

Abstract
Neurofibroma (NF), a benign peripheral nerve sheath tumor, is very uncommon in the sinonasal tract, with only a few reported cases in the English literature. Cases within the files of the authors’ institutions confined to the sinonasal tract were compared to cases reported in the English literature (Medline 1966–2014). The 12 patients included 6 females and 6 males, aged 26–75 years (mean 46.2 years). The patients usually presented clinically with a mass lesion (n = 11), obstruction (n = 4) or pain (n = 3), with an average symptom duration of 42.9 months. Two patients had neurofibromatosis (NF1). Tumors involved the nasal cavity alone (n = 8), maxillary sinus alone (n = 2), or mixed sites (n = 2), with a range of 0.4–4.1 cm (mean 2.2 cm). The tumors were circumscribed, composed of spindled to wavy cells with curvilinear nuclei set in a background of collagenized stroma and mast cells. Nuclear palisading and perivascular hyalinization were not seen. Mitoses were scant. Pleomorphism, necrosis and increased cellularity were absent. By immunohistochemistry, the lesional cells were S100 protein, SOX10 and NFP positive, while CD34 highlighted the perineurium. INI1 was intact, with strong nuclear expression in all cases. All patients had surgical excision without recurrence (mean follow-up 8.6 years). The principle differential diagnoses include schwannoma, perineurioma, fibromatosis, and solitary fibrous tumor. NF of the sinonasal tract occurs in middle-aged patients without a gender predilection, usually with non-specific symptoms present for a long duration. Tumors are relatively large (mean 2.2), and usually affect one site only. Surgery is curative, with only 16.7 % NF1 associated. S100 protein, SOX10 and NFP highlight the Schwann cells, with CD34 highlighting the perineural fibroblasts.
Keywords: Neurofibroma, Sinonasal tract, Peripheral nerve sheath tumor, NF1, Immunohistochemistry
Introduction
Benign peripheral nerve sheath tumors are a relatively rare tumor arising primarily within the sinonasal tract and paranasal sinuses. Generally separated into schwannoma and neurofibroma (NF), NF are a benign tumor arising in intimate association with a peripheral nerve trunk, developing from Schwann cells, perineurites, and blended with fibroblastic cells. This benign tumor is usually a solitary lesion, although multiple tumors are seen more often in patients affected by neurofibromatosis 1 (NF1). NF are frequently considered within the differential diagnosis of other spindle cell tumors of the sinonasal tract (SNT). We undertook this study in order to more completely define the clinical, histologic, and immunophenotypic features of NF of the sinonasal tract with a comparison to cases reported in the English literature.
Materials and Methods
Twelve patients with sinonasal tract neurofibroma were identified in the files of the Departments of Pathology within Southern California Permanente Medical Group and Johns Hopkins Medical Institutions between 1997 and 2013 (Tables 1, 2). Materials were supplemented by a review of the patient demographics (gender, age); symptoms at presentation (including duration); and past medical history (specifically, NF1 history). In addition, we reviewed surgical pathology and operative reports and obtained follow-up information from the treating physician or the patient. Follow-up data included the exact location, size, treatment modalities, and current patient and disease status. Hematoxylin and eosin (H&E) stained slides from all cases were reviewed for morphologic assessment of the established diagnostic criteria for neurofibroma. This clinical investigation was conducted in accordance and compliance with all statutes, directives, and guidelines of an internal review board authorization (#5968) performed under the direction of Southern California Permanente Medical Group and the Code of Federal Regulations, Title 45, Part 46.
Table 1.
Case series of sinonasal tract neurofibroma
| Case | Sex | Race | Age (years) | Side | Symptoms | Symptom duration (months) | Location | Size (cm) | Treatment | F/U (years) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | B | 52 | R | Mass, obstruction, congestion | 4 | Maxillary sinus | 3.8 | Lateral rhinotomy and Caldwell-luc | A, NED, 17.1 |
| 2 | M | B | 31 | R | Mass | 2 | Nasal cavity (vestibule) | 0.9 | Excision | A, NED, 14.0 |
| 3 | F | W | 33 | R | Mass, obstruction | 8 | Maxillary sinus and ethmoid | 3.2 | FESS | A, NED, 13.6 |
| 4 | M | B | 54 | L | Mass, epistaxis | 4 | Nasal cavity (vestibule) | 0.4 | Excision | A, NED, 12.6 |
| 5 | F | W | 49 | R | Mass, epistaxis, pain | 24 | Nasal cavity (vestibule) | 0.5 | Excision | A, NED, 7.2 |
| 6 | M | W | 61 | L | Mass, obstruction | 40 | Nasal cavity | 4.1 | FESS | A, NED, 2.7 |
| 7 | F | W | 32 | L | Mass | 5 | Nasal cavity (vestibule) | 0.6 | Excision | A, NED, 1.2 |
| 8 | M | W | 75 | R | Mass, swelling, obstruction, pain, tenderness | 1 | Nasal cavity (vestibule) | 2.1 | Excision | A, NED, 0.8 |
| 9 | M | W | 34 | L | Mass, NF1 | N/R | Maxillary sinus | 4 | Excision | A, NED |
| 10 | F | A | 66 | L | Mass, pain | 24 | Maxillary sinus, nasal cavity, ethmoid, sphenoid, | 2 | Excision | A, NED |
| 11 | M | W | 26 | R | Mass | 120 | Nasal cavity | 3 | Excision | A |
| 12 | F | O | 41 | R | Mass, NF1 | 240 | Nasal cavity | 1.5 | Excision | A |
F/U follow-up, F female, M male, B black, W white, R right, L left, FESS functional endoscopic sinus surgery, A alive, NED no evidence of disease, N/R not reported, NF1 neurofibromatosis 1
Table 2.
