

Abstract
Pseudomonas aeruginosa is one of the most intractable human pathogens that pose serious clinical challenge due to extensive prevalence of multidrug-resistant clinical isolates. Armed with abundant virulence and antibiotic resistance mechanisms, it is a major etiologic agent in a number of acute and chronic infections. A complex and intricate network of regulators dictates the expression of pathogenicity factors in P. aeruginosa. Some proteins within the network play key roles and control multiple pathways. This review discusses the role of one such protein, AmpR, which was initially recognized for its role in antibiotic resistance by regulating AmpC β-lactamase. Recent genomic, proteomic and phenotypic analyses demonstrate that AmpR regulates expression of hundreds of genes that are involved in diverse pathways such as β-lactam and non-β-lactam resistance, quorum sensing and associated virulence phenotypes, protein phosphorylation, and physiological processes. Finally, ampR mutations in clinical isolates are reviewed to shed light on important residues required for its function in antibiotic resistance. The prevalence and evolutionary implications of AmpR in pathogenic and nonpathogenic proteobacteria are also discussed. A comprehensive understanding of proteins at nodal positions in the P. aeruginosa regulatory network is crucial in understanding, and ultimately targeting, the pathogenic stratagems of this organism.
Keywords: Pseudomonas aeruginosa virulence, global regulator, antibiotic resistance, quorum sensing, c-di-GMP, ser/thr protein phosphorylation
This is a timely and well-written review summarizing recent findings on the role of the global regulator AmpR on Pseudomonas aeruginosa virulence and physiology. The significance of this regulator has broadened from its established role in regulation of beta-lactam resistance to novel, unexpected, multiple regulatory functions including the switch between acute and chronic modes of infection.

This is a timely and well-written review summarizing recent findings on the role of the global regulator AmpR on Pseudomonas aeruginosa virulence and physiology. The significance of this regulator has broadened from its established role in regulation of beta-lactam resistance to novel, unexpected, multiple regulatory functions including the switch between acute and chronic modes of infection.
INTRODUCTION
Pseudomonas aeruginosa is a Gram-negative bacterium best known for its ability to cause opportunistic human infections. It is the primary cause of fatal lung infections among patients with cystic fibrosis (CF) (Doggett 1969; Lyczak et al., 2002) and the leading cause of secondary infections in immunocompromised patients such as those with AIDS, cancer, and burn wounds (Afessa et al., 1998; Vento et al., 2008; Branski et al., 2009; Hoiby 2011). Unfortunately, P. aeruginosa infections are associated with a poor prognosis and have high fatality rates (Aliaga et al., 2002; Hakki et al., 2007; Horino et al., 2012). A wide array of cell-associated and secreted virulence factors ensure the success of P. aeruginosa as a pathogen.
Pseudomonas aeruginosa infections are extremely difficult to treat due to its ability to switch from acute to chronic infection phenotype and develop multidrug resistance (Hogardt and Heesemann 2013). Currently, β-lactams alone or in combination with aminoglycosides form the first line of defense against P. aeruginosa (Foundation 2011). However, clinicians worldwide are now faced with P. aeruginosa strains that are resistant to most β-lactams, aminoglycosides, and quinolones (Lister et al., 2009). Antibiotic-resistant isolates of P. aeruginosa are selectively favored in vivo in patients with CF (Chen et al., 1995; Bonfiglio et al., 1998). The development of resistance to almost all clinically relevant antibiotics by P. aeruginosa has allowed its classification as an ESKAPE pathogen (Enterococcus faecium,Staphylococcus aureus,Klebsiella pneumoniae,Acinetobacter baumannii,Pseudomonas aeruginosa, and Enterobacter spp.), dreaded in the hospitals as they are capable of confounding any treatment strategy (Rice 2010; Pendleton et al., 2013). Addressing this public health threat will require a better understanding of the molecular mechanisms of antibiotic resistance.
Hyperexpression and presence of β-lactamases in the biofilm matrix, either free or in membrane vesicles, has been linked to the reduced effectiveness of β-lactam therapy (Ciofu et al., 2010; Hengzhuang et al., 2011). A major mechanism of β-lactam resistance in P. aeruginosa is the overproduction of the chromosomally encoded, inducible β-lactamase, AmpC (Lodge et al., 1990; Kong et al., 2005b). The LysR-type transcriptional regulator AmpR modulates expression of ampC encoding β-lactamase (Lodge et al., 1993; Kong et al., 2005b). Recent studies have shown that, apart from regulating antibiotic resistance, AmpR has an extensive regulon that encompasses several virulence and physiological factors (Balasubramanian et al., 2011, 2012, 2014; Kumari et al., 2014b). Furthermore, AmpR is a key player in the intricate network of regulators that is responsible for mediating virulence and antibiotic resistance in P. aeruginosa (Balasubramanian et al., 2013a).
This review summarizes the role of P. aeruginosa AmpR in regulating pathogenesis. We also discuss our current understanding of AmpR-mediated regulation of critical virulence and physiological determinants. Specifically, we focus on the role of AmpR in regulating antibiotic resistance and the switch between acute and chronic infection traits. Given that AmpR is also found in many other Gram-negative bacterial pathogens (Seoane et al., 1992; Proenca et al., 1993; Gould et al., 2006), inhibiting its function will likely be a viable option to deal with bacterial infections in the clinical setting.
AMPR IN ENTEROBACTERIA
The regulation of β-lactam resistance is controlled by ampR–ampC module (where the gene loci are linked, divergently transcribed, and functionally conserved) in many enterobacterial species (Fig. 1). Chromosomally encoded ampC is found in most of the enterobacterial species, albeit with a distinct regulatory pattern. In Escherichia coli and Shigella sonnei, low-level constitutive expression of ampC is directed by a promoter located within the coding sequence of the upstream fumarate reductase operon, frd (Grundstrom and Jaurin 1982; Bergstrom et al., 1983; Cole and Nicolas 1986). Resistance to modern β-lactams in these species occurs rather infrequently and is mostly mediated by promoter mutations, novel promoters, weakened attenuators, or multiple, tandem duplications of the ampC gene (Normark et al., 1977; Cole and Guest 1979; Edlund et al., 1979; Jaurin et al., 1982; Olsson et al., 1982). In contrast, expression of ampC in other organisms such as Citrobacter freundii,Enterobacter cloacae, and Serratia marcescens is induced by β-lactam antibiotics (Lindberg et al., 1985; Cole and Nicolas 1986; Nicolas et al., 1987; Mahlen et al., 2003). Such induction results in therapeutic failure of β-lactam treatment due to stable overproduction of the AmpC. The most significant genetic difference that results in inducible ampC is the presence of ampR, encoding a transcriptional regulator upstream of, and divergently transcribed from, the ampC gene (Fig. 1, Lindberg et al., 1985; Honore et al., 1986). In C. freundii and E. cloacae,ampC expression is repressed and induced by AmpR in the absence and presence of inducers, respectively (Lindberg et al., 1985; Lindberg and Normark 1987).
Figure 1.

Genetic locus of the ampR-ampC module. The open reading frames and operons surrounding ampR-ampC in Pseudomonas aeruginosa and different enterobacterial species are shown. The presence of a divergently transcribed ampR (P. aeruginosa, Enterobacter cloacae, Citrobacter freundii, and Serratia marcescens) indicates inducible β-lactamase production, whereas in Escherichia coli and Shigella sonnei, ampC expression is constitutively low.
The expression of C. freundii or E. cloacae ampC-ampR in E. coli results in the synthesis of inducible β-lactamase (Lindberg et al., 1985). Moreover, ampR from E. cloacae and C. freundii can cross-complement each other in E. coli, which typically lacks ampR (Lindberg and Normark 1987). Together, these findings indicate that all other factors required for ampC induction are present in the E. coli chromosome. Moreover, the close homology between the 3′-ends of E. coli and E. cloacae frd operons and the region downstream of the E. cloacae ampC promoter suggests that ampR may have been deleted from the ampC region of the E. coli chromosome following the divergence from a common ancestor (Honore et al., 1986).
AMPR-AMPC IN PSEUDOMONAS AERUGINOSA
The divergently transcribed ampR–ampC gene arrangement in P. aeruginosa is similar to that seen in other organisms including C. freundii and E. cloacae (Fig. 1; Lodge et al., 1993). Sequence analysis revealed that P. aeruginosa AmpR bears a high degree of homology to its counterparts in C. freundii (58%) and E. cloacae (62%) (Lodge et al., 1990). A high degree of homology is also seen in the helix-turn-helix (HTH) and the hydrophobic domains, whereas homology is lower in the effector-binding domains (EBDs) (Fig. 2). Preliminary experiments in our laboratory suggest that AmpR is possibly an inner membrane protein, but similar studies have not been performed on its counterparts in other bacteria. In a previous study, C. freundii AmpR was purified from an insoluble cellular fraction in E. coli, suggesting its membrane localization (Jacobs et al., 1997), in agreement with the sequence analysis (Fig. 2). It would be interesting to determine whether AmpR is indeed a membrane protein and raises exciting questions on the AmpR-mediated regulatory aspects in bacteria.
Figure 2.
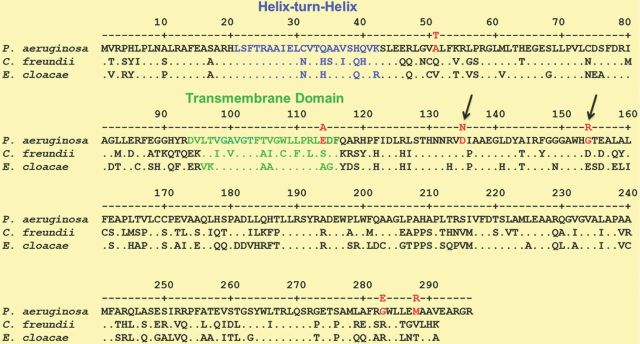
Pseudomonas aeruginosa AmpR sequence homology. The P. aeruginosa AmpR sequence from the Pseudomonas Database (Winsor et al., 2011) was used to determine similarity with its homologs in two other Enterobacteriaceae members using clustalw2. Mutations in the P. aeruginosa AmpR sequence identified in antibiotic-resistant clinical isolates are shown in red. The arrows indicate the mutations confirmed in the laboratory that contribute to enhanced resistance.
Protein modeling shows two C-terminal EBDs and an N-terminal HTH domain, separated by a hydrophobic helix (Fig. 3). The EBD of the C. freundii AmpR was recently crystallized and shown to be a dimer (Balcewich et al., 2010). The crystal structure revealed that each EBD has two subdomains that form a pocket between them. The groove on the surface of subdomain I of the EBD, along with the pocket, forms the putative effector-binding site (Balcewich et al., 2010). Recently, P. aeruginosa AmpR was also shown to be a dimer (Caille et al., 2014).
Figure 3.

