

Recessive mutations in NAGLU, which encodes α–N-acetylglucosaminidase, cause the severe childhood lysosomal disease mucopolysaccharidosis IIIB. Using whole-exome sequencing of four individuals, Tétreault et al. show that a NAGLU variant also segregates with a dominant late-onset painful sensory neuropathy, with affected individuals showing reduced α–N-acetylglucosaminidase activity.
Keywords: ataxia, axonal neuropathy, whole-exome sequencing, lysosomal disorder

Recessive mutations in NAGLU, which encodes α–N-acetylglucosaminidase, cause the severe childhood lysosomal disease mucopolysaccharidosis IIIB. Using whole-exome sequencing of four individuals, Tétreault et al. show that a NAGLU variant also segregates with a dominant late-onset painful sensory neuropathy, with affected individuals showing reduced α–N-acetylglucosaminidase activity.
Abstract
Late-onset painful sensory neuropathies are usually acquired conditions associated with common diseases. Adult presentations of known hereditary forms are often accompanied by other organ involvement. We recruited a large French-Canadian family with a dominantly inherited late-onset painful sensory neuropathy. The main clinical feature is recurrent leg pain that progresses to constant painful paraesthesias in the feet and later the hands. As it evolves, some patients develop a mild sensory ataxia. We selected four affected individuals for whole exome sequencing. Analysis of rare variants shared by all cases led to a list of four candidate variants. Segregation analysis in all 45 recruited individuals has shown that only the p.Ile403Thr variant in the α-N-acetyl-glucosaminidase (NAGLU) gene segregates with the disease. Recessive NAGLU mutations cause the severe childhood lysosomal disease mucopolysacharidosis IIIB. Family members carrying the mutation showed a significant decrease of the enzymatic function (average 45%). The late-onset and variable severity of the symptoms may have precluded the description of such symptoms in parents of mucopolysaccharidosis IIIB cases. The identification of a dominant phenotype associated with a NAGLU mutation supports that some carriers of lysosomal enzyme mutations may develop later in life much milder phenotypes.
Introduction
Late-onset polyneuropathies, and in particular painful sensory neuropathies, are a heterogeneous group of disorders often accompanying other systemic illnesses such as diabetes, and are rarely found to have a genetic basis (Hoeijmakers et al., 2012). On the other hand, dominant and recessive hereditary sensory and autonomic neuropathies (HSAN) have an early-onset and are usually painless (Axelrod and Gold-von Simson, 2007). The best characterized forms of HSAN are the group of HSANs, and the ones observed in familial amyloidosis, some lysosomal diseases (GM2 gangliosidosis; MIM 272800 and Fabry diseases; MIM 301500) and more rarely neuropathies associated with mitochondrial diseases such as in Charcot-Marie-Tooth disease type 2A (MIM 609260) due to MFN2 (MIM 608507) mutations (Sedel et al., 2007; Botez and Herrmann, 2010). Sensory ataxias are another group of sensory disorders where a peripheral or central sensory dysfunction will contribute to an ataxic gait (Spinazzi et al., 2010). The genetic work-up of late forms of painful adult-onset sensory neuropathy is limited. The advent of whole exome sequencing has opened the way to a rapid and cost-effective search for mutations for Mendelian disorders (Gilissen et al., 2011). In the case of consanguineous or very large families, exome sequencing has been particularly rewarding (Gilissen et al., 2011). The identification of a large family with many members affected by a dominant late-onset painful neuropathy prompted the use of exome sequencing to identify the mutated gene.
Material and methods
Patients
DNA was isolated from peripheral blood using Gentra Puregene blood kit (Qiagen) according to the manufacturer’s protocol. All participants signed an informed consent approved by the ethics board of the Centre de Recherche du CHUM (CRCHUM).