Summary of case series of sinonasal tract neurofibroma
| Characteristic | Number [n = 12] |
|---|---|
| Gender | |
| Females | 6 |
| Males | 6 |
| Age (years) | |
| Range | 26–75 |
| Mean | 46.2 |
| Median | 45 |
| Female (mean) | 45.5 |
| Male (mean) | 46.8 |
| Symptom duration (months) a | |
| Range | 1–240 |
| Mean | 42.9 |
| Female patients (mean) | 50.8 |
| Male patients (mean) | 33.4 |
| Left (mean) | 18.3 |
| Right (mean) | 57.0 |
| Anatomic side a | |
| Left | 5 |
| Right | 7 |
| Anatomic site a | |
| Nasal cavity only | 8 |
| Maxillary sinus only | 2 |
| Mixed site | 2 |
| Size (cm) a | |
| Range | 0.4–4.1 |
| Mean | 2.2 |
| Median | 2.1 |
| Female (mean) | 1.9 |
| Male (mean) | 2.4 |
| Left (mean) | 2.2 |
| Right (mean) | 2.1 |
| Patient follow-up (mean, years) a | |
| Alive, no evidence of disease (n = 10) | 8.6 |
aNot reported for all cases
Immunophenotypic analysis was performed by a standardized BenchMark-XT™ method employing 4 µm-thick, formalin fixed, paraffin embedded sections. Table 3 documents the pertinent, commercially available immunohistochemical antibody panel used. When required, cellular conditioning for antigen retrieval was performed by various standardized retrieval techniques, as standardized and validated in our laboratory. Standard positive controls were used throughout, with serum used as the negative control. The antibody reactions were described as either positive or negative; nuclear, cytoplasmic, membranous or combination; and a percentage reported for the Ki-67 antibody. Standard positive controls were used throughout, with serum used as the negative control.
Table 3.
Immunohistochemical panel and results
| Antigen/antibody/clone | Company | Dilution | Result |
|---|---|---|---|
| S-100 protein | Dako | 1:2,000 | 12/12 (100 %); strong, diffuse, nuclear and cytoplasmic positive |
| Glial fibrillary acidic protein (GFAP)(6F2) | Dako | 1:200 | 7/12 (58 %); weak to strong, focal cytoplasmic positive |
| SOX10 (EP268) | Epitomics | 1:250 | 7/12 (58 %); strong, focal, nuclear |
| NFP (2F11) | Dako | Neat | 9/12 (75 %); weak to strong, diffuse, cytoplasmic |
| Epithelial membrane antigen (EMA)(E29) | Ventana medical systems | Neat | 1/12 (8 %); strong, focal, cytoplasmic |
| CD34 (QBEnd/10) | Ventana medical systems | Neat | 11/12 (92 %); strong, diffuse, cytoplasmic (adjacent to neural cells) |
| bcl-2 | Dako | 1:40 | 11/12 (92 %); weak to strong, diffuse, cytoplasmic positive |
| INI-1 (BAF47) | Sigma-Aldrich | Neat | 12/12 (100 %); strong, diffuse, nuclear (intact) positive |
| p63 (7jul) | Leica microsystems | 1:40 | 1/12 (8 %); strong, focal, nuclear positive |
| Cytokeratin-pan (AE1/AE3:M3515) | Dako | 1:40 | 0/12 (0 %) |
| Smooth muscle actin (66.4.C2) | Leica microsystems, buffalo grove, IL | 1:200 | 0/12 (0 %) |
| CD117 (C-Kit) | Dako | 1:400 | 0/12 (0 %); positive in mast cells |
| β-Catenin | Cell signal | 1:1,800 | 10/12 (83 %); weak to strong, diffuse, cytoplasmic positive (no nuclear stain) |
| Calretinin | Zymed | Neat | 4/12 (33 %); strong, focal, nuclear (in addition to mast cells) positive |
A review of the English literature between 1966 and 2014 was performed. Clinical series of “head and neck soft tissue tumors” were selected if critical information about sinonasal tract neurofibroma were included (Table 4) [1–14]. Foreign language articles were only included if they were published alongside an English translation. Many articles with limited or lacking information, no histologic illustrations, confusing histology descriptions or duplicate publications were excluded [4, 5, 15–42].
Table 4.
