

Abstract
The diagnosis and treatment decisions in glomerular disease are principally based on renal pathology and nonspecific clinical laboratory measurements such as serum creatinine and urine protein. Using these classification approaches, patients have marked variability in rate of progression and response to therapy, exposing a significant number of patients to toxicity without benefit. Additionally, clinical trials are at risk of not being able to detect an efficacious therapy in relevant subgroups as patients with shared clinical-pathologic diagnoses have heterogeneous underlying pathobiology. To change this treatment paradigm, biomarkers that reflect the molecular mechanisms underlying the clinical-pathologic diagnoses are needed. Recent progress to identify such biomarkers has been aided by advances in molecular profiling, large-scale data generation and multi-scalar data integration, including prospectively collected clinical data. This article reviews the evolving success stories in glomerular disease biomarkers across the genotype-phenotype continuum and highlights opportunities to transition to precision medicine in glomerular disease.
Keywords: translational medicine, nephrotic syndrome, precision medicine, molecular taxonomy
Despite being considered rare diseases, primary glomerular diseases together generate a large individual, societal and economic burden, accounting for nearly 13% of prevalent end-stage renal disease patients in 2010 [1]. Even before reaching end-stage renal disease, patients with glomerular disease suffer significant morbidity from the disease itself, including devastating complications such as infections, reduced quality of life, thrombotic events, hypertension, cardiovascular complications and early mortality [2–5] and from side effects of prolonged exposure to immunomodulatory therapies (e.g. steroid toxicity, calcineurin nephrotoxicity, impaired fertility and bladder toxicity of cyclophosphamide, and infectious risk [6–9]).
The clinical presentation of these diseases is marked by a constellation of signs and symptoms that cross the distinct clinical-pathologic diagnoses, including hematuria, proteinuria, hyperlipidemia, edema and hypoalbuminemia. For many of these diseases, activity is defined by coarse surrogate markers such as level of proteinuria, hematuria and glomerular filtration rate (GFR) estimated by equations that have not been validated in glomerular disease. Conversely, although primary glomerular diseases are currently categorized as distinct histopathologic categories, they likely result from heterogeneous biological disorders given the dramatic variability in the presentation, rate of progression and response to therapy. A certain proportion of patients with membranous nephropathy (MN), for example, will undergo a spontaneous remission, whereas others with the same pathologic diagnoses will progress to end-stage kidney disease, despite aggressive immunosuppressive therapy.
Thus, the patient and clinician experience is marked by delays in diagnosis and uncertainty regarding prognostic and therapeutic decisions for many of these diseases. The nonspecific immunosuppressive therapies do not target the underlying disease mechanism and can often only be applied in a trial and error fashion, leaving patients exposed to significant treatment toxicity without the expected benefit. The most recent Kidney Disease: Improving Global Outcomes clinical practice guidelines on glomerular disease noted a ‘paucity of robust data from RCTs [Randomized Controlled Trials] to support the treatment recommendations and suggestions that have been made’, highlighting the specific challenge that many variants and subtypes of glomerular disease are rare [10]. Participants with common clinical-pathologic diagnoses are grouped together in clinical trials, despite their likely heterogeneous underlying pathobiology, limiting the ability to detect efficacy in relevant subgroups, even if they are present.
THE GOAL: PRECISION MEDICINE : IN GLOMERULAR DISEASE
Recent advances in molecular medicine allow comprehensive analysis of molecular profiles in clinical samples. Integrating the clinical molecular information with novel discoveries in the pathobiology of glomerular disease, however, has provided hope that the current treatment paradigm can be transformed. The concept of ‘precision medicine’ aims to combine the clinical-pathologic diagnoses with molecular disease definitions to allow a patient's diagnosis, prognosis and treatment to be tailored to their individual needs (Figure 1) [11]. While a comparatively novel approach in the field of nephrology, it has been successfully deployed in oncology where tumors with a common histology are now analyzed at a genetic and molecular level to provide a mechanistic diagnosis, leading to accurate prognosis and tailored therapy, maximizing efficacy and limiting toxicity. These include success stories such as epidermal growth factor receptor-directed therapy in EGFR-mutant lung adenocarcinoma [12] and now standard-of-care cytogenetic profiling in acute myeloid leukemia to define risk and inform post-induction therapies [13].
