

Abstract
The ability to differentiate mouse embryonic stem cells (ESC) to neural progenitors allows the study of the mechanisms controlling neural specification as well as the generation of mature neural cell types for further study. In this protocol we describe a method for the differentiation of ESC to neural progenitors using serum-free, monolayer culture. The method is scalable, efficient and results in production of ~70% neural progenitor cells within 4 - 6 days. It can be applied to ESC from various strains grown under a variety of conditions. Neural progenitors can be allowed to differentiate further into functional neurons and glia or analyzed by microscopy, flow cytometry or molecular techniques. The differentiation process is amenable to time-lapse microscopy and can be combined with the use of reporter lines to monitor the neural specification process. We provide detailed instructions on media preparation and cell density optimization to allow the process to be applied to most ESC lines and a variety of cell culture vessels.
Keywords: Developmental Biology, Issue 99, Embryonic stem cells, differentiation, screening, cell culture, N2B27, in vitro, monolayer, neural specification
Introduction
Embryonic stem cells are pluripotent cells derived from the early embryo with the capacity to proliferate indefinitely in vitro while retaining the ability to differentiate into all adult cell types following reintroduction into an appropriate stage embryo (by forming a chimaera), injection into syngeneic or immunocompromised hosts (by forming a teratoma) or in vitro subject to appropriate cues1. The in vitro differentiation of mouse embryonic stem cells into neural lineages was first described in 1995 and involved the formation of multicellular suspension aggregates (embryoid bodies, EBs) in serum-containing media supplemented with the morphogen retinoic acid2-4. Since then a variety of protocols have been developed to allow neural differentiation5. Many still utilize aggregation, others co-culture with inducing cell types and several involve the use of serum-free media. All protocols have advantages and disadvantages and the precise nature of neural or neuronal cells produced also varies according to the protocol used.
The ideal protocol would be robust, scalable and make use of fully defined media and substrates, be amenable to non-invasive monitoring of the differentiation process and result in the generation of pure populations of neural progenitors able to be patterned by external cues and to differentiate into all neuronal and glial subtypes with high efficiency and yield in a relatively short time. In the last dozen years we have been using a method for generating neural progenitors and neurons from mouse ESC in a low density, serum-free adherent monolayer culture6-10. This protocol fulfils many of the criteria set out above: in our hands the efficiency of differentiation has been quite consistent over many years and a variety of cell lines, it can be scaled up or down (we successfully use vessels from 96-well plates to 15 cm diameter dishes) and the media used are well-defined. The process is amenable to timelapse microscopy for the monitoring of the differentiation and a variety of patterning cues can be added to induce the generation of distinct types of neuronal subtypes (e.g., Shh and Fgf8 for dopaminergic neurons6).
Nevertheless, there are some challenges to the successful application of this protocol. One of the key aspects is careful preparation of the media. We always prepare the media in-house despite the availability of commercial alternatives. One of the supplements used (N2; see Protocol) has modifications over the standard commercially available versions. Finally, one of the most important steps for successful application of this method is the optimal cell density at plating. This is mainly because while the autocrine nature of one of the inducing signals (Fgf411) requires that sufficient cells are present to allow optimal viability and differentiation, at too high densities differentiation is impaired (possibly in part due to autocrine production of LIF12). It is therefore important that both media preparation and cell plating are performed carefully and consistently to ensure optimal results.
Protocol
1. Media Preparation
NOTE: The protocol relies on the use of a mix of two separate media: DMEM/F12 supplemented with modified N2 supplement and Neurobasal supplemented with B27 supplement, typically in a 1:1 ratio.
- Prepare the modified N2 supplement by mixing the components in a 15 ml tube. Do not vortex or filter this; mix by inverting the tube until the solution is clear.
- Start by pipetting 7.2 ml of DMEM/F12, then add 1 ml of 25 mg/ml insulin (made up in 0.01 M HCl) and mix well by inverting the tube. It takes a couple of minutes until the solution is clear.