English literature review of sinonasal tract neurofibroma cases (1–14)
| Characteristic | Number [n = 24] |
|---|---|
| Gender a | |
| Females | 11 |
| Males | 12 |
| Age (years) | |
| Range | 6–69 |
| Mean | 46 |
| Median | 45 |
| Female (mean) | 47 |
| Male (mean) | 48.6 |
| Symptom duration (months) a | |
| Range | 1–108 |
| Mean | 24.4 |
| Female patients (mean) | 11.6 |
| Male patients (mean) | 26.2 |
| Left (mean) | 9.8 |
| Right (mean) | 31.8 |
| Bilateral (mean) | 9 |
| Laterality a | |
| Left | 8 |
| Right | 14 |
| Bilateral | 2 |
| Syndrome associated (NF1) | 2 |
| Anatomic site a | |
| Nasal cavity alone | 15 |
| Maxillary sinus alone | 4 |
| Frontal sinus alone | 2 |
| Mixed Sites | 3 |
| Size (cm) a | |
| Range | 1–6 |
| Mean | 3.8 |
| Median | 4 |
| Female (mean) | 4.1 |
| Male (mean) | 3.5 |
| Left (mean) | 3.5 |
| Right (mean) | 4.1 |
| Bilateral (mean) | 3.5 |
| Patient follow-up (mean, years) a | |
| Alive or dead, no evidence of disease (n = 20) | 3.3 |
| Alive, with disease (n = 1) | 0.5 |
aNot reported for all cases
Results
Patient Demographics and Clinical Presentation
The patients included 6 females and 6 males, whose ages ranged from 26 to 75 years of age, with a mean of 46.2 years. Patients presented clinically with a mass lesion (n = 12), along with obstruction (n = 3), pain (n = 3) and epistaxis (n = 2). The duration of symptoms ranged from 1 to 240 months, with a mean of 42.9 months, without any difference between the genders. Tumors affected the right (n = 7) or left (n = 5), without any bilateral cases. Curiously, left-sided tumors had a shorter mean duration of symptoms (18.3 months) than right-sided tumors (57 months), but this was not statistically significant (p = 0.432).
Treatment and Follow-Up
All patients were treated surgically, with or without complete removal of the lesion. Of these patients, 10 were alive at last follow-up without evidence of local or recurrent disease or disease progression (malignant transformation), with a mean follow-up of 8.6 years (range 0.8–17.1 years; median 9.9 years). The two patients with NF1 were lost to follow-up.
Pathology
Macroscopic Features
The tumors affected the nasal cavity alone (n = 8), with the nasal vestibule the most common subsite (n = 5). Two tumors affected only the maxillary sinus, while the remaining two affected more than one site (nasal cavity and maxillary sinus; maxillary sinus and infratemporal fossa; Fig. 1). The majority of lesions were received as multiple, irregular fragments, described as soft or firm, white, tan-pale, lesions. The excised specimens measured 0.4–4.1 cm in greatest dimension (Table 2), with a mean of 2.2 cm, without any gender or side differences. The largest tumors (4.1 cm, nasal cavity; 3.8 cm, maxillary sinus) did not involve multiple or contiguous sites.
Fig. 1.
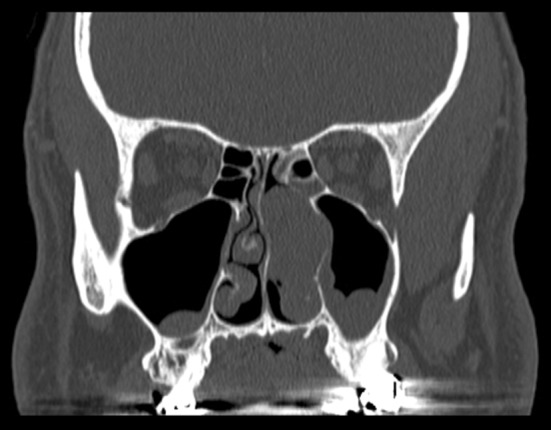
A coronal computed tomography scan demonstrates a 3.5 cm mass occupying the left nasal cavity, with focal extension into the maxillary sinus. There is no bone destruction
Microscopic Features
The tumors were identified below an intact squamous or respiratory mucosa (Fig. 2). There was no encapsulation or well formed periphery. Nerve twigs were identified within the tumor as residual axons, primarily at the periphery (Fig. 2), but also noted within the proliferation. The lesional cells were arranged in irregular interlacing fascicles, bundles or single spindled cells (Fig. 3). The cells were separated by variable proportions of coarse collagen bundles, which give the characteristic “shredded carrot” appearance (Fig. 4). A myxoid to loose connective tissue stroma was seen in most cases, with a mucopolysaccharide-rich stroma. The cells were ovoid to spindled with wavy, undulating to pointy nuclei surrounded by scant cytoplasm (Figs. 4, 5). The cytoplasm shows thin processes or wisps extending outward into the stroma. Perineurial cells cannot be reliably identified on standard H&E stained slides. Occasional Pacinian corpuscles could be seen, but well developed Wagner–Meissner bodies or even pseudomeissnerian corpuscles were not prominent. Mast cells are seen throughout the proliferation. Nuclear pleomorphism was not a conspicuous feature, although isolated nuclear enlargement was noted. Two tumors showed a more tortuous multinodular appearance with slightly increased matrix material, features associated with plexiform NF (Fig. 6). Nerve fibers were noted within enlarged fascicles that had significant myxoid stroma. One tumor showed a mixture of Schwann cells arranged in Verocay bodies with focal perivascular hyalinization along with the more haphazard distribution seen in the remaining tumor, suggesting a composite of schwannoma and neurofibroma (Fig. 6). Epithelioid cells, skeletal muscle and glandular epithelium were not identified. Pigmentation (melanin) was not identified. Finally, in comparison to malignant peripheral nerve sheath tumors, there was no increased cellularity, no necrosis and >4 mitoses/10 high power fields was not seen.