FIGURE 1:
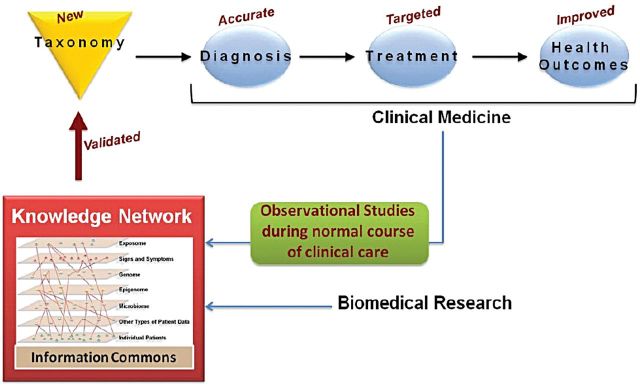
Towards precision medicine of renal disease. Data capturing all aspects of the disease are combined with biomedical research to create a knowledge network, available to the research community to define disease in functional terms (Reprinted from Toward Precision Medicine: Building a Knowledge Network for Biomedical Research and a New Taxonomy of Disease, with permission from the National Academy of Sciences).
The success of precision medicine in glomerular disease will depend on our ability to define mechanistic homogeneous disease groups inside our current syndromic disease classes. This will require the detection and validation of classifiers, i.e. biomarkers, including those that adequately categorize the patient's disease (diagnostic biomarkers), those that provide information about a patient's response to therapy (predictive biomarker) and those that provide information about the patient's long-term outcome (prognostic biomarkers). For many glomerular diseases, with complex and multifactorial underlying pathology, the best diagnostic, prognostic or predictive biomarker may, in fact, be a combination of clinical characteristics linked to genetic, tissue, serum and/or urine biomarkers.
In the case of glomerular disease, the challenges to discovery and validation of biomarkers arise principally from the fact that they are rare diseases, often requiring long follow-up duration to reach clinically meaningful outcomes. Additionally, surrogate outcomes are not comprehensively available for all glomerular diseases, animal models have variable degree of biologic similarity to human disease [14, 15] and the underlying pathobiology is not yet fully elucidated in the majority of glomerular diseases. While acknowledging these hurdles, there are distinct advantages to the development of biomarkers in these diseases compared with other organ systems. The kidney biopsy tissue, not unlike tumor tissue, offers a unique window into the organ currently under attack and allows personal molecular profiling of an individual patient's biopsy. Additionally, urine offers an opportunity to frequently and repeatedly measure noninvasive biomarkers reflecting, in this liquid kidney biopsy, the state of the renal tissue with a higher degree of precision than blood biomarkers for many other diseases.
EVOLVING SUCCESS STORIES IN : GLOMERULAR DISEASE BIOMARKERS ACROSS THE GENOTYPE-PHENOTYPE CONTINUUM
Leveraging these unique opportunities for biomarkers in kidney disease as well as advances in large-scale data generation and multi-scalar data integration has allowed for some early success in the development of biomarkers across the genotype-phenotype continuum of glomerular disease. Using these techniques, it is possible to link molecular and clinical phenotype, integrating multiple layers of data, including genetic, proteomic, metabolomics, morphologic with environmental exposures and clinical outcomes (Figure 2).
FIGURE 2:

Integration of multiple layers of data across the genotype-phenotype continuum to identify glomerular disease biomarkers.
Genetic
Multiple success stories have emerged in understanding the genetic causes and risk profiles associated with glomerular disease. Monogenetic diseases with high penetrance of disease represent one example of a gene mutation serving as a diagnostic, prognostic and predictive biomarker. Familial segregation of a known disease gene can establish causality, disease trajectory and even therapeutic implications. A recent study sequenced 27 genes in 1783 families (2016 affected individuals) with steroid-resistant nephrotic syndrome and found a single gene cause in 29.5% of individuals, which was inversely correlated with age of onset [16]. Interestingly, 1% of these individuals had a coenzyme Q10 biosynthesis pathway mutation, raising the possibility of a therapeutic target. While the familial segregation of a variant with disease most clearly supports a single mutation as a biomarker for precision medicine, the implications of sporadic mutations are not yet clear. Population studies will need to define approaches to establish causality and sensitivity and specificity in this very different context.