AmpR is a global regulator in Pseudomonas aeruginosa. AmpR regulates resistance to different classes of clinically relevant antibiotics, either positively (β-lactams, aminoglycosides) or negatively (quinolones). AmpR plays a key role in determining P. aeruginosa virulence and physiology by regulating expression of transcriptional and post-transcriptional regulators that feed into critical networks, such as QS, Gac-Rsm, iron uptake, and stress response pathways. The data in the figure were obtained from gene expression/proteomic/phenotypic assays (Balasubramanian et al., 2012, 2014; Kumari et al., 2014a,b). Only the gene expression confirmed by qPCR and/or phenotypes confirmed by assays are shown here. Arrow colors indicate positive (green) and negative (red) regulation. No distinction has been made between direct and indirect regulations. AmpR model based on the Protein Model Portal (Tacconelli et al., 2014) is shown in the center. The model shows the DNA-binding helix-turn-helix motif (blue), a hydrophobic domain (green), and the C-terminal effector-binding domain (yellow and cyan).
In C. freundii, AmpR recognizes the 15-bp DNA sequence 5′ TCTGCTGCTAAATTT 3′ in PampC (Lindquist et al., 1989; Lodge et al., 1990). In P. aeruginosa,in silico analysis of microarray data revealed an A-T-rich putative AmpR-binding site (5′ TCTGCTCCAAATTT 3′) in the ampR–ampC intergenic region (Zeng et al., 2007; Balasubramanian et al., 2012). The binding site was further refined using ChIP Seq data to show that the bases that are critical to AmpR binding are As and Ts at positions 1, 6, 9, 10, and 13 (Balasubramanian et al., 2014).
The HTH motif in C. freundii AmpR has been shown to be important in DNA binding (Lindquist et al., 1989). Studies in our laboratory have shown that in P. aeruginosa, the third helix of the HTH motif is critical to DNA binding (Caille et al., 2014). Further, amino acid residues in this helix that are important for DNA binding have been identified using mutation analysis (Caille et al., 2014).
AmpR belongs to the LysR family of transcriptional regulators that typically autorepress their own expression (Schell, 1993; Maddocks and Oyston, 2008). Autoregulation has been demonstrated in C. freundii (Lindquist et al., 1989) but not in E. coli mini cells expressing C. freundii AmpR (Lindberg et al., 1985), suggesting that there are exceptions to the autoregulatory process. The ampR–ampC intergenic region in P. aeruginosa is only 149 bp, and the putative AmpR-binding site overlaps promoters of both ampR and ampC (Lindquist et al., 1989). This suggests that AmpR binding to this region would allow negative autoregulation, in addition to modulating ampC expression (Lindquist et al., 1989). However, in P. aeruginosa, autoregulation occurs only under sub-MIC antibiotic exposure in the alginate constitutively producing strain PDO300 and not in the isogenic parent, PAO1 (Kong et al., 2005; Balasubramanian et al., 2011). AmpR autoregulation in P. aeruginosa thus appears to be dependent on the presence of the alginate master regulator AlgT/U and β-lactam stress (Balasubramanian et al., 2011).
AMPR AS GLOBAL REGULATOR IN P. AERUGINOSA
Even though the role of AmpR in β-lactam resistance was known since 1990s, recent studies demonstrated its role in controlling disparate but important pathogenic determinants (Kong et al., 2005b; Balasubramanian et al., 2011, 2012, 2014; Kumari et al., 2014a,b). The major facets of AmpR-mediated virulence regulation that likely impact the clinical success of P. aeruginosa are discussed below.
Role of P. aeruginosa AmpR in antibiotic resistance
Antibiotic resistance is a major concern when dealing with P. aeruginosa infections in the hospitals. The current treatment regimen for P. aeruginosa infections is a combination therapy of an antipseudomonal β-lactam in association with either an aminoglycoside or a fluoroquinolone (preferably ciprofloxacin) (Mesaros et al., 2007). Having been identified as a positive regulator of the chromosomal AmpC β-lactamase, the role of AmpR in β-lactam resistance has been established in different bacterial species (Lindberg and Normark 1986; Normark et al., 1986). Not surprisingly, loss of AmpR function in P. aeruginosa was found to render the strain sensitive to β-lactam antibiotics (Kong et al., 2005b; Balasubramanian et al., 2012; Kumari et al., 2014a).
In many of the Enterobacteriaceae members, the AmpR-AmpC system is tightly linked to other amp genes whose purported natural function is in the recycling of cell wall/peptidoglycan. In P. aeruginosa, these genes include ampG and ampP encoding permeases; ampD, ampDh2, and ampDh3 encoding amidases; and nagZ encoding a hydrolase (Juan et al., 2006; Asgarali et al., 2009; Kong et al., 2010; Zamorano et al., 2010; Zhang et al., 2010). The cell wall degradation products produced by the action of these genes are presumed to be effectors of AmpR, resulting in activation/repression of ampC expression (Jacobs et al., 1994, 1997). In addition to regulating ampC, AmpR is required for the expression of all the above genes (Balasubramanian et al., 2012). Pseudomonas aeruginosa AmpR thus plays a central role in the cell wall recycling as well as AmpC-mediated β-lactam resistance.
Interestingly, the regulation of β-lactam resistance by P. aeruginosa AmpR seems to involve more than one pathway. AmpR positively regulates the expression of MexR, a repressor of the MexAB-OprM efflux pump that is involved in the efflux of β-lactam antibiotics (Balasubramanian et al., 2012). Although it is counterintuitive to AmpR being a positive regulator of β-lactam resistance, it also suggests that we are far from understanding β-lactam resistance. Moreover, deleting ampC in PAO1 (Kumari et al., 2014a) or in PAOΔampR (D. Zincke, unpublished data) abolishes β-lactam resistance in spite of having a functional MexAB-OprM system. These observations suggest that AmpC is the major resistance determinant and MexAB pump by itself is not enough to confer β-lactam resistance.
Resistance to β-lactams is also regulated by the CreBCD system (Moya et al., 2009; Zamorano et al., 2010). CreBC forms a two-component system that positively regulates expression of an inner membrane protein CreD (Avison et al., 2001); together, they regulate β-lactam resistance (Avison et al., 2004). AmpR regulates CreD in a CreB-independent manner, possibly affecting β-lactam resistance via CreBCD (Balasubramanian et al., 2012). Furthermore, AmpR functions as a negative regulator of a second chromosomal β-lactamase, an oxacillinase termed OxaB (PoxB, PA5514), whose activity spectrum is limited primarily to carbapenems (D. Zincke, unpublished data; Kong et al., 2005). The physiological significance of negative regulation of oxaB expression by AmpR is unclear. Electrophoretic mobility shift assays suggest indirect regulation of oxaAB operon (PA5513-5514) by AmpR under the conditions tested (O. Caille, unpublished data), although it is possible that recognition and binding of Poxa by AmpR requires as yet unidentified signals.
In addition to regulating β-lactam resistance positively, AmpR also regulates quinolone resistance (Balasubramanian et al., 2012). Gene expression and phenotypic assays indicate that AmpR negatively regulates transcription and, ultimately, function of the mexEF-oprN efflux system, by modulating expression of mexT, encoding the positive regulator (Balasubramanian et al., 2012). Repression of mexT expression by AmpR is β-lactam independent (Balasubramanian et al., 2012). It is important to note that AmpR positively and negatively regulates resistance to β-lactams and quinolones, respectively.
In response to β-lactam antibiotic stress, AmpR also negatively regulates the expression of proteins involved in aminoglycoside resistance such as MexXY efflux pump and Aph, an aminoglycoside 3′-phosphotransferase (Kumari et al., 2014b). The regulation of proteins involved in aminoglycosides resistance in response to β-lactam stress is not expected. Furthermore, phenotypic microarray data suggest that AmpR influences resistance to multiple antibiotics (Balasubramanian et al., 2012). Thus, AmpR is among the very few transcriptional regulators in P. aeruginosa that modulates resistance to different classes of antibiotics (Fig. 3).
AmpR mediates antibiotic cross-resistance
Currently, combination therapy with two or more antibiotics of different classes is used to tackle P. aeruginosa infections (Paul and Leibovici, 2005; Tamma et al., 2012). In recent years, however, the advantages of combination therapy have been questioned in light of data suggesting adverse effects of drugs when combined (Paul et al., 2004, 2006; Boyd and Nailor 2011; Johnson et al., 2011; Tamma et al., 2012). Specifically, while much research is focused on studying bacterial resistance to high antibiotic doses, response to subinhibitory concentrations (SICs) of antibiotics remains largely unexplored. For instance, SIC of carbapenems, specifically imipenem, leads to clinical resistance in P. aeruginosa (Livermore, 1987; Kumari et al., 2014a). Often, resistance to multiple classes of antibiotics is mediated by the same mechanism or regulator (MexAB, AmpR), raising the real possibility of antibiotic cross-resistance.
Pre-exposure of P. aeruginosa PAO1 cells to SIC of imipenem (as low as 3 ng μL−1) induces transient cross-resistance to other clinically relevant β-lactams, such as piperacillin, ceftazidime, ticarcillin, and aztreonam (Kumari et al., 2014a). This is possibly because imipenem is the most proficient inducer of ampC and ampR expression compared to other clinically used antibiotics (Kumari et al., 2014a) and residual amounts of imipenem can prime the cells to resist subsequently used β-lactams. Imipenem-mediated cross-resistance is completely dependent on AmpR (Kumari et al., 2014a).
AmpR regulates β-lactam and non-β-lactam resistance in both AmpC-dependent and independent manner. AmpR is a negative regulator of MexXY (Kumari et al., 2014b), suggesting that the ampR mutants would exhibit enhanced resistance to aminoglycosides. However, phenotypic analyses show that loss of ampR results in enhanced aminoglycoside susceptibility, which can be further enhanced upon pre-exposure to SIC of various β-lactams and non-β-lactams (Kumari et al., 2014a). Thus, although the MexXY proteins are made in higher quantities, the ampR mutants are susceptible to aminoglycosides, suggesting the existence of post-translational modification. Moreover, resistance toward aminoglycosides in P. aeruginosa clinical isolates is mostly dependent on horizontally acquired enzymes or membrane alterations (Lister et al., 2009), highlighting the multilayered control of antibiotic resistance.
The ampR mutant is not only sensitive to many β-lactam antibiotics; it regulates critical virulence factors as discussed in the following sections. Targeting AmpR will thus render the strain less virulent and enhance sensitivity to β-lactams, and possibly aminoglycosides without affecting their efficacy at SIC pre-exposure to antibiotics, making it an attractive drug candidate. The findings by our group and others on development of cross-resistance (Masuda et al., 2000; Kumari et al., 2014a) warrant further studies looking into this very relevant clinical phenomenon.
AmpR regulates QS in P. aeruginosa
The pathogenic potential of P. aeruginosa is determined by a large number of both cell-associated (flagella, pili, lipopolysaccharide) and secreted virulence determinants such as pyocyanin, exotoxin A, cyanides, proteases, elastases, and rhamnolipids among others. The expression of a vast majority of the secreted virulence factors is under the control of a population-density-dependent gene expression process called quorum sensing (Williams and Camara, 2009). Population density is sensed by small, diffusible molecules, either acyl homoserine lactones (AHLs) or quinolones (PQSs), which are produced by the bacteria (Jimenez et al., 2012). The las and rhl AHL-dependent systems together affect about 10% of the P. aeruginosa transcriptome (Schuster and Greenberg, 2006). The master transcriptional regulator LasR is at the top of the QS hierarchy in P. aeruginosa and controls expression of the Las, Rhl, and PQS systems (Latifi et al., 1996; Pesci et al., 1997; McGrath et al., 2004). LasR itself is regulated by many different transcriptional regulators (reviewed in Balasubramanian et al., 2013a).