Fig. 2.

a An intact squamous epithelium (vestibule skin) is noted overlying an unencapsulated NF. b A nerve twig (lower right) is seen at the periphery of this NF
Fig. 3.

a There are irregular interlacing fascicles and bundles of spindled cells separated by collagen bundles. b Small whorls are noted giving the impression of pseudomeissnerian corpuscles in this NF
Fig. 4.

a Dense collagen fibrils can be seen separating these Schwann and perineurial cells. b More of a haphazard arrangement to these Schwann and perineurial cells and fibroblasts. Note the mast cells (arrows)
Fig. 5.

a The nuclei are wavy with pointy edges arranged in a syncytium. b Small bundles of nuclei and cytoplasm give the appearance of Wagner–Meissner bodies, but they are not well formed or classical (pseudomeissnerian corpuscles)
Fig. 6.

a The stroma is more myxoid in this plexiform neurofibroma, showing wavy cells and nuclei within the Schwann cell compartment. b This area shows characteristic features of a schwannoma, part of a hybrid tumor
Immunohistochemical Findings
The immunophenotypic findings are reported in Table 3. The various components of the tumors, including Schwann cells, perineurial cells and fibroblasts, stain to a variable degree with the various markers tested. Subpopulations were positive with S100 protein (Fig. 7), GFAP, CD34 (Fig. 8) and bcl-2 (Fig. 9), while SOX10 (Fig. 9), NFP and calretinin (Fig. 9) highlighted the axons. CD34 often highlighted nerve twigs and pseudomeissnerian corpuscles (Fig. 8). β-Catenin showed a cytoplasmic reaction only; there was an intact INI-1 (Fig. 9). EMA showed only focal reactivity in one case. Other markers tested were negative.
Fig. 7.

S100 protein shows both positive and negative cells, highlighting the multicellular components of a NF
Fig. 8.

CD34 yielded several different patterns: a a strong intercellular pattern highlighting the neurial-like and fibroblastic cells; b highlighting the nerve twigs within the proliferation; c the staining outlines the pseudomeissnerian corpuscles
Fig. 9.

The various cellular compartments were highlighted with a calretinin staining rare cells and mast cells, b an intact INI1, c bcl-2, and d SOX10
Discussion
Benign peripheral nerve sheath tumors of the sinonasal tract are quite uncommon, with Schwannoma seemingly more frequent than NF [11, 29]. We could only find isolated case reports of sinonasal tract NF in a review of the English literature. Thought to arise from peripheral nerves, NF are more common around the nasal vestibule than other sites within the sinonasal tract. The nasal cavity alone is most commonly affected (64 %), followed by the maxillary sinus (17 %), or mixed sites. There is no gender predilection, with a mean age at presentation of 46 years. Patients usually present with symptoms that have been present for 2–3 years duration, usually presenting with a mass lesion that is often associated with obstruction, bleeding (epistaxis) or pain. The tumors show an overall mean of 3.1 cm (a combination of this case series and the cases reported in the literature), suggesting that they are quite slow growing in light of the number of months of symptoms. The vast majority of lesions are unilateral, although occasional bilateral tumors can be seen [3, 8]. They tend to be solitary when presenting in the sinonasal tract. In general, NF1 association is uncommon, although 4 of 36 total cases (11.1 %) were syndrome associated, with these tumors showing a plexiform histology. The syndrome associated patients were 35 years old (mean age at presentation), with one female and three males. Overall, only one patient showed malignant transformation of a NF [11], although malignant PNST (neurogenic sarcoma, malignant Schwannoma) arising de novo or from schwannoma are well documented [19, 20]. Overall, for sinonasal tract NF in general, the patients were alive without evidence of disease at last follow-up without development of recurrence, with the exception of two patients [11, 12].
Pathologic Features
The histologic features of NF in the sinonasal tract are morphologically identical to other sites. Curiously, none of our cases nor those reported in the literature showed glandular morphology, or had pigmentation (melanin), findings reported in other anatomic sites. The tumors are often associated with nerves, showing a centripetal distribution of the unencapsulated tumor cells. The tumors show a characteristic blending of Schwann cells, perineurial cells and fibroblasts with collagen and myxoid stroma, along with a few mast cells. There is generally a lack of palisading, which is more common in schwannoma. The Schwann cell nucleus tends to be more tear drop or ovoid, curved at one end and pointed at the other. Retrogressive changes are not usually a feature of neurofibroma, but are seen in schwannoma. As Schwann cells can be seen in schwannoma and neurofibroma, hybrid peripheral nerve sheath tumors can be seen. The hybrid morphology may include schwannoma, neurofibroma and/or perineurioma [8, 12, 13]. These tumors show the characteristic features of schwannoma with Antoni A and B patterns, Verocay bodies, perivascular hyalinization along with areas of cystic degeneration and histiocyte infiltrate. The areas of neurofibroma could be plexiform type (associated with a number of nerve twigs and a loose myxoid-like stroma) or more characteristic with short wavy fascicles, perineurial or stellate endoneurial cells, and fibroblasts with a collagenous background. The parallel arrangement of interconnected cords of cells in a lace-like growth pattern can be seen in perineurioma. Therefore, even though definitive separation of schwannoma from neurofibroma and perineurioma is desirable, a morphologic spectrum with overlap and blending between tumors can be seen.