More common allele variants, with lower disease phenotype penetrance, have also been recently demonstrated to represent opportunities to risk stratify patients. Risk loci for several glomerular diseases have been identified and not only help to define disease mechanism, but also show promise as biomarkers. Homozygosity for risk alleles in both HLA-DQA1 and PLA2R-1 in idiopathic MN showed an odds ratio of 78.5 in a cohort of Caucasian patients [17]. The diagnostic and prognostic utility of these loci will require additional validation work, but indicate a role of PLA2R not only as an antigen for a biomarker, but also as an element in the pathogenesis of MN.
In addition to identifying risk alleles for the development of a specific glomerular disease, successful identification of genes associated with progression of kidney disease has been found to be more broadly applicable across distinct histopathologic diagnoses. High-risk variants in Apolipoprotein A1 (APOL1), initially discovered as a risk for the development of focal segmental glomerulosclerosis (FSGS) in patients of African descent [18], has more recently been shown to be a risk factor for progression of kidney disease in two large cohort studies, independent of cause of the chronic kidney disease [19]. The significant risk for progression with APOL1 risk variants are at a level that ongoing studies might indeed show diagnostic clinical utility in the near future, i.e. in the setting of living donor transplant evaluation. In contrast, panels of genetic risk loci identified by large-scale CKD GWAS studies have provided mechanistic insights into CKD [20, 21], but currently do not provide effect sizes suitable for direct diagnostic deployment [22].
Epigenetic and gene expression
Mechanistic discovery work using epigenetics and gene expression profiling has provided a complementary approach to identification of new biomarkers and potential therapeutic targets. An advantage of these studies is the possibility of integrating candidate genes from GWAS studies into a functional context by combining GWAS, gene expression and clinical information. This approach was used in a recent study that identified 97 pathways associated with CKD across a broad spectrum of glomerular diseases. These pathways included both known and newly associated CKD pathways, many of which were shared across histopathologic diseases, highlighting the disease mechanisms common to CKD [21]. Additionally, a study in kidney transplant patients found that inclusion of molecular profile with clinical risk factors improved the ability to identify patients at high risk for antibody-mediated rejection [23]. These examples of molecular profiling provide opportunities to further understand disease mechanism, but also to identify targets for more mechanistic therapies.
Analysis of cell lineage-specific transcript offers the opportunity to define the state of a critical cell type in glomerular disease. An elegant series of studies by Wickman et al. describes how urinary podocin mRNA can serve as a marker for podocyte depletion in animal models and a wide spectrum of glomerular diseases [24]. Across disease categories, high urinary transcript levels of this marker were associated with current glomerular disease activity as well as overall progression arguing for a potential future use to monitor podocyte health in therapeutic interventions targeting podocyte damage.
Urine and serum protein biomarkers
Certainly in clinical practice, the urinary biomarker of proteinuria does provide information regarding risk of progression and currently serves as the benchmark against all novel strategies that have to be compared. Proteinuria has been shown within glomerular disease [25] and in other causes of chronic kidney disease a nonspecific predictor for future decline in renal function [26]. Its lack of specificity and mechanistic insight and the close interaction with glomerular hemodynamics, however, limits its ability as an ideal glomerular disease biomarker. As new biomarkers are discovered and validated, they will be compared against proteinuria to determine the amount of additional information provided beyond the current, albeit imperfect, clinical standards of eGFR and urine protein to creatinine ratio.
Perhaps the most exciting success story in the development of a multi-purpose glomerular disease biomarker is the M-type phosolipase A2 Receptor antibody (PLA2R) in MN [27]. It has now been robustly replicated as a diagnostic biomarker with impressive specificity of >90% in multiple cohorts [28–31] and has received FDA approval for this indication [32]. There is strong plausibility for its implication in the causal pathway from GWAS identifying the PLA2R as a risk loci [17]. And preliminary results suggest its potential use as prognostic biomarker, waxing and waning with disease activity [33]. Further understanding of its pathogenic role may lead to uniquely targeted therapies, personalized to the subset of MN patients with this marker.