AmpR regulates expression of the major QS regulators LasR, RhlR, MvfR, and QscR, thereby controlling expression of the entire QS regulatory cascade (Balasubramanian et al., 2014). Recent studies in our laboratory identified a putative AmpR-binding site in PlasR (5′ TTGGTTAATAGTTT 3′) and demonstrated direct AmpR binding to the promoter (Balasubramanian et al., 2014). Consequently, loss of AmpR results in a significant loss in the production of QS-regulated acute virulence factors, such as the proteases LasA and LasB, and pyocyanin (Balasubramanian et al., 2012). AmpR-mediated QS regulation is also required for full pathogenicity of the bacterium, as demonstrated by reduced virulence of ampR mutants in the Caenorhabditis elegans acute infection model (Balasubramanian et al., 2012). AmpR thus lies at the heart of the P. aeruginosa pathogenesis network by regulating QS.
AmpR regulates physiological processes and metabolism in P. aeruginosa
Given the diverse phenotypes regulated by AmpR, it is not surprising that some genes involved in metabolic pathways are included in its regulon (Balasubramanian et al., 2012, 2014). Phenotypic microarray analysis showed that utilization of citrulline, histidine, leucine, serine, shikimic acid, spermidine, and pyridoxal is negatively regulated by AmpR (Balasubramanian et al., 2012). The physiological implications of this negative regulation are as yet unclear. Also, the ampR mutant is more sensitive to many agents that affect cell growth (those belonging to the BIOLOG sensitivity panel), suggesting that a functional AmpR is critical to robust survival of P. aeruginosa (Balasubramanian et al., 2012).
AmpR is involved in regulating critical physiological processes such as iron acquisition (Balasubramanian et al., 2014). As iron is a limiting factor for growth in the host, bacteria have evolved siderophores to chelate extracellular iron (Meyer et al., 1996; Martin et al., 2011). AmpR positively regulates expression of the major P. aeruginosa siderophore genes encoding pyoverdine and pyochelin (Balasubramanian et al., 2014). Loss of ampR results in impaired growth under iron-limiting conditions, which can be rescued by making conditions iron replete. This observation and the fact that siderophore genes are downregulated suggest that AmpR affects iron uptake and not utilization in P. aeruginosa (Balasubramanian et al., 2014). AmpR also positively regulates expression of the small RNA rgPrrF1, which is involved in iron uptake regulation. However, the master repressor of iron uptake, Fur, is not regulated by AmpR (Balasubramanian et al., 2014), suggesting an AmpR-mediated, Fur-independent regulation mechanism.
AmpR-mediated regulation of the stress response system
Bacterial cells have evolved elaborate mechanisms to counteract various stress conditions. The major stationary phase sigma factor affecting bacterial stress response is RpoS, which also regulates virulence (Suh et al., 1999; Schuster et al., 2004; Potvin et al., 2008). The P. aeruginosa RpoS regulon has previously been identified to include 772 genes (Schuster et al., 2004). Gene expression studies in P. aeruginosa show an AmpR-dependent positive regulation of RpoS (Balasubramanian et al., 2012). This suggests that AmpR may regulate the stress response via RpoS in P. aeruginosa.
A functional AmpR is also required for survival of P. aeruginosa upon exposure to heat shock (Balasubramanian et al., 2014). AmpR affects heat tolerance in P. aeruginosa by positively regulating genes of the DnaJ-DnaK-GrpE Hsp70 system and the small RNA rgP32, which is part of the dnaJ-dapB-p32 operon (Stover et al., 2000; Balasubramanian et al., 2014). Due to positive regulation of rpoS expression by AmpR, the temperature sensitivity of PAOΔampR is more enhanced in the stationary phase compared with log phase (Balasubramanian et al., 2014).
RpoS, along with GacA, regulates expression of a small RNA rgRgsA, which contributes to hydrogen peroxide resistance (Gonzalez et al., 2008). AmpR positively regulates expression of the major catalase katA, rgRgsA, and 68 other genes involved in P. aeruginosa oxidative stress response, including the master regulator OxyR, either directly or indirectly (Balasubramanian et al., 2014). Besides, loss of ampR in PAO1 increases its susceptibility to H2O2, suggesting a weakened oxidative stress response (Balasubramanian et al., 2014). These findings indicate that AmpR is an integral part of the stress response system in P. aeruginosa.
Regulation of secondary-messenger-mediated signaling
The central role of cyclic-di-GMP in several critical bacterial processes such as virulence, stress survival, motility, biofilm formation, and dispersion is well established (Romling et al., 2013; Ryan 2013). Given the importance of this messenger molecule, intracellular levels of c-di-GMP are tightly regulated by diguanylate cyclases and phosphodiesterases, and some proteins have both these domains (Ryan 2013). The P. aeruginosa PAO1 genome encodes 39 proteins that contain these domains and are thus capable of modulating intracellular c-di-GMP levels (Stover et al., 2000; Kulasakara et al., 2006). AmpR positively regulates three of these phosphodiesterase-domain-containing proteins, BifA, CdpA, and PA4781 (Kumari et al., 2014). This suggests that AmpR potentially negatively regulates c-di-GMP level in the cells by positively regulating phosphodiesterase gene expression (Kumari et al., 2014) and needs further investigation. Interestingly, the gene upstream of ampR,PA4108, codes for a phosphodiesterase (Ryan et al., 2009) but was not identified in our transcriptomic or proteomic analyses.
Serine/Threonine/Tyrosine phosphorylation
Ser/Thr/Tyr phosphorylation plays a critical role in determining eukaryotic protein function (Cohen 2000; Hunter 2000). This process has now been demonstrated in prokaryotes also, albeit at much lower levels (Kannan et al., 2007; Macek et al., 2007). Additional studies addressing Ser/Thr/Tyr phosphorylation in bacteria are needed to understand their role in critical regulatory processes.
Previous studies have identified post-translational modifications to play a role in important virulence processes, such as motility and the HCP1-mediated type 6 secretion system in P. aeruginosa (Kelly-Wintenberg et al., 1993; Mougous et al., 2007). Phosphoproteome analysis identified AmpR to be a major negative regulator of P. aeruginosa protein phosphorylation (Kumari et al., 2014b). The study identified phosphorylation of 45 proteins to be negatively regulated by AmpR, either in the presence or in the absence of β-lactam stress (Kumari et al., 2014b). These include major virulence determinants such as the anaerobic growth regulator Anr, the outer membrane component of the MexAB efflux pump, OprM, a transcriptional activator of the MexEF-OprN efflux system, MexT, and the penicillin-binding proteins MrcB and MurD (Kumari et al., 2014b). Given the important role of these proteins in P. aeruginosa physiology and pathogenesis, the effect of phosphorylation on protein function and the role of AmpR in this process need further elucidation.
ROLE OF AMPR IN ACUTE–CHRONIC INFECTION SWITCH
One of the major features of P. aeruginosa is its ability to cause both acute and chronic infections. The physiology of the cells is widely different between these two infection phases and is characterized by opposing phenotypes. The infection process is initiated by planktonic cells that express a wide variety of acute virulence factors, including expression of flagella and pili (Vallet et al., 2001; Ma et al., 2009); QS-regulated virulence factors such as proteases, elastases, phenazines, and toxins (Williams and Camara 2009); and type III secretion system (Hauser, 2009). Cells in this stage of infection are typically sensitive to antibiotics (Hogardt and Heesemann 2013), unless initial infection was by an antibiotic-resistant strain. Expression of these acute virulence factors is designed to aid in establishment of infection.
Upon transitioning to chronic infection in patients with CF, chronic obstructive pulmonary disease, emphysema, or otitis media, P. aeruginosa forms biofilms that indicate a poor prognosis for patient health (Harmsen et al., 2010). Formation of biofilms is probably the most critical factor that allows P. aeruginosa survival in the CF lung, and is associated with acquisition of niche-specific adaptive mutations and diversification (Boles and Singh 2008; Harmsen et al., 2010; Yang et al., 2011; Lopez-Causape et al., 2013). Extensive research over the years has identified some critical determinants that trigger and support the transition from acute to chronic infection. The CF airways are a complex environment that is extensively compartmentalized based on differences in the local inflammatory processes and antibiotic penetration (Bjarnsholt et al., 2009; Hoiby et al., 2010). The host immune response-mediated oxidative stress, inflammation, and antibiotic treatment have been identified as triggers for P. aeruginosa diversification in the CF lung biofilms (Mathee et al., 1999; Ciofu et al., 2005; Kohanski et al., 2007; Boles and Singh, 2008; Driffield et al., 2008). In addition to biofilm formation, other major changes associated with the chronic infection process include hypermutability, conversion to mucoidy, and acquisition of antibiotic resistance (Doggett 1969; Oliver et al., 2000). Hypermutability is a determining feature of chronic lung infections. CF lung isolates acquire mutations early on in lasR and mucA, and later in the antimutator genes mutS,mutT,mutL,mutY,mutM, and uvrD, resulting in many of the phenotypes associated with chronic infections (Ciofu et al., 2010). Even though mutY and mutM are overexpressed in ampR mutants (>2.0-fold), there is no significant difference in mutation frequencies to rifampicin and streptomycin (Balasubramanian et al., 2014). However, given that MutY and MutM are weak antimutators compared with MutS (Ciofu et al., 2010), it is possible that loss of ampR in the CF lung potentially alters mutation frequencies, affecting survival in chronic infections. The occurrence and frequency of ampR mutations in CF lungs remains to be determined.
Previous studies have demonstrated that the RetS-LadS-GacSA-Rsm regulatory cascade plays a central role in the acute-chronic switch (Lapouge et al., 2008). The hybrid sensor kinases RetS and LadS have opposing effects on the GacS sensor kinase (Laskowski and Kazmierczak 2006; Ventre et al., 2006). RetS forms dimers with GacS inhibiting its function, whereas LadS phosphorylates GacS (Goodman et al., 2009). GacS, through GacA, activates expression of the small regulatory RNAs, rgRsmY and rgRsmZ, which sequester and block activity of the negative regulator RNA-binding protein RsmA (Brencic et al., 2009). RsmA inactivation by rgRsmY/rgRsmZ activates transcription of genes involved in biofilm formation and represses genes involved in acute virulence and motility (Jimenez et al., 2012). RsmA mutants show reduced colonization in the initial infection stages, but ultimately favored chronic infection in a mouse model of acute pneumonia (Mulcahy et al., 2008). AmpR negatively regulates RsmA activity by upregulating the expression of ladS and rgRsmZ (Balasubramanian et al., 2014), thus feeding into the acute-chronic regulatory switch.
