

Abstract
Motivation: Discerning genetic contributions to diseases not only enhances our understanding of disease mechanisms, but also leads to translational opportunities for drug discovery. Recent computational approaches incorporate disease phenotypic similarities to improve the prediction power of disease gene discovery. However, most current studies used only one data source of human disease phenotype. We present an innovative and generic strategy for combining multiple different data sources of human disease phenotype and predicting disease-associated genes from integrated phenotypic and genomic data.
Results: To demonstrate our approach, we explored a new phenotype database from biomedical ontologies and constructed Disease Manifestation Network (DMN). We combined DMN with mimMiner, which was a widely used phenotype database in disease gene prediction studies. Our approach achieved significantly improved performance over a baseline method, which used only one phenotype data source. In the leave-one-out cross-validation and de novo gene prediction analysis, our approach achieved the area under the curves of 90.7% and 90.3%, which are significantly higher than 84.2% (P < e−4) and 81.3% (P < e−12) for the baseline approach. We further demonstrated that our predicted genes have the translational potential in drug discovery. We used Crohn’s disease as an example and ranked the candidate drugs based on the rank of drug targets. Our gene prediction approach prioritized druggable genes that are likely to be associated with Crohn’s disease pathogenesis, and our rank of candidate drugs successfully prioritized the Food and Drug Administration-approved drugs for Crohn’s disease. We also found literature evidence to support a number of drugs among the top 200 candidates. In summary, we demonstrated that a novel strategy combining unique disease phenotype data with system approaches can lead to rapid drug discovery.
Availability and implementation: nlp.case.edu/public/data/DMN
Contact: rxx@case.edu
1 Introduction
Identifying the genetic basis for human diseases plays an important role in elucidating disease mechanisms and discovering targets of drug treatments (Hurle et al., 2013; Plenge et al., 2013). For computational strategies to predict disease-associated genes, integrating new data may lead to new discoveries (Barabási et al., 2011; Piro and Di Cunto, 2012; Tiffin et al., 2009; Tranchevent et al., 2011; Wang et al., 2011). Traditional approaches exploited genomic data and prioritized genes for a disease if the genes are functionally similar to the known disease genes (Aerts et al., 2006; Franke et al., 2006; Köhler et al., 2008; Xu and Li, 2006). Recent studies incorporated clinical phenotype data to increase the ability of identifying new disease-associated genes (Hwang et al., 2012; Lage et al., 2007; Li and Patra, 2010; Vanunu et al., 2010; Wu et al., 2008, 2009), assuming that similar disease phenotypes reflect overlapping genetic causes (Brunner and Van Driel, 2004; Houle et al., 2010; Oti et al., 2008, 2009).
However, most current disease gene prediction approaches (Hwang et al., 2012; Lage et al., 2007; Li and Patra, 2010; Vanunu et al., 2010; Wu et al., 2008, 2009) used only one single data source of human disease phenotypes. Phenotypic similarity databases were usually obtained by extracting phenotype knowledge from texts, such as biomedical literature (Korbel et al., 2005) and the phenotype descriptions in Online Mendelian Inheritance in Man (OMIM) (Lage et al., 2007; Robinson et al., 2008; Van Driel et al., 2006). Among them, mimMiner (Van Driel et al., 2006) and human phenotype ontology (Robinson et al., 2008) are based on OMIM and have been widely used in disease gene prediction studies (Hoehndorf et al., 2011; Hwang et al., 2012; Li and Patra, 2010; Natarajan and Dhillon, 2014; Vanunu et al., 2010). Recently, we explored a different database containing phenotypic knowledge— the semantic network in Unified Medical Language System (UMLS)—and constructed a new phenotype network called Disease Manifestation Network (DMN) (Chen et al., 2015). We demonstrated that DMN not only reflects genetic relationships among diseases, but also contains different knowledge compared with the existing database (Chen et al., 2015). We hypothesize that integrating this new phenotype network with the widely used disease phenotype data will improve the prediction of disease genetics.
In this study, we developed a novel and generic approach to combine multiple different data sources on human disease phenotype, and predict disease-associated genes from seamlessly integrated phenotypic and genomic data. To demonstrate the approach, we integrated DMN, mimMiner, a protein interaction network and known disease–gene associations. We predicted new disease-associated genes from the heterogeneous network, and demonstrated the benefit of incorporating an additional phenotype network DMN by comparing with a baseline approach, which is also based on network analysis but only used mimMiner.
We demonstrated that the disease–gene associations predicted by our approach, in combination with the drug target data, may guide the discovery of new candidate drugs. We used Crohn’s disease as an example, which has increasing worldwide prevalence (Molodecky et al., 2012) and is currently incurable (Atreya et al., 2014; Cosnes et al., 2011). We predicted candidate genes for Crohn’s disease, and prioritized candidate drugs based on the rank of drug target genes. We validated the result with the Food and Drug Administration (FDA)-approved therapies for Crohn’s disease. Our result provides empirical evidence that our disease genetics prediction strategy, which combined unique data and a novel system approach, can lead to rapid drug discovery.