Other protein biomarkers are at various stages of investigation. Several urinary biomarkers have been identified, but not robustly validated. Urinary cytokines and chemokines hold particular promise given their role in inflammation and fibrosis. For example, urinary monocyte chemoattractant protein-1 (MCP-1) has been shown to correlate with lupus nephritis activity and may be a useful noninvasive marker of disease activity [34].
Pathology and morphometric markers
Histological evaluation of the renal biopsy remains the benchmark in diagnosis of glomerular diseases. The amount of information and the experience from over a century of evaluating structural alterations and their association with clinical presentation and outcome make this an essential and critical part of the diagnostic workup of a patient with glomerular disease. Subclassification based on pathology and particularly assessment of the degree of tubule-interstitial damage has been shown to provide robust prognostic information [35, 36]. FSGS classification schemes provide an example of specific pathologic features associating well with outcomes [37]. Specifically, tip lesions associating with improved response to therapy and better clinical outcomes while collapsing lesions, such as those in the setting of HIV infection, associating with worse clinical outcomes. Similarly, the Oxford classification of IgA nephropathy identified reproducible biopsy features, including the mesangial hypercellularity score, segmental glomerulosclerosis, endocapillary hypercellularity and tubular atrophy/interstitial fibrosis, which associated with renal outcome, independent of initial GFR and proteinuria [38, 39].
Additionally, there are novel approaches to the analysis of pathology material which aim to maximize this unique window into the diseased organ. Morphometry has been used to quantify the pathologic features of a biopsy and allow for quantitative assessment of those features against clinical outcomes. Measurements of interstitial fibrosis, glomerular size and podocyte number have been shown to be important markers of risk of disease progression across disease categories [24, 40, 41]. A descriptor-based pattern of injury scoring system of kidney biopsy tissue has been deployed in the nephrotic syndrome study network (NEPTUNE) cohort, where descriptors are assigned at the level of the individual glomerulus, providing granular data in addition to the clinical-pathologic diagnosis [42]. And, as noted above, as new markers are developed, the opportunity to identify the specific tissue distribution of these molecules (i.e. PLA2R) allows linking the noninvasive biomarkers with intra-renal pathophysiology.
THE PATH FORWARD TO MOVE : BIOMARKERS INTO CLINICAL CARE AND THE DEVELOPMENT OF TARGETED THERAPIES
Prospective, standardized data collection
Large observational cohort studies, with prospective clinical data collection paired with standardized biospecimen banking, are essential for the validation of candidate biomarkers. The challenge for rare diseases, such as glomerular disease, is to implement such studies in a broad range of populations to assess both validity and generalizability. Additionally, for many glomerular diseases, long follow-up times are needed to reach clinically meaningful end points to validate surrogate outcomes. Such studies are resource intensive, but an invaluable asset for impactful biomarker studies. Several examples from chronic kidney disease have demonstrated the success of this approach. These include well-known worldwide studies such as the Chronic Renal Insufficiency Cohort (CRIC) [43], Chronic Kidney Disease in Children (CKid) [44], Canadian study of prediction of death, dialysis and interim cardiovascular events (CaNPREDDICT) [45], NEFRONA [46], the French CKD renal epidemiology and information Network cohort study (CKD-REIN) [47], and Mild to Moderate Kidney Disease study (MMKD) [48], enrolling large numbers of patients and controls and serve as effective platforms for biomarker discovery and validation. Additionally, consortia such as the CKD Biomarkers Consortium (www.ckdbiomarkersconsortium.org) and the IMI Summit (www.imi-summit.eu) bring together researchers and data sources to maximize biomarker discovery.
Similar efforts are underway in the glomerular disease community to establish the infrastructure of prospective cohort studies. The Nephrotic Syndrome Study Network (NEPTUNE, www.neptune-study.org) is a prospective multicenter cohort study that enrolls nephrotic syndrome patients at the time of clinically indicated kidney biopsy and collects clinical data and bio samples prospectively with a target recruitment of 450 patients with minimal change disease (MCD), FSGS and MN [49]. CureGN (www.curegn.org) is a multicenter prospective cohort study that will enroll 2400 prevalent and incident patients with MCD, FSGS, MN and IgA nephropathy. Similar studies are maturing in other countries as well, including NEPTUNE China, EURenOmics (www.eurenomics.eu) with Podonet (www.podonet.org), UK registry for rare renal diseases (RADAR, www.renalradar.org) and the European Renal cDNA bank (ERCB). These studies allow for the multi-dimensional data integration needed for the development of precision medicine and novel biomarkers.
