

Whole-genome sequencing of Shiga toxin–producing Escherichia coli O157 isolates allowed identification of linked cases with unprecedented sensitivity and specificity, with twice as many clusters identified compared with current methods.
Keywords: public health, whole-genome sequencing, Shiga toxin–producing Escherichia coli O157, national surveillance
Abstract
Background. National surveillance of gastrointestinal pathogens, such as Shiga toxin–producing Escherichia coli O157 (STEC O157), is key to rapidly identifying linked cases in the distributed food network to facilitate public health interventions. In this study, we used whole-genome sequencing (WGS) as a tool to inform national surveillance of STEC O157 in terms of identifying linked cases and clusters and guiding epidemiological investigation.
Methods. We retrospectively analyzed 334 isolates randomly sampled from 1002 strains of STEC O157 received by the Gastrointestinal Bacteria Reference Unit at Public Health England, Colindale, in 2012. The genetic distance between each isolate, as estimated by WGS, was calculated and phylogenetic methods were used to place strains in an evolutionary context.
Results. Estimates of linked clusters representing STEC O157 outbreaks in England and Wales increased by 2-fold when WGS was used instead of traditional typing techniques. The previously unidentified clusters were often widely geographically distributed and small in size. Phylogenetic analysis facilitated identification of temporally distinct cases sharing common exposures and delineating those that shared epidemiological and temporal links. Comparison with multi locus variable number tandem repeat analysis (MLVA) showed that although MLVA is as sensitive as WGS, WGS provides a more timely resolution to outbreak clustering.
Conclusions. WGS has come of age as a molecular typing tool to inform national surveillance of STEC O157; it can be used in real time to provide the highest strain-level resolution for outbreak investigation. WGS allows linked cases to be identified with unprecedented specificity and sensitivity that will facilitate targeted and appropriate public health investigations.
Gastrointestinal disease is an important public health problem in England, with up to 20% of the population experiencing at least 1 episode of acute gastroenteritis each year [1]. An effective national surveillance program for gastrointestinal diseases is imperative to identify cases with linked exposures; this is especially pertinent for pathogens that may enter nationally distributed food networks. Although conventional epidemiological investigation using detailed questionnaires and contact tracing is vital, to achieve optimal surveillance we must complement these activities with a rapid and robust molecular typing method to accurately discriminate between linked cases and sporadic infections.
With >1000 presumptive isolates submitted to the Gastrointestinal Bacteria Reference Unit (GBRU) annually [2], infections with Shiga toxin–producing Escherichia coli O157 (STEC O157) continue to exert a public health burden in England, both economically and in terms of morbidity and mortality. Symptoms of STEC infection range from mild to severe, but typically include bloody diarrhea. Approximately 6% of cases develop hemolytic uremic syndrome (HUS) [3].
The main reservoir of STEC O157 in England is cattle, although it is carried by other animals, mainly ruminants [4, 5]. Transmission to humans occurs through direct or indirect contact with animals or their environments, consumption of contaminated food or water, and person-to-person contact [6–8]. Contamination of the food supply can cause large-scale national and multinational outbreaks [9–11].
Outbreaks, involving ≥2 cases in different households or residential institutions, vary in number annually but since 2009 have contributed between 9% and 25% of isolates in England and Wales (GBRU/Department of Gastrointestinal Emerging and Zoonotic Infections, in-house data), with the majority of cases being apparently sporadic. All isolates received by GBRU are routinely phage typed [12], but in England, the majority (60%) of isolates are either PT8 or PT21/28, and so the ability of this method to discriminate between cases resulting from separate exposures is very low. Multi locus variable number tandem repeat analysis (MLVA) is used to provide higher levels of strain discrimination.
The utility of whole-genome sequencing (WGS) for the investigation of outbreaks has already been demonstrated for several bacterial pathogens [13, 14], and there is increasing evidence in the literature for the positive contribution of WGS to outbreaks involving gastrointestinal pathogens [15–19]. The aim of this study was to expand the use of WGS by evaluating a WGS approach to inform national surveillance of a major pathogen. By validating the WGS approach using clearly defined outbreak and sporadic cases of STEC O157 and by investigating the findings, WGS can provide additional insights into outbreak definition, transmission networks, and other aspects of the underlying epidemiology of this pathogen.
