI] [PubMed] [Google Scholar]
- 7.Boulware DR, Hullsiek KH, Puronen CE, et al. Higher levels of CRP, D-dimer, IL-6, and hyaluronic acid before initiation of antiretroviral therapy (ART) are associated with increased risk of AIDS or death. J Infect Dis 2011; 203:1637–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sandler NG, Wand H, Roque A, et al. Plasma levels of soluble CD14 independently predict mortality in HIV infection. J Infect Dis 2011; 203:780–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuller LH, Tracy R, Belloso W, et al. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med 2008; 5:e203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woollard KJ, Geissmann F. Monocytes in atherosclerosis: subsets and functions. Nat Rev Cardiol 2010; 7:77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Funderburg NT, Zidar DA, Shive C, et al. Shared monocyte subset phenotypes in HIV-1 infection and in uninfected subjects with acute coronary syndrome. Blood 2012; 120:4599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kelesidis T, Kendall MA, Yang OO, Hodis HN, Currier JS. Biomarkers of microbial translocation and macrophage activation: association with progression of subclinical atherosclerosis in HIV-1 infection. J Infect Dis 2012; 206:1558–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burdo TH, Lo J, Abbara S, et al. Soluble CD163, a novel marker of activated macrophages, is elevated and associated with noncalcified coronary plaque in HIV-infected patients. J Infect Dis 2011; 204:1227–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giorgi JV, Hultin LE, McKeating JA, et al. Shorter survival in advanced human immunodeficiency virus type 1 infection is more closely associated with T lymphocyte activation than with plasma virus burden or virus chemokine coreceptor usage. J Infect Dis 1999; 179:859–70. [DOI] [PubMed] [Google Scholar]
- 15.Akinkuolie AO, Buring JE, Ridker PM, Mora S. A novel protein glycan biomarker and future cardiovascular disease events. J Am Heart Assoc 2014; 3:e0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Asmuth DM, Ma ZM, Mann S, et al. Gastrointestinal-associated lymphoid tissue immune reconstitution in a randomized clinical trial of raltegravir versus non-nucleoside reverse transcriptase inhibitor-based regimens. AIDS 2012; 26:1625–34. [DOI] [PubMed] [Google Scholar]
- 17.Massanella M, Negredo E, Puig J, et al. Raltegravir intensification shows differing effects on CD8 and CD4 T cells in HIV-infected HAART-suppressed individuals with poor CD4 T-cell recovery. AIDS 2012; 26:2285–93. [DOI] [PubMed] [Google Scholar]
- 18.Asmuth DM, Ma ZM, Hayes T, et al. Raltegravir (RAL) therapy is associated with reduced microbial translocation (MT) and monocyte activation in HIV infected subjects naive to antiretroviral therapy (ART). AIDS 2014 [abstract WEPE013] In: 20th International AIDS Conference 20–25 July 2014 Melbourne, 2014. [Google Scholar]
- 19.Hatano H, Hayes TL, Dahl V, et al. A randomized, controlled trial of raltegravir intensification in antiretroviral-treated, HIV-infected patients with a suboptimal CD4+ T cell response. J Infect Dis 2011; 203:960–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lake J, McComsey G, Hulgan T, et al. Switch to raltegravir decreases soluble CD14 in virologically suppressed overweight women: the Women, Integrase and Fat Accumulation Trial. HIV Med 2014; 15:431–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lennox JL, Landovitz RJ, Ribaudo HJ, et al. Efficacy and tolerability of 3 nonnucleoside reverse transcriptase inhibitor-sparing antiretroviral regimens for treatment-naive volunteers infected with HIV-1: a randomized, controlled equivalence trial. Ann Intern Med 2014; 161:461–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown TT, Chen Y, Currier JS, et al. Body composition, soluble markers of inflammation, and bone mineral density in antiretroviral therapy-naive HIV-1-infected individuals. J Acquir Immune Defic Syndr 2013; 63:323–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stein JH, Brown TT, Ribaudo HJ, et al. Ultrasonographic measures of cardiovascular disease risk in antiretroviral treatment-naive individuals with HIV infection. AIDS 2013; 27:929–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Rl Stat Soc Ser B 1995; 57:289–300. [Google Scholar]