Rigorous biostatistics
Bringing a biomarker from discovery to clinical practice is a multi-step process, requiring statistical analyses at each stage. Several excellent review articles describe these approaches and their potential pitfalls [50–52]. Initial assay development includes understanding external modifiers of a marker's level (e.g. diurnal variation, effect of medication or diet) as well as measurement validity (e.g. variability, precision, limits of assay detection). An ideal experimental biomarker to move forward in clinical testing will have an association with disease classification or outcome as well as biologic plausibility. Once an assay can be implemented in a clinical cohort, there are multiple statistical approaches to fully understand the clinical utility of the marker. An assessment of correlation between a marker and disease classification or outcome of interest is an initial descriptive step. While an important plausibility check to move it forward, further analyses are needed. The frequently used ROC curves and c-statistics provide information about discrimination, the ability of a marker to differentiate between patients with and without an outcome of interest. Sensitivity and specificity are used to construct this curve, but positive predictive value and negative predictive value have more clinical relevance to patients and clinicians. While discrimination may be most helpful in the research setting, calibration, the agreement between predicted and observed outcome, is critical when applying a marker to the clinical setting. Other approaches are necessary to describe calibration, such as the Hosmer–Lemeshow goodness-of-fit test [53]. To understand the contribution of a new biomarker to a panel of established markers or clinical characteristics, methods such as R 2, Brier score and likelihood ratio tests are used. Newer methods can also be applied in this situation such as the Net Reclassification Improvement and the Integrated Discrimination Improvement, which capture information more relevant to a clinical setting [54, 55]. Importantly, separate discovery and validation cohorts are needed to understand generalizability of a novel biomarker. Finally, clinical testing of the impact of the biomarker on outcome is an essential step for validation mandated prior to registration with the FDA. In the renal field, the most recent example of a biomarker from discovery using genome-wide screening approaches to FDA approval can be seen in AKI with NGAL [56].
CONCLUSION
The discovery and validation of ‘useful’ biomarkers in glomerular disease are critical not only for research purposes and stratification for clinical trials, but also ultimately for the ability to practice precision medicine for patients with this disease. Glomerular disease patients have dramatically heterogeneous disease trajectories and response to therapies, despite their overlapping pathologic diagnosis and clinical presentation. To be able to develop a panel of biomarkers across the genotype-phenotype continuum will allow for mechanistically targeted therapies. Successful biomarkers already exist in this field, from the cross-disease utility of urine protein to disease-specific markers such as anti-glomerular basement membrane antibody. Several exploratory studies are already transitioning into clinical validation, including anti-PLA2R, and monogenetic mutations. Discovery studies and replication cohorts are being assembled across the globe to facilitate such work. As a result, biomarker-based diagnosis, prognosis and therapeutic decisions will be possible in glomerular disease.
CONFLICT OF INTEREST STATEMENT
None declared.
(See related article by De Vriese and Fervenza. Con: Biomarkers in glomerular diseases: putting the cart before the wheel? Nephrol Dial Transplant 2015; 30: 885–890; See related article by Ronco. Moderator’s view: Biomarkers in glomerular diseases-translated into patient care or lost in translation? Nephrol Dial Transplant 2015; 30: 899–901.)
ACKNOWLEDGEMENTS
This work has been supported in part to M.K. by U54 DK083912. NEPTUNE (U54 DK083912) is part of NCATS Rare Disease Clinical Research Network (RDCRN). RDCRN is an initiative of the Office of Rare Disease Research (ORDR), NCATS. NEPTUNE is funded through collaboration between NCATS, the NIDDK, the NephCure Kidney International and the Halpin Foundation.