2 Methods
We integrated DMN, mimMiner and a genetic network based on protein–protein interactions (PPIs), and constructed a heterogeneous network in Figure 1. Given a disease, we prioritized the genes using a ranking algorithm extended from the random walk model. We validated our approach using well-studied disease–gene associations from OMIM and compared the performance with a baseline disease gene prediction method that used only one phenotype network. We also evaluated our approach in predicting genes for diseases of different classes. Finally, we identified candidate drug therapies for Crohn’s disease based on gene prediction results, and demonstrated the translational potential of our newly predicted genes.
Fig. 1.
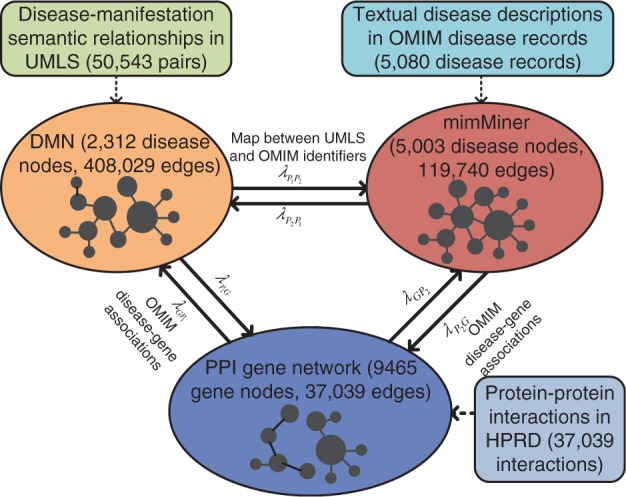
Network integration
2.1 Integrate networks
We first constructed the DMN, mimMiner and the PPI network. To construct DMN, we extracted 50 543 disease-manifestation pairs from UMLS and calculated pairwise disease similarities based on disease manifestations (Chen et al., 2015). Then we downloaded mimMiner (Van Driel et al., 2006) and built the PPI network using 37 039 binary interactions among 9465 genes in the Human Protein Reference Database, which has high coverage and accuracy (Kann, 2010; Moreau and Tranchevent, 2012; Prasad et al., 2009) and has been used in many disease gene discovery studies (Köhler et al., 2008; Li and Patra, 2010; Vanunu et al., 2010; Wu et al., 2008, 2009).
We connected the three networks as shown in Figure1. We linked the disease nodes with the same semantic meanings in DMN and mimMiner using 1313 pairwise mappings between UMLS and OMIM identifiers from the UMLS Metathesaurus. We also connected 1188 disease nodes in DMN and 1542 in mimMiner to the gene nodes in the PPI network based on the disease–gene associations in OMIM. Note that our approach can easily incorporate more phenotypic or genetic networks in the same way, given that the new networks contain different knowledge from the existing ones.
The adjacency matrix of the heterogeneous network is given as follows:
| (1) |
where P1, P2 and G represent DMN, mimMiner and the genetic network, respectively, and the diagonal sub-matrices AG, and are their adjacency matrices. The off-diagonal and are the adjacency matrices of the bipartite graphs connecting each pair of the three networks, and and represent their transposes.
2.2 Predict disease-associated genes from the integrated network
Our prediction model was based on random walk with restart, which is a network-based ranking algorithm. The random walk model avoids over-emphasizing the connections through high-degree nodes and has been useful in biomedical applications (Berger et al., 2010; Köhler et al., 2008; Li and Patra, 2010). It simulates a random walker starting from a set of seed nodes and calculated the ranking scores for all the nodes as the probability of being reached by the random walker after convergence. We set certain disease nodes as the seeds and ranked all the gene nodes to predict their association with the given diseases.
We extended the algorithm by regulating the movements of the random walker between any two networks among DMN, mimMiner and the PPI network with the jumping probabilities (Fig. 1). For example, if the random walker stands on a node in DMN, which is connected with both mimMiner and the genetic network, it has the option to walk to mimMiner with the probability , to the PPI network with the probability or stay within DMN with the probability .
We calculated the ranking scores for all nodes as follows. Assume p0 is a vector of initial scores for each node, pk is the score vector at step k and was iteratively updated by
| (2) |
where γ is the probability that the random walker restarts from the seeds at each step, and M is the transition matrix defined based on the adjacency matrix in (1). We assumed the update converges if the difference between scores in adjacent iterations was smaller than 1 × e−8. The transition matrix consists of three intra-network transition matrices on the diagonal, and six inter-network transition matrices off-diagonal:
| (3) |
We calculated the inter-network transition matrices in (4), which first normalized the adjacency matrices of the bipartite network , and then weighted them with the jumping probabilities between networks Ni and Nj.
| (4) |
The intra-network transition matrices were calculated in (5), which normalized the adjacency matrix of a network Ni, and weighted the matrix with the probability that the random walker jumps within the same network.
| (5) |
In (5), ‘·’ represents dot product and is an indicator function, whose value is 1 if the kth row of contains at least one non-zero element. For the generic case, where N phenotype networks were incorporated, the transition matrix M is defined as follows:
| (6) |
The inter-network transition matrices (off-diagonal) and intra-network transition matrices (diagonal) can still be calculated with (4) and (5), respectively.