Immunohistochemistry
The nuclei and cytoplasm of the schwannoma cells were strongly and diffusely immunoreactive with S100 protein, with weak and focal to patchy reactivity with GFAP, SOX10 and bcl-2 in these same cells. SOX10 transcription factor is expressed in Schwannian and melanocytic lineages, along with myoepithelial cells. Therefore, the expression in a subpopulation of tumor cells in this series, highlights the Schwannian cell population. Bcl-2 is known to be expressed in Schwann cells, although in general it has been reported that malignant PNST tend to show a higher percentage of cells positive and a stronger intensity of staining [43]. As can be seen from the illustration (Fig. 9), there is focal to diffuse positive staining with bcl-2 in this clinical series. The CD34 highlights the fibroblasts, which can be quite a dominant finding (as noted in several of our cases), while also highlighting nerve twigs and giving an outline around pseudomeissnerian corpuscles. Calretinin, a calcium-binding protein is in the same family as S100 protein, is present in a diffuse and strong fashion in schwannoma, while highlighting only isolated or rare cells in NF, along with mast cells. Calretinin may help in separating between schwannoma and NF, since S100 protein is positive in both tumors [44, 45]. Claudin-1 and GLUT-1 are well known to be observed highlighting perineurial cells, especially along the cytoplasmic processes, and would be expected to highlight these cells in sinonasal tract NF [46, 47]. β-Catenin is a part of the Wnt pathway, known to be dysregulated or deregulated in NF1. Interestingly, in a different study, β-catenin was strongly expressed and showed increased protein expression in NF1-silenced cell lines, supporting the finding of β-Catenin expression by immunohistochemistry [48].
None of the cellular compartments were positive with pan-cytokeratin, smooth muscle actin, or CD117 (although mast cells were highlighted by the latter). INI-1 was intact, showing a strong and diffuse nuclear reaction in all of the tumors.
Differential Diagnosis
NF of the sinonasal tract is frequently misdiagnosed. The most frequent misdiagnoses for the sinonasal tract neurofibroma cases were (in order of frequency) schwannoma, dermatofibrosarcoma protuberans, fibrosarcoma, meningioma, leiomyoma, solitary fibrous tumor, leiomyosarcoma, malignant fibrous histiocytoma, low-grade sinonasal sarcoma with neural and myogenic features, proliferative fasciitis, “inflammatory pseudotumor”, fibromatosis, and fibrous histiocytoma.
The lack of nuclear atypia, a lack of a “herringbone” fascicular growth pattern, and an absence of atypical mitotic figures should assist in separating NF from malignant tumors. Although myofibroblasts may occasionally be positive for desmin, we have not observed this finding in this series of neurofibroma. Despite the fact that some smooth muscle tumors may be negative for desmin, one can generally use desmin, especially when it is strongly staining, in addition to the morphology and strength of the actins staining, to separate smooth muscle tumors from NF. The recently described “Low Grade Sinonasal Sarcoma with Neural and Myogenic features” [49, 50], shows a cellular spindle cell neoplasm with uniform, bland, elongate nuclei, an infiltrative growth pattern, invaginated respiratory gland but with inconspicuous mitoses and a positive reaction with S100 protein and actin.
NF lacks the purposeful direction of myofibroblasts, elongate vessels, and infiltrative growth pattern of fibromatosis. Further, the elongated spindle cell population and collagen deposition makes this lesion distinctly different from NF. However, β-catenin is positive in fibromatosis, showing a nuclear expression. Solitary fibrous tumor shows a bland spindle cell population arranged in a patternless to fascicular architecture, with heavy, wiry, keloid-like collagen deposition in association with a rich vascular plexus can help to confirm the diagnosis with the addition of a limited, pertinent and focused immunohistochemistry panel, including CD34, bcl-2 and S100 protein, with the latter negative [51–53]. A myoepithelioma or cellular pleomorphic adenoma may occasionally yield a monomorphic population, but would show reactions with S100 protein, cytokeratins, p63 and GFAP, while negative with CD34. EMA highlights meningioma, which would also show a reaction with CK7, while S100 protein and CD34 should be negative. Melanoma would react with S100 protein, but also shows HMB45, Melan-A, and tyrosinase reactions, and usually demonstrate significant pleomorphism and increased mitoses. Synovial sarcoma would be positive with epithelial markers, as well as TLE1. Up to 18 % of cutaneous fibrous histiocytoma (dermatofibroma) will show CD34 immunoreactivity [54], but usually the pattern of growth and collagen deposition will be different.
If there is more than one distinct pattern present, then a hybrid tumor can be diagnosed. In general there is a neurofibroma combined with either a schwannoma and/or a perineurioma. The growth pattern of perineurioma is usually quite distinctive, while GLUT-1 and claudin-1 would strongly react with the tumor cells.
