

Abstract
Listeria monocytogenes is a Gram-positive facultative intracellular pathogen that is capable of causing serious invasive infections in immunocompromised patients, the elderly, and pregnant women. The most common manifestations of listeriosis in humans include meningitis, encephalitis, and fetal abortion. A significant but much less documented sequelae of invasive L. monocytogenes infection involves the heart. The death rate from cardiac illness can be up to 35% despite treatment, however very little is known regarding L. monocytogenes colonization of cardiac tissue and its resultant pathologies. In addition, it has recently become apparent that subpopulations of L. monocytogenes have an enhanced capacity to invade and grow within cardiac tissue. This protocol describes in detail in vitro and in vivo methods that can be used for assessing cardiotropism of L. monocytogenes isolates. Methods are presented for the infection of H9c2 rat cardiac myoblasts in tissue culture as well as for the determination of bacterial colonization of the hearts of infected mice. These methods are useful not only for identifying strains with the potential to colonize cardiac tissue in infected animals, but may also facilitate the identification of bacterial gene products that serve to enhance cardiac cell invasion and/or drive changes in heart pathology. These methods also provide for the direct comparison of cardiotropism between multiple L. monocytogenes strains.
Keywords: Infection, Issue 99, Bacterial pathogenesis, intracellular pathogen, tissue tropism, bacterial invasion, cardiac infection, listeriosis
Introduction
Listeria monocytogenes is a Gram-positive intracellular pathogen capable of causing severe disease in susceptible populations, including the elderly, pregnant women, people with HIV/AIDS, and persons receiving chemotherapy1. Infection in these populations is frequently the result of ingesting contaminated food products, and most infections are associated with large-scale food-borne outbreaks2,3. In humans and other mammals, L. monocytogenes is capable of translocating across the epithelial border of the small intestine, thereafter being transported to the liver4,5. Animal models suggest that ingested bacteria replicate within the intestinal villi and transit to the liver through the portal vein or spread via the mesenteric lymph nodes into the blood stream, leading to hematogenous dissemination to the liver and spleen6,7. In the liver and spleen, the bacterium is capable of mediating uptake into both professional phagocytes as well as resident parenchymal cells, and quickly establishes infections within these organs. As bacterial load increases, numerous bacteria are dispersed back into the blood, where they are capable of further colonizing susceptible tissues including the central nervous system and placenta (where present). Colonization of these sites precludes most common manifestations of listeriosis in humans, including meningitis, encephalitis, and fetal abortion2.
Selected subpopulations of L. monocytogenes have been recently shown to have an enhanced capacity to invade and replicate within cardiac tissue8. Manifestations of heart involvement are varied, and range from endocarditis and pericarditis, to fulminant myocarditis complete with conduction abnormalities9-13. The overall number of L. monocytogenes cardiac cases per year is low but may be under estimated as this facet of infection is not generally well recognized. Colonization of the heart by pathogens often requires host predispositions such as pre-existing valvular damage or artificial heart valves. There are, however, isolates of L. monocytogenes that have been identified that are notable for their capacity to colonize the hearts of infected animals in the absence of any cardiac damage and/or abnormalities8.
Herein are described in vitro and in vivo methods for assessing bacterial colonization of cardiac tissue within infected animals using invasion assays in tissue culture as well as live animal infections. These methods have proven useful in not only for identifying strains with the potential to colonize cardiac tissue in infected animals, but should also be useful for the identification of bacterial gene products that serve to enhance cardiac cell invasion and/or result in changes in heart pathology. These methods facilitate the comparison of cardiotropism between multiple strains. For the methods described here, L. monocytogenes 10403S is used as a well-studied representative of a non-cardiotropic strain and the clinical isolate 07PF0776 is used as a representative example of a cardiotropic strain. These two strains were chosen to provide a comparison for bacterial invasion of cardiac cells in vitro and colonization of hearts of infected mice in vivo. The isolate 07PF0776 is a clinical isolate recovered from an interventricular abscess that caused a fatal arrhythmia in an HIV+ patient8. L. monocytogenes isolates may vary in their virulence potential, and given the propensity for Listeria to infect persons with immunosuppression and pregnant women, persons within these populations should exercise caution while assessing different clinical isolates.
Protocol
1. Storage and Culture Conditions for L. monocytogenes Strains
Prepare solid media by autoclaving brain-heart infusion agar (BHI) and pouring the molten media into petri plates. Allow the plates to dry overnight at room temperature, then overnight again at 37 °C.
Using a sterile loop, obtain a small sample of L. monocytogenes from either previously made freezer stocks (bacteria suspended in 20% glycerol in BHI liquid media, stored at -80 °C) or from an agar plate containing colonies previously struck for isolation.