Exome sequencing
Whole exome sequencing was performed at Perkin Elmer sequencing facilities. Exons were captured using the Agilent SureSelect 50Mb capture kit (v4) and the sequencing was performed on an Illumina HiSeq2000. Data analysis was performed via Perkin Elmer webportal. Whole exome sequencing results were aligned to the Human reference genome (UCSC genome browser Hg19) using Burrows-Wheeler Alignment Tool (BWA v.0.6.2) (Li and Durbin, 2009). Duplicates were marked using Picard tools and Genome Analysis Toolkit (GATK v.1.6) (McKenna et al., 2010) was used for base-quality recalibration, local sequence realignment for indel optimization, and variant filtering to minimize base calling and mapping errors. Variant calling was performed using GATK UnifiedGenotyper. Finally variants were annotated using GATK. We obtain high quality sequencing results, with an average of 97.5% of the reads mapping to the reference sequence and 72% of the nucleotides covered at 30× or more (Supplementary Table 2).
Sanger sequencing
Variants were validated by Sanger sequencing at McGill University and Genome Quebec Innovation Centre (Montreal, Quebec, Canada).
Enzymatic assay
Enzyme activity was measure on isolated leucocytes from 38 individuals from this family (18 affected cases) by dosage of a fluorigenic substrate. The method was standardized in the Clinical Medical Genetics laboratory of the CHU-Sainte-Justine.
Results
We recruited a large French-Canadian family from the Saguenay region of Québec (45 individuals; 21 affected and possible cases) presenting a late-onset sensory painful neuropathy (Fig. 1A). The pedigree supports a dominant mode of transmission with intra-familial variability in age of onset and severity. None accepted a sural nerve biopsy. The main clinical features are summarized in Table 1. Cases where considered affected if they had evidence on examination of a peripheral neuropathy, such as decreased vibration sense or a loss of some deep tendon reflexes, whereas possible cases only had suggestive painful symptoms often accompanied by perturbed sleep. All the tested cases where later found to have a decreased NAGLU enzymatic function. The first symptoms are lower extremity pains, that are dysaesthetic and are not necessarily crampiform, that start between the ages of 18 and 61 years (mean 41). These symptoms evolve over decades into continuous painful feet, usually leading to medical consultation. Many years’ later distal upper extremity paraesthesias also appear. Poor sleep related to cramps and painful paraesthesias increases with age. Dysautonomic symptoms were only observed in Patient XX. This patient presented negative autonomic testing except for one documented hypotension on the Tilt-Table Test. The majority have stable blood pressure even late in the course of the condition. Distal weakness or muscle atrophy is not observed, even in the six cases older than 65 (Table 1). Deep tendon reflexes are preserved initially but with age are lost, starting distally. There is a stalking glove decreased sensation to touch and pinprick that progresses with age but never becomes severe enough to lead to ulcerations. Decreased vibration sense also evolves with time into a complete loss at the level of the toes. Position sense is largely preserved. There is no nystagmus or cerebellar signs on examination. Tandem walking becomes increasingly difficult in older patients. Nerve conduction studies and EMG in the younger cases are normal. With time they may develop decreasing sensory and later motor nerve amplitudes suggestive of a predominantly sensory axonal neuropathy. Electrophysiological changes appear late in the course and are not universally present in some of the older patients (Table 1). Case II was the only patient who had serial electrophysiological studies over 10 years. These show absent sural nerve conductions at age 55. At age 65, he presented progressive decreasing amplitudes and slowing of sensory and motor conductions accompanied with distal denervation in the feet and tibialis anterior (Supplementary Table 3).
Figure 1.

Family pedigrees. (A) Pedigree of Family 1, a large French-Canadian family originating from the Saguenay Lac-Saint-Jean region. The red circles indicate the unaffected individuals sequenced in the segregation analysis. (B) Pedigree for Family 2.
Table 1.