Our gene prediction model allows accumulating evidences from different disease phenotype networks and preserves the unique information in each network. For example, if a pair of diseases is connected in both DMN and mimMiner, the random walker can reach one disease node from the other with a strengthened probability; if the diseases are connected in only one network, the random walker may still reach one disease from the other through the links between networks, but with a relatively lower probability.
2.3 Evaluate gene prediction in cross-validation analyses
We first performed a leave-one-out cross-validation analysis and compared our approach with a baseline method (Li and Patra, 2010), which only used one phenotype network. We removed one disease–gene association each time, set the disease as the seed and tested the rank of the retained gene. If the same disease appeared in both phenotype networks (diseases from the two networks have the same semantic meaning) and were connected to the same gene, the redundant disease–gene association was also removed.
We evaluated the ranks of the tested genes with two metrics: (i) we calculated the percentage of successful prioritizations, in which the retained genes were ranked in top 1 (excluding the other known disease genes) and (ii) we generated a receiver operating characteristic (ROC) curve for each method and calculated the area under the curve (AUC). To generate the ROC, we followed the definitions in Aerts et al. (2006), Köhler et al. (2008) and Li and Patra (2010): sensitivity refers to the percentage of tested genes that are ranked above a particular threshold among all prioritizations, and specificity refers to the percentage of genes ranked below this threshold. For instance, a sensitivity/specificity value of 70/90 indicates that the correct disease gene was ranked among the top 10% of genes in 70% of the prioritizations. The ROC shows the plot of sensitivity against 1−specificity when varying the rank threshold from the top to bottom. The two metrics are complimentary: the AUC evaluates the entire rank of genes, while the success ratio is more strict and evaluates the top-ranked genes.
Currently, the causal genes for over 1500 genetic disorders remain unknown (Antonarakis and Beckmann, 2006). A primary advantage of phenotype-driven gene prediction approaches, compared with the conventional gene function-driven approaches, is that they can predict genes for diseases without known genetic basis. Therefore, we further conducted a de novo gene prediction analysis to evaluate our approach. In de novo gene prediction, we removed all disease-gene links for a query disease each time. If the disease appeared in both phenotype networks, we removed all its gene associations through both phenotype networks. Then we set the disease as the seed, ranked all the genes and compared the AUCs between different approaches. In this experiment, we have different settings from the leave-one-out cross-validation and tested multiple retained genes in each prioritization. We generated an ROC curve for each prioritization following the definitions in Chen et al. (2011) and Hwang et al. (2012) and averaged AUCs across all prioritizations. For each ROC, sensitivity is the percentage of retained genes that are ranked above a threshold among all the retained genes in one prioritization, and specificity is the percentage of negative genes (genes that are not known disease genes) ranked below the threshold among all the negative genes. Because the top-ranked genes are more important than the lower ranked genes, we highlighted a set of false positive cutoffs for the ROC curves and compared the corresponding average AUCs between methods. A better method will rank more true positive genes above the false positives, resulting in larger average AUCs at smaller cutoffs.
2.4 Evaluate gene prediction for different disease classes
The degree that phenotypic associations reflect genetic overlaps varies for different disease classes. Thus phenotype-driven gene predictions may have varying performance. We classified diseases into nine groups based on International Classification of Diseases (10th edition), and repeated the two cross-validation experiments within each group to evaluate the performance variance of our method.
2.5 Drug discovery for Crohn’s disease based on predicted disease-associated genes
We used Crohn’s disease as an example to demonstrate that our gene prediction method has the translation potential to guide drug discovery. Crohn’s disease is a chronic and relapsing inflammatory disorder that affects millions of people and has an increasing prevalence (Molodecky et al., 2012). It involves genetic abnormalities that lead to overly aggressive responses to commensal enteric bacteria (Sartor, 2006). Current treatment options, such as systemic anti-inflammatory drugs, targeted drugs and surgeries, may be effective for only a subset of patients or lead to severe side effects (Baumgart and Sandborn, 2007). Therefore, discovering new drug therapies for Crohn’s disease is of great interests.
We first predicted genes for Crohn’s disease using our approach. Then we compared the result with the disease-associated genes in genome-wide association studies (GWAS) catalog (Hindorff et al., 2009). We also evaluated the ranks of drug genes extracted from DrugBank (Law et al., 2014). We hypothesized that if the predicted genes are useful for guiding drug discovery, the top-ranked candidate genes would be enriched for the disease-associated genes in GWAS and drug target genes.
Then we extracted 1190 drugs targeting on the genes in our PPI network using the drug target data from DrugBank. We ranked these candidate drugs based on the sum of the random walk scores for their target genes. We validated our rank of candidate drugs with seven FDA-approved Crohn’s disease drugs (extracted from the drug-indication data in DrugBank), and further investigated the literature evidence for the top 200 candidate drugs.