Using aseptic technique, streak the sample for single colony isolation. Be sure to sterilize the loop between cross streaks to prevent excess carry-over and ensure the isolation of individual colonies of the strain being assessed. Incubate the plate(s) overnight at 37 °C.
Using a sterile loop, remove one colony of the isolate and inoculate 2 ml of BHI broth (without agar) in a 14 ml polystyrene tube.
For assays and infections, incubate the broth culture statically (without shaking) overnight in a 37 °C incubator. For stocking of cultured isolates, incubate the broth culture at 37 °C with shaking at 180-200 rpm overnight. Cultures can be stocked by adding glycerol to a final concentration of 20% and storing sealed tubes at -80 °C indefinitely.
2. Storage and Culturing Conditions of H9c2 Cardiac Myoblast-like Cells
Purchase or obtain a frozen stock of H9c2 cells, as well as Dubelco’s Modified Eagle’s Medium (DMEM) containing high glucose and pyruvate, Fetal Bovine Serum (FBS), L-glutamine (Glut), and a Penicillin-Streptomycin-Glutamine mixtures (PSG). FBS, Glut, and PSG should be stocked at -20 °C, while DMEM can be stored at 4 °C. It is helpful to aliquot FBS into 50 ml conical tubes prior to freezing.
In a laminar flow hood, thaw two aliquots of FBS. Add one aliquot to one 500 ml bottle of DMEM, making a 10% FBS/DMEM mixture. To this, add 6 ml of Glut and stock the resulting solution at 4 °C. This solution can be labeled “DMEM without Antibiotics.” (DMEM –Ab)
To a second 500 ml bottle of DMEM, add the other 50 ml aliquot of FBS. To this solution, add 6 ml of PSG and stock at 4 °C. This solution can be labeled “DMEM with Antibiotics.” (DMEM +Ab)
While in the laminar hood, remove 10 ml of DMEM +Ab and place in a 25 ml tissue culture flask.
Leaving the flask in the laminar hood, remove a frozen aliquot of H9c2 cells from storage in liquid nitrogen and quickly place the tube in a 37 °C water bath until the culture has thawed.
Spray the tube with 70% ethanol to sanitize, then return to the hood. Working quickly to minimize cell time in concentrated DMSO, add the full aliquot of cells to the 10 ml of DMEM +Ab. This dilutes the DMSO sufficiently for overnight cell viability and adherence.
Place the flask in a tissue-culture incubator at 37 °C, 5% C02, and 95% humidity overnight.
The following morning, check the cell layer to ensure adherence. Cells will likely be between 80-100% confluent by this time.
In the tissue-culture hood, remove the media contained within the flask using the vacuum, leaving only the cells within the flask.
Add approximately 2 ml of 0.25% Trypsin-EDTA to the cells and rock gently for approximately 3 sec.
Invert the flask such that the trypsin solution is no longer in contact with the monolayer, and remove the trypsin by vacuum.
Add a second 2 ml aliquot of trypsin to the cells, and move the flask to an inverted microscope. Observe the cells while gently rocking the flask to ensure the monolayer disperses.
Once the monolayer is dispersed, immediately return to the tissue culture hood and add 4 ml of DMEM +Ab to the cell suspension.
Remove a 2 ml portion of the cell suspension, and add it to a 50 ml tissue culture flask containing 23 ml of DMEM +Ab. Rock the flask gently, then place it in the tissue-culture incubator in the conditions listed in step 2.5.
Check the cells daily until they reach approximately 80% confluence (approximately 2.0-4.0 x 105 cells per ml). It is very important that cells do not become too confluent, as they may differentiate into skeletal muscle myotubes when starved of FBS for prolonged periods of time14 and this differentiation can alter bacterial invasion. Monitor the cells carefully and start with a new tube of cells if a culture becomes confluent.
Once the cells reach 80% confluence, they can either be passaged into another 50 ml flask as just described, or prepared for invasion assay.
3. Preparing H9c2 Cells for Invasion Assay
In the tissue culture hood, remove the media from a 50 ml flask of H9c2 cells that have reached 80% confluence using vacuum aspiration.
Add 4 ml of 0.25% Trypsin-EDTA solution to the monolayer and remove immediately using vacuum aspiration.
Add another 4 ml portion of 0.25% Trypsin-EDTA to the flask and move the flask to an inverted microscope to observe the dispersion of the monolayer.
Once the cells have dispersed, return to the hood and add 4 ml of DMEM –Ab to the cells. Pipetting the solution up and down ensures complete removal of cells from the bottom of the flask.
Following resuspension of the monolayer, remove all of the suspension and place it in a 50 ml conical tube.