- 25.Duprez DA, Neuhaus J, Kuller LH, et al. Inflammation, coagulation and cardiovascular disease in HIV-infected individuals. PLoS One 2012; 7:e44454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Triant VA, Meigs JB, Grinspoon SK. Association of C-reactive protein and HIV infection with acute myocardial infarction. J Acquir Immune Defic Syndr 2009; 51:268–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patterson KB, Prince HA, Stevens T, et al. Differential penetration of raltegravir throughout gastrointestinal tissue: implications for eradication and cure. AIDS 2013; 27:1413–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buzon MJ, Massanella M, Llibre JM, et al. HIV-1 replication and immune dynamics are affected by raltegravir intensification of HAART-suppressed subjects. Nat Med 2010; 16:460–5. [DOI] [PubMed] [Google Scholar]
- 29.Hatano H, Strain MC, Scherzer R, et al. Increase in 2-long terminal repeat circles and decrease in D-dimer after raltegravir intensification in patients with treated HIV infection: a randomized, placebo-controlled trial. J Infect Dis 2013; 208:1436–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martinez E, D'Albuquerque PM, Llibre JM, et al. Changes in cardiovascular biomarkers in HIV-infected patients switching from ritonavir-boosted protease inhibitors to raltegravir. AIDS 2012; 26:2315–26. [DOI] [PubMed] [Google Scholar]
- 31.Hicks C, Gulick RM. Raltegravir: the first HIV type 1 integrase inhibitor. Clin Infect Dis 2009; 48:931–9. [DOI] [PubMed] [Google Scholar]
- 32.Murray JM, Emery S, Kelleher AD, et al. Antiretroviral therapy with the integrase inhibitor raltegravir alters decay kinetics of HIV, significantly reducing the second phase. AIDS 2007; 21:2315–21. [DOI] [PubMed] [Google Scholar]
- 33.Scopelliti F, Pollicita M, Ceccherini-Silberstein F, et al. Comparative antiviral activity of integrase inhibitors in human monocyte-derived macrophages and lymphocytes. Antiviral Res 2011; 92:255–61. [DOI] [PubMed] [Google Scholar]
- 34.Vallejo A, Gutierrez C, Hernandez-Novoa B, et al. The effect of intensification with raltegravir on the HIV-1 reservoir of latently infected memory CD4 T cells in suppressed patients. AIDS 2012; 26:1885–94. [DOI] [PubMed] [Google Scholar]
- 35.Tenorio AR, Zheng Y, Bosch RJ, et al. Soluble markers of inflammation and coagulation but not T-cell activation predict non-AIDS-defining morbid events during suppressive antiretroviral treatment. J Infect Dis 2014; 210:1248–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baker JV, Neuhaus J, Duprez D, et al. Changes in inflammatory and coagulation biomarkers: a randomized comparison of immediate versus deferred antiretroviral therapy in patients with HIV infection. J Acquir Immune Defic Syndr 2011; 56:36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van SJ. Interleukin-6: an overview. Annu Rev Immunol 1990; 8:253–78. [DOI] [PubMed] [Google Scholar]
- 38.Neuhaus J, Jacobs DR, Jr, Baker JV, et al. Markers of inflammation, coagulation, and renal function are elevated in adults with HIV infection. J Infect Dis 2010; 201:1788–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gupta SK, Mi D, Moe SM, Dube MP, Liu Z. Effects of switching from efavirenz to raltegravir on endothelial function, bone mineral metabolism, inflammation, and renal function: a randomized, controlled trial. J Acquir Immune Defic Syndr 2013; 64:279–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hunt PW, Sinclair E, Rodriguez B, et al. Gut epithelial barrier dysfunction and innate immune activation predict mortality in treated HIV infection. J Infect Dis 2014; 210:1228–38. [DOI] [PMC free article] [PubMed] [Google Scholar]