3 Results
3.1 Integrating DMN with mimMiner significantly improves the performance of disease gene predictions
We compared our gene prediction approach with a baseline method, which integrated mimMiner and the PPI network used in our approach, and predicted disease gene associations with a random walk model (Li and Patra, 2010). We chose parameters for both the methods to achieve optimal performance in the cross-validations and ensure fair comparison, but different parameter values only slightly affect the results. For our method, the jumping probabilities and were set to 0.1; and were set to 0.7 and and were set to 0.4. For the baseline method, the jumping probability between mimMiner and the PPI network was set to 0.9. The probability of restarting from seeds (γ is (2)) was set to 0.7 for both methods
3.1.1 Leave-one-out cross–validation
Our approach achieved significantly better success ratios and AUCs than the baseline method. The integrated network in our approach contains a total of 2397 unique disease–gene associations. If one disease appeared in the two phenotype networks and were connected to a same gene, the two disease-gene links were counted only once. In 1100 of the 2397 validation runs (45.89%), our approach successfully ranked the retained genes in top 1. The success ratio is significantly higher (P < e−4) than 10.36% for the baseline method (Table 1). In addition, Figure 2 compares the ROC curves for gene prediction methods. Our approach achieved an AUC of 90.65%, which is significantly higher (P < e−4) than 84.2% for the baseline approach.
Table 1.
Ratios of successful disease–gene association predictions in the leave-one-out cross-validation experiment
| Phenotype networks | Success number | Success ratio (%) |
|---|---|---|
| mimMiner | 219 | 10.36 |
| DMN and mimMiner | 1100 | 45.89 |
Note: All diseases were included in the experiment.
Fig. 2.
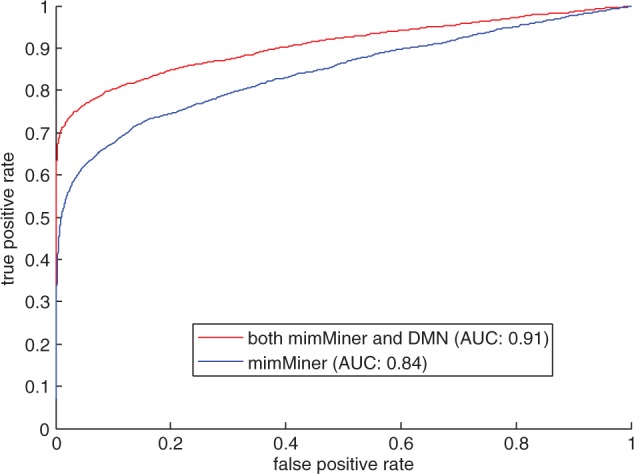
The ROC curves and AUCs for our method (red) and the baseline method (blue) in the leave-one-out cross-validation analysis
3.1.2 De novo gene prediction
Our approach is effective in de novo gene predictions, and outperforms the baseline method by boosting the phenotype knowledge. Specifically, our method achieves an average AUC of 90.33%, which is significantly higher than 81.28% for the baseline method using mimMiner alone (P < e−12). Figure 3 shows that at six false positive cutoffs, integrating DMN and mimMiner achieves significantly higher AUCs (P < e−18) than using only mimMiner. For example, at the cutoff of 10, we achieve an average AUC of 59.19%, while that for the baseline method is 24.17% (P < e−95). For the diseases that only have one associated gene in OMIM, our method successfully predicted the tested genes in top 1 for 52.12% of diseases, while the baseline method succeeded in 11.47% prioritizations (P < e−4). These results show that de novo gene prediction highly depends on disease phenotype relationships, and our method successfully took the advantage of more comprehensive knowledge in multiple phenotypic networks to achieve better performance.
Fig. 3.
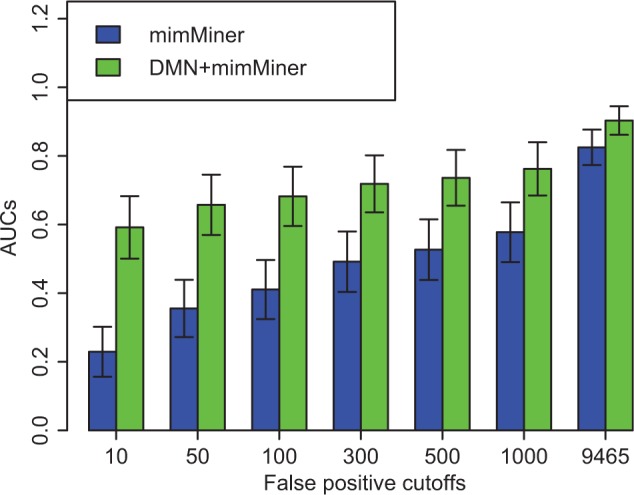
Average AUCs of de novo gene prediction for our approach (green) and the baseline approach (blue). We compared overall AUCs, as well as the AUCs when the numbers of false positive genes are up to 10, 50, 100, 300, 500 and 1000
3.2 Our method achieves high but varying performance for different disease classes
We evaluated the approach for nine disease classes. In the leave-one-out cross-validation, 93.4% retained genes was ranked within top 100, and the AUCs for all disease classes are close and above 90%. But the ranks of the retained genes vary up and down within the top 100 for different disease classes. Figure 4 shows the top part of ROC curves for each disease class. The corresponding AUC is the highest for ‘congenital malformations and deformations’, and lowest for ‘mental diseases’ and ‘malignant neoplasms’. Table 2 (the column of ‘All diseases’) compares the success ratio for all diseases between disease classes, and shows that our approach ranked 78% retained genes for congenital malformations and deformations in top 1, while prioritized 26% and 27% retained genes for malignant neoplasms and mental diseases, respectively.
