

Abstract
Luminescent Identification of Functional Elements in 3’UTRs (3’LIFE) allows the rapid identification of targets of specific miRNAs within an array of hundreds of queried 3’UTRs. Target identification is based on the dual-luciferase assay, which detects binding at the mRNA level by measuring translational output, giving a functional readout of miRNA targeting. 3’LIFE uses non-proprietary buffers and reagents, and publically available reporter libraries, making genome-wide screens feasible and cost-effective. 3’LIFE can be performed either in a standard lab setting or scaled up using liquid handling robots and other high-throughput instrumentation. We illustrate the approach using a dataset of human 3’UTRs cloned in 96-well plates, and two test miRNAs, let-7c and miR-10b. We demonstrate how to perform DNA preparation, transfection, cell culture and luciferase assays in 96-well format, and provide tools for data analysis. In conclusion 3'LIFE is highly reproducible, rapid, systematic, and identifies high confidence targets.
Keywords: Molecular Biology, Issue 99, microRNA, luciferase assay, 3' untranslated region, high-throughput, transfection, post-transcriptional gene regulation, cancer
Introduction
The overall goal of this method is to detect and precisely map microRNA (miRNA) targets in high-throughput. MiRNAs are endogenous non-coding RNAs ~22 nucleotides in length. Following transcription and processing, mature miRNAs are incorporated in a protein complex called the RNA induced silencing complex (RISC). Each miRNA guides the RISC to target elements located primarily in the 3’untranslated regions (3’UTRs) of messenger RNAs (mRNAs), resulting in either translation repression or mRNA cleavage 1. MiRNA recognize target sites based on standard Watson-Crick and G:U wobble base pairing, and are degenerate in nature, containing multiple mismatched base pairs and bulged regions. Many miRNAs are broadly conserved from plants to humans 2,3, where they play a diverse range of biological roles. In metazoans miRNAs can influence multiple biological processes including cell fate decisions 4, developmental timing 5, and frequently exhibit tissue specific expression patterns 6,7. MiRNA misexpression can also result in aberrant gene regulation, which can have substantial influence on cell behaviour based solely on the function of target genes. As such, miRNAs are linked to a wide range of diseases, including neurodegeneration 8,9, diabetes 10 and cancer 11. Bioinformatic and wet-bench approaches suggest that each miRNA may be capable of targeting hundreds to thousands of distinct mRNAs 12-14, indicating that high-throughput or genome wide approaches are required to probe this large pool of potential interactions.
Identifying target genes is a critical component of mechanistically defining miRNA function, and to do so researchers must be able to reveal targets on a large scale. Several approaches have been developed to identify miRNA targets, including bioinformatic prediction algorithms, high-throughput sequencing of targeted mRNAs, and reporter based assays. Each of these approaches has inherent strengths and weaknesses. Given that miRNA targeting is guided by sequence specificity, most notably of nucleotides 2-6 of the miRNA (termed the seed region), several algorithms have been developed to predict miRNA targets throughout the genome of many organisms. These algorithms are trained using the observed base-pairing motifs of validated miRNA targets, and frequently utilize parameters such as stringent seed pairing, site conservation, and/or thermodynamic stability 15. While these filters refine the large number of putative targets with sufficient complementarity to only high confidence targets, they may exclude species specific and non-canonical miRNA target sites, which recent evidence suggests are widespread 16-24. Furthermore, these predictions do not take into account mechanisms of mRNA processing that exclude miRNA target sites, such as alternative polyadenylation 25, RNA editing 26, RNA methylation 27, and cooperative binding. As such, high false positive and false negative rates have been reported for many algorithms 22,24,28. While these algorithms are useful to identify candidate miRNA targets for subsequent experimental validation, these high error rates limit the efficacy of bioinformatic approaches for systematic miRNA target detection.
To systematically probe for interactions between a given miRNA and potentially targeted 3’UTRs we have developed a high-throughput assay called Luminescent Identification of Functional Elements in 3’UTRs (3’LIFE) 24. This assay measures direct interactions and translational repression of the test 3’UTR by a query miRNA using a dual luciferase reporter system. In this system, the 3’UTR of a gene of interest is cloned downstream of the firefly luciferase (fluc) reporter reading frame. The reporter construct is cotransfected with a query miRNA in HEK293T cells. MiRNA targeting is determined by measuring the relative change between the test fluc::3’UTR reporter and a second non-specific Renilla luciferase reporter. Importantly, luciferase assays detect functional miRNA/mRNA interactions that influence the translational output of the reporter. This is a key advantage over traditional methods to detect miRNA regulation, such as RT-qPCR and Western blots, in that this bypasses differences in mRNA degradation and translational repression, as well as changes in protein abundance independent of 3’UTR based regulation.
