

Abstract
Immunohistochemistry is a widely used technique for detecting the presence, location, and relative abundance of antigens in situ. This introductory level protocol describes the reagents, equipment, and techniques required to complete immunohistochemical staining of rodent brain tissue, using markers for microglia and neuronal elements as an example. Specifically, this paper is a step-by-step protocol for fluorescent visualization of microglia and neurons via immunohistochemistry for Iba1 and Pan-neuronal, respectively. Fluorescence double-labeling is particularly useful for the localization of multiple proteins within the same sample, providing the opportunity to accurately observe interactions between cell types, receptors, ligands, and/or the extracellular matrix in relation to one another as well as protein co-localization within a single cell. Unlike other visualization techniques, fluorescence immunohistochemistry staining intensity may decrease in the weeks to months following staining, unless appropriate precautions are taken. Despite this limitation, in many applications fluorescence double-labeling is preferred over alternatives such as 3,3'-diaminobenzidine tetrahydrochloride (DAB) or alkaline phosphatase (AP), as fluorescence is more time efficient and allows for more precise differentiation between two or more markers. The discussion includes troubleshooting tips and advice to promote success.
Keywords: Neuroscience, Issue 99, Immunohistochemistry, Iba1, Pan-Neuronal, Fluorescence, Double-Labeling, Microglia, Neuron, Rat, Antibody
Introduction
Immunohistochemistry is a process for detecting antigens (i.e. proteins) in tissue sections by using primary antibodies which bind specifically to the antigens of interest. Immunohistochemistry was pioneered by J.R. Marrack in 1934 when he determined that antibodies could localize antigens with great specificity1. Beginning in 1942, some of the first in vitro studies using fluorescent antibodies to visualize immunohistochemistry were published2,3, after which the first in vivo histochemical study was published 4. During the 1960’s, three decades after the inception of immunohistochemical methods, enzyme-conjugated antibodies began to be used as secondary reagents. These methods were simultaneously and independently developed in France and the U.S.5,6. Today, a wide array of antibodies provides endless possibilities for immunohistochemistry studies7.
The overall goal of this correspondence is to provide a brief introduction into immunohistochemical staining; it is not meant to be a comprehensive and exhaustive review of this technique. In the method outlined, immunohistochemical techniques for two antigens are presented (markers for microglia and neurons) for staining of paraformaldehyde perfused, sucrose cryoprotected, cryosectioned rat brain. Immunohistochemical staining begins with blocking nonspecific antigen binding to reduce background staining. Next, incubation with primary antibody allows for binding to a specific antigen in the tissue. Following the primary antibody, another antibody, termed secondary antibody, is applied to link the primary antibody to a conjugated visualization signal8. The secondary antibody targets the immunoglobulin G (IgG) domain specific to the species in which the primary antibody was raised. The secondary antibody amplifies the signal of the primary antibody since the Fab regions of the secondary antibody bind to multiple sites on the IgG domain of the primary antibody. Either enzymes or fluorescent molecules conjugated to the Fc regions of the secondary antibody enable visualization. For example, a rabbit anti-Iba1 primary antibody is a rabbit IgG molecule specific for Iba1. When donkey anti-rabbit IgG is applied as a secondary antibody, it will recognize and bind to multiple regions of the rabbit anti-Iba1 IgG (see Figure 1). The donkey antibody can be visualized by various methods. This correspondence focuses on detection of a fluorophore conjugated to the secondary antibody, which recognizes the primary antibody, for visualization by fluorescent microscopy. In fluorescent immunohistochemistry, a nuclear stain such as Hoechst or DAPI can be used to visualize all nuclei.
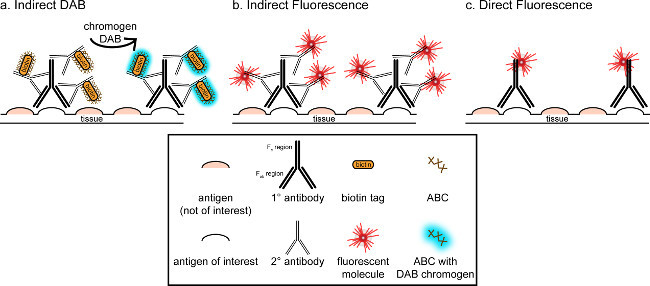 Figure 1: Schematic representation of direct vs. indirect antibody labeling techniques. Antibodies bind to the antigen of interest and can be amplified by secondary antibodies raised against the species of the primary antibodies. This technique can be performed using avidin-biotin complex (ABC) for amplification and DAB for visualization (A), or a directly conjugated fluorescent secondary antibody (B). Alternatively, primary antibodies can be directly conjugated with many different tags, including biotin or a fluorophore (C). Please click here to view a larger version of this figure.