Conclusion
In conclusion, NF of the sinonasal tract occurs infrequently, but should be considered in the differential diagnosis of a sinonasal tract mass. The tumors are most common in middle-aged patients who usually present with a mass present for a long duration. NF1 is seen in about 10 % of cases, although sinonasal tract tumors are usually solitary lesions. Awareness of the rather distinctive microscopic features of NF will help to distinguish it from other benign and malignant entities within the sinonasal tract. Complete local excision is curative.
Acknowledgments
The authors thank to Ms. Hannah B. Herrera for her conscientious research assistance.
Conflict of interest
There is no financial conflict of interest.
Footnotes
The opinions or assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting the views of the Southern California Permanente Medical Group or The Permanente Medical Group.
References
- 1.Agarwal MK, Gupta OP, Samant HC, et al. Neurofibroma of the maxillary antrum. Oral Surg Oral Med Oral Pathol. 1979;48:150–152. doi: 10.1016/0030-4220(79)90053-7. [DOI] [PubMed] [Google Scholar]
- 2.Annino DJ, Jr, Domanowski GF, Vaughan CW. A rare cause of nasal obstruction: a solitary neurofibroma. Otolaryngol Head Neck Surg. 1991;104:484–488. doi: 10.1177/019459989110400410. [DOI] [PubMed] [Google Scholar]
- 3.Biswas D, Mal R. Bilateral solitary maxillary sinus neurofibroma. Ear Nose Throat J. 2010;89:E1–E2. [PubMed] [Google Scholar]
- 4.Bradley PJ, Singh SD. Congenital nasal masses: diagnosis and management. Clin Otolaryngol Allied Sci. 1982;7:87–97. doi: 10.1111/j.1365-2273.1982.tb01568.x. [DOI] [PubMed] [Google Scholar]
- 5.Hillstrom RP, Zarbo RJ, Jacobs JR. Nerve sheath tumors of the paranasal sinuses: electron microscopy and histopathologic diagnosis. Otolaryngol Head Neck Surg. 1990;102:257–263. doi: 10.1177/019459989010200309. [DOI] [PubMed] [Google Scholar]
- 6.Kim YD, Bai CH, Suh JS, et al. Transnasal endoscopic excision of an isolated neurofibroma of the nasal septum. Rhinology. 1997;35:89–91. [PubMed] [Google Scholar]
- 7.Kohli PS, Goel K. Endoscopic excision of solitary neurofibroma arising from posterior nasal septum. Clin Rhinol. 2011;4:43–46. doi: 10.5005/jp-journals-10013-1068. [DOI] [Google Scholar]
- 8.Kuroda N, Kazakov DV, Hes O, et al. Hybrid peripheral nerve sheath tumor of the nasal cavity showing schwannomatous, neurofibromatous, and perineuriomatous areas. Med Mol Morphol. 2010;43:82–85. doi: 10.1007/s00795-008-0418-7. [DOI] [PubMed] [Google Scholar]
- 9.Manganaris A, Tsompanidou C, Manganaris T. A peripheral nerve sheath tumour as a cause of nasal obstruction. J Laryngol Otol. 2006;120:e44. doi: 10.1017/S0022215106004440. [DOI] [PubMed] [Google Scholar]
- 10.Mori H, Kakuta S, Yamaguchi A, et al. Solitary intraosseous neurofibroma of the maxilla: report of a case. J Oral Maxillofac Surg. 1993;51:688–690. doi: 10.1016/S0278-2391(10)80271-X. [DOI] [PubMed] [Google Scholar]
- 11.Perzin KH, Panyu H, Wechter S. Nonepithelial tumors of the nasal cavity, paranasal sinuses and nasopharynx. A clinicopathologic study. XII: schwann cell tumors (neurilemoma, neurofibroma, malignant schwannoma) Cancer. 1982;50:2193–2202. doi: 10.1002/1097-0142(19821115)50:10<2193::AID-CNCR2820501036>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 12.Robitaille Y, Seemayer TA, El DA. Peripheral nerve tumors involving paranasal sinuses: a case report and review of the literature. Cancer. 1975;35:1254–1258. doi: 10.1002/1097-0142(197504)35:4<1254::AID-CNCR2820350433>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 13.Stevens DJ, Kirkham N. Neurofibromas of the paranasal sinuses. J Laryngol Otol. 1988;102:256–259. doi: 10.1017/S0022215100104669. [DOI] [PubMed] [Google Scholar]
- 14.Vaideeswar P, Madiwale CV, Kathpal D, et al. Sinonasal neurogenic tumours. Indian J Pathol Microbiol. 2002;45:161–163. [PubMed] [Google Scholar]
- 15.Chakravarti A, Vishwakarma SK, Arora VK. Solitary neurofibroma causing nasal obstruction. J Indian Med Assoc. 1999;97:526. [PubMed] [Google Scholar]