- Add 1 ml of 100 mg/ml apo-transferrin (made up in water), 33 µl of 0.6 mg/ml progesternone (made up in ethanol), 100 µl of 160 mg/ml (1 M) putrescine (made up in water), 10 µl of 3 mM sodium selenite (made up in water) and 666 µl of 7.5% bovine serum albumin and mix the tube well. Aliquot into 2.5 ml and store at -20 °C for up to 2 months.
- Prepare the media.
- To 250 ml of DMEM/F12 add 2.5 ml of N2. Mix well but do not shake or filter.
- To 250 ml of Neurobasal media add 5 ml of B27 supplement. Mix well but do not shake or filter.
- Mix the media from steps 1.2.1 and 1.2.2 in a 1:1 ratio. In some cases a 1:3 mix may be required (supplemented as the 1:1 mix in step 1.2.4).
- To the mix add 0.5 ml of L-glutamine (final concentration 0.2 mM) and 3.5 µl of 2-mercaptoethanol (final concentration 0.1 mM) and mix well without shaking. Store the media at 4 °C for up to 3 weeks and avoid exposing to light. This final mix is called N2B27.
- Prepare the gelatin solution.
- Prepare a 1% gelatin solution in ultrapure water and autoclave at 121 °C, 15 psi, for 15 min. It is important that the bottle used for this is completely clean from detergents or disinfectants, so it is recommended that a new bottle is used for this and is only ever rinsed in ultrapure water between uses. Aliquot the 1% solution into 50 ml aliquots and keep at 4 °C for months.
- Warm an aliquot of 1% gelatin in a 37 °C water bath until dissolved and add to 500 ml of warm phosphate-buffered saline (PBS). Mix well and pipet enough to cover the bottom of each well or plate. Allow the gelatin to coat the surface for at least 30 min.
2. Plating the Cells
NOTE: This protocol applies equally to mouse ESC grown in 10% serum with leukemia inhibitory factor (LIF), serum replacement with LIF or serum-free media with LIF and bone morphogenetic protein 4 (BMP4) or MEK and GSK3 inhibitors (2i media) with or without LIF. However, the timing and efficiency of the differentiation may vary depending on the media and cells (see discussion). For the experiments shown here we used the 46C mouse ESC line (that has an EGFP reporter knocked into one of the endogenous Sox1 alleles), grown in GMEM with 10% serum and LIF. For optimal results it is important that cells are dissociated and replated in the N2B27 media; simply changing of media from GMEM/serum/LIF to N2B27 always results in a reduced differentiation efficiency compared to replating the cells.
Grow the cells in GMEM with 10% serum, 1 mM sodium pyruvate, 1x non-essential amino acids, 0.1 M 2-mercaptoethanol, and 100 units/ml LIF13. To get a subconfluent culture of mouse ESC, plate 1 x 106 cells into a T25 culture flask which will take 2 - 3 days.
Observe cells under bright-field microscope. When the cells are subconfluent and ready to passage, rinse them twice in PBS. Add 1 ml of cell dissociation reagent and return to the incubator for 2 - 5 min. Although trypsin and other enzymes can also be used for cell dissociation, they can have a negative impact on cell attachment so the plating densities will need to be adjusted.
Once the cells are beginning to lift from the plate, tap the vessel to dislodge them into a single cell suspension. Collect the cells into a total of 10 ml of media and cell dissociation reagent and pipet into a 15 ml centrifuge tube.
Spin the cells at 300 x g for 3 min at RT.
Carefully aspirate the supernatant and resuspend the pellet in 10 ml of pre-warmed N2B27 by pipetting up and down 3 - 5 times. When pipetting the suspension down, pipet against the side of the tube to avoid creating bubbles. Count the cells with an automated cell counter or a haemocytometer and record the concentration.
Prepare a cell suspension containing the desired number of cells per unit of volume to be plated per well in pre-warmed N2B27. A density of 10,500 cells per cm2 in a 6-well dish (i.e., 1 x 105 cells per well of a 6-well dish) is optimal. See Table 1 for suggested number of cells per cm2 and plating media volumes for a variety of cell culture vessels.