METHODS
Strain Selection
A total of 572 isolates were selected for sequencing: 334 isolates were randomly selected from 1002 STEC O157 culture-positive isolates received by GBRU from cases in England, Wales, and Northern Ireland during 2012; 147 isolates were randomly selected from 939 STEC O157 culture-positive isolates from cases in England, Wales, and Northern Ireland received by GBRU in 2013; and an additional 91 English historical isolates received between 1990 and 2011 were selected based on phage type diversity to provide context as a sample of the background population. The total collection contained strains from known outbreaks, household clusters, serial strains isolated from the same patient, and strains from apparently sporadic cases. A total of 18 phage types [20] were represented.
Genome Sequencing and Sequence Analysis
Genomic DNA was fragmented and tagged for multiplexing with Nextera XT DNA Sample Preparation Kits (Illumina) and sequenced using the Illumina GAII platform with 2 × 150 bp reads. Short reads were mapped to the reference STEC O157 strain Sakai [21] using BWA-SW [22]. The sequence alignment map output from BWA was sorted and indexed to produce a binary alignment map (BAM) using Samtools [23]. GATK2 [24] was used to create a variant call format file from each of the BAMs, which were further parsed to extract only single-nucleotide polymorphism (SNP) positions that were of high quality in all genomes (Mapping Quality >30, Depth >10, Genotype Quality >30, Variant Ratio >0.9). An alignment of polymorphic positions was used to create approximate maximum likelihood trees using FastTree [25] under the Jukes–Cantor model of nucleotide evolution. Pairwise SNP distances between the genomes of each strain were calculated. FASTQ sequences were deposited in the National Center for Biotechnology Information Short Read Archive under bioproject PRJNA248042.
Data Handling
STEC enhanced surveillance questionnaires (SESQs) are administered to all cases of STEC O157 in England. The SESQ collects demographic details; risk status; clinical condition (including progression to HUS); household or other close contact details; exposures including travel, food, and water consumption, contact with animals, and environmental factors; epidemiological case classification; and outbreak /cluster status. SESQ data were reviewed for each selected strain and strains classified in respect to known outbreak status, known household cluster status, or whether multiple isolates originated from the same patient. Any strains fulfilling these criteria were designated as having a known epidemiological link.
Pairwise SNP distances were calculated for all strains in this study. In previously reported outbreaks, onset of illness in cases occurs a median of 39 days from another linked case with a mode of 1 (Public Health England). Using specimen dates of isolates, temporality between isolates of different genetic distances were compared. The pairwise SNP distribution and temporal links between known linked cases was examined and a relatedness threshold determined accordingly. As related strains are likely to originate from a common source, the threshold was termed the common source threshold (CST). This threshold was then applied to all other strains in the dataset and evaluated for epidemiological context.
Related strains within the CST were classified into clusters on the basis of having at least 1 SNP distance within the CST to another isolate in the dataset. Clusters not previously identified were designated WGS linked clusters. Temporal and geographic links between cases in clusters were examined and comparisons made between epidemiologically identified and WGS linked clusters.
Deeper phylogenetic relationships were also investigated to ascertain whether they provided epidemiologically useful information or associations. Clusters of 25 SNP genetic distance were constructed (herby referred to as phylogenetic clusters [PCs]) and those with >1 CST cluster within each PC were investigated for shared epidemiological associations.
All STEC O157 isolates reported between 1 May 2012 and 31 December 2013 that have been typed through both MLVA and WGS were used to investigate clustering dynamics for each method. Survival analysis was used to test the null hypothesis that there is no difference in timeliness and completeness of clustering-related isolates using the 2 methods. For survival analysis, an isolate clustering with another isolate based on ≤1 locus variant for MLVA or ≤CST for WGS represented a failure. Across the study period, isolates will enter at various time points based on laboratory report date. At that point, the isolate is at risk of clustering with other isolates already in the study population or isolates entering the study at a later date. Kaplan–Meier estimates of the survivor function were estimated for both methods and displayed as cumulative survival curves with accompanying tables presenting those at risk at specific time points. The proportional hazards assumption was tested by plotting the log cumulative hazard in both groups. Where the proportional hazards assumption applied, the survival function in the 2 groups was compared by calculating a hazard ratio using Cox regression.