The loss of ampR results in many phenotypes resembling a chronic infection strain. These include loss of QS-dependent (proteases, elastases, pyocyanin) and QS-independent (downregulation of T3SS genes) acute virulence factors, increased fluoroquinolone resistance, and enhanced biofilm formation (Balasubramanian et al., 2012, 2014; Kumari et al., 2014a). Many of these effects of AmpR could be accounted for by the fact that AmpR directly regulates LasR, the QS master regulator (Balasubramanian et al., 2014). Recent proteomic analyses have demonstrated that AmpR positively regulates phosphodiesterases that reduce c-di-GMP levels (Kumari et al., 2014b). High levels of c-di-GMP enhance biofilm formation and promote chronic infection by P. aeruginosa (Jimenez et al., 2012). Therefore, modulation of intracellular c-di-GMP levels by regulating phosphodiesterase gene expression is one potential explanation for how AmpR controls biofilm formation. Moreover, AmpR also negatively regulates expression of AlgT/U (Balasubramanian et al., 2011), which controls alginate production, an important component of P. aeruginosa biofilms. Thus, AmpR-mediated negative regulation of algT/U expression could be an additional biofilm control mechanism. Although alginate itself is not required for biofilm formation (Stapper et al., 2004), copious amounts are typically found in P. aeruginosa CF biofilms (Harmsen et al., 2010). As RsmA negatively regulates biofilm formation, one would expect a lower biofilm formation in the ampR mutant, due to sequestration of RsmA by rgRsmZ. This is contrary to the negative regulation of biofilm formation by AmpR (Balasubramanian et al., 2012). However, given the complex, multitiered gene regulation in P. aeruginosa (Jimenez et al., 2012; Balasubramanian et al., 2013a,b), the relative contributions of the individual regulator signals in determining the final outcome (e.g. a phenotype) remain largely unexplained.
AMPR MUTATIONS IN CLINICAL ISOLATES
The infecting clonal types of P. aeruginosa undergo many changes upon infection to adapt and colonize, a process driven by mutations (Folkesson et al., 2012; Wong et al., 2012; Behrends et al., 2013). Several recent studies have identified genes that are mutated in either clinical isolates of P. aeruginosa or strains that have been subjected to CF-like growth conditions (Hoffman et al., 2009; Cramer et al., 2010; Feliziani et al., 2010; Chung et al., 2012; Wong et al., 2012; Hogardt and Heesemann, 2013). As part of the adaption process in the CF lung, the isolates lose their ability to produce acute virulence factors and overexpress chronic virulence traits, as discussed earlier. This is facilitated by mutations in mucA and lasR early on in the infection (Smith et al., 2006; Ciofu et al., 2010), resulting in alginate overproduction (Martin et al., 1993) and downregulation of QS-regulated virulence factors (Venturi 2006).
Being a regulator of several important pathways in P. aeruginosa (Balasubramanian et al., 2012, 2014), acquiring mutations in ampR to alter antibiotic resistance will likely disturb the balance of the regulatory network in the organism. Thus, the mode of ampC de-repression in clinical isolates is often through mutations in accessory genes that are AmpR-regulated (Balasubramanian et al., 2012), such as the ampD alleles encoding amidase and its homologs (Juan et al., 2006; Schmidtke and Hanson, 2008), nagZ encoding hydrolase (Zamorano et al., 2010), or genes that are outside of the AmpR regulon, such as dacB encoding penicillin-binding protein 4 (Moya et al., 2009). Some strains that have been implicated in outbreaks harbor more than one mutation, resulting in multidrug-resistant (MDR) and extensively drug-resistant (XDR) clones (Deplano et al., 2005; Suarez et al., 2011). The incidence of MDR and XDR clones of P. aeruginosa in patients is on the rise and undermines treatment strategies (Mesaros et al., 2007; Pena et al., 2012). Genetic analysis of the molecular mechanisms contributing to enhanced resistance of the XDR clones revealed combinations of resistance to β-lactams (AmpC overproduction and inactivation of OprD), fluoroquinolone resistance (point mutations in GyrA), resistance to gentamycin and tobramycin (aadB gene acquired on a class I integron), and upregulation of aminoglycoside resistance (mutation in the mexZ repressor of the MexXY-OprM efflux pump) (Cabot et al., 2012).
Some ampR mutations in clinical isolates are associated with high levels of β-lactamase production in MDR and XDR high-risk clones of P. aeruginosa (Cabot et al., 2012) and are summarized in Fig. 2. Specifically, in a majority of the most prevalent P. aeruginosa ST175 high-risk XDR/MDR isolates analyzed, a novel mutation in AmpR (glycine 154-arginine) was the reason for constitutive activation of ampC expression (Cabot et al., 2012). In the sporadic XDR/MDR and moderately resistant strains, other ampR mutations were detected (E114A, G283E, M288R, A51T; Fig. 2), but these polymorphisms are also found in wild-type strains such as PA14 (Winsor et al., 2011; Cabot et al., 2012). Complementing an ampR deletion strain in trans with a plasmid harboring ampR-G154R enhanced ampC expression and resistance to ceftazidime (Cabot et al., 2012). In light of studies that demonstrate AmpR to be a positive regulator of acute virulence factors and antibiotic resistance (Balasubramanian et al., 2012, 2014), it is very possible that locking AmpR in an active conformation contributes to the success of high-risk XDR clones such as ST175. This, however, remains to be examined.
In C. freundii, AmpR becomes a constitutive activator of ampC expression upon amino acid substitutions R86C, G102E, and D135N (Kuga et al., 2000; Balcewich et al., 2010), of which only the D135N mutation has been found in a clinical isolate (Bagge et al., 2002). Studies in our laboratory have demonstrated that mutating the aspartic acid residue at position 135 to asparagine (D135N) in P. aeruginosa AmpR locks it in the constitutively active conformation (Caille et al., 2014). However, the G102E mutation in P. aeruginosa AmpR seems to destabilize the protein, leading to the loss of activity (Caille et al., 2014). These studies from our laboratory and elsewhere demonstrate that mutations in ampR play an important role in regulating antibiotic resistance in P. aeruginosa.
PREVALENCE OF AMPR IN PROTEOBACTERIA
Phylogenetic analysis reveals that AmpR homologs are found in many α-, β-, and γ-proteobacteria (Fig. 4). There appears to be two main branches in the phylogenetic tree (Fig. 4). The first branch contains human pathogens (P. aeruginosa and Enterobacteriaceae members such as Citrobacter freundii,Enterobacter cloacae,Klebsiella pneumoniae, E. coli, Morganella morganii, and Yersinia enterocolitica) and plant pathogens/symbionts (Erwinia sp, Agrobacterium sp., P. fluorescens, and Rhizobium sp). The second branch consists of different Burkholderia and Serratia species, Azorhizobium sp., Caulobacter sp., and Acidovorax sp., among others. The genus Burkholderia forms two different subclades, with the major human pathogens belonging to the B. cepacia complex such as B. cepacia,B. cenocepacia, and B. multivorans being part of the same subclade (Fig. 4). It is interesting to note that AmpR in Serratia, an Enterobacteriaceae member, bears a higher homology to the Burkholderia AmpR than to the other Enterobacteriaceae members.
Figure 4.

Prevalence and relatedness of AmpR in proteobacteria. The precomputed blast data for the AmpR (PA4109) amino acid sequence from the Pseudomonas Genome Database (Winsor et al., 2011) were used to identify homologs in other bacteria. The cutoff score was set at 850 corresponding to 56% protein identity. For the sake of clarity, only the top hit identified in the sequenced genomes of each species was considered for further analysis. The matches that conform to these criteria were aligned using NCBI Constraint-Based Multiple Alignment Tool (Papadopoulos and Agarwala 2007), and the resulting alignment file was used to generate a phylogenetic tree (Dereeper et al., 2008, 2010). The GI protein IDs in the Newick format of the tree were replaced with organism names (identified using the NCBI Batch Entrez), and the tree was visualized using the Interactive Tree of Life (Letunic and Bork 2007, 2011).
In many of these bacteria, as in P. aeruginosa, AmpR plays an important role in conferring β-lactam resistance (Seoane et al., 1992; Naas et al., 1995; Trepanier et al., 1997; Weng et al., 2004; Okazaki and Avison, 2008). Considering the global regulatory role of AmpR in P. aeruginosa, it would be interesting to see whether it plays a similar role in other pathogenic/nonpathogenic bacteria.
Several members of these genera have previously been identified as emerging human pathogens, especially in the CF lung (Davies and Rubin 2007; Raso et al., 2008), while others are important plant pathogens and are related to Pseudomonas. It is therefore not surprising that they all harbor the ampR gene, suggesting that they acquired it early on in the evolutionary process. Moreover, given the shared habitat (rhizhosphere) for many of these bacteria, it is likely that ampR was acquired by horizontal gene transfer.
CONCLUSIONS
Treatment for P. aeruginosa infections poses an immense clinical challenge due to its potent virulence arsenal, ability to establish persistent chronic infections, and extensive drug resistance. Research over the years has generated much information about regulation of its pathogenesis, but as the function of nearly half of the genome is unknown (Winsor et al., 2011), we still have a long way to go in understanding the mechanisms involved. The large genome of P. aeruginosa allows it to dedicate a huge portion toward the regulation of various virulence determinants. At the same time, there is extensive cross talk between the regulators of various pathways, resulting in a challenging network of systems controlling various aspects of pathogenesis (Balasubramanian et al., 2013a).
AmpR is one of 434 transcriptional regulators identified in the PAO1 genome, many of which remain uncharacterized (Stover et al., 2000; Balasubramanian et al., 2013b). With a large complement of regulatory proteins and accessory metabolic genes, it is no surprise that P. aeruginosa is able to adapt and thrive in a wide range of habitats. Analyses of potential and empirically demonstrated gene regulatory networks reveal wide gaps in our current knowledge of the system (Balasubramanian et al., 2013a,b). Understanding how AmpR and other regulators orchestrate the virulence and metabolic processes in P. aeruginosa in response to external signals is critical in dealing with infections caused by this successful opportunistic pathogen.
The microarray, RNA-Seq, and proteomic analysis of PAOΔampR mutant sufficiently established the role of AmpR as a key regulator of antibiotic resistance as well as acute and chronic infections (Fig. 3). Apart from the known pathways, AmpR regulon also includes small RNAs. rgRNAs have been shown to be extensively involved in gene regulation in P. aeruginosa and other bacteria (Wilderman et al., 2004; Brencic and Lory 2009; Brencic et al., 2009; Sonnleitner et al., 2009, 2011; Wiedenheft et al., 2012). The interplay between rgRNAs and transcriptional regulators in controlling critical functions in bacteria is being increasingly appreciated.
Although recent studies have identified over 500 novel sRNAs in P. aeruginosa (Dotsch et al., 2012; Gomez-Lozano et al., 2012), their function and regulation have not been elucidated. Given the important regulatory role of AmpR in P. aeruginosa virulence and metabolism, it is not surprising that rgRNAs, such as rgRsmZ, asPrrF1, rgP32, and rgRgsA, are AmpR-regulated (Fig. 3). It is possible that other sRNAs are AmpR-regulated. Future research on determining the AmpR-regulated sRNAs will provide valuable information to our current understanding. Given the many different ways in which sRNAs can modulate gene expression (Sonnleitner et al., 2012) and potentially undiscovered ones, we can look forward to exciting new discoveries in bacterial gene regulation in the coming years.