Luciferase assays are widely utilized to validate direct miRNA targets because of their relative simplicity and sensitivity, yet their use in high-throughput screens is limited by high costs associated with consumable reagents, the lack of 3’UTR libraries from public sources, and the absence of standardized luciferase protocols, leading to difficulties in comparing functional repression across multiple datasets. To facilitate the use of the 3’LIFE assay, we have placed emphasis on simplification of experimental design, utilization of non-commercial transfection 24 and luciferase reagents 29, creating a 3’UTR library which is regularly updated and expanded, and is available through a public plasmid repository 30.
The scalability of the 3’LIFE assay allows screening of a large 3’UTR library for targeting by a given miRNA without biasing the screen towards bioinformatically identified genes. In addition to testing canonical and predicted interactions, this systematic approach allows the identification of novel targets driven via non-canonical and/or species-specific interactions. Importantly, the effect of miRNA targeting on protein production is generally understood to result in modest translational repression 15,31, suggesting that a primary role of miRNA regulation is to fine-tune protein output, protect against aberrant levels of gene expression, and provide robustness to cell specific programs 32,33. The sensitivity of the luciferase assay combined with the inherently large number of negative miRNA/mRNA interactions in the 3’LIFE screen allows the detection of subtle effects of miRNA targeting on a large number of genes, and the identification of multiple components of gene networks that are regulated by a given miRNA 24.
Here we describe the 3'LIFE protocol, and demonstrate it’s feasibility by screening two well characterized miRNAs, miR-10b and let-7c against a panel of 275 human 3'UTRs (Figure 1).
Protocol
1. Cell Culture (24-48 hr prior to transfection)
24-48 hr prior to transfection seed a sufficient quantity of HEK293T cells based on the number of 96-well plates being transfected. NOTE: For consistent transfections, plate cells at a sufficient density to favor rapid division, yet not be at more than 70-90% confluency at the time of transfection.
Each 96-well plate requires 9 x 106 cells (75,000 cells per well, and 120 wells per plate to account for use of reservoir and multichannel pipette). Calculate the doubling time of HEK293 cells (typically ~20 hr), and seed the appropriate number of cells to obtain at least 9 x 106 cells at the time of transfection. A 145 mm circular culture plate is typically sufficient for 3 96-well transfections when grown to ~90% confluency, with ~10% of cells remaining to reseed a new plate.
2. Preparation Prior to Transfection
NOTE: The preparation of the buffers and plasmid DNA in step 2.0-2.2 should be performed in the days prior to transfection since the preparation of these reagents may be time consuming.
Human 3'UTR clones that are compatible with the 3'LIFE assay are available through a public plasmid repository 30. Purify DNA plasmid manually or with liquid handling robots. Use a transfection grade alkaline lysis mini-prep 96-well kit and follow the manufacturer’s instructions. Resuspend the purified vectors to ~100 ng/μl per well. NOTE: Insufficient luciferase signal will result if plasmid concentration falls below 40 ng/μl.
Obtain the pLIFE-miRNA vectors 30 or clone using the pipeline in Figure 1B 24. Resuspend the vectors at a concentration of 500 ng/μl for each miRNA and Blank control plasmids.
Owing to the sensitivity of the nucleofection buffer conditions, ensure that the total volume of transfected materials (including cells and plasmids) does not exceed 10% of the total liquid in each well of the 96-well transfection plate. To achieve this, concentrate the pLIFE-miRNA plasmid stock at a concentration of least 500 ng/μl.
Prepare 10x firefly luciferase buffer reagents (Table 1), and the 1x Renilla luciferase buffer reagents (Table 2), which can be stored for up to 6 months. NOTE: The DTT in the firefly luciferase buffer must to be stored in solution at -20 oC in single use aliquots.
3. Prepare Following Items Immediately Prior to Transfection
Prepare transfection buffer containing PBS, 1.5% HEPES, pH 7.0. Prepare this fresh, although it may be stored for up to 1 month at 4 °C without noticeable decreases in transfection efficiency. In formulating buffer and plasmid DNA volumes, assume 120 reactions for each 96-well plate to sufficiently account for errors in pipetting and volume lost using liquid reservoirs and multichannel pipettes. Aliquot 18 μl per well transfection buffer (120 wells/96-well plate = 2.16 ml per plate), and set aside. NOTE: The cell-electroporation device is extremely sensitive to the buffer conditions used to transfect cells. Accuracy when preparing buffers will ensure consistent performance of the equipment. Extra care must be taken when performing the assay to prevent evaporation of buffers, specifically by minimizing the time that buffer is left uncovered in microcentrifuge tubes, 96-well plates, reservoirs, and electrode plates.
Reserve four wells for the following controls, no pLIFE-3’UTR (to measure background of luciferase assay), pLIFE-SV40 3’UTR (negative target control), positive control for miRNA #1, positive control for miRNA #2. NOTE: These vectors are publically available 30. Alternatively, any previously validated target can be used as a positive control.
Warm media, trypsin (0.25%) to 37 °C.
To each well of 96-well cell culture plate, add 200 μl of DMEM supplemented with 10% FBS, 1% Pen/Strep, and placed in a 37 °C incubator for use following transfection.