RESULTS
Distribution of Pairwise Distance Between Closely Related Isolates
For 183 of 425 strains used in this study, an epidemiological link to at least 1 other case was known. This included 16 cases where multiple isolates were sequenced from the same person, 43 isolates that were part of 26 separate household clusters, and 124 cases that were part of 14 known outbreaks. The remaining 242 strains had no common link previously identified. The pairwise SNP distance distribution revealed that no pair of epidemiologically linked isolates had >5 SNP differences with a mean of 1 SNP in isolates from the same household (standard deviation [SD], 0.99) or known common source (SD, 1.04) and 0.3 SNPs (SD, 0.60) from isolates from the same person (Figure 1).
Figure 1.

Histogram showing proportion of pairs against single-nucleotide polymorphism (SNP) distance of cases with a known epidemiological link.
One hundred thirty-six cases with no known epidemiological link were within ≤5 SNPs to another case. The majority (87%) of pairs that fell within the 5 SNP threshold comprised strains isolated within 30 days of each other with a mean interval between pairs of samples being 11 days. Between a genetic distance of 5 and 10 SNPs, the mean interval between pairs of samples (n = 24) increased to 258 days (Figure 2). As all previously linked isolates fell within a 5 SNP threshold and the majority of pairs of cases within this threshold were temporally linked, we hypothesize a threshold of 5 SNPs to categorize isolates as related. In this context, the term “related” alludes to a common source of infection and strains that are within 5 SNPs of another are referred to as falling within the CST.
Figure 2.

Histogram showing frequency of pairs against single-nucleotide polymorphism (SNP) distance. Each bar is colored as a proportion of pairs isolated within <7 days, 7–30 days, and >30 days.
Applying the CST
One hundred sixty strains isolated during 2012 fell within the CST. These strains can be formed into 53 clusters where members of the cluster must share at least 1 link within the CST. Twenty of the clusters (46 strains) represented either household outbreaks or multiple strains from the same patient. The remaining 33 clusters comprised 114 strains representing 34% of the dataset. Routine public health investigation previously undertaken had not identified 20 of 33 clusters, and these were designated WGS linked clusters. Of the 20 WGS linked clusters, 18 comprised between 2 and 4 cases, while 2 larger clusters comprised 12 and 7 cases. Overall, if we conclude that all cases within the CST are part of epidemiologically linked clusters, this corresponds to an increase in sensitivity of >50% in detecting linked cases outside the household setting when using WGS to supplement the current approach.
Epidemiology of WGS Linked Clusters
The 20 WGS linked clusters were statistically more geographically dispersed than the 13 epidemiologically linked clusters (Figure 3A), with a mean residential distance of 169 km (SD, 111 km) for the former and 29 km (SD, 34 km) for the latter (P = .04 [5], 1-tailed t test). Strains of STEC O157 associated with a large national foodborne PT8 outbreak from 2011 [9] and a petting farm PT21/28 outbreak [26] were included for context (Figure 3B). The geographical dispersal of cases linked by WGS mirrors the distribution of the national PT8 outbreak as well as encompassing the distribution of a geographically restricted outbreak. Conversely, the epidemiologically linked clusters most closely mirrored the geographically restricted outbreak, highlighting the difficulty in recognizing national distributed cases without high-resolution strain discrimination such as WGS.
Figure 3.