Figure 1: Schematic representation of direct vs. indirect antibody labeling techniques. Antibodies bind to the antigen of interest and can be amplified by secondary antibodies raised against the species of the primary antibodies. This technique can be performed using avidin-biotin complex (ABC) for amplification and DAB for visualization (A), or a directly conjugated fluorescent secondary antibody (B). Alternatively, primary antibodies can be directly conjugated with many different tags, including biotin or a fluorophore (C). Please click here to view a larger version of this figure.
An alternative method for visualization of immunohistochemical staining uses 3,3'-diaminobenzidine tetrahydrochloride (DAB; see Figures 1 and 2). This differs from fluorescence by using a biotinylated or horse-radish peroxidase (HRP) conjugated secondary antibody, which provides an enzyme to convert DAB to a precipitate that is visible under bright field microscopy. In instances where a single antigen is of interest or staining is required to be long lasting, DAB may be more appropriate than fluorescent staining. However, DAB staining is not well-suited for differentiation between multiple markers, especially if two nuclear antigens are of interest. For information on DAB materials and protocol modifications, consult Table 1. Alternately, nitro blue tetrazolium chloride/5-Bromo-4-chloro-3-indolyl phosphate (NBT/BCIP) can be used to visualize an alkaline phosphatase (AP) conjugated secondary antibody.
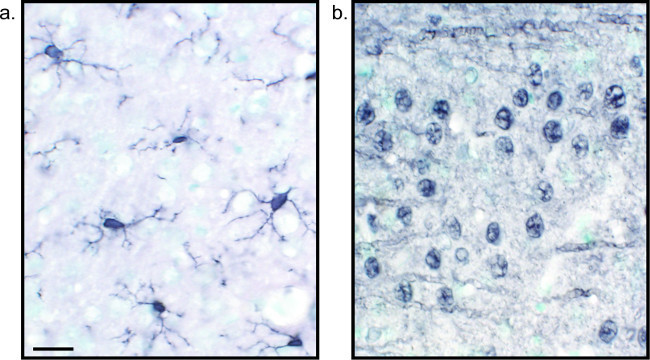 Figure 2: Representative images of nickel-enhanced DAB single-labeled rat brain tissue sections. Rat brain sections which are labeled with nickel-enhanced DAB for Iba1 (A) and Pan-neuronal (B) allow for long-lasting analysis of microglia or neurons alone. Scale bar 20 µm. Please click here to view a larger version of this figure.
Figure 2: Representative images of nickel-enhanced DAB single-labeled rat brain tissue sections. Rat brain sections which are labeled with nickel-enhanced DAB for Iba1 (A) and Pan-neuronal (B) allow for long-lasting analysis of microglia or neurons alone. Scale bar 20 µm. Please click here to view a larger version of this figure.
One must consider the estimated abundance of the antigen of interest within the tissue being analyzed. Indirect methods (as described above) are useful for targets with low abundance. When the antigen of interest is in high abundance, direct methods can be applied. Direct methods involve a primary antibody that is directly conjugated to a visualization signal, and thus no secondary antibody is required. This method simplifies the staining process, but eliminates the amplification achieved by indirect methods. Using a directly conjugated primary antibody also eliminates cross-reactivity of secondary antibodies when double-labeling.
This communication details the protocol for double-labeling with Iba1 and Pan-neuronal (details in Table 1). Iba1 stains microglia in many activation states, including ramified, hyper-ramified, activated, amoeboid, and rod. Pan-neuronal stains neuronal axons, dendrites, and soma. Since Iba1 stains most microglia and Pan-neuronal targets the neuron, this combination of stains is useful in gaining a broad understanding of microglia-neuron interactions.
In sum, immunohistochemical staining relies on the careful selection of antibodies. As the research question becomes more specific, antibodies raised to alternate antigens may be desired. To target a specific microglial activation state, one may elect to use CD45 or CD68 antibodies, rather than Iba1. Further, in working with mice, F4/80 may provide the necessary results. Similarly, neuronal elements can be specifically targeted with antibodies raised against the nucleus, synapse (pre- or post-), axon, and growth cone. Additionally, there are other markers which differentiate the age of the neuron (Double-cortin, NeuN), and neuronal regeneration (GAP-43).
Protocol
NOTE: All procedures were carried out in compliance with The Institutional Animal Care and Use Committee (IACUC) of the University of Arizona. A list of recommended materials and equipment can be found in Table 1.
1. Tissue Preparation
- Perfusion
- Euthanize rodent with an overdose of sodium pentobarbital (25 mg/kg, IP), and perfuse transcardially with phosphate buffered saline (PBS) until completely exsanguinated (3-5 min) at a flow rate of 8 ml/min. For in-depth perfusion instructions, see Gage et al 20129.