Using a combination approach of transcriptomic, proteomic, and phenotypic assays, AmpR was determined to affect the expression of 2121 genes, 363 of which overlapped in at least two analyses (Balasubramanian et al., 2012, 2014; Kumari et al., 2014b). As AmpR occupies a nodal position in the regulatory network of P. aeruginosa that affects expression of diverse phenotypes (Balasubramanian et al., 2013a), it makes for an attractive therapeutic target to combat the antibiotic resistance problem. Loss of ampR, in addition to rendering P. aeruginosa sensitive to many β-lactam antibiotics, also results in reduced production of many acute virulence factors (Balasubramanian et al., 2012). Small molecule inhibitors of AmpR can be potential therapeutic agents against P. aeruginosa in the clinical setting, thus reducing virulence and rendering the cells sensitive to β-lactam antibiotics, without causing selective pressure. Although PAOΔampR displays fluoroquinolone resistance, targeting AmpR is still a good proposition because the strains will become susceptible to β-lactams and have reduced virulence factor production. Moreover, use of the major fluoroquinolone ciprofloxacin in the clinical setting is on the decline owing to the development of high-level resistance (Hidron et al., 2008).
In conclusion, understanding the regulatory network of P. aeruginosa in a holistic manner is imperative to compete with the evolving bacterial strategies against antibiotic use. With fewer new antibiotics being discovered, the focus should also be on developing new therapeutic strategies involving important players of resistance and virulence, such as AmpR.
Acknowledgments
This study was in part supported by National Institutes of Health – Minority Biomedical Research Support SCORE (SC1AI081376; KM and HK), and National Science Foundation IIP-1237818 [PFI-AIR: CREST-I/UCRC-Industry Ecosystem to Pipeline Research] (KM). The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Author contribution
D.B. and H.K. contributed equally to this work.
Conflict of interest statement. None declared.
REFERENCES
- Afessa B, Green W, Chiao J, Frederick W. Pulmonary complications of HIV infection: autopsy findings. Chest. 1998;113:1225–9. doi: 10.1378/chest.113.5.1225. [DOI] [PubMed] [Google Scholar]
- Aliaga L, Mediavilla JD, Cobo F. A clinical index predicting mortality with Pseudomonas aeruginosa bacteraemia. J Med Microbiol. 2002;51:615–9. doi: 10.1099/0022-1317-51-7-615. [DOI] [PubMed] [Google Scholar]
- Asgarali A, Stubbs KA, Oliver A, Vocadlo DJ, Mark BL. Inactivation of the glycoside hydrolase NagZ attenuates antipseudomonal beta-lactam resistance in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2009;53:2274–82. doi: 10.1128/AAC.01617-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avison MB, Horton RE, Walsh TR, Bennett PM. Escherichia coli CreBC is a global regulator of gene expression that responds to growth in minimal media. J Biol Chem. 2001;276:26955–61. doi: 10.1074/jbc.M011186200. [DOI] [PubMed] [Google Scholar]
- Avison MB, Niumsup P, Nurmahomed K, Walsh TR, Bennett PM. Role of the ‘cre/blr-tag’ DNA sequence in regulation of gene expression by the Aeromonas hydrophila beta-lactamase regulator, BlrA. J Antimicrob Chemother. 2004;53:197–202. doi: 10.1093/jac/dkh077. [DOI] [PubMed] [Google Scholar]
- Bagge N, Ciofu O, Hentzer M, Campbell JI, Givskov M, Hoiby N. Constitutive high expression of chromosomal beta-lactamase in Pseudomonas aeruginosa caused by a new insertion sequence (IS1669) located in ampD. Antimicrob Agents Chemother. 2002;46:3406–11. doi: 10.1128/AAC.46.11.3406-3411.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian D, Kong KF, Jayawardena SR, Leal SM, Sautter RT, Mathee K. Co-regulation of {beta}-lactam resistance, alginate production and quorum sensing in Pseudomonas aeruginosa. J Med Microbiol. 2011;60:147–56. doi: 10.1099/jmm.0.021600-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian D, Schneper L, Merighi M, Smith R, Narasimhan G, Lory S, Mathee K. The regulatory repertoire of Pseudomonas aeruginosa AmpC β-lactamase regulator AmpR includes virulence genes. PLoS ONE. 2012;7:e34067. doi: 10.1371/journal.pone.0034067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian D, Schneper L, Kumari H, Mathee K. A dynamic and intricate regulatory network determines Pseudomonas aeruginosa virulence. Nucleic Acids Res. 2013a;41:1–20. doi: 10.1093/nar/gks1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian D, Murugapiran SK, Silva-Herzog E, et al. Transcriptional regulatory network in Pseudomonas aeruginosa. In: (Madan Babu M, editor. Bacterial Gene Regulation and Transcriptional Networks. Norfolk, UK: Caiser Academic Press; 2013b. pp. 199–222. [Google Scholar]
- Balasubramanian D, Kumari H, Jaric M, et al. Deep sequencing analyses expands the Pseudomonas aeruginosa AmpR regulon to include small RNA-mediated regulation of iron acquisition, heat shock and oxidative stress response. Nucleic Acids Res. 2014;42:979–98. doi: 10.1093/nar/gkt942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balcewich MD, Reeve TM, Orlikow EA, Donald LJ, Vocadlo DJ, Mark BL. Crystal structure of the AmpR effector binding domain provides insight into the molecular regulation of inducible ampC beta-lactamase. J Mol Biol. 2010;400:998–1010. doi: 10.1016/j.jmb.2010.05.040. [DOI] [PubMed] [Google Scholar]
- Behrends V, Ryall B, Zlosnik JE, Speert DP, Bundy JG, Williams HD. Metabolic adaptations of Pseudomonas aeruginosa during cystic fibrosis chronic lung infections. Environ Microbiol. 2013;15:398–408. doi: 10.1111/j.1462-2920.2012.02840.x. [DOI] [PubMed] [Google Scholar]
- Bergstrom S, Lindberg FP, Olsson O, Normark S. Comparison of the overlapping frd and ampC operons of Escherichia coli with the corresponding DNA sequences in other gram-negative bacteria. J Bacteriol. 1983;155:1297–1305. doi: 10.1128/jb.155.3.1297-1305.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjarnsholt T, Jensen PO, Fiandaca MJ, et al. Pseudomonas aeruginosa biofilms in the respiratory tract of cystic fibrosis patients. Pediatr Pulmonol. 2009;44:547–58. doi: 10.1002/ppul.21011. [DOI] [PubMed] [Google Scholar]
- Boles BR, Singh PK. Endogenous oxidative stress produces diversity and adaptability in biofilm communities. P Natl Acad Sci USA. 2008;105:12503–08. doi: 10.1073/pnas.0801499105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfiglio G, Laksai Y, Franchino L, Amicosante G, Nicoletti G. Mechanisms of beta-lactam resistance amongst Pseudomonas aeruginosa isolated in an Italian survey. J Antimicrob Chemother. 1998;42:697–702. doi: 10.1093/jac/42.6.697. [DOI] [PubMed] [Google Scholar]
- Boyd N, Nailor MD. Combination antibiotic therapy for empiric and definitive treatment of gram-negative infections: insights from the Society of Infectious Diseases Pharmacists. Pharmacotherapy. 2011;31:1073–84. doi: 10.1592/phco.31.11.1073. [DOI] [PubMed] [Google Scholar]
- Branski LK, Al-Mousawi A, Rivero H, Jeschke MG, Sanford AP, Herndon DN. Emerging infections in burns. Surg Infect. 2009;10:389–97. doi: 10.1089/sur.2009.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brencic A, Lory S. Determination of the regulon and identification of novel mRNA targets of Pseudomonas aeruginosa RsmA. Mol Microbiol. 2009;72:612–32. doi: 10.1111/j.1365-2958.2009.06670.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brencic A, McFarland KA, McManus HR, Castang S, Mogno I, Dove SL, Lory S. The GacS/GacA signal transduction system of Pseudomonas aeruginosa acts exclusively through its control over the transcription of the RsmY and RsmZ regulatory small RNAs. Mol Microbiol. 2009;73:434–45. doi: 10.1111/j.1365-2958.2009.06782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabot G, Ocampo-Sosa AA, Dominguez MA, et al. Genetic markers of widespread extensively drug-resistant Pseudomonas aeruginosa high-risk clones. Antimicrob Agents Chemother. 2012;56:6349–57. doi: 10.1128/AAC.01388-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caille O, Zincke D, Merighi M, et al. Structural and functional characterization of Pseudomonas aeruginosa global regulator AmpR. J Bacteriol. 2014. (provisionally accepted) [DOI] [PMC free article] [PubMed]
- Chen HY, Yuan M, Livermore DM. Mechanisms of resistance to beta-lactam antibiotics amongst Pseudomonas aeruginosa isolates collected in the UK in 1993. J Med Microbiol. 1995;43:300–9. doi: 10.1099/00222615-43-4-300. [DOI] [PubMed] [Google Scholar]
- Chung JC, Becq J, Fraser L, et al. Genomic variation among contemporary Pseudomonas aeruginosa isolates from chronically infected cystic fibrosis patients. J Bacteriol. 2012;194:4857–66. doi: 10.1128/JB.01050-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciofu O, Riis B, Pressler T, Poulsen HE, Hoiby N. Occurrence of hypermutable Pseudomonas aeruginosa in cystic fibrosis patients is associated with the oxidative stress caused by chronic lung inflammation. Antimicrob Agents Chemother. 2005;49:2276–82. doi: 10.1128/AAC.49.6.2276-2282.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciofu O, Mandsberg LF, Bjarnsholt T, Wassermann T, Hoiby N. Genetic adaptation of Pseudomonas aeruginosa during chronic lung infection of patients with cystic fibrosis: strong and weak mutators with heterogeneous genetic backgrounds emerge in mucA and/or lasR mutants. Microbiology. 2010;156:1108–19. doi: 10.1099/mic.0.033993-0. [DOI] [PubMed] [Google Scholar]
- Cohen P. The regulation of protein function by multisite phosphorylation-a 25 year update. Trends Biochem Sci. 2000;25:596–601. doi: 10.1016/s0968-0004(00)01712-6. [DOI] [PubMed] [Google Scholar]
- Cole ST, Guest JR. Production of a soluble form of fumarate reductase by multiple gene duplication in Escherichia coli K12. Eur J Biochem. 1979;102:65–71. doi: 10.1111/j.1432-1033.1979.tb06263.x. [DOI] [PubMed] [Google Scholar]
- Cole ST, Nicolas MH. Beta-lactam resistance mechanisms in Gram-negative bacteria. Microbiol Sci. 1986;3:334–9. [PubMed] [Google Scholar]
- Cramer N, Wiehlmann L, Tummler B. Clonal epidemiology of Pseudomonas aeruginosa in cystic fibrosis. Int J Med Microbiol. 2010;300:526–33. doi: 10.1016/j.ijmm.2010.08.004. [DOI] [PubMed] [Google Scholar]