Clinical features of cases affected by a late-onset painful sensory polyneuropathy
| Family | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 2 | 2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Case number | I | II | III | IV | V | VI | VII | IX | X | XI | XII | XIII | XIV | XV | XVIII | XIX | XX | XXI | XXII | XXIII | XXIV |
| Status | A | A | A | P | A | A | P | A | A | P | P | P | P | A | P | A | A | P | A | A | A |
| Sex | M | F | F | M | F | F | F | F | F | M | M | F | M | M | M | F | F | F | M | M | F |
| NAGLU enzyme function | 0.99 | NT | 1.49 | 0.95 | 1.52 | 0.87 | 0.7 | 1.37 | 0.96 | 1.3 | 1.12 | 1.03 | 0.83 | 1.32 | 0.64 | 2.21 | 0.85 | 0.97 | NT | NT | NT |
| Year of birth | 1947 | 1948 | 1935 | 1986 | 1939 | 1944 | 1965 | 1942 | 1946 | 1957 | 1980 | 1955 | 1991 | 1938 | 1960 | 1973 | 1972 | 1979 | 1941 | 1937 | 1964 |
| Age exam | 61 | 60 | 75 | 24 | 74 | 69 | 47 | 70 | 67 | 55 | 32 | 57 | 21 | 75 | 53 | 40 | 41 | 34 | 60 | 62 | 43 |
| Age onset of daily leg pain | 52 | 48 | 60 | 20 | 35 | 61 | 44 | 61 | 49 | NDP | 30 | NDP | 19 | 50 | 45 | 18 | 30 | NDP | 12 | 18 | <18 |
| Lower leg pains | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | NT | + | + |
| Sleep disturbances | + | + | + | + | + | + | + | + | + | − | + | + | + | − | + | + | + | − | NT | NT | + |
| Sensory ataxia (age onset) | − | 50 | 65 | − | 65 | + | − | + | 64 | − | − | − | − | − | − | 34 | 38 | − | − | − | 35 |
| Loss of DTR | + | + | − | − | + | + | − | + | + | − | − | − | − | + | − | + | − | − | − | + | + |
| Decreased vibration sense | + | + | + | − | + | + | − | + | + | − | − | − | − | + | − | − | + | − | + | + | + |
| Pinprick level | + | + | + | − | + | − | − | + | + | − | − | − | − | − | − | − | + | − | − | NT | + |
| Nystagmus | − | − | − | − | − | − | − | − | − | NT | − | NT | NT | − | NT | − | − | − | − | − | − |
| Muscle weakness | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| Age of last EMG | 60 | 65 | 75 | 24 | 38 | 42 | |||||||||||||||
| Motor conduction | Norm | ↓ amp. | Norm | Norm | Norm | Norm | |||||||||||||||
| Sensory conduction | ↓ amp. | ↓ amp. | Norm | Norm | Norm | Norm | |||||||||||||||
| EMG | Norm | Denerv. | Norm | Norm | Norm | Norm |
A = affected; amp. = amplitude; Denerv = denervation; DTR = deep tendon reflexes; P = possible; Norm = normal; NT = not tested.