Turn on all cell electroporation devices followed by the supporting software. Use Pulse code FF120 for HEK293T cells and PBS/HEPES buffer.
4. Preparation of Plasmid DNA and Cell Mixture
NOTE: The following protocol assumes transfecting three 96-well plates in one experiment for a screen with two miRNAs (miRNA-#1 and miRNA-#2). Each plate will correspond to the same 96-well plate of pLIFE-3’UTR plasmids, and be treated three times with pLIFE-miRNA-blank, pLIFE-miRNA-#1, or pLIFE-miRNA-#2.
Prepare 3 stocks of pLIFE-miRNA + transfection buffer for each miRNA. This stock should account for 50% (10 μl) of total volume of each well, multiplied by 120 wells. Thus, each stock should contain 1.08 ml buffer + 120 μl plasmid DNA (pLIFE-miRNA).
Remove cells from 145 mm culture plate by eluting media, washing gently with PBS, and treating with ~5 ml 0.25% trypsin for 5 min at 37 °C. Neutralize trypsin with an equal volume of media, and pellet cells at 300 x g for 5 min.
Remove trypsin/media, and resuspend pellet in ~5-10 ml media (depending on cell density and accurate range of cell counter).
Count cells using a cell counter. Ensure that cells are >95% viable and within accurate range of machine. NOTE: An inaccurate cell count can result from extremely high cell concentrations (>6.0x 106/ml). Transfecting too many cells can drastically reduce efficiency of miRNA targeting by reducing plasmid:cell ratio and/or decreasing transfection efficiency.
Aliquot three tubes each containing 9 x 106 cells, corresponding to the cells required for transfection of one 96-well plate. Spin cells at 300 x g for 3 min.
Remove media. Be sure to remove as much media as possible with minimal disturbance of the pellet as excess media can impact transfection efficiency.
Resuspend cells in 1.2 ml transfection buffer/miRNA plasmid mixture, and set aside.
- The following steps detail resuspension of pLIFE-3’UTR plasmid in transfection buffer. As this occurs in 96 well plates, take care to avoid evaporation of buffer by covering plates at all times.
- Using a multichannel pipette, move 32.4 μl transfection buffer into each well of a 96 well PCR plate (9 μl [per transfection] * 3 [plates] * 1.2 [to account for pipette error]).
- Add 3.6 μl (~100 ng/μl) of mini-prepped pLIFE-3’UTR plasmid to each well and mix thoroughly.
- Pipette 10 μl of this mixture into each well of the 96-well transfection plate and cover.
5. Transfection
Move 1.2 ml of the first cell/buffer/pLIFE-miRNA plasmid mixture into reservoir. Mix well.
Add 10 μl of this mixture into the first 96-well transfection plate already containing 10 μl transfection buffer/pLIFE-3’UTR. Mix well by pipetting up and down several times. NOTE: Equal suspension of cells in the buffer will ensure even and thorough passage of the electrical current through the cuvette and maximize transfection efficiency.
Place 96-well transfection plate on the cell electroporation device and initiate transfection.
Once transfection is complete, add 100 μl of pre-warmed media from 96-well culture plate to each well of the 96-well transfection plate and mix well. Move 100 μl from each well into the 96-well culture plate.
Mix cells in culture plate with pipette positioned vertically in the center of the well, as cells will tend to aggregate on the sides of the well unless mixed properly.
Repeat 5.1-5.5 for the remaining two plates.
- Cleaning 96-well transfection plate
- The 96-well transfection plates can be recycled by washing with 70% EtOH to ensure no carry over of nucleic acids between experiments. Perform two 70% EtOH washes using a spray bottle to completely fill each well, followed by wiping down excess EtOH on the electrode strips (bottom side) and allowing the transfection plates to completely dry in the culture hood. NOTE: We have tested for carry-over DNA contamination by transfecting 12 wells with 2 μg pmaxGFP plasmid each into HEK293T cells, followed by a single wash with 70% EtOH, and a second transfection with no plasmid DNA. With this extremely high plasmid concentration, extremely bright reporter, and a single wash, there was no observable fluorescence in any of the 12 replicate transfections.
Culture cells for 48-72 hr at 37 °C, followed by the dual luciferase assay.
6. Cell Lysate Preparation for Luciferase Assay
Dilute lysis buffer with 4 parts water, 1 part 5x passive lysis buffer in a reservoir. Calculate 26 μl/well, adding ~20 extra volumes to account for loss in the reservoir. NOTE: Buffer is stored at -20 ºC and can be extremely viscous, thus prior to allowing the 5x buffer to approach room temperature will improve pipetting accuracy.
Analyze each well for transfection efficiency using fluorescence microscopy. Note any inconsistencies in wells that did not transfect efficiently (>90% transfection efficiency), or are expressing low levels of RFP indicative of overcrowding or media exhaustion. Remove these wells from the analysis.
Completely remove the media from the cells, being careful not to elute too quickly which will cause cells to detach. NOTE: Remaining media will dilute the lysate and cause fluctuations in values across experiments.
