

Abstract
The RUNX2 gene is a physiological regulatory gene implicated in the development of cleidocranial dysplasia (CCD). A 13-month-old child presented with clinical features of CCD. At the age of 3 years the diagnosis was corroborated by clinical genetic assessment and DNA analysis, revealing a missense mutation p.R131C (c.391C>T) in RUNX2. At the age of 8 years the child was found to have a unique dental phenotype, represented by lack of supernumerary teeth and congenital absence of one tooth. A simple therapeutic approach was adopted, consisting of interceptive orthodontic treatment. The presence of this specific missense mutation in RUNX2, associated with the lack of typical supernumerary teeth may suggest a phenotype–genotype association.
Background
Cleidocranial dysplasia (CCD) is a rare autosomal dominant skeletal disorder characterised by hypoplastic or absent clavicles, increased head circumference, large fontanelles, short stature, hand malformation and orodental anomalies. The most significant dental manifestations include: (1) anomalies of eruption, delayed or absent eruption of deciduous/permanent teeth with ectopic position, (2) shape anomalies and (3) number anomalies: multiple supernumerary teeth in the permanent dentition. Most of the supernumerary teeth fail to erupt due to lack of space and failure of bone resorption. Supernumerary teeth often have an aberrant shape related to impaction and crowding. The diagnosis is based on clinical and radiological findings. Mutations are generally found in the Runt-related transcription factor 2 (RUNX2) gene, which acts as a master regulator of osteoblastic differentiation. So far, mutations in RUNX2 have been found in 75% of CCD cases, representing the major genetic cause of this disorder.1 In a large recent survey2 48 new heterozygous mutations were identified, one corresponding to c.391C>T (p.R131C). A patient from our series of CCD carrying the same heterozygous missense mutation of the RUNX2 gene has turned out to display during development as an yet unreported dental phenotype, that is, lack of supernumerary teeth and congenital absence of one incisor. This finding prompted us to report on this unusual dental phenotypic variant in CCD.
Case presentation
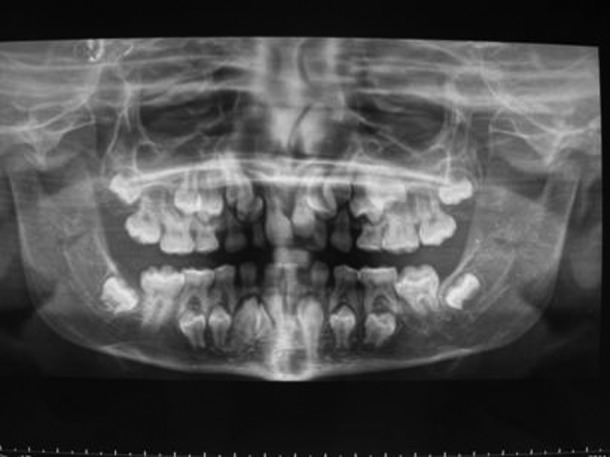
The proband, an 11-year-old Caucasian girl, is the first born of healthy unrelated Italian parents. The family history was negative for skeletal disorders. She was born at term by physiological delivery; birth weight was 3.5 kg, length 50 cm (50th–75th percentile) and head circumference 34 cm (50th percentile). Physical examination at 13 months of age revealed aplastic clavicles, mandibular hypoplasia, brachycephaly, frontal bossing, large fontanelles and delayed dental eruption. An audiometric test showed a mild bilateral sensorineural hearing loss. On the basis of clinical and imaging features, a diagnosis of CCD was suggested and a genetic molecular investigation for RUNX2 mutations was performed. At 8 years of age the patient was admitted to Department of Paediatric Dentistry of Children's Hospital “Burlo Garofalo”, IRCCS, Trieste, Italy. She presented with a unilateral crossbite, delayed dental eruption and no permanent teeth in the mouth. To verify the number of supernumerary teeth, an orthopantomogram (OPT) was carried out. Surprisingly, the OPT showed no supernumerary teeth, but agenesis of the left mandibular lateral incisor (figure 1). To correct the left crossbite and guide the eruption of the permanent teeth, a removable orthodontic rapid palate expansor was applied. A proper orthodontic treatment was established, impressions and study models were carried out and appropriate educational hygiene plan was prescribed to prevent dental caries and periodontal complications. Whereas it is known that the extraction of deciduous teeth does not promote eruption of underlying permanent teeth in CCD,3 the maxillary deciduous central incisors were removed under local anaesthesia to favour a normal positioning of the central maxillary permanent incisors. At 11 years of age, a successful correction of the crossbite was gained.