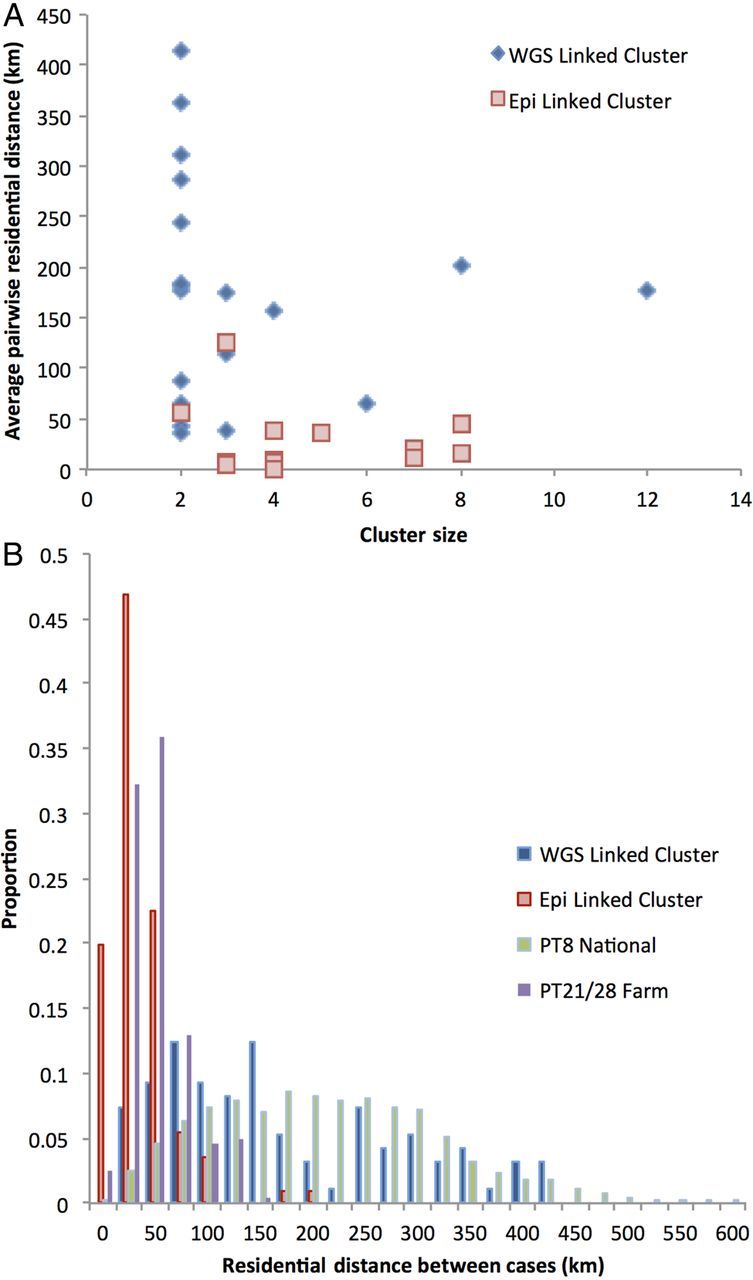
A, Scatter diagram showing the average pairwise residential distance of each close contact cluster against the size in number of cases. The coloring represents whether the cluster was already identified through epidemiological investigation or if identified by whole-genome sequencing (WGS) alone. B, Histogram showing the distribution of residential distance for WGS linked clusters and epidemiologically linked clusters. PT8 National and PT21/28 Farm represent distributed foodborne and point source outbreaks, respectively.
Retrospective epidemiological follow-up was undertaken for cases in the 2 larger WGS clusters. Cluster 1 comprised 12 nationally distributed cases with onset dates all within 15 days of each other. Following reinvestigation, the only common exposure identified was the consumption of a specific prepacked leafy salad from different branches of 1 major supermarket chain. Cluster 2 contained 7 cases, of which 4 cases were from separate English public health regions with onset dates spanning a 2-week period. Following reinvestigation, it was identified that 3 cases had visited the same village, where another case was resident, within the incubation period. All 4 cases had been hiking in the same national park, putting them at risk of environmental exposure. The remaining 3 cases shared no obvious exposures, suggesting that the cases were exposed to the same source of infection but via different routes and/or vehicles, highlighting the importance of using all available epidemiological investigations when interpreting WGS for outbreak investigations.