- Immediately following PBS flush, fix tissue by perfusing with 4% paraformaldehyde in PBS for 15-20 min at a flow rate of 8 ml/min.
- Remove brain and place in 4% paraformaldehyde for 24 hr, followed by graded sucrose solutions (15%, 30%, 30%, in sequence; prepared in tris buffered saline) at 4 °C. Transfer the brain to the subsequent sucrose solution only after the brain has sunk in each solution. Note: Usually, 5 days in each solution is sufficient time for tissue to sink.
- Tissue Freezing and Cryosectioning
- Place the brain in embedding medium, such as O.C.T. compound and submerge in isopentane at a temperature of -35 °C. Allow the brain to freeze for a minimum of 10 min, and then store at -80 °C. Problems can arise if diligence to temperature is not taken; please see discussion for troubleshooting information.
- Cut serial coronal sections at a thickness of 20 µm and a temperature of -20 °C. Collect tissue onto positively charged slides. Brain sections may be placed in a slide box wrapped in foil in a zip-top bag and stored long-term at -80 °C. This method of storage creates a double boundary to prevent exposure to air and frost.
2. Tissue Processing
NOTE: Example equipment and materials required for staining are shown in Figure 3. Alternatives are available, however, these images will aid those new to immunohistochemical staining to visualize appropriate items prior to purchasing.
 Figure 3: Example items required for immunohistochemical staining. The black box shown in (A) is an ideal humidity chamber for immunofluorescence, as slides are protected from light without the need to wrap the box on foil. Following sectioning, slides can be stored in a box such as the yellow box shown in (B). Wrapping the box tightly in foil and placing in a zip-top bag prior to the freezer helps protect the tissue samples from freezer burn. An example of slides is given in (C), with different staining dishes depicted in (D) and (E). Coverslips can vary in size and thickness (F), however 1.2 thick coverslips provide nice imaging results on most upright and confocal microscopes. A pencil such as that shown in (G) can be used to label slides. Permanent markers should be avoided as the ink can run, affecting both staining and the ability to determine what the sample is. A mini pap pen such as that shown in (H) enables a repellent border to be drawn on slides.
Figure 3: Example items required for immunohistochemical staining. The black box shown in (A) is an ideal humidity chamber for immunofluorescence, as slides are protected from light without the need to wrap the box on foil. Following sectioning, slides can be stored in a box such as the yellow box shown in (B). Wrapping the box tightly in foil and placing in a zip-top bag prior to the freezer helps protect the tissue samples from freezer burn. An example of slides is given in (C), with different staining dishes depicted in (D) and (E). Coverslips can vary in size and thickness (F), however 1.2 thick coverslips provide nice imaging results on most upright and confocal microscopes. A pencil such as that shown in (G) can be used to label slides. Permanent markers should be avoided as the ink can run, affecting both staining and the ability to determine what the sample is. A mini pap pen such as that shown in (H) enables a repellent border to be drawn on slides.
- Slide preparation
- Remove slides from freezer and thaw at room temperature.
- Optional: If sections have previously floated from slides, place thawed slides in an oven at 60 °C for no more than 4 hrs to help prevent tissue sections from floating off slides.
- Place slides in a slide rack and corresponding dish.
- Wash slides three times in PBS for 5 min each, changing solution between washes. From this step forward, avoid having sections without liquid for extended periods of time. Note: If sections dry out, background staining is increased and meaningful data cannot reliably be obtained.
- Tissue Staining
- In a light-tight staining box, create a “humidity chamber” with lint-free tissues soaked with deionized water.
- Dry the edges of the slide with a lint-free tissue, use a mini pap pen to make a liquid repellent border at the very edge of the slide, away from tissue sections. This border should ensure ample space between the meniscus of the liquid and the edge of the tissue so that surface tension does not affect staining. NOTE: The pap pen repellent border can be applied prior to 2.1.3 if the antibodies of interest do not require microwave antigen retrieval. If the pap pen has been applied prior to washing in PBS, the integrity of the liquid-repellent border must be checked at this step. Use a mini-pap pen to fill in any gaps in the border.
- With slides laid horizontally, block nonspecific antigen binding by incubating in 4% v/v serum in PBS (block solution). Pipette 300 µl of block solution per slide for 1hr at room temperature. Make sure the block solution extends to the pap pen at the edge of the slide and completely covers the tissue to avoid uneven staining caused by surface tension near the tissue.
- Use serum from the same species in which secondary antibody is made. Note: In this procedure, the secondary antibodies are made in donkey, and thus donkey serum is used. If secondary antibodies from two or more different species are used, include serum from each species.