We selected four affected individuals (Cases I, II, III and IV) for whole exome sequencing analysis. No genotyping was performed prior to the sequencing. We first looked at genes known to be associated with a phenotype similar to our patients. No mutations were uncovered in the genes for HSAN1–5, all known CMTs, Fabry’s α-galactosidase gene nor the hexoaminidase A and B of GM2 gangliosidoses. We compared the sequencing results of all four affected cases focusing only on variants shared by all affected and inherited in a dominant manner. As familial hereditary painful neuropathies are rare diseases, we filtered against variant databases (dnSNP132, 1000genomes) to exclude common variants in humans and an in-house collection of 10 regional Saguenay exomes from cases recruited for other projects. We identified 21 non-synonymous and indel variants present in coding sequences and donor or acceptor splice-sites. These variants were manually filtered against dbSNP137 and the exome variant server database (EVS, NHBLI exome sequencing project) to further eliminate other common variants (MAF ≥ 0.5%). This last step of filtering led to a short list of four candidate variants (Supplementary Table 1). Among them, only the missense variant (c.1208T>C; p.Ile403Thr) in the α-N-acetyl-glucosaminidase (NAGLU) gene (MIM 609701, ReSeq accession number NM_000263.3) was predicted to be pathogenic by the bioinformatics programs. CADD score (Kircher et al., 2014) went in the same direction; we obtained a PHRED score of 19.41 predicting this variant to be in the 1% most deleterious. As in silico software prediction for pathogenicity should be taken with caution, we performed validation by Sanger sequencing for all four candidate variants. Segregation analysis in the family (45 individuals) showed that only the p.Ile403Thr mutation in exon 6 of NAGLU was segregating with the disease, with all 21 affected and possible cases carrying the missense mutation (Supplementary Fig. 1A). This variant has not previously been observed in any available databases or in over 50 French-Canadian control subjects. The missense mutation is present in a conserved region of the protein (Supplementary Fig. 1B). Sequencing of the exons and intron-exon boundaries of NAGLU in an additional 50 French-Canadian cases affected by a sensory ataxia or a painful neuropathy of unknown subtypes did not detect any other NAGLU variants.
To find additional cases affected by a neuropathy with a dominant variant in NAGLU, we searched the Genomes Management Application (GEM.app) at the University of Miami (Gonzalez et al., 2013). Using this database we identified, in a CMT2 patient (Case XXII) (Fig. 1B), a nonsense variant (p.Glu123X) creating a premature stop codon (Supplementary Fig. 1C). The whole exome analysis of this North-American family of European descent showed 38 autosomal-dominant candidate genes, including NAGLU and a heterozygous change in the known recessive CMT gene GDAP1 (MIM 606598) (p.Val243Met, c.727G>A). Segregation analysis on available DNA from this family confirmed the presence of the NAGLU p.Glu123X in all three available affected members (Cases XXII, XXIII and XXIV) (Fig. 1B). This second family showed an age of onset between 12 and 18 years (Table 1). Affected family members had mild muscular weakness, but diminished reflexes in the neurological examination in their early 40s. Sense for vibration was moderately reduced and pinprick and positioning of extremities mildly reduced. Several patients complained of pain as a leading symptom. Decreased visual acuity and hearing loss since childhood were inconsistent features. Case XXIV reported intermittent balance issues and a resting tremor in her arms for ‘several years’. All family members were lost for re-contact or not available for a more recent follow-up examination.
The NAGLU gene encodes the enzyme α-N-acetyl-glucosaminidase, which is involved in the degradation of the glycosaminoglycan compound heparan sulphate (Selmer et al., 2012). More than 100 recessive mutations in this gene have previously been associated with mucopolysaccharidosis IIIB disorder (MPS IIIB; MIM 252920), a severe childhood-onset neurodegenerative disease. Most patients affected by MPS IIIB have a complete loss of enzyme activity (<3%) (Selmer et al., 2012). Impaired function of the enzyme leads to the accumulation of partially degraded mucopolysaccharides in lysosomes, and increased excretion of heparan sulphate in urine (Yogalingam and Hopwood, 2001; Beesley et al., 2005; Selmer et al., 2012). Because of the association of NAGLU mutations with a recessive disease, we searched for a second mutation in Family 1. Sequencing of the entire cDNA and 93% of the intronic regions did not uncover any other putative pathological changes in the family. To confirm the biological impact of the p.Ile403Thr variant, we performed an enzymatic assay using a previously described method, consisting in the dosage of a fluorogenic substrate (Pollard et al., 2013), standardized in the Clinical Medical Genetics laboratory of the CHU-Sainte-Justine Hospital on isolated leucocytes of 42 individuals from the French-Canadian family (19 affected cases). A significant 36–54% (mean 45%, P > 0.001) decrease in enzyme activity was observed for all carriers of the mutation (Fig. 2) when compared with unaffected individuals from the family and external controls, confirming the biological impact of the p.Ile403Thr variant. No enzymatic assay was performed on Family 2.