Add 26 μl of lysis buffer to each well, and place on a plate shaker/rocker at low/moderate speed for ~20-30 min. Use this time to prepare luciferase buffers, wash and prime the luminometer(s), and transfer lysate to opaque measurement plates.
7. Dual Luciferase Assay
NOTE: If multiple plates are being measured sequentially on one luminometer, create buffer master mixes with everything except ATP and substrates, adding these reagents followed by pH adjustment immediately before use with each plate. ATP and substrates may degrade over time; consistency in the amount of time these reagents are in the buffer will improve consistency across multiple plates.
- Prepare 1x luciferase buffers (Table 1):
- Wrap two tubes (typically 15/50 ml centrifuge tubes) containing the firefly and Renilla buffers with aluminum foil, as substrates may be light sensitive.
- Prepare 1x Firefly luciferase buffer. Add 1 ml of each the five 10x firefly luciferase reagents, adding EGTA last, to 5 ml H20 to a final 1x concentration.
- Add 0.025 g ATP to 10 ml 1x firefly buffer. Mix by inverting several times. Keep ATP on ice at all times. ATP will degrade, so if measuring more than one plate sequentially, buffer must be made fresh beginning at this step for each additional plate.
- Add 100 μl of 100 beetle luciferin (substrate) (Table 1) Buffer should change to yellowish color based on pH.
- 1x Renilla luciferase buffer reconstitution: Per 96-well plate, aliquot 10 ml of 1 “Renilla buffer”.
- Add 100 μl of BSA (44 mg/ml stock).
- If screening more than one plate, separate master mix into 10 ml aliquots.
- Add 100 μl of coelenterazine to buffer (previously aliquoted and stored at 100x conc.)
- Adjust pH of 1x firefly buffer to 8.0, followed by the 1x Renilla buffer to 5.0 using NaOH and HCl. NOTE: The activity of each buffer, and the ability of the Renilla buffer to quench the Firefly luciferase activity is highly dependent on pH. For consistent results be extremely accurate in this step.
- Bring volume of each buffer (corresponding to 1 96-well plate) to 10.5 ml to accommodate for luminometer priming.
Transfer lysate to opaque white plates: At this point the cells should be in lysis buffer for ~20 min. Take 25 μl from each well using a multichannel pipette, be sure to pipet up and down thoroughly to break up the clumps of cells and homogenize the lysate.
- Prepare the luminometer. Turn on the luminometer and select the protocol in the DLR folder, called “DLR with two injections”. Other formats are not compatible with the data analysis pipeline (below).
- Select the wells to be tested (all wells is the default).
- Extend the 'Delay before measurement' setting to 5 seconds, with a 10 sec measurement time (see 29 for explanation).
- Capillary wash steps: water 3x, EtOH 3x, water 3x, dry 3x. Prime buffers once into the waste, and then prime a second time back into the buffer tubes to ensure mixing. Inject the firefly buffer first and prime it in the left capillary, followed by Renilla in the right capillary.
- Initiate the luciferase assay. Each plate should take ~48 min to read. After completion save the file first, and then repeat the wash steps and shut off the luminometer. NOTE: Multiple plates can be read and data stored on the same excel file, however issues may be encountered with multiple plate reads where the luminometer program will crash. Be sure to save all data between measurements and take screenshots if program crashes before save is possible.
- Replace old buffers with new, being sure to prime at least twice with new buffers before starting the new plate.
8. Data Analysis
Utilize excel tables “3’LIFE - single plate analysis” and “3’LIFE – multiplate analysis” available from www.mangonelab.com. Copy raw data for firefly and Renilla luciferase measurements from luminometer output file into locations corresponding to the negative condition, miRNA #1, and miRNA #2 into the “3’LIFE – single plate analysis” spreadsheet.
- The spreadsheet will automatically calculate firefly/Renilla ratio and normalize each miRNA to the appropriate negative control, and normalize repression values across each plate. NOTE: This spreadsheet will automatically identify wells with low luciferase signal, highlight significantly repressed wells, and provide measures of repression across the entire plate. See 24 for detailed explanation of statistical analysis.
- Copy the values from the “Normalized RI Index” box, into corresponding cells in the “3’LIFE – multiplate analysis” spreadsheet at the position corresponding to replicate #1 for each miRNA tested. Repeat for all biological replicates performed on different days.
- If desired, insert the gene names and target prediction status in columns B and D, respectively.
- Allow the spreadsheet to automatically calculate the average of all plate. Observe the data in 96 well format as a heat map (Figure 2). The file also arranges the data in list format (Figure 2). NOTE: The repression index is the measure used to identify putative miRNA targets. Different stringency parameters can be used, based on individual preferences. On average we consider likely hits below repression index of 0.8 and statistically significant by t test (p-value <0.05). As shown in Figure 3 potential targets based on these criteria will be highlighted in red.