Outbreak Detection MLVA Versus WGS
Clustering based on the WGS defined CST increased sensitivity in identifying linked cases; however, it was also necessary to compare this approach to other fine-typing methods deployed for STEC O157 (eg, MLVA). Using a survival analysis of 481 samples typed by both methods in 2012–2013, survival (ie, not clustering with another isolate) showed no significant difference with MLVA vs WGS CST based on clustering a single isolate with another (log-rank test for equality of survivor function: P = .101; Cox hazard ratio = 0.89, P = .198) (Figure 4). This indicates there is no difference in timeliness of clustering between the 2 methods. However, when we consider the time to cluster completion (all cases of a cluster are clustered) from the initial cluster event (any 2 cases of a cluster are clustered), there is a significant speed increase in time to completion of clusters with WGS CST as opposed to MLVA (log-rank test for equality of survivor function: P = .0006; Cox hazard ratio = 1.44, P = .001) (Figure 5).
Figure 4.
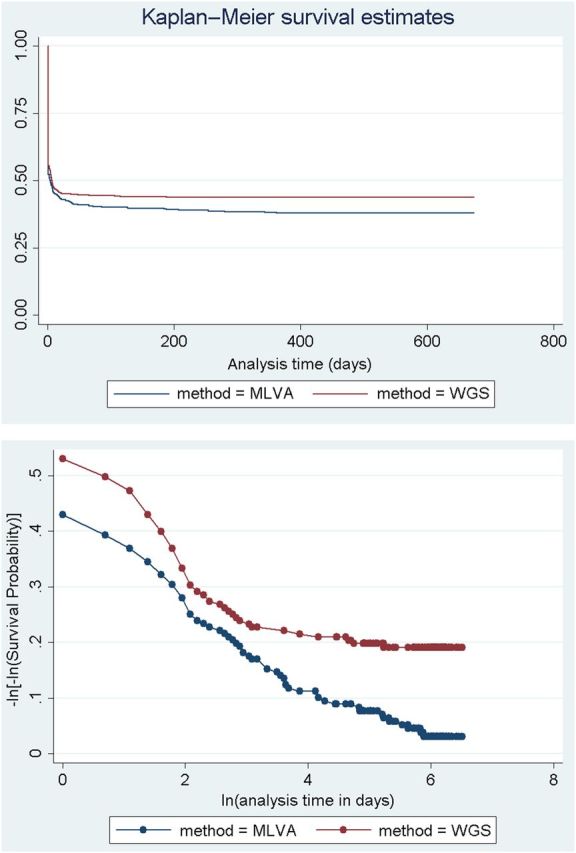
Kaplan–Meier failure estimates and proportional hazards assumption test showing that there is no difference in timeliness of clustering between whole-genome sequencing (WGS) and multi locus variable number tandem repeat analysis (MLVA).
Figure 5.
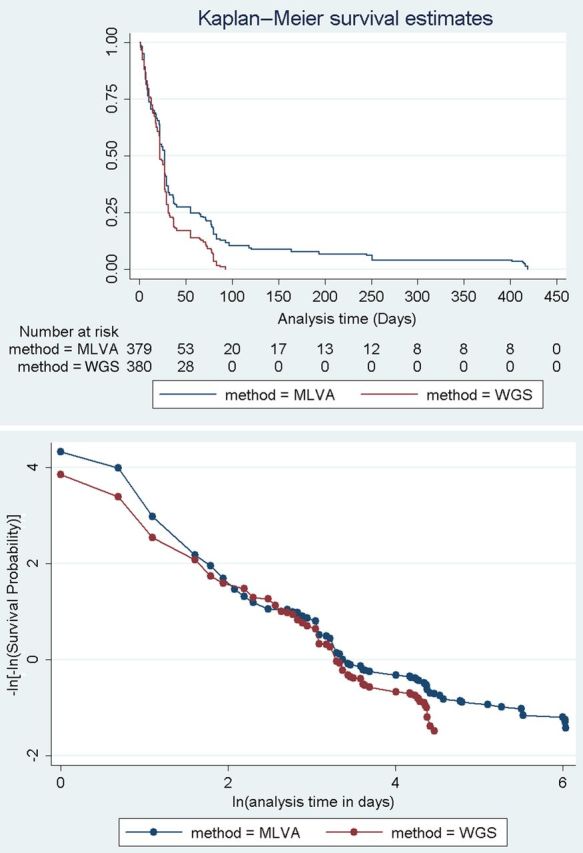
Kaplan–Meier survival estimates and proportional hazards assumption test showing that after isolates have clustered, time to completion of that cluster is significantly faster with whole-genome sequencing (WGS) than with multi locus variable number tandem repeat analysis (MLVA).