- 16.Chitale AR, Rege SR, Karnik PP, et al. Uncommon polypoid tumours of nasal cavity. Indian J Cancer. 1974;11:244–254. [PubMed] [Google Scholar]
- 17.Chua CN, Alhady M, Ngo CT, et al. Solitary nasal neurofibroma presenting as compressive optic neuropathy. Eye (Lond) 2006;20:1406–1408. doi: 10.1038/sj.eye.6702261. [DOI] [PubMed] [Google Scholar]
- 18.Heffner DK. Problems in pediatric otorhinolaryngic pathology, III. Teratoid and neural tumors of the nose, sinonasal tract, and nasopharynx. Int J Pediatr Otorhinolaryngol. 1983;6:1–21. doi: 10.1016/S0165-5876(83)80100-1. [DOI] [PubMed] [Google Scholar]
- 19.Hellquist HB, Lundgren J. Neurogenic sarcoma of the sinonasal tract. J Laryngol Otol. 1991;105:186–190. doi: 10.1017/S0022215100115312. [DOI] [PubMed] [Google Scholar]
- 20.High AS, Kay NJ. Solitary neurogenic sarcoma of the nose. J Laryngol Otol. 1985;99:1151–1159. doi: 10.1017/S0022215100098339. [DOI] [PubMed] [Google Scholar]
- 21.Hirao M, Gushiken T, Imokawa H, et al. Solitary neurofibroma of the nasal cavity: resection with endoscopic surgery. J Laryngol Otol. 2001;115:1012–1014. doi: 10.1258/0022215011909639. [DOI] [PubMed] [Google Scholar]
- 22.Holsinger FC, Hafemeister AC, Hicks MJ, et al. Differential diagnosis of pediatric tumors of the nasal cavity and paranasal sinuses: a 45-year multi-institutional review. Ear Nose Throat J. 2010;89:534–540. [PubMed] [Google Scholar]
- 23.Ijaduola TG, Ademiluyi SA. Otorhinolaryngological manifestations of von Recklinghausen’s disease in Nigerians. J Laryngol Otol. 1986;100:115–123. doi: 10.1017/S0022215100098819. [DOI] [PubMed] [Google Scholar]
- 24.Kimberling WJ, Lynch HT, Grush ML. Concurrent von Recklinghausen’s neurofibromatosis, hereditary spherocytosis and fronto nasal dysplasia. Nebr Med J. 1982;67:151–154. [PubMed] [Google Scholar]
- 25.Knight WA, III, Murphy WK, Gottlieb JA. Neurofibromatosis associated with malignant neurofibromas. Arch Dermatol. 1973;107:747–750. doi: 10.1001/archderm.1973.01620200061017. [DOI] [PubMed] [Google Scholar]
- 26.Krammer U, Wimmer K, Wiesbauer P, et al. Neurofibromatosis 1: a novel NF1 mutation in an 11-year-old girl with a giant cell granuloma. J Child Neurol. 2003;18:371–373. doi: 10.1177/08830738030180051901. [DOI] [PubMed] [Google Scholar]
- 27.Lee JH, Bae JH, Kim KS. A case of solitary neurofibroma of the nasal dorsum: resection using an external rhinoplasty approach. Eur Arch Otorhinolaryngol. 2005;262:813–815. doi: 10.1007/s00405-005-0915-4. [DOI] [PubMed] [Google Scholar]
- 28.Macomber WB, Wang MK. Congenital neoplasms of the nose. Plast Reconstr Surg. 1946;1953(11):215–229. doi: 10.1097/00006534-195303000-00006. [DOI] [PubMed] [Google Scholar]
- 29.Mondal AR, Rashid MA, Bera SP, et al. Neurofibroma of paranasal sinuses—a case report. Indian J Otolaryngol Head Neck Surg. 2004;56:40–42. doi: 10.1007/BF02968772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moreno PM, Meseguer DH. Solitary neurofibroma of the inferior nasal turbinate. Auris Nasus Larynx. 1998;25:329–331. doi: 10.1016/S0385-8146(98)00031-5. [DOI] [PubMed] [Google Scholar]
- 31.Oi H, Watanabe Y, Shojaku H, et al. Nasal Septal Neurinoma Acta Otolaryngol Suppl. 1993;504:151–154. doi: 10.3109/00016489309128144. [DOI] [PubMed] [Google Scholar]
- 32.Pantazopoulos PE. Schwannomas of nose, oral cavity and pharynx. Acta Otolaryngol. 1965;60:97–104. doi: 10.3109/00016486509126992. [DOI] [PubMed] [Google Scholar]
- 33.Pasic TR, Makielski K. Nasal schwannoma. Otolaryngol Head Neck Surg. 1990;103:943–946. doi: 10.1177/019459989010300610. [DOI] [PubMed] [Google Scholar]
- 34.Putney FJ, Moran JJ, Thomas GK. Neurogenic tumors of the head and neck. Laryngoscope. 1964;74:1037–1059. doi: 10.1288/00005537-196408000-00002. [DOI] [PubMed] [Google Scholar]
- 35.Rakshit BK, Sundareshwar B, Reddy CR. Nerve sheath tumors. Int Surg. 1973;58:557–560. [PubMed] [Google Scholar]
- 36.Rengahary SS, McMahon M, Bigongiari LR, et al. Neurofibroma of the infratemporal fossa: case report and technical note. Neurosurgery. 1982;11:43–47. doi: 10.1227/00006123-198207010-00009. [DOI] [PubMed] [Google Scholar]