Fig. 4.
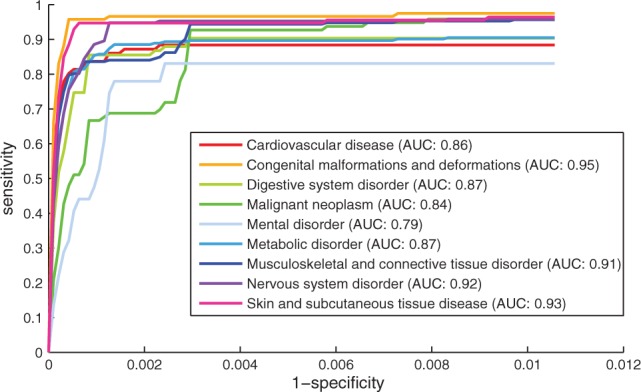
The ROC curves for each disease class in de novo gene prediction. We compared the top part of ROC curves and AUC scores based on the top 100 genes in each validation run
Table 2.
Success ratios of disease–gene association predictions for all diseases and monogenetic diseases in the nine disease classes
| Disease classes | All diseases (%) | Monogenetic diseases (%) |
|---|---|---|
| Congenital malformations and deformations | 77.97 | 90.48 |
| Skin and subcutaneous tissue disease | 70.80 | 81.58 |
| Nervous system disorder | 66.67 | 89.89 |
| Musculoskeletal and connective tissue disorder | 65.09 | 84.06 |
| Digestive system disorder | 65.06 | 80.00 |
| Metabolic disorder | 61.67 | 75.33 |
| Cardiovascular disease | 48.84 | 84.09 |
| Mental disorder | 27.12 | 71.43 |
| Malignant neoplasm | 26.04 | 50.00 |
In the de novo gene prediction, we observed similar performance variance among the nine disease classes. Figure 5 shows that the averaged AUC is the highest for congenital malformations and deformations and lowest for malignant neoplasms at all cutoffs. Table 2 (the column of ‘Monogenetic diseases’) shows that for monogenetic diseases, which have only one gene in OMIM, 90% predictions ranked the disease genes for congenital malformations and deformations in top 1, while 50% predictions succeeded for malignant neoplasms.
Fig. 5.

The ROC curves for each disease class in leave-one-out cross-validation. We compared the top part of ROC curves and AUC scores based on the top 100 genes in each validation run
We traced the disease phenotype features to explain the performance variance. The congenital malformations and deformations often have specific phenotypic features. For example, otospondylomegaepiphyseal dysplasia (OSMED) has manifestations such as ‘sensorineural hearing loss’ and ‘Pierre Robin syndrome’. These features link OSMED to phenotypically similar diseases in the network, such as Stickler syndrome and Marshall syndrome, which are also genetically related to OSMED. On the other hand, malignant neoplasms usually have non-specific manifestations, such as pain, fever and ascites, which are common in cancers with different genetic causes. Therefore, although our approach achieves high performance for all disease classes, building disease-specific models and introducing prior knowledge of disease phenotypes may further improve the accuracy of disease gene predictions.
3.3 Our gene prediction method has the potential to guide the drug discovery for Crohn’s disease
We ranked the 9465 genes in the PPI network for Crohn’s disease and compared the result with 70 genes associated with Crohn’s disease from GWAS catalog. These 70 genes also appeared in our gene rank, and have no overlap with the data in OMIM. Figure 6A1 shows that the number of GWAS genes drops when the rank based on our approach changes from the top to the bottom, while this number distributes evenly among random ranks (Fig. 6A2). Among the top 10% in our rank, we found 19 overlaps with the GWAS genes, which is a 2.5-fold enrichment (P < e−4) compared with the average of 50 random gene ranks. The result shows that our approach can prioritize the disease-associated genes obtained through statistical analysis on large-scale patient data.
Fig. 6.

(A1, A2) Evaluate our gene rank with the genes associated with Crohn’s disease from GWAS. (B1, B2) Evaluate our gene rank with the drug target genes. (C1, C2) Evaluate our drug rank with the FDA-approved drugs
Among the top genes in our rank, we found RIPK2, NLRC4 and ERBIN, which have substantial literature supports on their roles in Crohn’s disease (Gerard et al., 2013; Jostins et al., 2012; Kufer et al., 2006; Lupfer et al., 2013; Philpott et al., 2014; Standaert-Vitse et al., 2009; Tomalka et al., 2011) and directly interact with NOD2 (a Crohn’s disease gene in OMIM). In addition, we also found literature evidence to support a few top-ranked genes that are not directly interacting with the disease genes from OMIM and were not identified in GWAS. For example, NLRP3 (ranked top 32), CASP1 (ranked top 45) and BCL10 (ranked top 46) are associated with the innate immune responses to the intestinal microbiota, which has been linked with the pathogenesis of Crohn’s disease (Borthakur et al., 2007; Hirota et al., 2011; Netea et al., 2010; Villani et al., 2008).