- Pipette primary antibody onto slides. Note: Antibody concentrations for this staining have been optimized at 1:5,000 and 1:500 for Iba1 and Pan-neuronal, respectively. These concentrations have been found to show meaningful staining with an absence of background staining.
- Dilute block solution to 1% serum in PBS and add primary antibodies. Pipette 300 µl of primary antibody solution in 1% serum per slide. Again, ensure the fluid is to the edge of the pap pen. Incubate overnight at 4 °C.
- Include three control slides: one that contains neither Iba1 nor Pan-neuronal antibodies, one with Iba1 without Pan-neuronal antibody, and one with Pan-neuronal antibody without Iba1. Stain these slides in the same run with the same solutions, however omit the primary antibodies to test the non-specific binding of the secondary antibodies.
- The following morning, wash slides three times in PBS for 5 min each, changing solution between washes.
- Fluorescent antibodies are light-sensitive, therefore, from this step forward, minimize light exposure by ensuring wash containers are wrapped in foil and hybridization boxes are either black or incubated in the dark. Pipette the appropriate secondary antibodies on all slides and incubate for 60 min at room temperature at a concentration of 1:250 in block solution (see step 2.2.3) in a light-tight “humidity chamber” (see step 2.2.1).
- Use secondary antibodies of different wavelengths. Here, for the primary antibody rabbit anti-Iba1, use donkey anti- rabbit 594 as the appropriate secondary antibody. For the primary antibody mouse anti-Pan-neuronal, use donkey anti-mouse 488 as the appropriate secondary antibody. Alternately, use anti-rabbit 488 and anti-mouse 594.
- Wash slides three times in PBS for 5 min each.
- Optional: perform nuclear staining.
- Place in Hoechst (or other nuclear stain) at a concentration of 0.03 µg/ml in double distilled H20 for exactly 60 sec.
- Wash slides three times in PBS for 5 min each.
- Wash in ddH20.
- Coverslipping
- Coverslip slides with an aqueous mounting medium, such as Fluoromount-G or ProlongGold. Take care to remove all bubbles using a cotton-tipped applicator. Note: Other mounting agents could be used, however high bleed-through between dyes has been noted by some within days of coverslipping.
- Use clear nail polish to seal the edges, preventing the sections from drying out due to evaporation. Allow nail polish to dry in a light-tight container while slides remain flat and at room temperature, and then store in a light-tight container wrapped in foil at 4 °C.
3. Imaging the Stained Tissue
- Microscopy
- Allow the nail polish to dry for at least one hour before commencing microscopy, which should take place in a darkened room.
- Acquire photomicrographs using a confocal or research microscope with a fluorescent light source and a digital camera attachment. Using the accompanying software, set the exposure for each wavelength – 405, 488, and 594 – separately. Note: in-depth imaging instructions should be available online from the microscope manufacturer.
- Acquire photomicrographs in each channel without moving the sections or adjusting the focus. Take images in color, or alternately in grayscale and convert to color afterward. Note: Color or grayscale images from each channel can be collated in post-processing.
- Ensure that tissue sections are not exposed to ambient light or microscopic light for long periods of time, as photo-bleaching of the sections will occur. To avoid this, increase exposure time rather than increasing light/laser intensity.
- Do not turn off the fluorescent light source within 30 min of being turned on. Note: Switching the source on and off in quick succession may decrease the life span of the fluorescent bulb.
Representative Results
This staining protocol results in rat brain tissue sections that have microglia fluorescently labeled in the 594 channel (red) and neurons labeled in the 488 channel (green; see Figure 4). If a nuclear stain has been done, it will show in the 405 channel (blue). Images can be taken in different channels and overlaid for direct comparison of the three channels, or between any two channels. Many digital acquisition software suites include this functionality. Double-labeling with Iba1 and Pan-neuronal marker shown here demonstrates mostly ramified microglia, with long fine neuronal projections evident across cortical layers.