Figure 2.
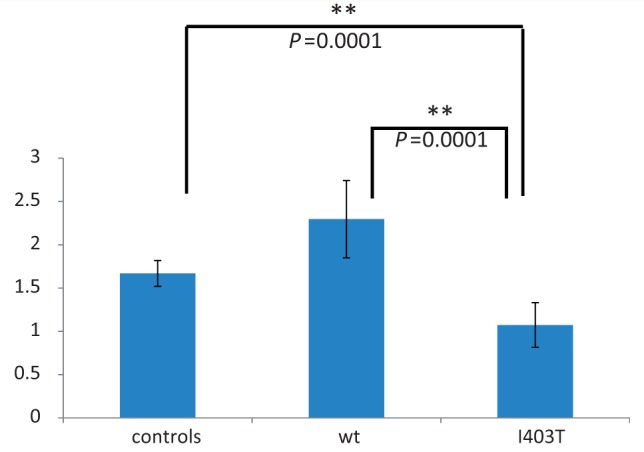
NAGLU enzymatic activity in leucocytes of affected and unaffected family members and controls. A significant decrease in activity is observed in affected cases when compared with those unaffected from the same family and external controls. We observe a 54% decrease when compared to unaffected cases (wt) and a 36% decrease compared to external controls (P > 0.001). Standard deviation and P-values were calculated using Graph Pad.
Recessive mutations causing MPS IIIB have been mostly associated with severe childhood phenotypes although attenuated phenotypes have been described (Valstar et al., 2010). Peripheral nerve involvement has not been described in severe cases or in the few published autopsies (Hadfield et al., 1980). However, sensory changes, ataxia and sleep disturbance have been described in some of the milder cases in a Norwegian family (Selmer et al., 2012). It has been suggested that missense mutations causing severe phenotype cluster in important functional domains (Meiyappan and Norton, 2012). The location of the p.Ile403Thr and p.Glu123X mutations on a linear protein representation shows that it maps to a regional cluster of mutations that are associated with severe MPS IIIB phenotypes (Fig. 3A). Four distinct domains are present in the NAGLU protein (Fig. 3A). The amino acid at position 403 is not part of the catalytic domain, but is present in the Tim-barrel domain. This domain is important for the formation of the catalytic site as well as the formation of the NAGLU homotrimer (Ochoa-Leyva et al., 2011; Meiyappan and Norton, 2012). Although until recently NAGLU was thought to function as a monomer, there is now structural evidence that it forms a trimeric active enzyme (Meiyappan and Norton, 2012). The location of the p.Ile403Thr mutation on the protein structure (Fig. 3B), as well as bioinformatics analysis (IMutant and MuPro), support an indirect impact of the mutation on enzyme activity by disturbing the protein’s stability as a homotrimer leading to either degradation of the protein in the lysosome or failure to exit the endoplasmic reticulum (Meiyappan and Norton, 2012). The nonsense variant in Family 2 truncates the protein in the N-terminal domain before the catalytic domain (Fig. 3A) eliminating any possibility of residual enzymatic activity.
Figure 3.

Location of mutations within the NAGLU protein. (A) Linear representation of the four domains (N-terminal, catalytic, Tim-barrel, and alpha-helical) of the NAGLU protein showing the position of the p.Glu123* variant in the N-terminal domain and the p.Ile403Thr variant in the Tim-barrel domain. Blue arrows represent the position of mutations associated with attenuated phenotypes and red arrows the ones causing severe phenotypes. (B) 3D structure of the human NAGLU protein showing the p.Ile403Thr mutation (dark blue) and severe mutations clusters (light blue). The red arrow indicates the catalytic site.