We also investigated the distribution of 1502 drug target genes (from DrugBank) among our gene rank. Figure 6B1 and B2 shows that our rank is more likely to prioritize druggable genes than the random ranks. The top 10% genes in our rank contain 331 drug target genes, which is a 2.1-fold enrichment (P < e−21) compared with the average of random cases. The result shows that our top-ranked predicted genes are enriched for druggable genes associated with Crohn’s disease, and offer the opportunities to detect candidate drugs for Crohn’s disease.
We ranked 1190 candidate drugs (from DrugBank) based on the sum of the random walk scores for their target genes. Figure 6C1 and C2 shows that our approach can prioritize the approved Crohn’s disease therapies. The top 200 in our rank contains four FDA-approved drugs, which is a 3.3-fold enrichment (P < e−3) compared with the average of random cases. Note that these four approved drugs, including Sulfasalazine, Mesalazine, Adalimumab and Natalizumab, do not directly target on the Crohn’s disease genes in OMIM, and were detected through the prioritized genes using our approach. We further investigated the other candidate drugs in top 200 in our rank, and found that a number of them are supported by literature evidence as candidate Crohn’s disease treatments. Table 3 shows a few examples of candidate drugs and their supports. Among them, the efficacy of tocilizumab has recently been tested in a randomized clinical trial (Lazzerini et al., 2013) and showed positive results in clinical remission.
Table 3.
Drug candidates for Crohn’s disease that are supported by literature
| Rank | Drugs | Current drug indications | References |
|---|---|---|---|
| 3 | Tocilizumab | Rheumatoid arthritis | Nishimoto and Kishimoto (2008) and Gergis et al. (2010) |
| 11 | Sargramostim | Myeloid reconstitution | Korzenik et al. (2005) and Roth et al. (2011) |
| 31 | Minocycline | Infections | Margolis et al. (2010) |
| 78 | Amitriptyline | Depression | Rahimi et al. (2012) |
| 80 | Desipramine | Depression | Rahimi et al. (2009) |
| 86 | Mecasermin | Growth failure | Rosenbloom (2009) and Puche and Castilla-Cortzar (2012) |
| 194 | Thalidomide | Erythema nodosum leprosum | Lazzerini et al. (2013) |
4 Conclusion and discussions
Incorporating clinical phenotype data can improve the prediction power of disease gene discovery methods. In this study, we developed a disease gene prediction framework leveraging multiple different human phenotype data sources. We explored a unique phenotype data source and constructed a new phenotype network called DMN. We designed an innovative strategy to predict disease-associated genes from the heterogeneous network combining DMN with mimMiner (a widely used phenotype database) and a genetic network. Comparing with the gene prediction approach using only one phenotype network, our approach significantly improved the performance through boosting phenotypic knowledge. Using Crohn’s disease as an example, we demonstrated that our gene prediction result has translational potentials to guide drug discovery.
As more human disease phenotype data become available, our approach can be further improved by integrating new disease phenotype networks, given that the new networks contain different knowledge. For example, our approach in this study included many Mendelian diseases. Adding phenotypic associations involving common complex diseases may offer novel insights. Also, the phenotypic relationships in this study are primarily based on disease-manifestation pairs. Other kinds of disease phenotype data, such as disease co-morbidities and gene expression profiles, may also reflect different aspects of genetic mechanisms. In the future, we will develop new approaches to rationally integrate heterogeneous phenotype data. For common complex diseases, we will also incorporate multiple different types of genetic associations besides the PPI network, such as the gene regulatory network into the approach.
In addition, phenotype-driven disease gene prediction approaches are effective at different degrees for disease classes (as we have demonstrated) and among different patients. Building disease-specific and patient-specific computational models may further improve the quality of disease gene predictions. We recently studied cancer-specific comorbidities and analyzed the variation of comorbidity patterns among stratified patients in different age and gender brackets (Chen and Xu, 2014a, b). Based on these results, we plan to build a cancer-specific gene prediction model.
Currently, we directly used disease–gene associations in drug discovery. The method to identifying candidate drugs can be further enhanced if more detailed information is available, including drug actions and disease pathogenesis, such as the direction of the genetic abnormality. For example, if a disease results from the loss of function, agonists will be potential drugs, whereas antagonists will lead to side effects. In the future work, we will develop rational drug discovery approach on the basis of our result and more data on both diseases and drugs.
Acknowledgements
R.X. conceived the study. Y.C. and R.X. designed the experiment. Y.C. performed the experiment and wrote the manuscript. All authors have participated in study discussion and manuscript preparation.
Funding
Y.C. and R.X. are funded by the Eunice Kennedy Shriver National Institute of Child Health & Human Development of the National Institutes of Health under the NIH Directors New Innovator Award number DP2HD084068 and the National Cancer Institute Training grant in Computational Genomic Epidemiology of Cancer (R25 CA094186-06). We would like to thank our funding and the reviewers for their invaluable comments and suggestions.
Conflict of Interest: none declared.