Epidemiological Context of Phylogenetic Clusters
Cases within the CST represent temporally linked cases, and these have been shown to include cases with common epidemiological exposures. Although the temporal relationship between pairs quickly dissipated as the genetic distance moved outside the CST, we investigated whether deeper phylogenetic relationships also provided epidemiologically useful information or associations. Nineteen PCs (see “Methods” section) were identified, and 10 had no geographical association or common exposures between the CST clusters within as assessed through the SESQ. One PC contained 3 CST clusters sharing a common exposure to a national park in the Midlands (Figure 6); each cluster correlated with year of isolation, highlighting the potential to identify the persistence of strains in the environment over time.
Figure 6.

Maximum likelihood phylogeny of 15 isolates representing 27 single-nucleotide polymorphisms (SNPs) across 25 coding DNA sequence (2 noncoding SNPs) with a total core genome size of 4 915 463 bp associated with cases that visited the same national park. The clusters represent 3 different common source threshold clusters, colored red, blue, and green within a single phylogenetic cluster. The level of resolution allows the delineation of strains from different years. The strain in red was temporally related to the strains in blue but significantly different genomically to suggest a different source of Shiga toxin–producing Escherichia coli exposure.
Two PCs contained CST clusters where the majority of strains were of Northern Irish provenance. Those patients who were not resident in Northern Ireland reported travel to various parts of the province in their SESQ. Similarly, PCs were identified with cases associated with Wales and travel to the Middle East (Figure 7).
Figure 7.

Maximum likelihood phylogeny of 374 isolates, representing 7756 single-nucleotide polymorphisms (SNPs), across 2902 coding DNA sequence (1166 noncoding SNPs) with a total core genome size of 3 808 948 bp. Common source threshold clusters identified through whole-genome sequencing (WGS) alone are colored red, and those identified through traditional methods are colored blue. Phylogenetic clusters that contained strains with related exposures are shaded green.
DISCUSSION
In this study, the potential of WGS in national surveillance of STEC O157 was assessed for its ability to improve outbreak detection and provide additional insights over conventional epidemiological investigations. WGS confirmed that strains from the same patient, from cases within the same household, and from cases with known epidemiological links had little or no difference in their core genomes. These cases fell within a 5 SNP threshold within which we found strong temporal correlations suggestive of epidemiological linkage. Using this empirically observed cutoff of 5 SNPs, we could determine with unprecedented clarity which strains of STEC O157 were likely to be epidemiologically linked. WGS detected linked cases of STEC O157 in 334 representative strains from an annual season with twice the sensitivity of current methods. This suggests that current outbreak detection is highly specific, but comparatively insensitive, and that the previous estimate of outbreaks, involving ≥2 cases in different households or residential institutions, contributing between 9% and 25% of isolates in England and Wales, is conservative. Previously elusive clusters were often more geographically dispersed than those identified using the traditional approach. It is suggested that these geographically dispersed outbreaks with no obvious common exposures are foodborne. This type of outbreak profiling will facilitate outbreak investigations through focusing hypothesis generation on foodborne exposures at an early stage.