Discussion
Although recessive mutations in NAGLU have usually been associated with a severe lysosomal storage disorder affecting the CNS, in some milder cases evidence of a sensory polyneuropathy with ataxia has been documented (Selmer et al., 2012). Furthermore, diminished tactile and thermal perception accompanied by abnormal lysosomal storage in the dorsal root ganglion was documented in NAGLU knockout mice encouraging the authors to suggest that such peripheral involvement must also be present in patients (Fu et al., 2012). In cases with MPS IIIB the more severe neuropsychiatric symptoms have precluded the exploration of a subtle peripheral sensory neuropathy. Whether a pain syndrome contributes to the explosive behaviour of some patients would need to be explored. In the case of parents or other carriers in families of a severe mutation it is likely that they were lost to follow-up following the death of the affected case at a young age. Based on our results, dominant mutations in NAGLU can lead to a milder late-onset largely sensory axonal neuropathy that evolves into a sensory-motor CMT2 neuropathy. Other lysosomal disorders such as hexosaminidase enzyme A and B deficiency, that cause GM2 gangliosidosis diseases Tay-Sachs (MIM 272800) and Sandhoff (MIM 268800), respectively, can also be accompanied by a painful sensory polyneuropathy (Sedel et al., 2007).
To date, more than 100 recessive mutations have been associated with MPS IIIB, most of which were observed in single families (Selmer et al., 2012). Some mutations were shown to be associated with a severe phenotype but others were responsible for milder symptoms depending on the residual enzymatic level. Even in healthy individuals, the enzyme level seems to vary, and some polymorphisms have been associated with a high enzymatic activity (Pollard et al., 2013). This heterogeneity in enzyme activity could explain in part the phenotype variability observed within and between our families. Deleterious variants in NAGLU are observed in public databases such as ExAC. We believe that a proportion of these individuals could develop a painful neuropathy phenotype later in life. The presence and severity of symptoms will in part depend on the enzyme activity level conferred by the normal allele. The diagnosis of such diseases is difficult due to the late onset.
There is growing evidence that heterozygote carriers of some recessive mutations may also develop a phenotype. This is the case in another lysosomal storage disorder, Gaucher disease, where heterozygote carriers of acid beta-glucosidase (GBA, MIM 230800) mutations have an increased risk of developing Parkinson’s disease (Bras et al., 2012). In HSAN2A (MIM 201300) heterozygote carriers of WNK1/HSN2 (MIM 605232) mutations are more sensitive to detect temperature changes (Loggia et al., 2009); while in HSAN4 (MIM 256800) and NTRK1 (MIM 191315) mutation carriers are predisposed to familial carpal tunnel syndrome (Minde et al., 2004). The best example of an intermediate milder phenotype in heterozygous carriers is in HSAN5 (MIM 608654) caused by mutations in the nerve growth factor beta (NGF, MIM 162030) (Rotthier et al., 2012). HSAN5 is a rare severe childhood sensory neuropathy predominantly affecting small-diameter fibres. Recessive patients respond normally to touch, pressure, and vibration, but have a selective loss of pain and temperature sensation leading to painless fractures, bone necrosis and neuropathic joint destruction. In heterozygote carriers, the phenotype presents in adulthood with variable symptoms of neuropathy but without complete insensitivity to pain (Minde et al., 2004).
This study reports that carriers from two families of a severe pathogenic mutation in NAGLU develop a late dominant painful axonal sensory neuropathy that evolves into a mild sensory ataxia with late electrophysiological signs of a concomitant motor polyneuropathy that together would allow it to be referred to as a CMT2. The late dominant phenotype could be caused by a very slow neurotoxicity due to mucopolysaccharide (GAG) accumulation with secondary accumulation of tau, GM2 and GM3 in lysosomes, as in MPS IIIB. The only PNS manifestations in these patients, as opposed to the major CNS disease observed in MPS IIIB, may reflect that they are masked in the recessive form by the extensive neurodegenerative involvement as observed in the knockout mice (Fu et al., 2012). As few genetic causes of late-onset sensory neuropathies have been uncovered (Hoeijmakers et al., 2012), this report raises the possibility that others will also be dominant lysosomal storage diseases in carriers of single gene defects. Comparing variants uncovered by whole exome sequencing in large undiagnosed cohorts may accelerate the identification of such dominant mutations. NAGLU should be sequenced in all predominantly late-onset sensory CMT2 cases with other family members with painful sensory symptoms. This emphasizes that dominant mutations in genes causing severe recessive metabolic disorders leading to CNS and PNS disorders should not be passed over when sieving through next generation sequencing data in cohorts with milder neurological phenotypes.