References
- Aerts S., et al. (2006) Gene prioritization through genomic data fusion. Nat. Biotechnol., 24, 537–544. [DOI] [PubMed] [Google Scholar]
- Antonarakis E.S., Beckmann S.J. (2006) Mendelian disorders deserve more attention. Nat. Rev. Genet., 7, 277–282. [DOI] [PubMed] [Google Scholar]
- Atreya R., et al. (2014) In vivo imaging using fluorescent antibodies to tumor necrosis factor predicts therapeutic response in Crohn’s disease. Nat. Med., 20, 313–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabási A.L., et al. (2011) Network medicine: a network-based approach to human disease. Nat. Rev. Genet., 12, 56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgart C.D., Sandborn J.W. (2007) Inflammatory bowel disease: clinical aspects and established and evolving therapies. Lancet, 369, 1641–1657. [DOI] [PubMed] [Google Scholar]
- Berger I.S., et al. (2010) Systems pharmacology of arrhythmias. Sci. Signal, 3, ra30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borthakur A., et al. (2007) Carrageenan induces interleukin-8 production through distinct Bcl10 pathway in normal human colonic epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol., 292, G829–G838. [DOI] [PubMed] [Google Scholar]
- Brunner H.G., Van Driel A.M. (2004) From syndrome families to functional genomics. Nat. Rev. Genet., 5, 545–551. [DOI] [PubMed] [Google Scholar]
- Chen Y., Xu R. (2014a) Network analysis of human disease comorbidity patterns based on large-scale data mining. In: Proceedings of the International Symposium on Bioinformatics Research and Applications, Zhangjiajie, China, June 28–30, 2014. pp. 243–254. [Google Scholar]
- Chen Y., Xu R. (2014b) Mining cancer-specific disease comorbidities from a large observational database. Cancer Inform., 13(Suppl. 1), 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., et al. (2011) Uncover disease genes by maximizing information flow in the phenome-interactome network. Bioinformatics, 27, i167–i176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., et al. (2015) Comparative analysis of a novel disease phenotype network based on clinical manifestations. J. Biomed. Inform., 53, 113–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosnes J., et al. (2011) Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology, 140, 1785–1794. [DOI] [PubMed] [Google Scholar]
- Franke L., et al. (2006) Reconstruction of a functional human gene network, with an application for prioritizing positional candidate genes. Am. J. Hum. Genet., 78, 1011–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerard R., et al. (2013) An immunological link between Candida albicans colonization and Crohn’s disease. Crit. Rev. Microbiol., doi:10.3109/1040841X.2013.810587. [DOI] [PubMed] [Google Scholar]
- Gergis U., et al. (2010) Effectiveness and safety of tocilizumab, an anti-interleukin-6 receptor monoclonal antibody, in a patient with refractory GI graft-versus-host disease. J. Clin. Oncol., 28, e602–e604. [DOI] [PubMed] [Google Scholar]
- Hindorff A.L., et al. (2009) Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA, 106, 9362–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota A.S., et al. (2011) NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm. Bowel Dis., 17, 1359–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoehndorf R., et al. (2011) PhenomeNET: a whole-phenome approach to disease gene discovery. Nucleic Acids Res., 39, e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houle D., et al. (2010) Phenomics: the next challenge. Nat. Rev. Genet., 11, 855–866. [DOI] [PubMed] [Google Scholar]
- Hurle R.M., et al. (2013) Computational drug repositioning: from data to therapeutics. Clin. Pharmacol. Ther., 93, 335–341. [DOI] [PubMed] [Google Scholar]
- Hwang T., et al. (2012) Co-clustering phenome-genome for phenotype classification and disease gene discovery. Nucleic Acids Res., 40, e146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jostins L., et al. (2012) Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature, 491, 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kann G.M. (2010) Advances in translational bioinformatics: computational approaches for the hunting of disease genes. Brief Bioinform., 11, 96–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler S., et al. (2008) Walking the interactome for prioritization of candidate disease genes. Am. J. Hum. Genet., 82, 949–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbel O.J., et al. (2005) Systematic association of genes to phenotypes by genome and literature mining. PLoS Biol., 3, e134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzenik R.J., et al. (2005) Sargramostim for active Crohn’s disease. N. Engl. J. Med., 352, 2193–2201. [DOI] [PubMed] [Google Scholar]
- Kufer A.T., et al. (2006) Role for erbin in bacterial activation of Nod2. Infect. Immun., 74, 3115–3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lage K., et al. (2007) A human phenome-interactome network of protein complexes implicated in genetic disorders. Nat. Biotechnol., 25, 309–316. [DOI] [PubMed] [Google Scholar]
- Law V., et al. (2014) DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res., 42, D1091–D1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzerini M., et al. (2013) Effect of thalidomide on clinical remission in children and adolescents with refractory Crohn disease: a randomized clinical trial. J. Am. Med. Assoc., 310, 2164–2173. [DOI] [PubMed] [Google Scholar]