Representative Results
The luminometer output file contains raw measurements for both firefly and Renilla luciferase proteins. This raw format is compatible with the “3’LIFE – single plate analysis” and “3’LIFE – multiplate analysis” spreadsheets available from the Mangone lab website (www.mangonelab.com). The single plate analysis spreadsheet automatically calculates firefly/Renilla ratio, normalizes each miRNA to the appropriate negative control, and normalizes repression values across each plate. This spreadsheet automatically identifies wells with low Renilla luciferase signal, highlights wells that exhibit repression compared to the negative control, and provides measures of repression across the entire plate (Figure 2). See 24 for detailed explanation of statistical analysis.
Multiple replicates can be analysed using the “multiplate analysis” spreadsheet. Each replicate is compared side by side, and statistical measures of the data are automatically calculated (Figure 3). In addition to comparing replicates with the “Normalized Repression” columns, the user can compare repression between the two miRNAs under the “miRNA#1/miRNA#2” columns. This measure divides the repression index for each miRNA for each replicate. This measure can indicate erroneous values from the luciferase assay (for example abnormally high or low readings with the negative control, see Figure 3, row A9, Rep #3), and wells where the repression index may not indicate substantial repression, but that do exhibit significant differences between the miRNAs. While this measure may not be used directly to indicate a miRNA target, it is useful for identifying outliers, problematic wells, or patterns in the data that are not solely attributed to direct miRNA regulation.
The Repression Index (RI) is used to call a putative miRNA target, with lower values corresponding to higher relative repression. The threshold for calling putative targets is based on the level of stringency required by the researcher, but combining the RI with 3’UTRs that display statistically significant p-values (p <0.05) will indicate high confidence targets (see Figure 2 Rows B8 and B12).
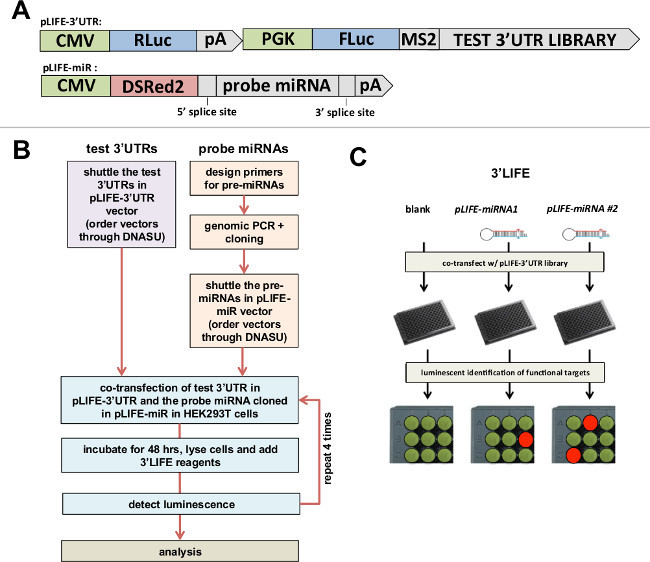 Figure 1: 3’LIFE Assay. (A)Gateway-compatible vectors used in the 3’LIFE assay. Top: The luciferase gene (FLuc) is fused to the test 3’UTR, while the Renilla luciferase gene (RLuc) is fused to the unspecific SV40 pA 3’UTR as control. Bottom: The RFP- miRNA-intron vector - The probe pre-miRNA, plus ~400 nucleotides within its genomic locus (to recapitulate endogenous miRNA processing), is cloned within an intron to allow its co-expression with DSRed2 fluorochrome. Both vectors are publically available (Seiler et al., 2013). (B) Flow chart of the 3’LIFE Assay. (C) 3’LIFE Pipeline: The dual-luciferase vector containing the test 3’UTR with or without the miRNA vectors are co-transfected into HEK293 cells in 96-well plates. The interaction between the miRNA and a bona fide 3’UTR target will lower the relative luminescence in specific wells (exemplified by the orange spot in the experimental plate).
Figure 1: 3’LIFE Assay. (A)Gateway-compatible vectors used in the 3’LIFE assay. Top: The luciferase gene (FLuc) is fused to the test 3’UTR, while the Renilla luciferase gene (RLuc) is fused to the unspecific SV40 pA 3’UTR as control. Bottom: The RFP- miRNA-intron vector - The probe pre-miRNA, plus ~400 nucleotides within its genomic locus (to recapitulate endogenous miRNA processing), is cloned within an intron to allow its co-expression with DSRed2 fluorochrome. Both vectors are publically available (Seiler et al., 2013). (B) Flow chart of the 3’LIFE Assay. (C) 3’LIFE Pipeline: The dual-luciferase vector containing the test 3’UTR with or without the miRNA vectors are co-transfected into HEK293 cells in 96-well plates. The interaction between the miRNA and a bona fide 3’UTR target will lower the relative luminescence in specific wells (exemplified by the orange spot in the experimental plate).