Supplementary Material
Acknowledgements
We would like to thank all the participants who partook in this study. We also thank Dr Edward Szekeres at Perkin Elmer for bioinformatics support.
Glossary
Abbreviations
- CMT
Charcot-Marie-Tooth
- HSAN
hereditary sensory and autonomic neuropathies
- MPS IIIB
mucopolysaccharidosis IIIB
Funding
This work was supported by the Monaco Foundation, the Fondation GO, the Association de la neuropathie sensitive, the NIH (R01NS075764 and U54NS065712 SZ), the CMT Association and the Muscular Dystrophy Association. M.T. received a Frederick Banting and Charles Best Doctoral Fellowship from the Canadian Institutes of Health Research (CIHR).
Supplementary material
Supplementary material is available at Brain online.
Web resources
EVS: http://evs.gs.washington.edu/EVS/
ExAC: Exome Aggregation Consortium (ExAC), Cambridge, MA (URL: http://exac.broadinstitute.org) [December 2014]
Genomes Management Application: https://genomics.med.miami.edu
Graph Pad: http://www.graphpad.com
IMutant: gpcr2.biocomp.unibo.it/∼emidio/I-Mutant/I-Mutant_help.html
Mutations Taster: www.mutationtaster.org
Mupro: www.ics.uci.edu/∼baldig/mutation.html
OMIM: http://omim.org
Polyphen: genetics.bwh.harvard.edu/pphz
UCSC: genome.ucsc.edu
References
- Axelrod FB, Gold-von Simson G. Hereditary sensory and autonomic neuropathies: types II, III, and IV. Orphanet J Rare Dis. 2007;2:39. doi: 10.1186/1750-1172-2-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beesley CE, Jackson M, Young EP, Vellodi A, Winchester BG. Molecular defects in Sanfilippo syndrome type B (mucopolysaccharidosis IIIB) J Inherit Metab Dis. 2005;28:759–67. doi: 10.1007/s10545-005-0093-y. [DOI] [PubMed] [Google Scholar]
- Botez SA, Herrmann DN. Sensory neuropathies, from symptoms to treatment. Curr Opin Neurol. 2010;23:502–8. doi: 10.1097/WCO.0b013e32833c7a19. [DOI] [PubMed] [Google Scholar]
- Bras J, Guerreiro R, Hardy J. Use of next-generation sequencing and other whole-genome strategies to dissect neurological disease. Nat Rev Neurosci. 2012;13:453–64. doi: 10.1038/nrn3271. [DOI] [PubMed] [Google Scholar]
- Fu H, Bartz JD, Stephens RL, McKarty DM. Peripheral nervous system neuropathology and progressive sensory impairments in a mouse model of Mucopolysaccharidosis IIIB. PLoS One. 2012;7:e45992. doi: 10.1371/journal.pone.0045992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilissen C, Hoischen A, Brunner HG, Veltman JA. Unlocking Mendelian disease using exome sequencing. Genome Biol. 2011;12:228. doi: 10.1186/gb-2011-12-9-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez MA, Lebrigio RF, Van Booven D, Ulloa RH, Powell E, Speziani F, et al. GEnomes Management Application (GEM.app): a new software tool for large-scale collaborative genome analysis. Hum Mutat. 2013;34:842–6. doi: 10.1002/humu.22305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadfield MG, Ghatak NR, Nakoneczna I, Lippman HR, Myer EC, Constantopoulos G, et al. Pathologic findings in mucopolysaccharidosis type IIIB (Sanfilippo's sydnrome B) Arch Neurol. 1980;37:645–50. doi: 10.1001/archneur.1980.00500590069012. [DOI] [PubMed] [Google Scholar]