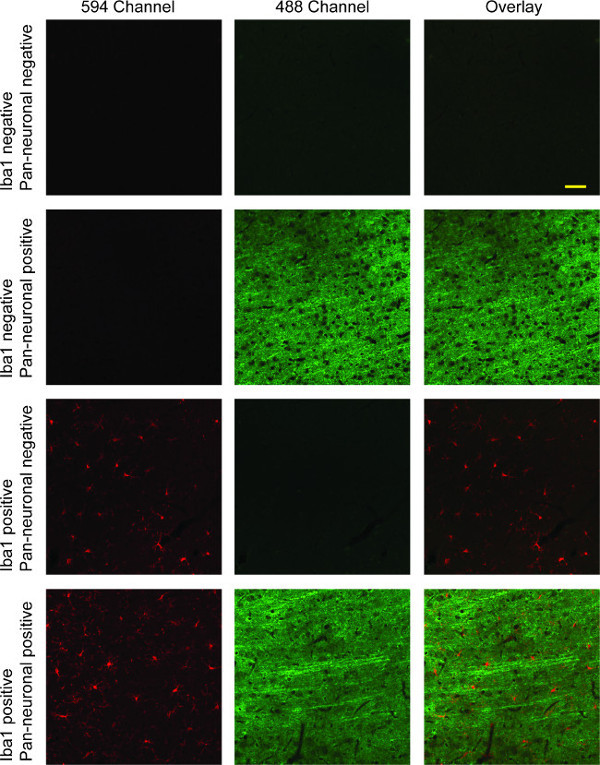 Figure 4: Representative confocal fluorescent images of Iba-1/Pan-neuronal double-labeling. Column 1 shows microglia stained with Iba1 (red). Neurons are shown in the second column in green, with the overlaid image of both red and green channels in the third column. In the first row, there is a complete lack of specific staining due to absence of both primary antibodies. The second row depicts specificity of Pan-neuronal primary antibody for neuronal staining, whereas the third row shows specificity of Iba1 for microglia, with a complete lack of cross-reactivity in both cases. The fourth row illustrates double-labeling with an overlay of the two channels showing both microglial and neuronal staining. Scale bar 50 µm.
Figure 4: Representative confocal fluorescent images of Iba-1/Pan-neuronal double-labeling. Column 1 shows microglia stained with Iba1 (red). Neurons are shown in the second column in green, with the overlaid image of both red and green channels in the third column. In the first row, there is a complete lack of specific staining due to absence of both primary antibodies. The second row depicts specificity of Pan-neuronal primary antibody for neuronal staining, whereas the third row shows specificity of Iba1 for microglia, with a complete lack of cross-reactivity in both cases. The fourth row illustrates double-labeling with an overlay of the two channels showing both microglial and neuronal staining. Scale bar 50 µm.
Poor staining results are also shown (Figure 5), as evidenced by lack of clearly defined cell bodies and fragmented microglial/neuronal processes. High background is indicative of poor quality staining, and can be seen by non-specific co-localization of antibodies (see Figure 5). Moreover, if the signal is weak, an amplification step with fluorescently bound streptavidin can be incorporated. Instead of using a directly tagged fluorescent secondary antibody in step 2.2.6, a biotinylated secondary is applied. Following washing in PBS, streptavidin (1:1000 in PBS) is incubated on the slides for 1 hr. Return to protocol at step 2.2.7 where slides are then washed and dipped in a counterstain or cover slipped.
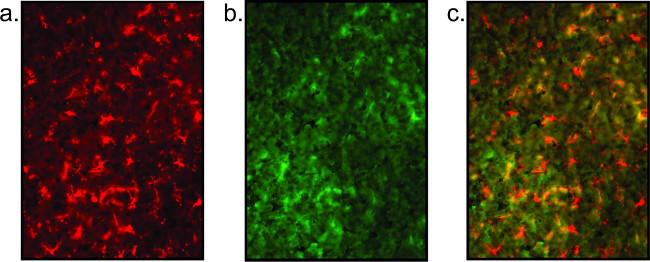 Figure 5: Representative fluorescent images of poor tissue quality. Shown here is an example of staining in poor tissue which results in a lack of clarity of microglial staining (a) and complete lack of neuronal staining (b). This neuronal staining is simply high background. The overlaid image (c) is not meaningful.
Figure 5: Representative fluorescent images of poor tissue quality. Shown here is an example of staining in poor tissue which results in a lack of clarity of microglial staining (a) and complete lack of neuronal staining (b). This neuronal staining is simply high background. The overlaid image (c) is not meaningful.