Remove a 20 ul portion of this suspension and count the number of cells per milliliter using a hemocytometer.
Once the concentration of cells in the solution is determined, adjust the concentration to 2.25 x 104 cells/ml by adding the appropriate volume of cell solution to fresh DMEM –Ab. The total volume of the adjusted solution should be 25 ml.
While in the hood, sterilize glass coverslips by dipping them in 90% Ethanol and briefly passing each coverslip through a flame. Add each coverslip to an individual well of a 24-well tissue culture treated plate directly after it is sterilized.
After all wells have an individual coverslip, use a 25 ml pipet to add 1 ml of the diluted cell solution to each of the wells in the plate, and push each coverslip down with a sterile needle.
Check each well using an inverted microscope to ensure similar amounts of cells are in each well. Put the 24 well plate in the tissue culture incubator overnight at 37 °C in 5% CO2 and 95% humidity.
The following morning, view each well to ensure that the coverslips have comparable amounts of adherent myoblasts and that the coverslips are not floating in the well.
4. Preparing Strains of L. monocytogenes for Invasion Assays
NOTE: All laboratory work is carried out in accordance with CDC Biosafety Level 2 guidelines.
After preparing the 24-well plate for assay, use a sterilized loop to remove single colonies of the strains to be examined for bacterial invasion and inoculate each into individual 14 ml tubes containing 2 ml of BHI broth.
Place the inoculated tubes into a 37 °C incubator tilted at 45° and incubate statically (without shaking) overnight.
The following morning, remove the culture tube from the incubator and vortex briefly to ensure uniform suspension of the bacteria.
Measure the optical density of the culture using a spectrophotometer at 600 nm wavelength. Be sure to zero the spectrophotometer using sterile BHI broth before reading the optical density.
For L. monocytogenes, the optical density is directly related to the concentration of bacteria per ml, such that a density of 1.000 = 1.0 x 109 CFU per ml.
Calculate the amount of culture needed to infect 2.25 x 104 cells with 2.25 x 106 (MOI = 100).
Remove the amount of culture needed and place it in a 1.5 ml centrifuge tube.
Spin the culture at 19,000 x g for 3 min, then discard the supernatant. Tap the open end of the tube against a paper towel in order to remove excess media stuck to the lip of the tube.
Resuspend the bacteria in 1 ml of sterile PBS, and vortex to ensure sufficient mixing. This mixture should contain approximately 2.25 x 106 CFU per 20 ul (1.125 x 108 CFU/ml).
5. Performing the Invasion Assay
The day before the invasion assay, prepare 24-well plates of H9c2 cells as described in Section 3, and also inoculate sterile BHI broth with the desired strain(s) of Listeria as described in Section 4.
Using the protocol in Section 4, prepare PBS-resuspended cultures of the strains to be assessed for invasion. Note: Infectious titer can be assessed at this point by plating dilutions of each sample onto solid media and enumerating colonies after overnight incubation.
Load at least 20 ul of the PBS-bacteria solution into individual wells of the 24-well plate. Note that at least three individual wells are needed for each strain in order to obtain results in triplicate, so up to 8 strains can be assessed in tandem.
After loading the wells with bacteria, gently rock the plate to encourage homogenous spread of each sample within the well, and place the 24-well plate back in the incubator for 45 min of incubation at 37 °C and 95% humidity, with 5% CO2.
During this incubation, prepare an aliquot of DMEM – Ab (Section 2) by simply removing 25 ml of DMEM –Ab and placing it in a 50 ml falcon tube (or equivalent).
Add gentamicin to the 25 ml DMEM –Ab aliquot to a concentration of 15 ug/ml (7.5 ul of a 50 mg/ml stock solution). Place the DMEM –Ab +Gent solution in a 37 °C waterbath for the duration of the 45 min incubation.
Aliquot 25 ml of PBS into a 50 ml falcon tube and place in the 37 °C waterbath during the 45 min incubation.
After the 45 min incubation is complete, remove the 24-well plate and place it in the hood. Remove the media containing bacteria from each well by vacuum aspiration, changing glass pipet tips between different strains.
Add approximately 1 ml of PBS to each well in order to wash loosely-adherent bacteria from the surface of the cells, then remove the PBS using vacuum aspiration.
Following complete removal of PBS, add 1 ml of DMEM –Ab +Gent to each well. The gentamicin will only kill those bacteria that remain outside of the cells, thus those which are intracellular will remain viable.
Place the 24-well plate back into the incubator and allow to incubate for an additional hour.
During the hour long incubation, prepare 14 ml polypropylene tubes by adding 1 ml of sterile ddH20 to each tube. One tube will be required for each coverslip, so for one 24-well plate, prepare 24 tubes total.