- Davies JC, Rubin BK. Emerging and unusual Gram-negative infections in cystic fibrosis. Semin Respir Crit Care Med. 2007;28:312–21. doi: 10.1055/s-2007-981652. [DOI] [PubMed] [Google Scholar]
- Deplano A, Denis O, Poirel L, et al. Molecular characterization of an epidemic clone of pan antibiotic-resistant Pseudomonas aeruginosa. J Clin Microbiol. 2005;43:1198–204. doi: 10.1128/JCM.43.3.1198-1204.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dereeper A, Guignon V, Blanc G, et al. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008;36:W465–9. doi: 10.1093/nar/gkn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dereeper A, Audic S, Claverie JM, Blanc G. BLAST-EXPLORER helps you building datasets for phylogenetic analysis. BMC Evol Biol. 2010;10:8. doi: 10.1186/1471-2148-10-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doggett RG. Incidence of mucoid Pseudomonas aeruginosa from clinical sources. Appl Microbiol. 1969;18:936–7. doi: 10.1128/am.18.5.936-937.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotsch A, Eckweiler D, Schniederjans M, et al. The Pseudomonas aeruginosa transcriptome in planktonic cultures and static biofilms using RNA sequencing. PLoS ONE. 2012;7:e31092. doi: 10.1371/journal.pone.0031092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driffield K, Miller K, Bostock JM, O'Neill AJ, Chopra I. Increased mutability of Pseudomonas aeruginosa in biofilms. J Antimicrob Chemother. 2008;61:1053–6. doi: 10.1093/jac/dkn044. [DOI] [PubMed] [Google Scholar]
- Edlund T, Grundstrom T, Normark S. Isolation and characterization of DNA repetitions carrying the chromosomal beta-lactamase gene of Escherichia coli K-12. Mol Gen Genet. 1979;173:115–25. doi: 10.1007/BF00330301. [DOI] [PubMed] [Google Scholar]
- Feliziani S, Lujan AM, Moyano AJ, et al. Mucoidy, quorum sensing, mismatch repair and antibiotic resistance in Pseudomonas aeruginosa from cystic fibrosis chronic airways infections. PLoS ONE. 2010;5:e12669. doi: 10.1371/journal.pone.0012669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkesson A, Jelsbak L, Yang L, Johansen HK, Ciofu O, Hoiby N, Molin S. Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: an evolutionary perspective. Nat Rev Microbiol. 2012;10:841–51. doi: 10.1038/nrmicro2907. [DOI] [PubMed] [Google Scholar]
- Cystic Fibrosis Foundation Patient Registry Annual Data Report. Bethesda, MD: Cystic Fibrosis Foundation; 2011. Foundation. [Google Scholar]
- Gomez-Lozano M, Marvig RL, Molin S, Long KS. Genome-wide identification of novel small RNAs in Pseudomonas aeruginosa. Environ Microbiol. 2012;14:2006–16. doi: 10.1111/j.1462-2920.2012.02759.x. [DOI] [PubMed] [Google Scholar]
- Gonzalez N, Heeb S, Valverde C, Kay E, Reimmann C, Junier T, Haas D. Genome-wide search reveals a novel GacA-regulated small RNA in Pseudomonas species. BMC Genomics. 2008;9:167. doi: 10.1186/1471-2164-9-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman AL, Merighi M, Hyodo M, Ventre I, Filloux A, Lory S. Direct interaction between sensor kinase proteins mediates acute and chronic disease phenotypes in a bacterial pathogen. Genes Dev. 2009;23:249–59. doi: 10.1101/gad.1739009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould VC, Okazaki A, Avison MB. Beta-lactam resistance and beta-lactamase expression in clinical Stenotrophomonas maltophilia isolates having defined phylogenetic relationships. J Antimicrob Chemother. 2006;57:199–203. doi: 10.1093/jac/dki453. [DOI] [PubMed] [Google Scholar]
- Grundstrom T, Jaurin B. Overlap between ampC and frd operons on the Escherichia coli chromosome. P Natl Acad Sci USA. 1982;79:1111–5. doi: 10.1073/pnas.79.4.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakki M, Limaye AP, Kim HW, Kirby KA, Corey L, Boeckh M. Invasive Pseudomonas aeruginosa infections: high rate of recurrence and mortality after hematopoietic cell transplantation. Bone Marrow Transplant. 2007;39:687–93. doi: 10.1038/sj.bmt.1705653. [DOI] [PubMed] [Google Scholar]
- Harmsen M, Yang L, Pamp SJ, Tolker-Nielsen T. An update on Pseudomonas aeruginosa biofilm formation, tolerance, and dispersal. FEMS Immunol Med Microbiol. 2010;59:253–68. doi: 10.1111/j.1574-695X.2010.00690.x. [DOI] [PubMed] [Google Scholar]
- Hauser AR. The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat Rev Microbiol. 2009;7:654–65. doi: 10.1038/nrmicro2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengzhuang W, Wu H, Ciofu O, Song Z, Hoiby N. Pharmacokinetics/pharmacodynamics of colistin and imipenem on mucoid and nonmucoid Pseudomonas aeruginosa biofilms. Antimicrob Agents Chemother. 2011;55:4469–74. doi: 10.1128/AAC.00126-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidron AI, Edwards JR, Patel J, Horan TC, Sievert DM, Pollock DA, Fridkin SK. NHSN annual update: antimicrobial-resistant pathogens associated with healthcare-associated infections: annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006–2007. Infect Control Hosp Epidemiol. 2008;29:996–1011. doi: 10.1086/591861. [DOI] [PubMed] [Google Scholar]
- Hoffman LR, Kulasekara HD, Emerson J, Houston LS, Burns JL, Ramsey BW, Miller SI. Pseudomonas aeruginosa lasR mutants are associated with cystic fibrosis lung disease progression. J Cyst Fibros. 2009;8:66–70. doi: 10.1016/j.jcf.2008.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogardt M, Heesemann J. Microevolution of Pseudomonas aeruginosa to a chronic pathogen of the cystic fibrosis lung. Curr Top Microbiol Immunol. 2013;358:91–118. doi: 10.1007/82_2011_199. [DOI] [PubMed] [Google Scholar]
- Hoiby N. Recent advances in the treatment of Pseudomonas aeruginosa infections in cystic fibrosis. BMC Med. 2011;9:32. doi: 10.1186/1741-7015-9-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoiby N, Bjarnsholt T, Givskov M, Molin S, Ciofu O. Antibiotic resistance of bacterial biofilms. Int J Antimicrob Agents. 2010;35:322–32. doi: 10.1016/j.ijantimicag.2009.12.011. [DOI] [PubMed] [Google Scholar]
- Honore N, Nicolas MH, Cole ST. Inducible cephalosporinase production in clinical isolates of Enterobacter cloacae is controlled by a regulatory gene that has been deleted from Escherichia coli. EMBO J. 1986;5:3709–14. doi: 10.1002/j.1460-2075.1986.tb04704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horino T, Chiba A, Kawano S, et al. Clinical characteristics and risk factors for mortality in patients with bacteremia caused by Pseudomonas aeruginosa. Intern Med. 2012;51:59–64. doi: 10.2169/internalmedicine.51.5698. [DOI] [PubMed] [Google Scholar]
- Hunter T. Signaling-2000 and beyond. Cell. 2000;100:113–27. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- Jacobs C, Huang LJ, Bartowsky E, Normark S, Park JT. Bacterial cell wall recycling provides cytosolic muropeptides as effectors for beta-lactamase induction. EMBO J. 1994;13:4684–94. doi: 10.1002/j.1460-2075.1994.tb06792.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs C, Frere JM, Normark S. Cytosolic intermediates for cell wall biosynthesis and degradation control inducible beta-lactam resistance in gram-negative bacteria. Cell. 1997;88:823–32. doi: 10.1016/s0092-8674(00)81928-5. [DOI] [PubMed] [Google Scholar]
- Jaurin B, Grundstrom T, Normark S. Sequence elements determining ampC promoter strength in E. coli. EMBO J. 1982;1:875–81. doi: 10.1002/j.1460-2075.1982.tb01263.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez PN, Koch G, Thompson JA, Xavier KB, Cool RH, Quax WJ. The multiple signaling systems regulating virulence in Pseudomonas aeruginosa. Microbiol Mol Biol Rev. 2012;76:46–65. doi: 10.1128/MMBR.05007-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SJ, Ernst EJ, Moores KG. Is double coverage of Gram-negative organisms necessary? Am J Health Syst Pharm. 2011;68:119–24. doi: 10.2146/ajhp090360. [DOI] [PubMed] [Google Scholar]
- Juan C, Moya B, Perez JL, Oliver A. Stepwise upregulation of the Pseudomonas aeruginosa chromosomal cephalosporinase conferring high-level beta-lactam resistance involves three AmpD homologues. Antimicrob Agents Chemother. 2006;50:1780–87. doi: 10.1128/AAC.50.5.1780-1787.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan N, Taylor SS, Zhai Y, Venter JC, Manning G. Structural and functional diversity of the microbial kinome. PLoS Biol. 2007;5:e17. doi: 10.1371/journal.pbio.0050017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly-Wintenberg K, South SL, Montie TC. Tyrosine phosphate in a- and b-type flagellins of Pseudomonas aeruginosa. J Bacteriol. 1993;175:2458–61. doi: 10.1128/jb.175.8.2458-2461.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. A common mechanism of cellular death induced by bactericidal antibiotics. Cell. 2007;130:797–810. doi: 10.1016/j.cell.2007.06.049. [DOI] [PubMed] [Google Scholar]
- Kong KF, Jayawardena SR, Del Puerto A, Wiehlmann L, Laabs U, Tummler B, Mathee K. Characterization of poxB, a chromosomal-encoded Pseudomonas aeruginosa oxacillinase. Gene. 2005a;358:82–92. doi: 10.1016/j.gene.2005.05.027. [DOI] [PubMed] [Google Scholar]
- Kong KF, Jayawardena SR, Indulkar SD, Del Puerto A, Koh CL, Hoiby N, Mathee K. Pseudomonas aeruginosa AmpR is a global transcriptional factor that regulates expression of AmpC and PoxB beta-lactamases, proteases, quorum sensing, and other virulence factors. Antimicrob Agents Chemother. 2005b;49:4567–75. doi: 10.1128/AAC.49.11.4567-4575.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong KF, Aguila A, Schneper L, Mathee K. Pseudomonas aeruginosa beta-lactamase induction requires two permeases, AmpG and AmpP. BMC Microbiol. 2010;10:328. doi: 10.1186/1471-2180-10-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuga A, Okamoto R, Inoue M. ampR gene mutations that greatly increase class C beta-lactamase activity in Enterobacter cloacae. Antimicrob Agents Chemother. 2000;44:561–7. doi: 10.1128/aac.44.3.561-567.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulasakara H, Lee V, Brencic A, et al. Analysis of Pseudomonas aeruginosa diguanylate cyclases and phosphodiesterases reveals a role for bis-(3′-5′)-cyclic-GMP in virulence. P Natl Acad Sci USA. 2006;103:2839–44. doi: 10.1073/pnas.0511090103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari H, Balasubramanian D, Zincke D, Mathee K. Role of Pseudomonas aeruginosa AmpR on beta-lactam and non-beta-lactam transient cross-resistance upon pre-exposure to subinhibitory concentrations of antibiotics. J Med Microbiol. 2014a;63:544–55. doi: 10.1099/jmm.0.070185-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari H, Murugapiran SK, Balasubramanian D, et al. LTQ-XL mass spectrometry proteome analysis expands the Pseudomonas aeruginosa AmpR regulon to include cyclic di-GMP phosphodiesterases and phosphoproteins, and identifies novel open reading frames. J Proteomics. 2014b;96C:328–42. doi: 10.1016/j.jprot.2013.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapouge K, Schubert M, Allain FH, Haas D. Gac/Rsm signal transduction pathway of gamma-proteobacteria: from RNA recognition to regulation of social behaviour. Mol Microbiol. 2008;67:241–53. doi: 10.1111/j.1365-2958.2007.06042.x. [DOI] [PubMed] [Google Scholar]