Aspirate the gelatin from the culture vessel and plate the cell suspension according to the suggested density and volume in Table 1. Do not swirl the plates as this will concentrate the cells to the center. Place the cultures in a humid incubator at 37 °C, 5% CO2.
Change media every 1 - 2 days by replacing all the media with fresh N2B27. Pipette gently as flushing the cells might affect density and lessen differentiation efficiency in the following days. Where initial viability after plating is poor, plate the cells in a 1:3 mix of the N2 and B27-supplemented media and change this to N2B27 after the first two days. Cells will begin to differentiate and should show Sox1 expression (visible by green fluorescence of the reporter in the 46C cell line used here) within 4 - 6 days.
3. Immunofluorescent Staining
NOTE: The protocol can be applied to any vessel type. The volume of each reagent is described per well of 6-well plate and can be scaled to any surface area. Replating is not recommended due to the low viability afterwards.
Remove differentiation medium and fix the cells by adding 500 µl of 4% (v/v) paraformaldehyde, leave for 15 min.
Wash and permeabilize the cells with 2 ml of PBS-T (phosphate buffered saline with 0.5% (v/v) tween-20) twice. Cells can be kept in PBS-T for up to 2 months at 4 °C.
Replace PBS-T with 1 ml of PBS-T with 10% (v/v) serum (from the same species as the secondary antibody). Incubate 1 hr on the plate rocker at RT to block any non-specific binding.
After 1 hr, wash the cells twice with 2 ml of PBS-T, and incubate in 500 µl mouse IgG against βIII-tubulin (diluted 1:1,000 in PBS-T with 10% serum). Put on the plate rocker, and leave O/N at 4 °C.
From this step, always protect the vessel from light. Wash the cells twice with PBS-T, then add 500 µl of fluorescence-labelled anti-mouse IgG antibody (diluted to 2 µg/ml in PBS-T with 10% serum). Put it on the plate rocker for 1 hr at RT.
Rinse the cells twice with PBS-T, then add 500 µl of DAPI (diluted to 0.5 µg/ml in PBS-T) and leave for 5 min.
Wash the cells again and keep them in PBS-T. Cells are ready to be photographed and can be kept for up to 2 weeks at 4 °C. Note that GFP signal fades over time so it is inappropriate to compare GFP intensity of fixed and live cells or of cells fixed at different times.
Representative Results
In this experiment, we used the 46C cell line14, mouse embryonic stem cells with an endogenous Sox1-GFP reporter, to track neural differentiation. By using this cell line, expression of Sox1, a marker for neural progenitor, can be detected by green fluorescence. Plating density is a critical factor to achieve neuronal differentiation. Mouse embryonic stem cells were plated in 6-well plate at different densities varying from 10,500 to 88,500 cells/cm2. Figure 1A shows differentiation efficiencies achieved by different plating densities in a 6-well plate. On day 6 of differentiation, cells were fixed in 4% paraformaldehyde and stained for the neuronal marker βIII-tubulin. At the optimal density (10,500 cells/cm2), cells differentiated to neural progenitors and neurons. This was observed by green fluorescent signal from Sox1-GFP reporter and immunofluorescent against the neuronal marker, βIII-tubulin. A lower plating density led to cell death within 3 days. However, plating at too high density (>36,500 cells/cm2) resulted in decreased proportion of green cells and βIII-tubulin positive cells. Figure 1B shows an experiment performed in a 96-well plate which required about 10 times higher cell density to reach the optimal (155,000 cells/cm2) and the too confluent (362,500 cells/cm2) level.
Optimal density can be different between media preparations, media component and cell batches, cell types, or previous culture conditions. It is recommended to optimize plating density for each experiment. Table 1 provides ranges of effective plating densities in various platforms that should be included in the optimization step. Within this range, it should be possible to get a density that gives a good differentiation efficiency within 4 - 6 days.