Investigations
Genetic analysis: At the age of 3 years molecular genetic analysis of RUNX2 was performed by direct sequencing (standard methods), demonstrating a heterozygous mutation (c.391C>T), resulting in the missense change p.Arg131Cys. This mutation has been previously detected in CCD, and is believed to be pathogenetic.4 The mutation was not detected in either the patient's mother or father.
Discussion
The present case was diagnosed with CCD at 13 months of age on the basis of clinical and radiological features. The diagnosis was subsequently molecularly confirmed when the child was 3 years old. The same missense mutation in the RUNX2 gene, c.391C>T (p.R131C) has been recently reported by Ott et al2 who had studied a large number of CCD patients and detected 48 new mutations. A mutation affecting the same arginine at position 131 (p.R131G) has been described,5 but neither in this case, nor in the case mentioned by Ott et al,2 the dental phenotype was well characterised.
The unexpected finding in a CCD patient, namely the absence of supernumerary teeth together with the missing of a single tooth apparent by the age of 9 years, has prompted us to report this unusual association. Supernumerary teeth are rather a constant feature in CCD. The lack of supernumerary teeth in CCD has been reported quite recently in two members of a family carrying a novel mutation of the RUNX2 gene, that is, a 2-basepair deletion in exon 5 (c.873_874delCA), leading to a premature protein termination (Q292fsX299).6 However, in this family no evidence was reported of a coexisting missing tooth. The molecular pathogenesis of CCD is related to the loss of function or haploinsufficiency of RUNX2,7 an osteoblastic-specific transcription factor belonging to the Runt domain gene family that promotes the differentiation of mesenchymal cells into osteoblasts.8 Mutations in the RUNX2 gene in humans cause CCD,9 and the patients present bone dysplasia and extra teeth.2 Interestingly, the supernumerary teeth in association with CCD develop as parts of a third dentition.3 Therefore, RUNX2 has been described as a positive regulator of the primary teeth but a negative regulatory of the permanent ones. The dental phenotypes are puzzling since the RUNX2 knockout mice show complete lack of teeth, which might suggest that oligodontia would be more likely in haploinsufficient humans. However, the mouse model is perhaps not ideally suited for modelling the human situation, since mice do not develop a secondary dentition. Nevertheless, recent data seem to support the role of RUNX2 as an inhibitor of tooth renewal, thus explaining the constant feature of extra teeth in CCD. The role of RUNX2 as an inhibitor of tooth renewal is further supported by recent experimental data on the development of a rudimentary SHH expression bud in the epithelium of RUNX2 mutant and heterozygous mice.10 In contrast to all the other mutations of the RUNX2 gene, both the mutation p.R131C found in our CCD patient and the mutation p.Q292fsX299 described by Bufalino et al,6 do not result in supernumerary teeth. It is noteworthy mentioning that the Q292fsX299 mutation is localised in the PST domain of the protein, while our p.R131C mutation is localised in the Runt domain where mutations leading to supernumerary teeth are generally detected. Moreover the mutation found in our patient is a missense mutation, substituting a cysteine for an arginine, and predictively exerts a dominant negative effect. This finding in our patient represents the first mutation in the Runt domain with the absence of supernumerary teeth. Moreover, the missing of a mandibular incisor in our case constitutes a peculiar CCD phenotype for the mutation p.R131C. RUNX2 proteins, in general, have an NLS (nuclear localisation signal) at the C-terminal border of the runt domain (PRRHRQKLD).11 In 2006, Kim et al5 investigated a new NLS and identified a stretch of nine amino acids (HWRCNKTLP) at the