- Laskowski MA, Kazmierczak BI. Mutational analysis of RetS, an unusual sensor kinase-response regulator hybrid required for Pseudomonas aeruginosa virulence. Infect Immun. 2006;74:4462–73. doi: 10.1128/IAI.00575-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latifi A, Foglino M, Tanaka K, Williams P, Lazdunski A. A hierarchical quorum-sensing cascade in Pseudomonas aeruginosa links the transcriptional activators LasR and RhIR (VsmR) to expression of the stationary-phase sigma factor RpoS. Mol Microbiol. 1996;21:1137–46. doi: 10.1046/j.1365-2958.1996.00063.x. [DOI] [PubMed] [Google Scholar]
- Letunic I, Bork P. Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics. 2007;23:127–8. doi: 10.1093/bioinformatics/btl529. [DOI] [PubMed] [Google Scholar]
- Letunic I, Bork P. Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 2011;39:W475–8. doi: 10.1093/nar/gkr201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg F, Normark S. Contribution of chromosomal beta-lactamases to beta-lactam resistance in enterobacteria. Rev Infect Dis. 1986;8(suppl 3):S292–S304. doi: 10.1093/clinids/8.supplement_3.s292. [DOI] [PubMed] [Google Scholar]
- Lindberg F, Normark S. Common mechanism of ampC beta-lactamase induction in enterobacteria: regulation of the cloned Enterobacter cloacae P99 beta-lactamase gene. J Bacteriol. 1987;169:758–63. doi: 10.1128/jb.169.2.758-763.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg F, Westman L, Normark S. Regulatory components in Citrobacter freundii ampC beta-lactamase induction. P Natl Acad Sci USA. 1985;82:4620–24. doi: 10.1073/pnas.82.14.4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindquist S, Lindberg F, Normark S. Binding of the Citrobacter freundii AmpR regulator to a single DNA site provides both autoregulation and activation of the inducible ampC beta-lactamase gene. J Bacteriol. 1989;171:3746–53. doi: 10.1128/jb.171.7.3746-3753.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister PD, Wolter DJ, Hanson ND. Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin Microbiol Rev. 2009;22:582–610. doi: 10.1128/CMR.00040-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livermore DM. Clinical significance of beta-lactamase induction and stable derepression in Gram-negative rods. Eur J Clin Microbiol. 1987;6:439–45. doi: 10.1007/BF02013107. [DOI] [PubMed] [Google Scholar]
- Lodge JM, Minchin SD, Piddock LJ, Busby JW. Cloning, sequencing and analysis of the structural gene and regulatory region of the Pseudomonas aeruginosa chromosomal ampC beta-lactamase. Biochem J. 1990;272:627–31. doi: 10.1042/bj2720627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge J, Busby S, Piddock L. Investigation of the Pseudomonas aeruginosa ampR gene and its role at the chromosomal ampC beta-lactamase promoter. FEMS Microbiol Lett. 1993;111:315–20. doi: 10.1111/j.1574-6968.1993.tb06404.x. [DOI] [PubMed] [Google Scholar]
- Lopez-Causape C, Rojo-Molinero E, Mulet X, et al. Clonal dissemination, emergence of mutator lineages and antibiotic resistance evolution in Pseudomonas aeruginosa cystic fibrosis chronic lung infection. PLoS ONE. 2013;8:e71001. doi: 10.1371/journal.pone.0071001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyczak JB, Cannon CL, Pier GB. Lung infections associated with cystic fibrosis. Clin Microbiol Rev. 2002;15:194–222. doi: 10.1128/CMR.15.2.194-222.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Conover M, Lu H, Parsek MR, Bayles K, Wozniak DJ. Assembly and development of the Pseudomonas aeruginosa biofilm matrix. PLoS Pathog. 2009;5:e1000354. doi: 10.1371/journal.ppat.1000354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macek B, Mijakovic I, Olsen JV, Gnad F, Kumar C, Jensen PR, Mann M. The serine/threonine/tyrosine phosphoproteome of the model bacterium Bacillus subtilis. Mol Cell Proteomics. 2007;6:697–707. doi: 10.1074/mcp.M600464-MCP200. [DOI] [PubMed] [Google Scholar]
- Maddocks SE, Oyston PC. Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology. 2008;154:3609–23. doi: 10.1099/mic.0.2008/022772-0. [DOI] [PubMed] [Google Scholar]
- Mahlen SD, Morrow SS, Abdalhamid B, Hanson ND. Analyses of ampC gene expression in Serratia marcescens reveal new regulatory properties. J Antimicrob Chemother. 2003;51:791–802. doi: 10.1093/jac/dkg133. [DOI] [PubMed] [Google Scholar]
- Martin DW, Schurr MJ, Mudd MH, Govan JR, Holloway BW, Deretic V. Mechanism of conversion to mucoidy in Pseudomonas aeruginosa infecting cystic fibrosis patients. P Natl Acad Sci USA. 1993;90:8377–81. doi: 10.1073/pnas.90.18.8377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LW, Reid DW, Sharples KJ, Lamont IL. Pseudomonas siderophores in the sputum of patients with cystic fibrosis. Biometals. 2011;24:1059–67. doi: 10.1007/s10534-011-9464-z. [DOI] [PubMed] [Google Scholar]
- Masuda N, Sakagawa E, Ohya S, Gotoh N, Tsujimoto H, Nishino T. Substrate specificities of MexAB-OprM, MexCD-OprJ, and MexXY-oprM efflux pumps in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2000;44:3322–7. doi: 10.1128/aac.44.12.3322-3327.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathee K, Ciofu O, Sternberg C, et al. Mucoid conversion of Pseudomonas aeruginosa by hydrogen peroxide: a mechanism for virulence activation in the cystic fibrosis lung. Microbiology. 1999;145:1349–57. doi: 10.1099/13500872-145-6-1349. [DOI] [PubMed] [Google Scholar]
- McGrath S, Wade DS, Pesci EC. Dueling quorum sensing systems in Pseudomonas aeruginosa control the production of the Pseudomonas quinolone signal (PQS) FEMS Microbiol Lett. 2004;230:27–34. doi: 10.1016/S0378-1097(03)00849-8. [DOI] [PubMed] [Google Scholar]
- Mesaros N, Nordmann P, Plesiat P, et al. Pseudomonas aeruginosa: resistance and therapeutic options at the turn of the new millennium. Clin Microbiol Infect. 2007;13:560–78. doi: 10.1111/j.1469-0691.2007.01681.x. [DOI] [PubMed] [Google Scholar]
- Meyer JM, Neely A, Stintzi A, Georges C, Holder IA. Pyoverdin is essential for virulence of Pseudomonas aeruginosa. Infect Immun. 1996;64:518–23. doi: 10.1128/iai.64.2.518-523.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mougous JD, Gifford CA, Ramsdell TL, Mekalanos JJ. Threonine phosphorylation post-translationally regulates protein secretion in Pseudomonas aeruginosa. Nat Cell Biol. 2007;9:797–803. doi: 10.1038/ncb1605. [DOI] [PubMed] [Google Scholar]
- Moya B, Dotsch A, Juan C, Blazquez J, Zamorano L, Haussler S, Oliver A. Beta-lactam resistance response triggered by inactivation of a nonessential penicillin-binding protein. PLoS Pathog. 2009;5:e1000353. doi: 10.1371/journal.ppat.1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulcahy H, O'Callaghan J, O'Grady EP, et al. Pseudomonas aeruginosa RsmA plays an important role during murine infection by influencing colonization, virulence, persistence, and pulmonary inflammation. Infect Immun. 2008;76:632–8. doi: 10.1128/IAI.01132-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naas T, Livermore DM, Nordmann P. Characterization of an LysR family protein, SmeR from Serratia marcescens S6, its effect on expression of the carbapenem-hydrolyzing beta-lactamase Sme-1, and comparison of this regulator with other beta-lactamase regulators. Antimicrob Agents Chemother. 1995;39:629–37. doi: 10.1128/AAC.39.3.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas MH, Honore N, Jarlier V, Philippon A, Cole ST. Molecular genetic analysis of cephalosporinase production and its role in beta-lactam resistance in clinical isolates of Enterobacter cloacae. Antimicrob Agents Chemother. 1987;31:295–9. doi: 10.1128/aac.31.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Normark S, Edlund T, Grundstrom T, Bergstrom S, Wolf-Watz H. Escherichia coli K-12 mutants hyperproducing chromosomal beta-lactamase by gene repetitions. J Bacteriol. 1977;132:912–22. doi: 10.1128/jb.132.3.912-922.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Normark S, Lindquist S, Lindberg F. Chromosomal beta-lactam resistance in enterobacteria. Scand J Infect Dis Suppl. 1986;49:38–45. [PubMed] [Google Scholar]
- Okazaki A, Avison MB. Induction of L1 and L2 beta-lactamase production in Stenotrophomonas maltophilia is dependent on an AmpR-type regulator. Antimicrob Agents Chemother. 2008;52:1525–8. doi: 10.1128/AAC.01485-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver A, Canton R, Campo P, Baquero F, Blazquez J. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science. 2000;288:1251–4. doi: 10.1126/science.288.5469.1251. [DOI] [PubMed] [Google Scholar]
- Olsson O, Bergstrom S, Normark S. Identification of a novel ampC beta-lactamase promoter in a clinical isolate of Escherichia coli. EMBO J. 1982;1:1411–6. doi: 10.1002/j.1460-2075.1982.tb01331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos JS, Agarwala R. COBALT: constraint-based alignment tool for multiple protein sequences. Bioinformatics. 2007;23:1073–9. doi: 10.1093/bioinformatics/btm076. [DOI] [PubMed] [Google Scholar]
- Paul M, Leibovici L. Combination antibiotic therapy for Pseudomonas aeruginosa bacteraemia. Lancet Infect Dis. 2005;5:192–3. doi: 10.1016/S1473-3099(05)70030-X. [DOI] [PubMed] [Google Scholar]