During differentiation ESC gradually change their morphology. Figure 2 shows cell and colony morphology from day 1 to day 6 after plating in 6-well plate at 10,500 cells/cm2. Cells were cultured in differentiation conditions and photographed every day. It is not unusual to see significant cell death on the first few days due to extreme change in culture condition. However, the remaining cells are still able to differentiate. On day 4, cells started to become neural progenitors as green fluorescent signal from Sox1-GFP reporter firstly appeared. However, even on day 6, not all the cells are Sox1-positive. Existence of some non-neural cells is expected, nonetheless, in the proper conditions, the majority of cells will be neural.
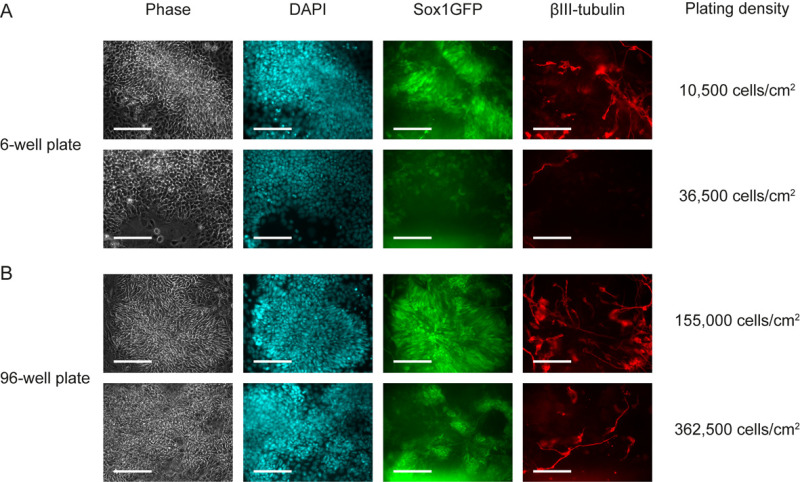 Figure 1. The effect of plating density on differentiation efficiency. Two different densities of Sox1-GFP reporter cells (46C) were plated and differentiated according to the protocol for 6 days. (A) Differentiation in a 6-well plate. (B) Differentiation in a 96-well plate. Scale bar: 100 µm. Please click here to view a larger version of this figure.
Figure 1. The effect of plating density on differentiation efficiency. Two different densities of Sox1-GFP reporter cells (46C) were plated and differentiated according to the protocol for 6 days. (A) Differentiation in a 6-well plate. (B) Differentiation in a 96-well plate. Scale bar: 100 µm. Please click here to view a larger version of this figure.
 Figure 2. Cell morphologies during the differentiation process. 46C ESC were differentiated at 10,500 cells per cm2 in a 6-well culture plate and photographed daily to observe changes in cell morphology. Scale bar: 300 µm. Please click here to view a larger version of this figure.
Figure 2. Cell morphologies during the differentiation process. 46C ESC were differentiated at 10,500 cells per cm2 in a 6-well culture plate and photographed daily to observe changes in cell morphology. Scale bar: 300 µm. Please click here to view a larger version of this figure.
| Platform | Range of effective density (cells/cm2) | Range of cell number to plate | Suggested initial media volume (µl) | Suggested media volume after day 1 (µl) |
| 6-well plate | 10,500 - 36,500 | 99,750 - 346,750 | 1,000 | 2,000 |
| 24-well plate | 15,600 - 46,800 | 29,640 - 88,920 | 500 | 1,000 |
| 96-well plate | 103,500 - 260,000 | 33,120 - 83,200 | 100 | 200 |
Table 1. Suggested plating densities and media volumes for different vessel sizes. The plating densities in this table were determined using the 46C cell line. For optimal results the plating density of each individual line may have to be adjusted.
Discussion
The monolayer neural differentiation protocol has been in use for over a decade6. The protocol is highly efficient, composed of defined medium, and done in a monolayer system which makes the system more applicable for preclinical (e.g., drug screening) uses. However, there are some critical factors that determine differentiation efficiency. This article points out those factors and the solution for each obstacle.