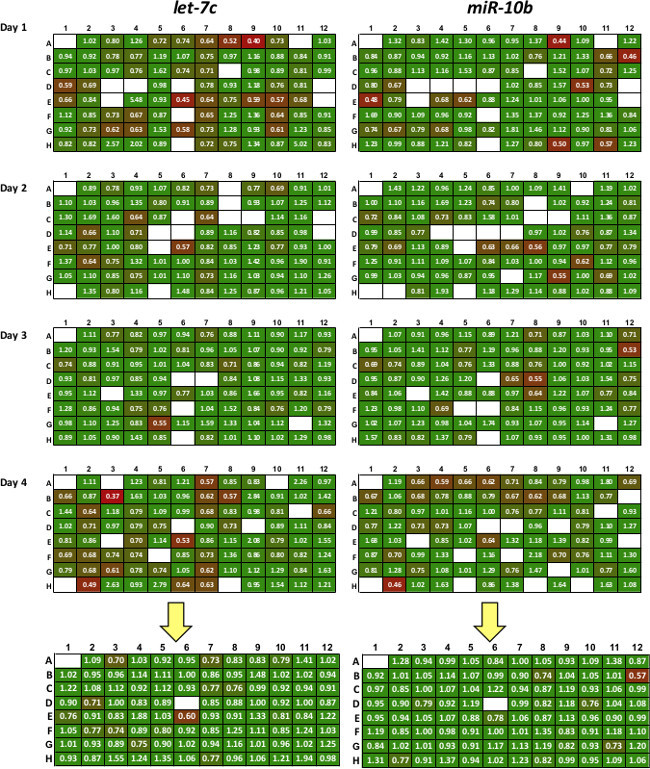 Figure 2: Sample of data produced with 3’LIFE assay. Each probe miRNA is tested in quadruplicate (replicates 1-4). Colors represent repression levels, with red colors indicating strong miRNA/3’UTR interaction. All replicates are averaged to produce high-quality putative targets shown in the summary plate below the yellow arrow. White box represent controls, failed transfections or wells with low transfection efficiency.
Figure 2: Sample of data produced with 3’LIFE assay. Each probe miRNA is tested in quadruplicate (replicates 1-4). Colors represent repression levels, with red colors indicating strong miRNA/3’UTR interaction. All replicates are averaged to produce high-quality putative targets shown in the summary plate below the yellow arrow. White box represent controls, failed transfections or wells with low transfection efficiency.
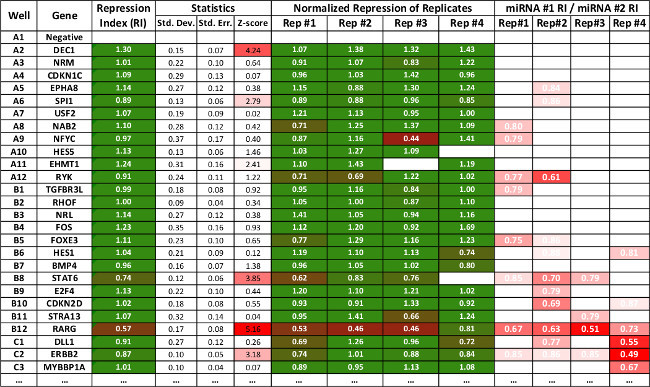 Figure 3: Table representing summary data of a subset of interactions produced using the 3’LIFE spreadsheet. The repression values are as in Figure 2. The software calculates standard deviation, standard error and z-score for each interaction. Statistically significant interactions are marked in red. The last four rows show relative repression of one miRNA to the other, and is used as secondary indicator to compare repression between two different miRNAs. The spreadsheet can be downloaded from www.mangonelab.com
Figure 3: Table representing summary data of a subset of interactions produced using the 3’LIFE spreadsheet. The repression values are as in Figure 2. The software calculates standard deviation, standard error and z-score for each interaction. Statistically significant interactions are marked in red. The last four rows show relative repression of one miRNA to the other, and is used as secondary indicator to compare repression between two different miRNAs. The spreadsheet can be downloaded from www.mangonelab.com
| Firefly luciferase buffer reagents | Final concentration (1x) |
| Glycylglycine | 25 mM |
| KxPO4 (pH 7.8) | 15 mM |
| MgSO4 | 15 mM |
| DTT (store at 4º) | 1 mM |
| EGTA | 4 mM |
| ATP* | 2 mM |
| Beetle luciferin* | 250 μM |
Table 1: Stock firefly luciferase reagents: 10x Stock solutions of Glycylglycine, KxPO4, MgSO4, DTT and EGTA can be prepared separately and stored prior to buffer reconstitution. 100x Beetle Luciferin (firefly luciferase substrate) can be stored by dissolving 50 mg luciferin in 7.134 ml H20 (25 mM). Aliquot 105 μl/plate of dissolved Beetle luciferin into tubes and store at -80 ºC. Per Promega technical support, this should be stable for >6 months, but may be light sensitive. NOTE: EGTA will not go into solution at neutral pH. Slowly add NaOH to EGTA until it dissolves completely.*Reagents added to final buffer immediately prior to the luciferase assay
| Renilla luciferase buffer reagents | Final concentration (1x) |
| NaCl | 1.1 M |
| Na2EDTA | 2.2 mM |
| KH2PO4 | .22 M |
| NaN3 | 1.3 mM |
| BSA* | .44 mg/ml |
| Coelenterazine* | 2.5 μM |
Table 2: Stock Renilla luciferase buffer reagents All the reagents except BSA and Coelenterazine can be mixed at a 1x concentration and stored at room temperature. Coelenterazine can be dissolved in acidified methanol and aliquoted per plate. Acidify methanol by adding HCl to final concentration of 5 mM (<3 pH). Dissolve 250 μg coelenterazine in 2.36 ml acidified methanol (250 μM) aliquot 105 ul/plate. The mix is stable for at least 6 months but may be light sensitive. *Reagents added to buffer immediately before luciferase assay.