| Name of Material/ Equipment | Company | Catalog Number | Comments/Description |
| Fisherbrand Superfrost Plus Glass Slides | Fisher Scientific | 22-034-979 | Used for tissue mounting (1.2.2) |
| Oven | Thermo Scientific | 51028112 | Used for tissue drying (2.1.1) |
| Mini Pap pen | Life Technologies | 00-8877 | Used in step 2.2.2 |
| Andwin Scientific Tissue-tek Slide Staining Dish | Fisher Scientific | 22-149-429 | Used for all washes during staining (2.2), as well as the Hoechst step (2.2.8) |
| Kimwipes | Fisher Scientific | 06-666-A | Used for drying slides (2.2) |
| Black Staining Box | Ted Pella | 21050 | Used for blocking and staining steps (2.2) |
| Normal Donkey Serum | Fisher Scientific | 50-413-253 | Used for block and antibody incubation (2.2) |
| Mouse α-Pan-neuronal | Millipore | MAB2300 | Used for primary antibody (2.2.4) |
| Rabbit α-Iba1 | Wako Chemical | 019-19741 | Used for primary antibody (2.2.4) |
| Donkey α-Rabbit 594 | Jackson ImmunoResearch | 711-585-152 | Used for secondary antibody (2.2.6) |
| Donkey α-mouse 488 | Jackson ImmunoResearch | 715-545-150 | Used for secondary antibody (2.2.6) |
| Caterer's foil | Any | N/A | Used in steps 1.2.2 and 2.3.2 |
| Fluoromount-G | Southern Biotech | 0100-01 | Used for coverslipping (2.2.8) |
| Coverslips | Fisher Scientific | 12544E | Used for coverslipping (2.2.8) |
| Clear Nail Polish | Any | N/A | Used for coverslipping (2.2.8) |
| Axio Observer.Z1 and LSM 710 (laser scanning, confocal) | Carl Zeiss | N/A | Used for imaging (3) |
| Axioskop A2 | Carl Zeiss | N/A | Used for imaging (3) |
| CitriSolv | FisherScientific | For DAB protocol | |
| ABC | Vector Laboratories | PK-6100 | For DAB protocol |
| DAB Peroxidase kit | Vector Laboratories | SK-4100 | For DAB protocol |
| Biotinylated horse α-rabbit IgG | Vector Laboratories | BA-1100 | For DAB protocol |
| Biotinylated horse α-mouse IgG | Vector Laboratories | BA-2001 | For DAB protocol |
| 30% Hydrogen Peroxide | FisherScientific | H325-500 | For DAB protocol |
| Wheaton slide racks and staining dishes | TedPella | 21043 | For DAB protocol |
| Masterflex perfusion pump and tubing | Cole-Parmer | Used for perfusion (1.1.1 and 1.1.2) | |
| Andwin scientific tissue-tek CRYO-OCT compound (case of 12) | Fisher Scientific | 14-373-65 | Used for tissue freezing (1.2.1) |
| Thermometer (-50 to 50 C) | Fisher Scientific | 15-059-228 | Used for tissue freezing (1.2.1) |
| Cryostat | Leica | CM3500S | Used for tissue sectioning (1.2.2) |
| Staining Dish, Plastic with 2 Lids | Grale Scientific | 353 | For antigen retrival |
| 20 Place Staining Rack, Slides Horizontal | Grale Scientific | 354 | For antigen retrival |
| Microwave | Any | N/A | For antigen retrival |
Table 1: Materials List.
Discussion
The overall goal of this communication was to introduce immunohistochemistry procedures to the reader. For this, the example of double-labeling with Iba1 and Pan-neuronal antigens to observe microglia and neurons in paraformaldehyde perfused, sucrose cryoprotected, cryosectioned rat brain was used.
This technique can be adapted to serve endless purposes. An array of different antigens in a variety of tissue types such as, but not limited to brain, lung, liver, kidney and intestine can be visualized immunohistochemically with minor adjustments to the protocol. Immunohistochemistry allows for the direct comparison of multiple markers and can encompass up to four different antibodies in a single sample. The number of antibodies used in one sample is limited by bleed-through, which describes overlapping of fluorescent wavelengths. There is a limited fluorescent light spectrum, which consequently limits the staining combinations within a single sample (see Figure 6).
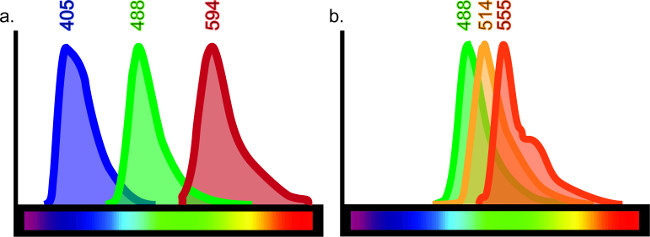 Figure 6: Color spectrum chart. Each fluorophore emits a specific wavelength of light when excited by a specific laser or filter. In order to effectively produce multi-labeled tissue, there should be minimal overlap between specific fluorophores conjugated to the antibodies. Although there are at least 10 different fluorophores available on the market, it is not possible to obtain clear results if all 10 are used. For example, triple-labeling with wavelength emissions at 405, 488 and 594 (A) would give clear labeling as there is minimal overlap, if any, between these wavelengths. However, if wavelength emissions at 488, 514 and 555 were used, the staining would not be easily interpreted as each antibody may be excited by the laser or filter meant for the other (B). This would give false co-labeling results. Online guides for fluorophore selection are available by searching for “fluorescent spectrum viewer”.