Following the hour long incubation, remove the 24-well plate from the incubator and place it in the hood, along with the tubes prepared in step 5.12.
Using sterile tweezers, remove each coverslip from its respective well and place it immediately in an individual tube. Be sure to dip the tweezers in ethanol and flame them between each coverslip in order to prevent contamination.
After all of the coverslips have been removed and placed into individual tubes, discard the 24-well plate.
Return to the bench and vortex each tube for 5-10 sec.
Remove a portion from each tube and place each sample in an individual well of a 96-well plate, then serially dilute each sample using 1:10 dilutions up to a 1:1,000 dilution.
Using prewarmed and dry LB plates, spot plate each of the dilution series onto the agar plates. A multichannel pipet can be used to plate 5-10 μl spots from each sample’s dilution series.
Also remove 20 μl samples from each undiluted well and spot plate this amount directly onto LB agar on a separate plate from the dilution series. (This larger volume can be used to enumerate poorly invasive strains).
After all of the samples have been spot plated, discard the 14 ml tubes containing the coverslips. Parafilm the 96-well plate and place it in a freezer box at -80 °C for replating, if necessary.
Allow the spots to dry completely. This process is hastened greatly by placing the plates in the hood and removing the lids.
After the spots have dried, place the plates in a 37 °C incubator overnight.
The following morning, count the number of colonies in each spot of the dilution series for which colony numbers can be easily assessed (between approximately 5 and 50 colonies) (Figure 1), and use the series to calculate the number of bacterial per coverslip for each strain assessed.
6. Preparing Strains of L. monocytogenes for Mouse Infections
The night before infection, use a sterilized loop to remove single colonies of the strains desired and inoculate each into individual 14 ml tubes containing 2 ml of BHI broth.
Place the inoculated tubes into a 37 ˚C incubator tilted at 45˚ and incubate statically (without shaking) overnight.
The following morning, remove the culture tube from the incubator and vortex briefly to ensure a uniform suspension of the bacteria.
Measure the optical density of the culture using a spectrophotometer at 600 nm wavelength. Be sure to zero the spectrophotometer using sterile BHI broth before reading the optical density. (For L. monocytogenes, the optical density is directly related to the concentration of bacteria per ml, such that a density of 1.000 = 1.0 x 109 CFU per ml)
Calculate the amount of culture needed to infect each animal. Injections typically contain 200 μl of PBS with 10,000 CFU of an individual strain, which translates to a desired concentration of 5.00 x 104 CFU/ml.
Remove a 5 μl aliquot of each final dilution and spread-plate it directly onto LB agar. Incubate overnight at 37 °C to confirm the concentration used for inoculation.
Inject each CFU suspension within 1 hr of diluting (see below).
7. Inoculating L. monocytogenes into Mice via Tail Vein Injection and Assessing Bacterial Burden within the Liver, Spleen, and Heart
Note: All animal work is carried out in accordance with CDC Biosafety Level 2 guidelines. Mice are typically ordered one week in advance and caged together with 5 animals per cage. Mice are allowed to acclimate to the new laboratory environment for four days before injection. These experiments used 6-8 week old female Swiss-Webster mice that were housed 5 to a cage in a barrier environment and fed a non-restricted diet.
Prepare one cage of mice at a time by removing the lid and food/water bins, and placing the cage under a heat lamp for five minutes. This process allows for the tail veins to dilate, making injections easier to perform.
Remove one animal and place it in a harness (or falcon tube with the end cut off) in order to restrain the animal during the injection.
Clean the injection site using an alcohol pad, and allow the alcohol to evaporate. This not only cleanses the site, but also further dilates the tail vein.
Using a 1 ml syringe equipped with a 27.5 gauge needle, gently inject the mouse with 200 μl of the desired culture into the tail vein.
Use a gauze to stop any bleeding which may occur as a result of the injection, and place the animal in a fresh cage.
Repeat this process for the remaining animals and strains. Be sure to label each cage of animals with the strain they have received.
Check on the animals daily, removing and sacrificing any animals which appear to be in ill. If no animals exhibit signs of illness, allow the infections to proceed for 72 hr before sacrificing all animals.
To sacrifice, use CO2 anesthesia from a bottled source until all respirations have stopped, then use cervical dislocation in order to assure that the animal is dead.
Following sacrifice, move the animals to a tissue culture hood for dissection.
Using a dissection block, pin each leg of the animal with large gauge needles and spray the animal with 70% ethanol until it’s saturated.
Cut the skin of the abdomen and thorax back using a Y-shaped incision that extends from the vaginal opening up to the xiphoid process, progressing then up to the axilla of each arm.