REFERENCES
- 1. U.S. Renal Data System, USRDS . Annual Data Report: Atlas of chronic kidney disease and end-stage renal disease in the United States. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, 2010. [Google Scholar]
- 2. Hingorani SR, Weiss NS, Watkins SL. Predictors of peritonitis in children with nephrotic syndrome. Pediatr Nephrol 2002; 17: 678–682 [DOI] [PubMed] [Google Scholar]
- 3. Kerlin BA, Ayoob R, Smoyer WE. Epidemiology and pathophysiology of nephrotic syndrome-associated thromboembolic disease. Clin J Am Soc Nephrol 2012; 7: 513–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ordonez JD, Hiatt RA, Killebrew EJ, et al. The increased risk of coronary heart disease associated with nephrotic syndrome. Kidney Int 1993; 44: 638–642 [DOI] [PubMed] [Google Scholar]
- 5. Ruth EM, Landolt MA, Neuhaus TJ, et al. Health-related quality of life and psychosocial adjustment in steroid-sensitive nephrotic syndrome. J Pediatr 2004; 145: 778–783 [DOI] [PubMed] [Google Scholar]
- 6. Cattran DC, Alexopoulos E, Heering P, et al. Cyclosporin in idiopathic glomerular disease associated with the nephrotic syndrome: workshop recommendations. Kidney Int 2007; 72: 1429–1447 [DOI] [PubMed] [Google Scholar]
- 7. Cigni A, Faedda R, Atzeni MM, et al. Hormonal strategies for fertility preservation in patients receiving cyclophosphamide to treat glomerulonephritis: a nonrandomized trial and review of the literature. Am J Kidney Dis 2008; 52: 887–896 [DOI] [PubMed] [Google Scholar]
- 8. Hahn D, Hodson EM, Willis NS, et al. Corticosteroid therapy for nephrotic syndrome in children. Cochrane Database Syst Rev 2007: CD001533 doi:10.1002/14651858.CD001533.pub5 [DOI] [PubMed] [Google Scholar]
- 9. Monach PA, Arnold LM, Merkel PA. Incidence and prevention of bladder toxicity from cyclophosphamide in the treatment of rheumatic diseases: a data-driven review. Arthritis Rheum 2010; 62: 9–21 [DOI] [PubMed] [Google Scholar]
- 10. Kidney Disease: Improving Global OUtcomes (KDIGO) Glomeulonephritis Work Group. KDIGO clinical practice guideline for glomerulonephritis. Kidney Int 2012; 2 (Suppl. 2): 139–274 [Google Scholar]
- 11. N.R. Council (ed). Toward Precision Medicine: Buildling a Knowledge Network for Biomedical Reserach for a New Taoxonomy of Disease. Washington, DC: National Academies Press, 2011, p. 128 [PubMed] [Google Scholar]
- 12. Buettner R, Wolf J, Thomas RK. Lessons learned from lung cancer genomics: the emerging concept of individualized diagnostics and treatment. J Clin Oncol 2013; 31: 1858–1865 [DOI] [PubMed] [Google Scholar]
- 13. Rampal R, Levine RL. Leveraging cancer genome information in hematologic malignancies. J Clin Oncol 2013; 31: 1885–1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hodgin JB, Nair V, Zhang H, et al. Identification of cross-species shared transcriptional networks of diabetic nephropathy in human and mouse glomeruli. Diabetes 2013; 62: 299–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Berthier CC, Bethunaickan R, Gonzalez-Rivera T, et al. Cross-species transcriptional network analysis defines shared inflammatory responses in murine and human lupus nephritis. J Immunol 2012; 189: 988–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sadowski CE, Lovric S, Ashraf S, et al. A single-gene cause in 29.5% of Cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol 2014. ; pii: ASN.2014050489 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stanescu HC, Arcos-Burgos M, Medlar A, et al. Risk HLA-DQA1 and PLA(2)R1 alleles in idiopathic membranous nephropathy. N Engl J Med 2011; 364: 616–626 [DOI] [PubMed] [Google Scholar]