Figure 6: Color spectrum chart. Each fluorophore emits a specific wavelength of light when excited by a specific laser or filter. In order to effectively produce multi-labeled tissue, there should be minimal overlap between specific fluorophores conjugated to the antibodies. Although there are at least 10 different fluorophores available on the market, it is not possible to obtain clear results if all 10 are used. For example, triple-labeling with wavelength emissions at 405, 488 and 594 (A) would give clear labeling as there is minimal overlap, if any, between these wavelengths. However, if wavelength emissions at 488, 514 and 555 were used, the staining would not be easily interpreted as each antibody may be excited by the laser or filter meant for the other (B). This would give false co-labeling results. Online guides for fluorophore selection are available by searching for “fluorescent spectrum viewer”.
When double-labeling, it is best to choose primary antibodies made in different species, as both antibodies can be applied simultaneously. Double-labeling with two antibodies from the same species is possible, although technically difficult. When both primary antibodies are made in the same species, staining must be done sequentially, by completing the staining for one antigen before the second. Fewer issues will arise if the primary antibodies from the same species have different Ig classes (i.e. mouse IgG and mouse IgM or different subclasses IgG1 and IgG2a). For more details consult additional references10,11. This protocol uses rabbit anti-Iba1 and mouse anti-Pan-neuronal. It is also necessary to include appropriate negative controls. For double-labeling with Iba1 and Pan-neuronal, one slide contains no primary antibodies, one slide contains anti-Iba1 only and one slide contains anti-Pan-neuronal only (see Figure 4). These negative controls confirm an absence of cross-reactivity and false staining. All other slides contain both primary antibodies. It is also important to avoid using secondary antibodies that may cross-react. For example, using donkey anti-rabbit 594 and rabbit anti-mouse 488 would result in donkey anti-rabbit 594 binding to both rabbit anti-Iba1 primary antibody and the donkey anti-rabbit 594 anti-mouse 488 secondary antibody. This would create false co-labeling.
Antibodies for a particular protein (antigen) are not always the same. For example, some antibodies are raised against antigens which are present in frozen tissue, whereas others will recognize an antigen which has been altered during the paraffin embedding process. For this reason, particular attention should be paid to the antibody specification sheet prior to purchase. If the intended application for the antibody is not listed, trouble-shooting may be required to obtain staining, or staining may not work at all. In addition, previously published data may offer “tricks of the trade” to obtain staining. Of importance to note is that subtle differences may be seen in different batches of antibodies; slight modifications may need to be made with each re-order of an antibody.
Most antibodies are good for 12 months from the date of receipt. Be sure to indicate the date of arrival on the product specification sheet. If antibody staining is not observed, the antibody may be too old or stored incorrectly. It is therefore also important to check each antibody’s specification sheet for proper storage.
Additionally, for many antibodies, different clones are available. Different clones recognize different parts of the antigen of interest. A given clone may be better for tissue subjected to different processing techniques (wax-embedded/frozen), target tissue type, or target species. Pay particular attention to the species in which the antibody was raised. When the antibody is raised in the same species as the tissue for analysis, high background may be observed. To combat this, additional blocking with commercially available kits such as a mouse on mouse kit, may be required for staining. This technique can also be adapted to cell culture. Fixation of cells, clone and specificity need to be checked on the specification sheet for applications.
Fixation with paraformaldehyde cross-links proteins, which could hide antigen binding sites in some conditions. Antigen retrieval can break protein cross-links and expose binding sites. If antigen retrieval is required, insert the following protocol after step 2.1.3: using a plastic staining dish and insert (see Table 1), microwave slides in citrate buffer on a low power setting for 10 minutes, taking care to not let the solution boil rigorously (specific power levels not included due to variability in settings between microwaves). Alternately, bring citrate buffer to 80 °C in a glass staining dish in an oven, then add slides for 10 min.
Poor tissue quality can also cause problems with staining (see Figure 5). There are a number of precautions one can take to avoid compromising tissue quality. First, be patient and diligent with sucrose changes in order to thoroughly displace water in the tissue before freezing. When freezing, keep the temperature constant. If the tissue freezes too slowly, water molecules will expand as they freeze, creating holes in the tissue. Alternately, freezing at too low a temperature will cause the tissue to crack.
Protocols will vary slightly for each antibody of interest. Upon receipt of a new antibody, one must optimize the protocol accordingly. If background is high (Figure 5), try blocking for a longer period of time. Alternately, increase the serum concentration of the block solution or add serum from multiple different species e.g. goat and rabbit serum. Additionally, casein can be used to reduce non-specific binding. Skim milk (made from dry powder) contains casein and can be used in a range of concentrations (typically, 1-5% w/v). Milk can be used alone, or in conjunction with serum. If an antigen of interest is an intracellular marker, detergents such at TritonX-100 or Tween-20 can be added to the blocking solution to permeabilize cell membranes. Thicker tissue sections can be processed similarly by floating the tissue sections in wells instead of mounting them onto slides. Paraffin-embedded tissue can also be processed similarly, but requires a de-waxing step prior to blocking. Feldengut et al.12 addresses both of these methods.