Pull back the skin and remove the needles from each leg to hold the dissection open.
Cut gently through the peritoneum, exposing the intestines, stomach, liver, and spleen.
By first cutting the hepatic vein and coronal ligament at the top of the liver, begin to remove the liver. Additional ligaments to be removed are found beneath the liver, connecting it to the first section of the small intestine, as well as to the musculature of the back.
Place the liver in 5 ml of sterile ddH20 in a 50 ml falcon tube.
Next, remove the spleen by isolating it and gently cutting away the vasculature and ligaments. The spleen will be removed more readily than the liver. Place the spleen in a separate falcon tube containing 5 ml sterile ddH20.
Locate the diaphragm and cut gently along the ribcage in order to visualize the thorax. Cut through the xiphoid process and sternum to the level of the neck in order to open the thorax, exposing the heart and lungs.
Using forceps, grab the heart gently by its apex, and lift it such that the aorta and pulmonary vessels are in tension. Cut these vessels to free the heart.
Place the heart into a 50 ml falcon tube containing 5 ml of sterile ddH20.
Discard the mouse carcass, and repeat this process for every animal, using clean falcon tubes containing 5 ml sterile ddH20 for each organ removed.
After all organs have been removed, clean the workstation and return to the bench.
Prepare a set of 5 tubes to be used for cleaning and sterilizing the homogenizer for each set of organs from five mice (i.e. one 5 tube set for 5 livers, 5 tubes for 5 spleens, and 5 tubes for 5 hearts). Four of each of the tubes should be filled with 30 ml sterile ddH20, and one should be filled with 30 ml 90% EtOH.
Using a TissueMaster (or equivalent homogenizer), dip the homogenizer (while running) into the tubes prepared in step 7.21 as follows to clean the probe:
Tube 1 (ddH2O) 5 sec in Tube 2 (ddH2O) 5 sec in Tube 3 (EtOH) 5 sec in Tube 4 (ddH2O) 5 sec in Tube 5 (ddH2O)
Homogenize one liver using the homogenizer for at least 2 min, or until no visible portions of organ remain. Use tweezers to remove any large debris from the probe.
Repeat the cleaning process described in step 7.22
Repeat the homogenization process for the other livers, making sure to wash the probe between each liver using the process described in step 7.22.
After all of the livers are completely homogenized, discard the wash tubes (1-5) for the liver.
Repeat the homogenize/clean process for both the spleen and hearts as well, making sure to use a new set of wash tubes for each series of organs. If at any point the wash tubes become turbid, discard them and replace with new tubes to finish the series.
After all of the organs have been homogenized, do one last cleaning cycle on the homogenizer and dry it well for storage.
Place approximately 200 μl of each organ into an individual well of a 96-well plate.
Perform serial dilutions on the organ samples by diluting 1:10 in a series up to a 1:10,000 dilution (liver and spleen) or up to a 1:1,000 dilution (heart).
Spot plate the dilution series onto pre-warmed LB agar. Also remove 20 ul of each undiluted organ and plate onto separate LB agar plates in order to enumerate the CFU from poorly colonized organs.
Discard the falcon tubes containing the homogenized organs, and wrap the 96-well plates containing the dilution series with parafilm and place it in a freezer box at -80 °C.
Allow the spots to dry. This process is quickened by placing the plates in the hood and removing the lids.
After the spots have dried, place the plates in a 37 °C incubator overnight.
Count the number of colonies in each spot the following morning and use the dilution series to calculate the total number of bacteria per organ.
Representative Results
Selected isolates of L. monocytogenes exhibit enhanced invasion of cardiac cells in cell culture and in mouse models of infection. Figure 1 shows an example of how bacterial colonies may appear following spot plating of suspensions on agar media. This method allows accurate assessment of CFUs within a sample without using large numbers of agar media plates. Figure 2 shows an example of a tissue culture-based assay comparing the ability of strain 10403S to invade heart cells with that of strain 07PF0776. More than twice as many 07PF0776 bacterial CFU can be recovered from infected H9c2 cardiac cells following gentamicin treatment in comparison to cells infected with 10403S. Differences of 2 to 4-fold are routinely observed for cardiotropic strains using this assay. Figure 3 shows an example of the recovery of bacteria from the livers, spleens, and hearts of infected mice at 3 days post-infection. The infection of mice with the cardiotropic strain 07PF0776 or strain 10403S results in comparable numbers of bacteria recovered from the livers and spleens of infected mice, however mice infected with 07PF0776 are more likely to yield detectable numbers of bacteria from the heart and to exhibit greater bacterial burdens in this organ.