- Li Y., Patra C.J. (2010) Genome-wide inferring gene-phenotype relationship by walking on the heterogeneous network. Bioinformatics, 26, 1219–1224. [DOI] [PubMed] [Google Scholar]
- Lupfer C., et al. (2013) Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat. Immunol., 14, 480–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis J.D., et al. (2010) Potential association between the oral tetracycline class of antimicrobials used to treat acne and inflammatory bowel disease. Am. J. Gastroenterol., 105, 2610–2616. [DOI] [PubMed] [Google Scholar]
- Molodecky A.N., et al. (2012) Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology, 142, 46–54. [DOI] [PubMed] [Google Scholar]
- Moreau Y., Tranchevent C.L. (2012) Computational tools for prioritizing candidate genes: boosting disease gene discovery. Nat. Rev. Genet., 13, 523–536. [DOI] [PubMed] [Google Scholar]
- Natarajan N., Dhillon S.I. (2014) Inductive matrix completion for predicting gene-disease associations. Bioinformatics, 30, i60–i68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea G.M., et al. (2010) IL-1β processing in host defense: beyond the inflammasomes. PLoS Pathog., 6, e1000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimoto N., Kishimoto T. (2008) Humanized antihuman IL-6 receptor antibody, tocilizumab. Handb. Exp. Pharmacol., 181, 151–160. [DOI] [PubMed] [Google Scholar]
- Oti M., et al. (2008) Phenome connections. Trends Genet., 24, 103–106. [DOI] [PubMed] [Google Scholar]
- Oti M., et al. (2009) The biological coherence of human phenome databases. Am. J. Hum. Genet., 85, 801–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpott J.D., et al. (2014) NOD proteins: regulators of inflammation in health and disease. Nat. Rev. Immunol., 14, 9–23. [DOI] [PubMed] [Google Scholar]
- Piro R.M., Di Cunto F. (2012) Computational approaches to disease-gene prediction: rationale, classification and successes. FEBS J., 279, 678–696. [DOI] [PubMed] [Google Scholar]
- Plenge M.R., et al. (2013) Validating therapeutic targets through human genetics. Nat. Rev. Drug Discov., 12, 581–594. [DOI] [PubMed] [Google Scholar]
- Prasad K.T., et al. (2009) Human protein reference database—2009 update. Nucleic Acids Res., 37(Suppl. 1), D767–D772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puche E.J., Castilla-Cortzar I. (2012) Human conditions of insulin-like growth factor-I (IGF-I) deficiency. J. Transl. Med., 10, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahimi R., et al. (2009) Efficacy of tricyclic antidepressants in irritable bowel syndrome: a meta-analysis. World J. Gastroenterol., 15, 1548–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahimi R.H., et al. (2012) Antidepressants can treat inflammatory bowel disease through regulation of the nuclear factor-B/nitric oxide pathway and inhibition of cytokine production: a hypothesis. World J. Gastrointest. Pharmacol. Ther., 3, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson P.N., et al. (2008) The Human Phenotype Ontology: a tool for annotating and analyzing human hereditary disease. Am. J. Hum. Genet., 83, 610–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbloom L.A. (2009) Mecasermin (recombinant human insulin-like growth factor I). Adv. Ther., 26, 40–54. [DOI] [PubMed] [Google Scholar]
- Roth L., et al. (2011) Sargramostim (GM-CSF) for induction of remission in Crohn’s disease. Cochrane Database Syst. Rev., CD008538. [DOI] [PubMed] [Google Scholar]
- Sartor B.R. (2006) Mechanisms of disease: pathogenesis of Crohn’s disease and ulcerative colitis. Nat. Clin. Pract. Gastroenterol. Hepatol., 3, 390–407. [DOI] [PubMed] [Google Scholar]
- Standaert-Vitse A., et al. (2009) Candida albicans colonization and ASCA in familial Crohn’s disease. Am. J. Gastroenterol., 104, 1745–1753. [DOI] [PubMed] [Google Scholar]
- Tiffin N., et al. (2009) Linking genes to diseases: it’s all in the data. Genome Med., 1, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomalka J., et al. (2011) A novel role for the NLRC4 inflammasome in mucosal defenses against the fungal pathogen Candida albicans. PLoS Pathog., 7, e1002379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tranchevent L.C., et al. (2011) A guide to web tools to prioritize candidate genes. Brief Bioinform., 12, 22–32. [DOI] [PubMed] [Google Scholar]
- Van Driel A.M., et al. (2006) A text-mining analysis of the human phenome. Eur. J. Hum. Genet., 14, 535–542. [DOI] [PubMed] [Google Scholar]
- Vanunu O., et al. (2010) Associating genes and protein complexes with disease via network propagation. PLoS Comput. Biol., 6, e1000641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villani C.A., et al. (2008) Common variants in the NLRP3 region contribute to Crohn’s disease susceptibility. Nat. Genet., 41, 71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., et al. (2011) Network-based methods for human disease gene prediction. Brief Funct. Genomics, 10, 280–293. [DOI] [PubMed] [Google Scholar]
- Wu X., et al. (2008) Network-based global inference of human disease genes. Mol. Syst. Biol., 4, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., et al. (2009) Align human interactome with phenome to identify causative genes and networks underlying disease families. Bioinformatics, 25, 98–104. [DOI] [PubMed] [Google Scholar]
- Xu J., Li Y. (2006) Discovering disease-genes by topological features in human protein-protein interaction network. Bioinformatics, 22, 2800–2805. [DOI] [PubMed] [Google Scholar]