- 18. Genovese G, Friedman DJ, Ross MD, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 2010; 329: 841–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Parsa A, Kao WH, Xie D, et al. APOL1 risk variants, race, and progression of chronic kidney disease. N Engl J Med 2013; 369: 2183–2196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kottgen A, Glazer NL, Dehghan A, et al. Multiple loci associated with indices of renal function and chronic kidney disease. Nat Genet 2009; 41: 712–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Martini S, Nair V, Keller BJ, et al. Integrative biology identifies shared transcriptional networks in CKD. J Am Soc Nephrol 2014; 25: 2559–2572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. O'Seaghdha CM, Yang Q, Wu H, et al. Performance of a genetic risk score for CKD stage 3 in the general population. Am J Kidney Dis 2012; 59: 19–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Loupy A, Lefaucheur C, Vernerey D, et al. Molecular microscope strategy to improve risk stratification in early antibody-mediated kidney allograft rejection. J Am Soc Nephrol 2014; 25: 2267–2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wickman L, Afshinnia F, Wang SQ, et al. Urine podocyte mRNAs, proteinuria, and progression in human glomerular diseases. J Am Soc Nephrol 2013; 24: 2081–2095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Troyanov S, Wall CA, Miller JA, et al. Focal and segmental glomerulosclerosis: definition and relevance of a partial remission. J Am Soc Nephrol 2005; 16: 1061–1068 [DOI] [PubMed] [Google Scholar]
- 26. Gansevoort RT, Matsushita K, van der Velde M, et al. Lower estimated GFR and higher albuminuria are associated with adverse kidney outcomes. A collaborative meta-analysis of general and high-risk population cohorts. Kidney Int 2011; 80: 93–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Beck LH, Jr, Bonegio RG, Lambeau G, et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med 2009; 361: 11–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hoxha E, Kneissler U, Stege G, et al. Enhanced expression of the M-type phospholipase A2 receptor in glomeruli correlates with serum receptor antibodies in primary membranous nephropathy. Kidney Int 2012; 82: 797–804 [DOI] [PubMed] [Google Scholar]
- 29. Dahnrich C, Komorowski L, Probst C, et al. Development of a standardized ELISA for the determination of autoantibodies against human M-type phospholipase A2 receptor in primary membranous nephropathy. Clinica Chimica Acta; Int J Clin Chem 2013; 421: 213–218 [DOI] [PubMed] [Google Scholar]
- 30. Zhou GY. Membranous glomerulonephritis associated with myeloperoxidase anti-neutrophil cytoplasmic antibody-associated glomerulonephritis. Nefrologia: Publicacion Oficial de la Sociedad Espanola Nefrologia 2012; 32: 548–551 [DOI] [PubMed] [Google Scholar]
- 31. Du Y, Li J, He F, et al. The diagnosis accuracy of PLA2R-AB in the diagnosis of idiopathic membranous nephropathy: a meta-analysis. PLoS ONE 2014; 9: e104936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. US Food and Drug Administration. Anti-Phospholipase A2 Receptor - FDA. http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpmn/pmn.cfm?ID=K132195 (9 November 2014, date last accessed) [Google Scholar]
- 33. Bech AP, Hofstra JM, Brenchley PE, et al. Association of anti-PLA(2)R antibodies with outcomes after immunosuppressive therapy in idiopathic membranous nephropathy. Clin J Am Soc Nephrol 2014; 9: 1386–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rovin BH, Song H, Birmingham DJ, et al. Urine chemokines as biomarkers of human systemic lupus erythematosus activity. J Am Soc Nephrol 2005; 16: 467–473 [DOI] [PubMed] [Google Scholar]
- 35. Fukagawa M, Noda M, Shimizu T, et al. Chronic progressive interstitial fibrosis in renal disease--are there novel pharmacological approaches? Nephrol Dial Transplant 1999; 14: 2793–2795 [DOI] [PubMed] [Google Scholar]
- 36. Bohle A, Muller GA, Wehrmann M, et al. Pathogenesis of chronic renal failure in the primary glomerulopathies, renal vasculopathies, and chronic interstitial nephritides. Kidney Int Suppl 1996; 54: S2–S9 [PubMed] [Google Scholar]
- 37. D'Agati VD, Kaskel FJ, Falk RJ. Focal segmental glomerulosclerosis. N Engl J Med 2011; 365: 2398–2411 [DOI] [PubMed] [Google Scholar]
- 38. Working Group of the International IgA Nephropathy Network and the Renal Pathology Society Cattran DC, Coppo R, et al. The Oxford classification of IgA nephropathy: rationale, clinicopathological correlations, and classification. Kidney Int 2009; 76: 534–545 [DOI] [PubMed] [Google Scholar]