In this study, we show that for identifying linked cases, the current threshold of ≤1 locus variant for clustering provides the same sensitivity as using the WGS CST. This is an important finding, as it not only gives confidence in the interpretation of MLVA to those public health laboratories not yet ready to adopt WGS methodologies but also allows cross-communication of results between practitioners of these 2 techniques. An important distinction between the 2 methods is the time it takes to resolve complete clusters of cases within an outbreak, with WGS CST completing clusters significantly faster than MLVA. This feature can be explained by the fact all linked cases tend to fall within the CST for all cases, whereas in a large MLVA cluster several isolates will only be joined via an intermediate isolate (ie, double-locus variants joined by a shared single-locus variant). This phenomenon has implications in accurately defining the microbiological case definition at the start of an outbreak investigation as outbreaks that resolve themselves to a single cluster may appear as multiple clusters until intermediate isolates are sampled.
The phylogenetic context of common source clusters was analyzed to see if there was any epidemiological signal between separate but related common source events. Several regional or travel-associated PCs were identified, highlighting the geographical isolation of STEC O157 even within the British Isles. The geographical signal observed in the WGS of STEC O157 has been described previously [27] and has obvious implications in facilitating outbreak investigations. For example, isolates could be linked to food sourced from specific regions of the world, or cases could be ruled out of a point source outbreak by confirming their strain originated from further afield, given adequate sampling of potential source populations.
The primary aims of gastrointestinal disease surveillance are to identify outbreaks, monitor long-term trends, and inform the effectiveness of policy and other public health interventions. WGS demonstrates unparalleled sensitivity and accuracy in identifying linked cases coupled with phylogenetic clustering of how strains are related over time and space. Its ability to accurately define sporadic cases over time enables better characterization of the population at risk and to assess the relative importance of exposures leading to sporadic infections, which may differ from those leading to outbreaks.
Timely analysis and interpretation of WGS data will inform public health interventions by identifying linked cases (ie, early warning of outbreaks) as well as inferring epidemiological context through evolutionary relationships. Furthermore, the ability to unambiguously rule out associations will prevent inappropriate public health actions from being taken, saving resources at the health protection and local authority level. Good communication and rapid sharing of real-time STEC O157 WGS data with colleagues working in the agriculture, veterinary, and food industries across international borders will allow evidence-based trace-back of isolates to their source and reveal specific risk factors in the food chain and environment, thus facilitating the targeting of resources and public health interventions to have maximum impact on reducing the burden of STEC O157 disease in England.
Notes
Acknowledgments. We would like to thank all the staff at Gastrointestinal Bacteria Reference Unit and Gastrointestinal Emerging and Zoonotic Infections who work on STEC O157.
Disclaimer. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health (NIH).
Financial support. This work was supported by the National Institute for Health Research scientific research development fund (grant number 108601). W. P. H. was supported by the National Institute of General Medical Sciences of the NIH (award number U54GM088558).
Potential conflicts of interest. J. W. is on the board of Discuva Ltd and has received funding from Hoffmann-La Roche. W. P. H. has received funds from Antigen Discovery Ltd. All other authors report no potential conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Wheeler JG, Sethi D, Cowden JM, et al. Study of infectious intestinal disease in England: rates in the community, presenting to general practice, and reported to national surveillance. The Infectious Intestinal Disease Study Executive. BMJ 1999; 318:1046–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jenkins C, Lawson A, Cheasty T, Bolton E, Smith G. Assessment of a real-time PCR for the detection and characterisation of verocytotoxigenic Escherichia coli. J Med Microbiol 2012; 61:1082–5. [DOI] [PubMed] [Google Scholar]
- 3.Lynn RM, O'Brien SJ, Taylor CM, et al. Childhood hemolytic uremic syndrome, United Kingdom and Ireland. Emerg Infect Dis 2005; 11:590–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pennington H. Escherichia coli O157. Lancet 2010; 376:1428–35. [DOI] [PubMed] [Google Scholar]