 Figure 1: Example of spot plating technique for determining bacterial CFUs. H9c2 cells grown on glass coverslips and infected with L. monocytogenes were lysed and suspensions were serially diluted using 1:10 dilutions up to a 1:1,000 dilution (left panel). A multichannel pipet was used to pipet 10 ul from each well directly onto a LB agar plate, and the plate was incubated overnight at 37 °C (right panel). The number of bacteria per coverslip are assessed by counting bacterial CFU associated with the appropriate dilution.
Figure 1: Example of spot plating technique for determining bacterial CFUs. H9c2 cells grown on glass coverslips and infected with L. monocytogenes were lysed and suspensions were serially diluted using 1:10 dilutions up to a 1:1,000 dilution (left panel). A multichannel pipet was used to pipet 10 ul from each well directly onto a LB agar plate, and the plate was incubated overnight at 37 °C (right panel). The number of bacteria per coverslip are assessed by counting bacterial CFU associated with the appropriate dilution.
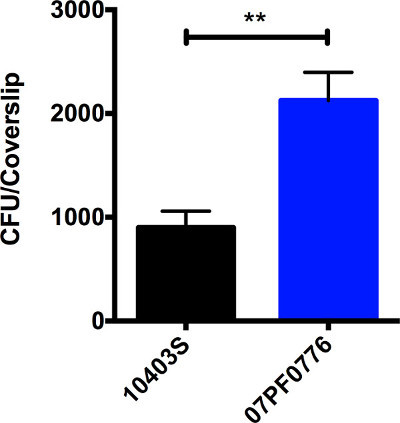 Figure 2: L. monocytogenes strain 07PF0776 exhibits enhanced invasion of cardiac cells in tissue culture. Invasion assays were performed in H9c2 cells using an MOI = 100. Graph depicts the average numbers of intracellular bacterial CFUs recovered +/- SE from cells infected with 10403S (black) versus the cardiotropic 07PF0776 strain (blue). ** indicates a significance of p <0.01.
Figure 2: L. monocytogenes strain 07PF0776 exhibits enhanced invasion of cardiac cells in tissue culture. Invasion assays were performed in H9c2 cells using an MOI = 100. Graph depicts the average numbers of intracellular bacterial CFUs recovered +/- SE from cells infected with 10403S (black) versus the cardiotropic 07PF0776 strain (blue). ** indicates a significance of p <0.01.
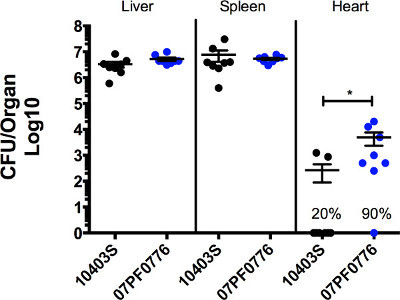 Figure 3: L. monocytogenes strain 07PF0776 exhibits enhanced invasion of the heart in mouse infection models. Animal were inoculated with 10,000 CFU via the tail vein. Infections were allowed to progress for 72 hr, at which point the animals were sacrificed and the livers, spleens, and hearts were collected and processed to determine bacterial CFU per organ. Solid circles represent the CFU obtained from individual mice, with the average value for all animals within a group indicated by a line +/- SE. The percentage values indicate the number of animals containing detectable bacterial CFU within the heart. * indicates a significance of p <0.05.
Figure 3: L. monocytogenes strain 07PF0776 exhibits enhanced invasion of the heart in mouse infection models. Animal were inoculated with 10,000 CFU via the tail vein. Infections were allowed to progress for 72 hr, at which point the animals were sacrificed and the livers, spleens, and hearts were collected and processed to determine bacterial CFU per organ. Solid circles represent the CFU obtained from individual mice, with the average value for all animals within a group indicated by a line +/- SE. The percentage values indicate the number of animals containing detectable bacterial CFU within the heart. * indicates a significance of p <0.05.
Discussion
L. monocytogenes is a widespread and well-characterized human pathogen, capable of causing a number of different disease manifestations15. The bacterium has been previously described for its ability to translocate across barriers, such as the blood-brain-barrier and placental-fetal barriers, in order to reach and colonize the central nervous system and developing fetus, respectively. The in vivo ability of the organism to colonize these tissues is often complemented by an in vitro ability to invade the representative cells in culture that make up the organs targeted. For instance, invasion of epithelial cells in the choroid plexus has been associated with the ability of the organism to colonize the CNS16; and villous trophoblast explants have been used to represent the maternal-fetal barrier17. In this protocol, methods been described that are useful for assessing bacterial invasion of heart cells in culture as well as for comparison of bacterial colonization of the heart for individual isolates relative to colonization of the liver and spleen.