- 39. Working Group of the International IgA Nephropathy Network and the Renal Pathology Society Roberts IS, Cook HT, et al. The Oxford classification of IgA nephropathy: pathology definitions, correlations, and reproducibility. Kidney Int 2009; 76: 546–556 [DOI] [PubMed] [Google Scholar]
- 40. Walsh M, Sar A, Lee D, et al. Histopathologic features aid in predicting risk for progression of IgA nephropathy. Clin J Am Soc Nephrol 2010; 5: 425–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lemley KV, Lafayette RA, Safai M, et al. Podocytopenia and disease severity in IgA nephropathy. Kidney Int 2002; 61: 1475–1485 [DOI] [PubMed] [Google Scholar]
- 42. Barisoni L, Nast CC, Jennette JC, et al. Digital pathology evaluation in the multicenter Nephrotic Syndrome Study Network (NEPTUNE). Clin J Am Soc Nephrol 2013; 8: 1449–1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lash JP, Go AS, Appel LJ, et al. Chronic Renal Insufficiency Cohort (CRIC) Study: baseline characteristics and associations with kidney function. Clin J Am Soc Nephrol 2009; 4: 1302–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Furth SL, Cole SR, Moxey-Mims M, et al. Design and methods of the Chronic Kidney Disease in Children (CKiD) prospective cohort study. Clin J Am Soc Nephrol 2006; 1: 1006–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Levin A, Rigatto C, Brendan B, et al. Cohort profile: Canadian study of prediction of death, dialysis and interim cardiovascular events (CanPREDDICT). BMC Nephrol 2013; 14: 121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Junyent M, Martinez M, Borras M, et al. Predicting cardiovascular disease morbidity and mortality in chronic kidney disease in Spain. The rationale and design of NEFRONA: a prospective, multicenter, observational cohort study. BMC Nephrol 2010; 11: 14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stengel B, Combe C, Jacquelinet C, et al. The French chronic kidney disease-renal epidemiology and information network (CKD-REIN) cohort study. Nephrol Dial Transplant 2014; 29: 1500–1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kronenberg F, Kuen E, Ritz E, et al. Apolipoprotein A-IV serum concentrations are elevated in patients with mild and moderate renal failure. J Am Soc Nephrol 2002; 13: 461–469 [DOI] [PubMed] [Google Scholar]
- 49. Gadegbeku CA, Gipson DS, Holzman LB, et al. Design of the Nephrotic Syndrome Study Network (NEPTUNE) to evaluate primary glomerular nephropathy by a multidisciplinary approach. Kidney Int 2013; 83: 749–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Buyse M, Sargent DJ, Grothey A, et al. Biomarkers and surrogate end points–the challenge of statistical validation. Nat Rev Clin Oncol 2010; 7: 309–317 [DOI] [PubMed] [Google Scholar]
- 51. Cook NR. Use and misuse of the receiver operating characteristic curve in risk prediction. Circulation 2007; 115: 928–935 [DOI] [PubMed] [Google Scholar]
- 52. Fassett RG, Venuthurupalli SK, Gobe GC, et al. Biomarkers in chronic kidney disease: a review. Kidney Int 2011; 80: 806–821 [DOI] [PubMed] [Google Scholar]
- 53. Hosmer DW, Taber S, Lemeshow S. The importance of assessing the fit of logistic regression models: a case study. Am J Public Health 1991; 81: 1630–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Leening MJ, Steyerberg EW, Van Calster B, et al. Net reclassification improvement and integrated discrimination improvement require calibrated models: relevance from a marker and model perspective. Stat Med 2014; 33: 3415–3418 [DOI] [PubMed] [Google Scholar]
- 55. Leening MJ, Vedder MM, Witteman JC, et al. Net reclassification improvement: computation, interpretation, and controversies: a literature review and clinician's guide. Ann Intern Med 2014; 160: 122–131 [DOI] [PubMed] [Google Scholar]
- 56. Parikh CR, Coca SG, Thiessen-Philbrook H, et al. Postoperative biomarkers predict acute kidney injury and poor outcomes after adult cardiac surgery. J Am Soc Nephrol 2011; 22: 1748–1757 [DOI] [PMC free article] [PubMed] [Google Scholar]