Discussion
The 3’LIFE assay identifies functional miRNA targets in 3’UTRs in high-throughput. This assay is useful for researchers who wish to experimentally identify a large number of putative targets for their miRNA of interest. The 3’LIFE assay is a powerful approach to query for 3’UTR driven regulation, in that the assay provides a functional measure of miRNA targeting, and the binary testing of a single reporter::3’UTR against a single miRNA can confidently address the targeting status of individual genes. To validate this approach, we screened a panel of 275 3’UTRs and against two miRNAs, let-7c and miR-10b, and included 10 previously validated target genes in this library. Eight of these ten genes exhibited repression 24. We also observed a significant enrichment of unvalidated bioinformatically predicted targets, and unpredicted 3’UTRs that contain canonical seed elements among our top hits, suggesting that 3’LIFE is capable of identifying bona fide miRNA targets.
A key indicator of the sensitivity of high-throughput screens is the false positive and false negative rates. While the false positive rate of this assay needs to be evaluated using additional alternative approaches to validate hits, eight of the ten positive controls included in our proof-of-principle screen exhibited repression, suggesting a false negative rate of 20%. However, many techniques are used to identify miRNA targets in different cellular contexts, and 3’UTR processing and regulation by trans-acting factors is known to be highly tissue specific. For example, the majority of 3’UTRs contain multiple polyadenylation sites, which control the length of the 3’UTR in the mature mRNA. In many cases the use of proximal polyadenylation sites is tissue specific, and may exclude miRNA target sites. Additionally, cooperative miRNA targeting, competition with RNA-binding proteins, and mRNA secondary structure may all impact the ability of 3’LIFE to detect miRNA targets in specific tissues. Because of this, the detection of targets by the 3’LIFE assay may vary based on the cellular context in which the assay is performed, complicating the evaluation of absolute error rates. This protocol is optimized for HEK293T cells, but alternative cell lines can be used if the researcher wishes to perform the assay in a specific biological context. However, the optimization of transfection efficiency and cell survival with each cell line will have to be optimized using multiple buffer conditions, pulse codes, and number of cells. An example of an optimization scheme can be found at Wolter et al. 24.
This protocol has been optimized in 96-well format and specifies the use of certain high-throughput instrumentation. In the case that the institution does not possess the equipment required for 96-well Nucleofection, alternative transfection reagents could be used to perform the 3’LIFE assay, as long as the transfection efficiency remains high. Additionally, the luciferase assay is the most time consuming aspect of the 3’LIFE assay. As such, the use of multiple luminometers is recommended for high-throughput screens.
Disclosures
This work is supported by funds from the College of Liberal Arts and Science and the Biodesign Institute at Arizona State University and NIH Exploratory/Developmental Research Grant 1R21CA179144-01A1.
Acknowledgments
We thank Stephen Blazie, Karen Anderson, Josh LaBaer for advice and discussion. Karen Anderson, John Chaput, and Josh LaBaer for sharing reagents and instrumentation, Michael Gaskin and Andrea Throop for technical advise and protocols. Justin Wolter is a Maher scholar and thanks the Maher family for their generous support.