This protocol contains a number of critical steps, but among the most critical are those involving the use of the correct bacterial CFU for either the infection of tissue culture cells grown on coverslips or the infection of animals. If bacterial CFU number is not correctly controlled between different wells and animals then direct comparisons between samples cannot be made. It is important to double check the dilution series used to generate the inocula by direct plating of the final dilutions on media plates in order to guarantee that the bacterial CFU number has been accurately estimated.
The H9c2 cells used in these assays are derived from the lower half of a 13 day embryonic rat heart which included mostly ventricular tissue18. The cells propagate as mononucleated myoblasts and upon reaching confluency in tissue culture flasks or dishes begin to form multinucleated tubular structures. The cells have a generation time of approximately 30 hr18. Serum starvation of H9c2 cells has been associated with differentiation of the cells into skeletal muscle cells, whereas treatment of the myoblasts with 10 nM all-trans retinoic acid has been associated with myoblast differentiation into cardiac myocytes14. This protocol was focused on L. monocytogenes myoblast cell invasion, however the H9c2 cell line is a versatile cell line that could be used to investigate the effects of controlled cell differentiation on bacterial invasion.
The L. monocytogenes strain 07PF0776 was originally isolated from an HIV-infected patient who had a non-resusitatable asystolic arrest due to an invasive L. monocytogenes infection of the heart8. Subsequent analysis of this strain in mouse infection models indicated that it had an enhanced capacity to target and invade cardiac tissue. A limited analysis of additional random isolates of L. monocytogenes suggests that sub-populations of bacterial isolates are capable of infecting the hearts of mice in the absence of any prior damage to cardiac tissue or heart valves8. Interestingly, two of the best characterized L. monocytogenes strains, 10403S and EGD, were found to be poor colonizers of cardiac cells and tissue. Genome sequencing of the 07PF0776 isolate did not reveal the presence of any novel pathogenicity islands or provide evidence of unique gene clusters19; this suggests that 07PF0776 targets cardiac cells for invasion through modification of its existing arsenal of virulence gene products. Preliminary histochemical analysis indicates that 07PF0776 forms abscesses within infected mouse hearts that appear similar to the abscess observed within the original infected human patient (data not shown). Whether other cardiotropic L. monocytogenes isolates induce similar cardiac abscess formation remains to be determined.
The assays presented here can be easily modified to facilitate the examination of different aspects of infection. Individual coverslips can be fixed and stained for either light or fluorescence-based microscopy, tissue culture incubation times can be increased for the measurement and comparison of bacteria intracellular growth rates, tissues and organs can be paraffin embedded and processed for microscopic examination to examine abscess formation and bacterial distribution at the cellular level. When assessing levels of bacterial invasion of tissue culture cells or colonization of host tissues, there are limitations in terms of the minimum amount of bacteria each assay can detect. In vitro invasion assays have a lower limit of detection of approximately 300 CFU per coverslip. Adjusting the volume of water used to vortex the coverslips may enhance detection of low numbers of bacteria, with cell lysis volumes as low as 500 μl proving useful to detect poorly invasive strains. Coverslips may be briefly dipped in sterile water to remove excess gentamicin prior to lysis of host cells in smaller volumes. Volume levels up to 5 ml or greater can be used to properly enumerate highly invasive strains. In the animals, organ homogenates can be generated in volumes of 5 ml or 10 ml of ddH20, with lower volumes being useful to detect lower levels of bacteria, and higher volumes to better enumerate heavily colonized organs. The minimum detection range for infected organs is approximately 100 CFU. If organs are consistently colonized at high levels, consider using a larger volume of water (10 ml) and performing more dilutions within the 96-well plate.
The methods described can be applied to organs and tissues outside of the heart. Invasion assays and mouse infections such as these have been used to assess colonization in a multitude of sites, including the placenta, brain, gallbladder, liver, and intestine. Modifications of the protocols described above can also be made in order to accommodate hyperinvasive and/or hypervirulent strains, and substitution of heart cells with other cell types may be done to assess invasion phenotypes at sites outside of the cardiovascular system. Since different cell types have altered susceptibilities to L. monocytogenes invasion, adjusting parameters such as MOI and incubation times may be necessary in order to accurately recover data from the assays.
Disclosures
The authors report no competing financial interests.
Acknowledgments
This work was supported by Public Health Service grants AI41816 and AI099339 (N.E.F.) and by F31AI094886-01 (P.D.M.) from NIAID. The contents are solely the responsibility of the authors and do not necessarily represent the official views of the funding sources.