Due to the light-sensitive nature of fluorescent antibodies, it is critical that fluorescent antibodies are stored in dark containers and avoid light exposure following secondary antibody incubation. In contrast, when using DAB as a developing agent, exposure to light is not detrimental. Fluorescent staining is limited in its longevity. Fluorescent-stained slides should be stored in a light tight box at 4 °C. Images should be captured within a month of staining. If immediate imaging is not possible, consider DAB or other alternative staining methods or delay staining until imaging is possible. Staining with DAB is usually limited to staining one target antigen, whereas fluorescence allows for double- and triple-labeling. Double- and triple-labeling can be achieved using DAB however this technique is prone to complications and difficulty in isolating specific antigen staining. DAB staining may be visualized under bright field microscopy, whereas imaging fluorescent staining requires a fluorescent microscope.
Another factor to take into account with DAB staining is that cells span multiple layers of tissue. It is therefore difficult to obtain clear images of cells such as microglia which have spanning processes. To overcome this issue, some investigators cut their sections thinner at 8-12 µm.
Imaging of fluorescently labeled tissue is best achieved on a confocal microscope. Confocal microscopes eliminate the haze normally seen with a research fluorescent microscope by isolating and capturing a single plane of focus within the sample. With confocal microscopy, finer details are sharper and bleed-through between fluorescent dyes is reduced. Thicker samples can be imaged one plane at a time (z-stack) to demonstrate true co-localization. When access to a confocal microscope is not available, imaging on a research microscope is possible, however exposure time and light intensity need to be locked between images.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors would like to thank Mr. Ryan Hart and Mr. Arriz Lucas for their invaluable feedback on this communication. This work was supported by NIH NINDS R01 NS065052 and Phoenix Children’s Hospital Mission Support Funds.
References
- Marrack JR. Chemistry of antigens and antibodies. Nature. 1934;134:468–469. [Google Scholar]
- Coons AH, Creech HJ, Jones RN, Berliner E. The demonstration of pneumococcal antigen in tissues by the use of fluorescent antibody. J Immunol. 1942;45:159–170. [Google Scholar]
- Marshall JM. Localization of adrenocorticotropic hormone by histochemical and immunochemical methods. The Journal of experimental medicine. 1951;94:21–30. doi: 10.1084/jem.94.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellors RC. Histochemical demonstration of the in vivo localization of antibodies: antigenic components of the kidney and the pathogenesis of glomerulonephritis. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society. 1955;3:284–289. doi: 10.1177/3.4.284. [DOI] [PubMed] [Google Scholar]
- Nakane PK, Pierce GB. Enzyme-labeled antibodies: preparation and application for the localization of antigens. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society. 1966;14:929–931. doi: 10.1177/14.12.929. [DOI] [PubMed] [Google Scholar]
- Avrameas S, Uriel J. Method of antigen and antibody labelling with enzymes and its immunodiffusion application. Comptes rendus hebdomadaires des seances de l'Academie des sciences. Serie D: Sciences naturelles. 1966;262:2543–2545. [PubMed] [Google Scholar]
- Cuello AC. Immunohistochemistry. Wiley; 1983. [Google Scholar]
- Junqueira LCU, Mescher AL. Junqueira's basic histology : text and atlas. Thirteenth edition / edn 2013.
- Gage GJ, Kipke DR, Shain W. Whole animal perfusion fixation for rodents. Journal of visualized experiments : JoVE. 2012. [DOI] [PMC free article] [PubMed]
- Christensen NK, Winther L. In: Education Guide: Immunohistochemical (IHC) staining methods. Kumar GL, Rudbeck L, editors. Carpinteria, California: DAKO North America; 2009. pp. 103–108. [Google Scholar]
- Wang G, Achim CL, Hamilton RL, Wiley CA, Soontornniyomkij V. Tyramide signal amplification method in multiple-label immunofluorescence confocal microscopy) Methods. 1999;18:459–464. doi: 10.1006/meth.1999.0813. [DOI] [PubMed] [Google Scholar]
- Feldengut S, Del Tredici K, Braak H. Paraffin sections of 70-100 mum: a novel technique and its benefits for studying the nervous system. Journal of neuroscience methods. 2013;215:241–244. doi: 10.1016/j.jneumeth.2013.03.010. [DOI] [PubMed] [Google Scholar]