- Paul M, Benuri-Silbiger I, Soares-Weiser K, Leibovici L. Beta lactam monotherapy versus beta lactam-aminoglycoside combination therapy for sepsis in immunocompetent patients: systematic review and meta-analysis of randomised trials. BMJ. 2004;328:668. doi: 10.1136/bmj.38028.520995.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul M, Silbiger I, Grozinsky S, Soares-Weiser K, Leibovici L. Beta lactam antibiotic monotherapy versus beta lactam-aminoglycoside antibiotic combination therapy for sepsis. Cochrane Database Syst Rev. 2006;1:CD003344. doi: 10.1002/14651858.CD003344.pub2. [DOI] [PubMed] [Google Scholar]
- Pena C, Suarez C, Gozalo M, et al. Prospective multicenter study of the impact of carbapenem resistance on mortality in Pseudomonas aeruginosa bloodstream infections. Antimicrob Agents Chemother. 2012;56:1265–72. doi: 10.1128/AAC.05991-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendleton JN, Gorman SP, Gilmore BF. Clinical relevance of the ESKAPE pathogens. Expert Rev Anti Infect Ther. 2013;11:297–308. doi: 10.1586/eri.13.12. [DOI] [PubMed] [Google Scholar]
- Pesci EC, Pearson JP, Seed PC, Iglewski BH. Regulation of las and rhl quorum sensing in Pseudomonas aeruginosa. J Bacteriol. 1997;179:3127–32. doi: 10.1128/jb.179.10.3127-3132.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potvin E, Sanschagrin F, Levesque RC. Sigma factors in Pseudomonas aeruginosa. FEMS Microbiol Rev. 2008;32:38–55. doi: 10.1111/j.1574-6976.2007.00092.x. [DOI] [PubMed] [Google Scholar]
- Proenca R, Niu WW, Cacalano G, Prince A. The Pseudomonas cepacia 249 chromosomal penicillinase is a member of the AmpC family of chromosomal beta-lactamases. Antimicrob Agents Chemother. 1993;37:667–74. doi: 10.1128/aac.37.4.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raso T, Bianco O, Grosso B, Zucca M, Savoia D. Achromobacter xylosoxidans respiratory tract infections in cystic fibrosis patients. APMIS. 2008;116:837–41. doi: 10.1111/j.1600-0463.2008.00995.x. [DOI] [PubMed] [Google Scholar]
- Rice LB. Progress and challenges in implementing the research on ESKAPE pathogens. Infect Control Hosp Epidemiol. 2010;31:S7–S10. doi: 10.1086/655995. [DOI] [PubMed] [Google Scholar]
- Romling U, Galperin MY, Gomelsky M. Cyclic di-GMP: the first 25 years of a universal bacterial second messenger. Microbiol Mol Biol Rev. 2013;77:1–52. doi: 10.1128/MMBR.00043-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan RP. Cyclic di-GMP signalling and the regulation of bacterial virulence. Microbiology. 2013;159:1286–97. doi: 10.1099/mic.0.068189-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan RP, Lucey J, O'Donovan K, McCarthy Y, Yang L, Tolker-Nielsen T, Dow JM. HD-GYP domain proteins regulate biofilm formation and virulence in Pseudomonas aeruginosa. Environ Microbiol. 2009;11:1126–36. doi: 10.1111/j.1462-2920.2008.01842.x. [DOI] [PubMed] [Google Scholar]
- Schell MA. Molecular biology of the LysR family of transcriptional regulators. Annu Rev Microbiol. 1993;47:597–626. doi: 10.1146/annurev.mi.47.100193.003121. [DOI] [PubMed] [Google Scholar]
- Schmidtke AJ, Hanson ND. Role of ampD homologs in overproduction of AmpC in clinical isolates of Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2008;52:3922–7. doi: 10.1128/AAC.00341-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster M, Greenberg EP. A network of networks: quorum-sensing gene regulation in Pseudomonas aeruginosa. Int J Med Microbiol. 2006;296:73–81. doi: 10.1016/j.ijmm.2006.01.036. [DOI] [PubMed] [Google Scholar]
- Schuster M, Hawkins AC, Harwood CS, Greenberg EP. The Pseudomonas aeruginosa RpoS regulon and its relationship to quorum sensing. Mol Microbiol. 2004;51:973–85. doi: 10.1046/j.1365-2958.2003.03886.x. [DOI] [PubMed] [Google Scholar]
- Seoane A, Francia MV, Lobo JM Garcia. Nucleotide sequence of the ampC-ampR region from the chromosome of Yersinia enterocolitica. Antimicrob Agents Chemother. 1992;36:1049–52. doi: 10.1128/aac.36.5.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EE, Buckley DG, Wu Z, et al. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. P Natl Acad Sci USA. 2006;103:8487–92. doi: 10.1073/pnas.0602138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnleitner E, Abdou L, Haas D. Small RNA as global regulator of carbon catabolite repression in Pseudomonas aeruginosa. P Natl Acad Sci USA. 2009;106:21866–71. doi: 10.1073/pnas.pnas.0910308106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnleitner E, Gonzalez N, Sorger-Domenigg T, et al. The small RNA PhrS stimulates synthesis of the Pseudomonas aeruginosa quinolone signal. Mol Microbiol. 2011;80:868–85. doi: 10.1111/j.1365-2958.2011.07620.x. [DOI] [PubMed] [Google Scholar]
- Sonnleitner E, Romeo A, Blasi U. Small regulatory RNAs in Pseudomonas aeruginosa. RNA Biol. 2012;9:364–71. doi: 10.4161/rna.19231. [DOI] [PubMed] [Google Scholar]
- Stapper AP, Narasimhan G, Ohman DE, et al. Alginate production affects Pseudomonas aeruginosa biofilm development and architecture, but is not essential for biofilm formation. J Med Microbiol. 2004;53:679–90. doi: 10.1099/jmm.0.45539-0. [DOI] [PubMed] [Google Scholar]
- Stover CK, Pham XQ, Erwin AL, et al. Complete genome sequence of Pseudomonas aeruginosa PA01, an opportunistic pathogen. Nature. 2000;406:959–64. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- Suarez C, Pena C, Arch O, et al. A large sustained endemic outbreak of multiresistant Pseudomonas aeruginosa: a new epidemiological scenario for nosocomial acquisition. BMC Infect Dis. 2011;11:272. doi: 10.1186/1471-2334-11-272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh SJ, Silo-Suh L, Woods DE, Hassett DJ, West SE, Ohman DE. Effect of rpoS mutation on the stress response and expression of virulence factors in Pseudomonas aeruginosa. J Bacteriol. 1999;181:3890–97. doi: 10.1128/jb.181.13.3890-3897.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacconelli E, Cataldo MA, Dancer SJ, et al. ESCMID guidelines for the management of the infection control measures to reduce transmission of multidrug-resistant Gram-negative bacteria in hospitalized patients. Clin Microbiol Infect. 2014;20:1–55. doi: 10.1111/1469-0691.12427. [DOI] [PubMed] [Google Scholar]
- Tamma PD, Cosgrove SE, Maragakis LL. Combination therapy for treatment of infections with gram-negative bacteria. Clin Microbiol Rev. 2012;25:450–70. doi: 10.1128/CMR.05041-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trepanier S, Prince A, Huletsky A. Characterization of the penA and penR genes of Burkholderia cepacia 249 which encode the chromosomal class A penicillinase and its LysR-type transcriptional regulator. Antimicrob Agents Chemother. 1997;41:2399–405. doi: 10.1128/aac.41.11.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallet I, Olson JW, Lory S, Lazdunski A, Filloux A. The chaperone/usher pathways of Pseudomonas aeruginosa: identification of fimbrial gene clusters (cup) and their involvement in biofilm formation. P Natl Acad Sci USA. 2001;98:6911–6. doi: 10.1073/pnas.111551898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vento S, Cainelli F, Temesgen Z. Lung infections after cancer chemotherapy. Lancet Oncol. 2008;9:982–92. doi: 10.1016/S1470-2045(08)70255-9. [DOI] [PubMed] [Google Scholar]
- Ventre I, Goodman AL, Vallet-Gely I, et al. Multiple sensors control reciprocal expression of Pseudomonas aeruginosa regulatory RNA and virulence genes. P Natl Acad Sci USA. 2006;103:171–6. doi: 10.1073/pnas.0507407103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venturi V. Regulation of quorum sensing in Pseudomonas. FEMS Microbiol Rev. 2006;30:274–91. doi: 10.1111/j.1574-6976.2005.00012.x. [DOI] [PubMed] [Google Scholar]
- Weng SF, Lin JW, Chen CH, Chen YY, Tseng YH. Constitutive expression of a chromosomal class A (BJM group 2) beta-lactamase in Xanthomonas campestris. Antimicrob Agents Chemother. 2004;48:209–15. doi: 10.1128/AAC.48.1.209-215.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedenheft B, Sternberg SH, Doudna JA. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012;482:331–8. doi: 10.1038/nature10886. [DOI] [PubMed] [Google Scholar]
- Wilderman PJ, Sowa NA, FitzGerald DJ, FitzGerald PC, Gottesman S, Ochsner UA, Vasil ML. Identification of tandem duplicate regulatory small RNAs in Pseudomonas aeruginosa involved in iron homeostasis. P Natl Acad Sci USA. 2004;101:9792–7. doi: 10.1073/pnas.0403423101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams P, Camara M. Quorum sensing and environmental adaptation in Pseudomonas aeruginosa: a tale of regulatory networks and multifunctional signal molecules. Curr Opin Microbiol. 2009;12:182–91. doi: 10.1016/j.mib.2009.01.005. [DOI] [PubMed] [Google Scholar]
- Winsor GL, Lam DK, Fleming L, et al. Pseudomonas Genome Database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res. 2011;39:D596–D600. doi: 10.1093/nar/gkq869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong A, Rodrigue N, Kassen R. Genomics of adaptation during experimental evolution of the opportunistic pathogen Pseudomonas aeruginosa. PLoS Genet. 2012;8:e1002928. doi: 10.1371/journal.pgen.1002928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Jelsbak L, Molin S. Microbial ecology and adaptation in cystic fibrosis airways. Environ Microbiol. 2011;13:1682–9. doi: 10.1111/j.1462-2920.2011.02459.x. [DOI] [PubMed] [Google Scholar]
- Zamorano L, Reeve TM, Deng L, et al. NagZ inactivation prevents and reverts beta-lactam resistance, driven by AmpD and PBP 4 mutations, in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2010;54:3557–63. doi: 10.1128/AAC.00385-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng E, Mathee K, Narasimhan G. IEM: an algorithm for iterative enhancement of motifs using comparative genomics data. Comput Syst Bioinformatics Conf. 2007;6:227–35. [PubMed] [Google Scholar]
- Zhang Y, Bao Q, Gagnon LA, Huletsky A, Oliver A, Jin S, Langaee T. ampG gene of Pseudomonas aeruginosa and its role in beta-lactamase expression. Antimicrob Agents Chemother. 2010;54:4772–9. doi: 10.1128/AAC.00009-10. [DOI] [PMC free article] [PubMed] [Google Scholar]