- Hoeijmakers JG, Faber CG, Lauria G, Merkies IS, Waxman SG. Small-fibre neuropathies–advances in diagnosis, pathophysiology and management. Nat Rev Neurol. 2012;8:369–79. doi: 10.1038/nrneurol.2012.97. [DOI] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–5. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loggia ML, Bushnell MC, Tétreault M, Thiffault I, Bhérer C, Mohammed NK, et al. Carriers of recessive WNK1/HSN2 mutations for hereditary sensory and autonomic neuropathy type 2 (HSAN2) are more sensitive to thermal stimuli. J Neurosci. 2009;29:2162–6. doi: 10.1523/JNEUROSCI.4633-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cubilskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiyappan MCM, Norton AW. Crystal structure of human alpha-N-acetylglucosaminidase. In: United States Patent Application publication. Vol. US2012/0021436A1. Shire Human Genetics Therapies, United States, 2012. [Google Scholar]
- Minde J, Toolanen G, Andersson T, Nennesmo I, Remahl IN, Svensson O, et al. Familial insensitivity to pain (HSAN V) and a mutation in the NGFB gene. A neurophysiological and pathological study. Muscle Nerve. 2004;30:752–60. doi: 10.1002/mus.20172. [DOI] [PubMed] [Google Scholar]
- Ochoa-Leyva A, Montero-Moran C, Saab-Rincon G, Breiba LG, Soberon X. Exploring the Structure-Function Loop Adaptability of a (beta/alpha)(8)-Barrel Enzyme through Loop Swapping and Hinge Variability. J Mol Biol. 2011;411:143–57. doi: 10.1016/j.jmb.2011.05.027. [DOI] [PubMed] [Google Scholar]
- Pollard LM, Jones JR, Wood TC. Molecular characterization of 355 mucopolysaccharidosis patients reveals 104 novel mutations. J Inherit Metab Dis. 2013;36:179–87. doi: 10.1007/s10545-012-9533-7. [DOI] [PubMed] [Google Scholar]
- Rotthier A, Baets J, Timmerman V, Janssens K. Mechanisms of disease in hereditary sensory and autonomic neuropathies. Nat Rev Neurol. 2012;8:73–85. doi: 10.1038/nrneurol.2011.227. [DOI] [PubMed] [Google Scholar]
- Sedel F, Turpin JC, Baumann N. [Neurological presentations of lysosomal diseases in adult patients] Rev Neurol (Paris) 2007;163:919–29. doi: 10.1016/s0035-3787(07)92635-1. [DOI] [PubMed] [Google Scholar]
- Selmer KK, Gilfillan GD, Strømme P, Lyle R, Hughes T, Hjorthaug HS, et al. A mild form of Mucopolysaccharidosis IIIB diagnosed with targeted next-generation sequencing of linked genomic regions. Eur J Hum Genet. 2012;20:58–63. doi: 10.1038/ejhg.2011.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinazzi M, Angelini C, Patrini C. Subacute sensory ataxia and optic neuropathy with thiamine deficiency. Nat Rev Neurol. 2010;6:288–93. doi: 10.1038/nrneurol.2010.16. [DOI] [PubMed] [Google Scholar]
- Valstar MJ, Bruggenwirth HT, Olmer R, Wevers RA, Verheijen FW, Poorthuis BJ, et al. Mucopolysaccharidosis type IIIB may predominantly present with an attenuated clinical phenotype. J Inherit Metab Dis. 2010;33:759–67. doi: 10.1007/s10545-010-9199-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yogalingam G, Hopwood JJ. Molecular genetics of mucopolysaccharidosis type IIIA and IIIB: Diagnostic, clinical, and biological implications. Hum Mutat. 2001;18:264–81. doi: 10.1002/humu.1189. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.