References
- Freitag NE. From hot dogs to host cells: how the bacterial pathogen Listeria monocytogenes regulates virulence gene expression. Future Microbiol. 2006;1:89–101. doi: 10.2217/17460913.1.1.89. [DOI] [PubMed] [Google Scholar]
- Drevets DA, Bronze MS. Listeria monocytogenes: epidemiology, human disease, and mechanisms of brain invasion. FEMS Immunol Med Microbiol. 2008;53:151–165. doi: 10.1111/j.1574-695X.2008.00404.x. [DOI] [PubMed] [Google Scholar]
- Farber JM, Peterkin PI. Listeria monocytogenes. a food-borne pathogen. Microbiol. Rev. 1991;55:476–511. doi: 10.1128/mr.55.3.476-511.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecuit M. Understanding how Listeria monocytogenes targets and crosses host barriers. Clin Microbiol Infect. 2005;11:430–436. doi: 10.1111/j.1469-0691.2005.01146.x. [DOI] [PubMed] [Google Scholar]
- Lecuit M. Human listeriosis and animal models. Microbes Infect. 2007;9:1216–1225. doi: 10.1016/j.micinf.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Bou Ghanem EN, et al. InlA promotes dissemination of Listeria monocytogenes to the mesenteric lymph nodes during food borne infection of mice. PLoS Pathog. 2012;8:e1003015. doi: 10.1371/journal.ppat.1003015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melton-Witt JA, Rafelski SM, Portnoy DA, Bakardjiev AI. Oral infection with signature-tagged Listeria monocytogenes reveals organ-specific growth and dissemination routes in guinea pigs. Infect Immun. 2012;80:720–732. doi: 10.1128/IAI.05958-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonzo F, 3rd, Bobo LD, Skiest DJ, Freitag NE. Evidence for subpopulations of Listeria monocytogenes with enhanced invasion of cardiac cells. J Med Microbiol. 2011. [DOI] [PMC free article] [PubMed]
- Adler A, et al. Inflammatory pseudotumor of the heart caused by Listeria monocytogenes infection. J Infect. 2009;58:161–163. doi: 10.1016/j.jinf.2008.12.007. [DOI] [PubMed] [Google Scholar]
- Antolin J, Gutierrez A, Segoviano R, Lopez R, Ciguenza R. Endocarditis due to Listeria: description of two cases and review of the literature. Eur J Intern Med. 2008;19:295–296. doi: 10.1016/j.ejim.2007.06.020. [DOI] [PubMed] [Google Scholar]
- Brouqui P, Raoult D. Endocarditis due to rare and fastidious bacteria. Clin Microbiol Rev. 2001;14:177–207. doi: 10.1128/CMR.14.1.177-207.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brusch JL. Cardiac infections in the immunosuppressed patient. Infect Dis Clin North Am. 2001;15:613–638. doi: 10.1016/s0891-5520(05)70162-8. [DOI] [PubMed] [Google Scholar]
- McCue MJ, Moore EE. Myocarditis with microabscess formation caused by Listeria monocytogenes associated with myocardial infarct. Hum Pathol. 1979;10:469–472. doi: 10.1016/s0046-8177(79)80052-0. [DOI] [PubMed] [Google Scholar]
- Menard C, et al. Modulation of L-type calcium channel expression during retinoic acid-induced differentiation of H9C2 cardiac cells. J Biol Chem. 1999;274:29063–29070. doi: 10.1074/jbc.274.41.29063. [DOI] [PubMed] [Google Scholar]
- Czuprynski CJ. Listeria monocytogenes: silage, sandwiches and science. Anim Health Res Rev. 2005;6:211–217. doi: 10.1079/ahr2005111. [DOI] [PubMed] [Google Scholar]
- Grundler T, et al. The surface proteins InlA and InlB are interdependently required for polar basolateral invasion by Listeria monocytogenes in a human model of the blood-cerebrospinal fluid barrier. Microbes Infect. 2013;15:291–301. doi: 10.1016/j.micinf.2012.12.005. [DOI] [PubMed] [Google Scholar]
- Zeldovich VB, Robbins JR, Kapidzic M, Lauer P, Bakardjiev AI. Invasive extravillous trophoblasts restrict intracellular growth and spread of Listeria monocytogenes. PLoS Pathog. 2011;7:e1002005. doi: 10.1371/journal.ppat.1002005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimes BW, Brandt BL. Properties of a clonal muscle cell line from rat heart. Experimental cell research. 1976;98:367–381. doi: 10.1016/0014-4827(76)90447-x. [DOI] [PubMed] [Google Scholar]
- McMullen PD, et al. Genome sequence of Listeria monocytogenes 07PF0776, a cardiotropic serovar 4b strain. J Bacteriol. 2012;194:3552. doi: 10.1128/JB.00616-12. [DOI] [PMC free article] [PubMed] [Google Scholar]