- 37.Sharma DK, Sohal BS, Parmar TL, et al. Schwannomas of head and neck and review of literature. Indian J Otolaryngol Head Neck Surg. 2012;64:177–180. doi: 10.1007/s12070-011-0248-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sharma SB, Hong P. Solitary neurofibroma of the frontal sinus. Case Rep Otolaryngol. 2012;2012:373808. doi: 10.1155/2012/373808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shugar JM, Som PM, Biller HF, et al. Peripheral nerve sheath tumors of the paranasal sinuses. Head Neck Surg. 1981;4:72–76. doi: 10.1002/hed.2890040114. [DOI] [PubMed] [Google Scholar]
- 40.Sinha R, Basu S, Ganguly S, et al. Trilobed intranasal neurofibroma—an unusual presentation. J Indian Med Assoc. 1997;95(192):196. [PubMed] [Google Scholar]
- 41.Vujevich JJ, Goldberg LH, Kimyai-Asadi A, et al. Recurrent nodule on the nasal columella: a good reason to re-biopsy. Int J Dermatol. 2008;47:728–731. doi: 10.1111/j.1365-4632.2008.03536.x. [DOI] [PubMed] [Google Scholar]
- 42.Wilson JA, McLaren K, McIntyre MA, et al. Nerve-sheath tumors of the head and neck. Ear Nose Throat J. 1988;67(103–7):110. [PubMed] [Google Scholar]
- 43.Cunha KS, Caruso AC, Faria PA, et al. Evaluation of Bcl-2, Bcl-x and cleaved caspase-3 in malignant peripheral nerve sheath tumors and neurofibromas. An Acad Bras Cienc. 2013;85:1497–1511. doi: 10.1590/0001-3765201320120026. [DOI] [PubMed] [Google Scholar]
- 44.Fine SW, McClain SA, Li M. Immunohistochemical staining for calretinin is useful for differentiating schwannomas from neurofibromas. Am J Clin Pathol. 2004;122:552–559. doi: 10.1309/AGBGTBRJ4W0BC7LN. [DOI] [PubMed] [Google Scholar]
- 45.Ohashi R, Wakayama N, Kawamoto M, et al. Solitary nasal schwannoma: usefulness of CD34 and calretinin staining for distinction from histological mimics. J Nippon Med Sch. 2013;80:300–306. doi: 10.1272/jnms.80.300. [DOI] [PubMed] [Google Scholar]
- 46.Folpe AL, Billings SD, McKenney JK, et al. Expression of claudin-1, a recently described tight junction-associated protein, distinguishes soft tissue perineurioma from potential mimics. Am J Surg Pathol. 2002;26:1620–1626. doi: 10.1097/00000478-200212000-00010. [DOI] [PubMed] [Google Scholar]
- 47.Yamaguchi U, Hasegawa T, Hirose T, et al. Sclerosing perineurioma: a clinicopathological study of five cases and diagnostic utility of immunohistochemical staining for GLUT1. Virchows Arch. 2003;443:159–163. doi: 10.1007/s00428-003-0849-4. [DOI] [PubMed] [Google Scholar]
- 48.Luscan A, Shackleford G, Masliah-Planchon J, et al. The activation of the WNT signaling pathway is a Hallmark in neurofibromatosis type 1 tumorigenesis. Clin Cancer Res. 2014;20:358–371. doi: 10.1158/1078-0432.CCR-13-0780. [DOI] [PubMed] [Google Scholar]
- 49.Lewis JT, Oliveira AM, Nascimento AG, et al. Low-grade sinonasal sarcoma with neural and myogenic features: a clinicopathologic analysis of 28 cases. Am J Surg Pathol. 2012;36:517–525. doi: 10.1097/PAS.0b013e3182426886. [DOI] [PubMed] [Google Scholar]
- 50.Wenig BM. Recently described sinonasal tract lesions/neoplasms: considerations for the new world health organization book. Head Neck Pathol. 2014;8:33–41. doi: 10.1007/s12105-014-0533-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ferreiro JA, Nascimento AG. Solitary fibrous tumour of the major salivary glands. Histopathology. 1996;28:261–264. doi: 10.1046/j.1365-2559.1996.d01-419.x. [DOI] [PubMed] [Google Scholar]
- 52.Hanau CA, Miettinen M. Solitary fibrous tumor: histological and immunohistochemical spectrum of benign and malignant variants presenting at different sites. Hum Pathol. 1995;26:440–449. doi: 10.1016/0046-8177(95)90147-7. [DOI] [PubMed] [Google Scholar]
- 53.Bauer JL, Miklos AZ, Thompson LD. Parotid gland solitary fibrous tumor: a case report and clinicopathologic review of 22 cases from the literature. Head Neck Pathol. 2012;6:21–31. doi: 10.1007/s12105-011-0305-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rudolph P, Schubert B, Wacker HH, et al. Immunophenotyping of dermal spindle cell tumors: diagnostic value of monocyte marker Ki-M1p and histogenetic considerations. Am J Surg Pathol. 1997;21:791–800. doi: 10.1097/00000478-199707000-00007. [DOI] [PubMed] [Google Scholar]