Density of the cells after plating in the differentiation condition is perhaps the most critical factor. Plating the cells at too high density reduces differentiation efficiency, while plating at too low density leads to cell death. This phenomenon is likely due to autocrine growth factor signaling. Fgf4 is an autocrine growth factor that activates the MAPK cascade and drives ESC differentiation11. LIF is another cytokine produced by ESC. It activates the STAT3 pathway and promotes self-renewal15. This protocol makes use of N2B27 to make neural cells. N2B27 contains various factors which permit ESC survival and promotes neural cell growth, and does not contain either LIF or BMP4 which inhibit neural differentiation6. At the proper density, a suitable balance of Fgf4 and LIF signaling is maintained that allows ESC to differentiate. At high density, excess amount of autocrine LIF and BMP inhibit differentiation. In contrast, the cells at too low density have impaired survival in these fairly minimal media. Thus, when large numbers of cells without differentiation appear the plating number should be reduced, whereas an increase of the plating cell number is required if significant cell death is observed.
In addition to the cell number there are some other factors that indirectly affect plating density. The first factor is the local cell concentration, dependent on the technique used to evenly distribute the cells over the surface area. We found that swirling the well is not ideal, since it concentrates the cells to the center of the well and makes the density at the center too high, while at the periphery the density becomes too low. Other mixing techniques, for example, mixing the media with the cells before plating, or rocking the plate side-to-side are recommended. Moreover, when a vessel is placed in the incubator, uneven heating of the medium causes a convection force that draws the cells to the center of the well, resulting in distribution of the cells in a concentric ring pattern. A lower media volume on the plating day is recommended to reduce this effect. Leaving the plate outside the incubator for 30 min to let the cells attach, as well as warming up the medium before mixing can also help achieve a homogeneous plating density.
Secondly, the vessel type also affects plating density. Figure 1 clearly shows that the number of cells/well cannot be linearly scaled to the surface area of different vessels (in other words, the same number of cells/surface area cannot be applied to every vessel type). A smaller vessel (with greater volume/area ratio) is affected more by the media surface tension which makes a concave surface of medium, the so-called meniscus. The surface tension pushes the cells to the edge of the well and decreases density at the center. In addition, since media volume in the small vessel is low, media warm up fast, reducing the convection force. Therefore, the cells in a small vessel will move to the edge by the meniscus effect, and will not move to the center by convection forces. Thus, the smaller the vessel, the higher the plating density required to get the optimal density at the center.
Thirdly, the condition of the cells prior to plating also indirectly affects plating density and differentiation efficiency. We found a variation of the optimal density in some experiments. The experiments with higher death rate on the first day needed higher density for efficient differentiation. ESC cultures in serum and LIF are composed of a heterogeneous population of naïve pluripotent cells, primed pluripotent cells, and even a small number of differentiated cells16. Cells of these types have different ability to survive and differentiate under differentiation conditions. Since the protocol dictates on the number, but not the state of the cells, some experiments might start with more differentiated cells which will die on the first few days, leading to a lower density than expected. To avoid this variation, consistent culture of ESC is required to make sure that the constant proportion between each state is maintained.
Apart from density, timing is another factor to achieve efficient differentiation. In this publication, we describe the generation of neural progenitors within 6 days. However, the time can be different depending on the state of the starting ESC culture. As described above, ESC cultures are mixed populations that can respond differently to the differentiation condition. Not only the cell survival but also the timing of differentiation will be affected by the culture composition. This protocol can be applied to most ESC culture conditions e.g., in N2B27 media containing LIF and BMP4, or 2i and LIF. However, if there are more naïve cells in the culture, a longer period might be required for efficient differentiation. This is because naïve cells will need more time to switch to a primed state and then differentiate to neural progenitors.
Disclosures
The authors have nothing to disclose.