References
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Pasquinelli AE, et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature. 2000;408(6808):86–89. doi: 10.1038/35040556. [DOI] [PubMed] [Google Scholar]
- Reinhart BJ, Weinstein EG, Rhoades MW, Bartel B, Bartel DP. MicroRNAs in plants. Genes Dev. 2002;16(13):1616–1626. doi: 10.1101/gad.1004402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivey KN, et al. MicroRNA regulation of cell lineages in mouse and human embryonic stem cells. Cell Stem Cell. 2008;2(3):219–229. doi: 10.1016/j.stem.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75(5):843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- Wienholds E, et al. MicroRNA expression in zebrafish embryonic development. Science. 2005;309(5732):310–311. doi: 10.1126/science.1114519. [DOI] [PubMed] [Google Scholar]
- Darnell DK, et al. MicroRNA expression during chick embryo development. Dev Dyn. 2006;235(11):3156–3165. doi: 10.1002/dvdy.20956. [DOI] [PubMed] [Google Scholar]
- Jin P, Alisch RS, Warren ST. RNA and microRNAs in fragile X mental retardation. Nat Cell Biol. 2004;6(11):1048–1053. doi: 10.1038/ncb1104-1048. [DOI] [PubMed] [Google Scholar]
- Kim J, et al. A MicroRNA feedback circuit in midbrain dopamine neurons. Science. 2007;317(5842):1220–1224. doi: 10.1126/science.1140481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poy MN, et al. A pancreatic islet-specific microRNA regulates insulin secretion. Nature. 2004;432(7014):226–230. doi: 10.1038/nature03076. [DOI] [PubMed] [Google Scholar]
- Calin GA, Croce CM. MicroRNA signatures in human cancers. Nature Reviews Cancer. 2006;6(11):857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19(1):92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paraskevopoulou MD, et al. DIANA-microT web server v5.0: service integration into miRNA functional analysis workflows. Nucleic Acids Res. 2013;41(Web Server issue):W169–W173. doi: 10.1093/nar/gkt393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krek A, et al. Combinatorial microRNA target predictions. Nat Genet. 2005;37(5):495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vella MC, Choi EY, Lin SY, Reinert K, Slack FJ. The C. elegans microRNA let-7 binds to imperfect let-7 complementary sites from the lin-41 3'UTR. Genes Dev. 2004;18(2):132–137. doi: 10.1101/gad.1165404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal A, et al. miR-24 Inhibits cell proliferation by targeting E2F2, MYC, and other cell-cycle genes via binding to 'seedless' 3'UTR microRNA recognition elements. Molecular Cell. 2009;35(5):610–625. doi: 10.1016/j.molcel.2009.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cevec M, Thibaudeau C, Plavec JNMR. NMR structure of the let-7 miRNA interacting with the site LCS1 of lin-41 mRNA from Caenorhabditis elegans. Nucleic Acids Res. 1093;38(21):7814–7821. doi: 10.1093/nar/gkq640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin C, et al. Expanding the microRNA targeting code: functional sites with centered pairing. Molecular Cell. 2010;38(6):789–802. doi: 10.1016/j.molcel.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzouzi I, et al. MicroRNA-96 directly inhibits gamma-globin expression in human erythropoiesis. PLoS One. 2011;6(7):e22838. doi: 10.1371/journal.pone.0022838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, et al. miR-193b Regulates Mcl-1 in Melanoma. Am J Pathol. 2011;179(5):2162–2168. doi: 10.1016/j.ajpath.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi SW, Hannon GJ, Darnell RB. An alternative mode of microRNA target recognition. Nat Struct Mol Biol. 1038;19(3):321–327. doi: 10.1038/nsmb.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao LR, et al. MicroRNAs targeting oncogenes are down-regulated in pancreatic malignant transformation from benign tumors. PLoS One. 2012;7(2):e32068. doi: 10.1371/journal.pone.0032068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolter JM, Kotagama K, Pierre-Bez AC, Firago M, Mangone M. 3'LIFE: a functional assay to detect miRNA targets in high-throughput. Nucleic Acids Res. 2014. [DOI] [PMC free article] [PubMed]
- Mangone M, et al. The landscape of C. elegans 3'UTRs. Science. 2010;329(5990):432–435. doi: 10.1126/science.1191244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekdahl Y, Farahani HS, Behm M, Lagergren J, Ohman M. A-to-I editing of microRNAs in the mammalian brain increases during development. Genome Res. 2012;22(8):1477–1487. doi: 10.1101/gr.131912.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Dominissini D, Rechavi G, He C. Gene expression regulation mediated through reversible m(6)A RNA methylation. Nat Rev Genet. 2014;15(5):293–306. doi: 10.1038/nrg3724. [DOI] [PubMed] [Google Scholar]
- Zhou P, et al. Large-scale screens of miRNA-mRNA interactions unveiled that the 3'UTR of a gene is targeted by multiple miRNAs. Plos One. 2013;8(7):e68204. doi: 10.1371/journal.pone.0068204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer BW, Ferrer FA, Klinedinst DK, Rodriguez R. A noncommercial dual luciferase enzyme assay system for reporter gene analysis. Anal Biochem. 2000;282(1):158–161. doi: 10.1006/abio.2000.4605. [DOI] [PubMed] [Google Scholar]
- Seiler CY, et al. DNASU plasmid and PSI:Biology-Materials repositories: resources to accelerate biological research. Nucleic Acids Res. 2013. [DOI] [PMC free article] [PubMed]
- Baek D, et al. The impact of microRNAs on protein output. Nature. 2008;455(7209):64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert MS, Sharp PA. Roles for MicroRNAs in Conferring Robustness to Biological Processes. Cell. 2012;149(3):515–524. doi: 10.1016/j.cell.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelaez N, Carthew RW. Biological robustness and the role of microRNAs: a network perspective. Current Topics in Developmental Biology. 2012;99:237–255. doi: 10.1016/B978-0-12-387038-4.00009-4. [DOI] [PMC free article] [PubMed] [Google Scholar]