- 5.Ferens WA, Hovde CJ. Escherichia coli O157:H7: animal reservoir and sources of human infection. Foodborne Pathog Dis 2011; 8:465–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Locking ME, O'Brien SJ, Reilly WJ, et al. Risk factors for sporadic cases of Escherichia coli O157 infection: the importance of contact with animal excreta. Epidemiol Infect 2001; 127:215–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gillespie IA, O'Brien SJ, Adak GK, Cheasty T, Willshaw G. Foodborne general outbreaks of Shiga toxin-producing Escherichia coli O157 in England and Wales 1992–2002: where are the risks? Epidemiol Infect 2005; 133:803–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pritchard GC, Smith R, Ellis-Iversen J, Cheasty T, Willshaw GA. Verocytotoxigenic Escherichia coli O157 in animals on public amenity premises in England and Wales, 1997 to 2007. Vet Rec 2009; 164:545–9. [DOI] [PubMed] [Google Scholar]
- 9.Perry N, Cheasty T, Dallman T, Launders N, Willshaw G. Application of multi-locus variable number tandem repeat analysis to monitor verocytotoxin-producing Escherichia coli O157 phage type 8 in England and Wales: emergence of a profile associated with a national outbreak. J Appl Microbiol 2013; 115:1052–8. [DOI] [PubMed] [Google Scholar]
- 10.Buchholz U, Bernard H, Werber D, et al. German outbreak of Escherichia coli O104:H4 associated with sprouts. N Engl J Med 2011; 365:1763–70. [DOI] [PubMed] [Google Scholar]
- 11.Bell BP, Goldoft M, Griffin PM, et al. A multistate outbreak of Escherichia coli O157:H7-associated bloody diarrhea and hemolytic uremic syndrome from hamburgers. The Washington experience. JAMA 1994; 272:1349–53. [PubMed] [Google Scholar]
- 12.Khakhria R, Duck D, Lior H. Extended phage-typing scheme for Escherichia coli O157:H7. Epidemiol Infect 1990; 105:511–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walker TM, Ip CL, Harrell RH, et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect Dis 2013; 13:137–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Didelot X, Eyre DW, Cule M, et al. Microevolutionary analysis of Clostridium difficile genomes to investigate transmission. Genome Biol 2012; 13:R118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gilmour MW, Graham M, Van DG, et al. High-throughput genome sequencing of two Listeria monocytogenes clinical isolates during a large foodborne outbreak. BMC Genomics 2010; 11:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mellmann A, Harmsen D, Cummings CA, et al. Prospective genomic characterization of the German enterohemorrhagic Escherichia coli O104:H4 outbreak by rapid next generation sequencing technology. PLoS One 2011; 6:e22751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Underwood AP, Dallman T, Thomson NR, et al. Public health value of next-generation DNA sequencing of enterohemorrhagic Escherichia coli isolates from an outbreak. J Clin Microbiol 2013; 51:232–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McDonnell J, Dallman T, Atkin S, et al. Retrospective analysis of whole genome sequencing compared to prospective typing data in further informing the epidemiological investigation of an outbreak of Shigella sonnei in the UK. Epidemiol Infect 2013; 141:2568–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Allard MW, Luo Y, Strain E, et al. High resolution clustering of Salmonella enterica serovar Montevideo strains using a next-generation sequencing approach. BMC Genomics 2012; 13:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Willshaw GA, Smith HR, Cheasty T, Wall PG, Rowe B. Vero cytotoxin-producing Escherichia coli O157 outbreaks in England and Wales, 1995: phenotypic methods and genotypic subtyping. Emerg Infect Dis 1997; 3:561–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayashi T, Makino K, Ohnishi M, et al. Complete genome sequence of enterohemorrhagic Escherichia coli O157:H7 and genomic comparison with a laboratory strain K-12. DNA Res 2001; 8:11–22. [DOI] [PubMed] [Google Scholar]
- 22.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010; 26:589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics 2009; 25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010; 20:1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Price MN, Dehal PS, Arkin AP. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One 2010; 5:e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ihekweazu C, Carroll K, Adak B, et al. Large outbreak of verocytotoxin-producing Escherichia coli O157 infection in visitors to a petting farm in South East England, 2009. Epidemiol Infect 2012; 140:1400–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mellor GE, Sim EM, Barlow RS, et al. Phylogenetically related Argentinean and Australian Escherichia coli O157 isolates are distinguished by virulence clades and alternative Shiga toxin 1 and 2 prophages. Appl Environ Microbiol 2012; 78:4724–31. [DOI] [PMC free article] [PubMed] [Google Scholar]