Acknowledgments
Work was funded by a Development and Promotion of Science and Technology scholarship from the Thai Ministry for Education to W.W. and grants from Tenovus and the Anonymous Trust to M.P.S.
References
- Smith AG. Embryo-derived stem cells: of mice and men. Annu Rev Cell Dev Biol. 2001;17:435–462. doi: 10.1146/annurev.cellbio.17.1.435. [DOI] [PubMed] [Google Scholar]
- Bain G, Kitchens D, Yao M, Huettner JE, Gottlieb DI. Embryonic stem cells express neuronal properties in vitro. Dev. Biol. 1995;168:342–357. doi: 10.1006/dbio.1995.1085. [DOI] [PubMed] [Google Scholar]
- Fraichard A, et al. In vitro. differentiation of embryonic stem cells into glial cells and functional neurons. J. Cell Sci. 1995;108:3181–3188. doi: 10.1242/jcs.108.10.3181. [DOI] [PubMed] [Google Scholar]
- Strubing C, et al. Differentiation of pluripotent embryonic stem cells into the neuronal lineage in vitro gives rise to mature inhibitory and excitatory neurons. Mech. Dev. 1995;53:275–287. doi: 10.1016/0925-4773(95)00446-8. [DOI] [PubMed] [Google Scholar]
- Stavridis MP, Smith AG. Neural differentiation of mouse embryonic stem cells. Biochem Soc Trans. 2003;31:45–49. doi: 10.1042/bst0310045. [DOI] [PubMed] [Google Scholar]
- Ying QL, Stavridis M, Griffiths D, Li M, Smith A. Conversion of embryonic stem cells into neuroectodermal precursors in adherent monoculture. Nat Biotechnol. 2003;21:183–186. doi: 10.1038/nbt780. [DOI] [PubMed] [Google Scholar]
- Ying QL, Smith AG. Defined conditions for neural commitment and differentiation. Methods Enzymol. 2003;365:327–341. doi: 10.1016/s0076-6879(03)65023-8. [DOI] [PubMed] [Google Scholar]
- Stavridis MP, Lunn JS, Collins BJ, Storey KG. A discrete period of FGF-induced Erk1/2 signalling is required for vertebrate neural specification. Development. 2007;134:2889–2894. doi: 10.1242/dev.02858. [DOI] [PubMed] [Google Scholar]
- Stavridis MP, Collins BJ, Storey KG. Retinoic acid orchestrates fibroblast growth factor signalling to drive embryonic stem cell differentiation. Development. 2010;137:881–890. doi: 10.1242/dev.043117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speakman CM, et al. Elevated O-GlcNAc Levels Activate Epigenetically Repressed Genes and Delay Mouse ESC Differentiation Without Affecting Naive to Primed Cell Transition. Stem Cells. 2014;32:2605–2615. doi: 10.1002/stem.1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunath T, et al. FGF stimulation of the Erk1/2 signalling cascade triggers transition of pluripotent embryonic stem cells from self-renewal to lineage commitment. Development. 2007;134:2895–2902. doi: 10.1242/dev.02880. [DOI] [PubMed] [Google Scholar]
- Dani C, et al. Paracrine induction of stem cell renewal by LIF-deficient cells: a new ES cell regulatory pathway. Dev Biol. 1998;203:149–162. doi: 10.1006/dbio.1998.9026. [DOI] [PubMed] [Google Scholar]
- Smith AG. Culture and differentiation of embryonic stem cells. Journal of Tissue Culture Methods. 1991;13:89–94. [Google Scholar]
- Aubert J, et al. Screening for mammalian neural genes via fluorescence-activated cell sorter purification of neural precursors from Sox1-gfp knock-in mice. Proc. Natl. Acad. Sci. U. S. A. 2003;100(1):11836–11841. doi: 10.1073/pnas.1734197100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H, Burdon T, Chambers I, Smith A. Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev. 1998;12:2048–2060. doi: 10.1101/gad.12.13.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva J, Smith A. Capturing pluripotency. Cell. 2008;132:532–536. doi: 10.1016/j.cell.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]