N-terminal border of the Runt domain. Our p.R131C mutation affects a highly conserved residue of the Runt domain and falls inside the new NLS. Presently one can only speculate about the apparently ‘aberrant’ sequelae of the mutation as it seems to interfere with the regular differentiation of mesenchymal cells into osteoblasts at the bone level,9 but not at the level of tooth development. In tooth development, the interaction of a number of transcription factors, and genes MSX1, PAX9, SSH and RUNX2, regulates the dental placode formation, the condensed dental mesenchyma and the bud cup transition as well.12 It would be of interest to compare, on clinical grounds, the dental phenotype in further cases carrying the mutation c.391C>T and, on molecular grounds, explore the function of the mutant protein by in vitro studies and molecule modelling. From a practical point of view, the absence of supernumerary teeth in our CCD patient has allowed an easy dental treatment in contrast to other CCD cases which are known to require complex treatment, that is, multiple extractions of the impacted teeth, subsequent orthodontic rehabilitation, new intervention of maxillofacial surgery and again orthodontic treatment.13
To the best of our knowledge, this is the second mutation in the RUNX2 gene which is not associated with supernumerary teeth, thus suggesting the possibility of a causative role. Further cases and studies will be required to shed new light on the pathogenetic mechanisms and manifesting spectrum of dental anomalies in CCD.
Learning points.
RUNX2 gene is a physiological regulatory gene implicated in the development of cleidocranial dysplasia (CCD).
The most significant dental manifestations include: (1) anomalies of eruption, delayed or absent eruption of deciduous/permanent teeth with ectopic position, (2) anomalies of shape, (3) anomalies of number: multiple supernumerary teeth in the permanent dentition. Most of the supernumerary teeth fail to erupt because of the absence of space and failure of bone resorption.
To make a correct examination and management of CCD patients an orthopantomogram is very important, to verify presence or absence of supernumerary teeth.
The absence of supernumerary teeth is uncommon in CCD, and can be related to the localisation of RUNX2 mutations in different domains of the protein.
Footnotes
Competing interests: None.
Patient consent: Obtained.
References
- 1.Ott CE, Hein H, Lohan S, et al. Microduplications upstream of MSX2 are associated with a phenocopy of cleidocranial dysplasia. J Med Genet 2012;49:437–41. [DOI] [PubMed] [Google Scholar]
- 2.Ott CE, Leschik G, Trotier F, et al. Deletions of the RUNX2 gene are present in about 10% of individuals with cleidocranial dysplasia. Hum Mutat 2010;31:E1587–93. [DOI] [PubMed] [Google Scholar]
- 3.Jensen BL, Kreiborg S. Development of the dentition in cleidocranial dysplasia. Joral Pathol Med 1990;19:89–93. [DOI] [PubMed] [Google Scholar]
- 4.Quack I, Vonderstrass B, Stock M, et al. Mutation analysis of core binding factor A1 in patients with cleidocranial dyspl. Am J Hum Genet 1999;65:1268–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim HJ, Nam SH, Park HS, et al. Four novel RUNX2 mutations including a spliced donor site result in the cleidocranial dyspasia. J Cell Physiol 2006;207:114–22. [DOI] [PubMed] [Google Scholar]
- 6.Bufalino A, Paranaiba LMR, Gouvea AF, et al. Cleidocranial dysplasia: oral features and genetic analysis of 11 patients. Oral Dis 2012;18:184–90.. [DOI] [PubMed] [Google Scholar]
- 7.Suda N, Hattori M, Kosaki K, et al. Correlation between genotype and supernumerary tooth formation in cleidocranial dysplasia. Orth Craniofac Res 2010;13:197–202. [DOI] [PubMed] [Google Scholar]
- 8.Mundlos S. Cleidocranial dysplasia: clinical and molecular genetics. J Molec Genet 1999;36:177–82. [PMC free article] [PubMed] [Google Scholar]
- 9.Ducy P, Zhang R, Geoffroy V, et al. Os/f2Cbfa:a transcriptional activator of osteoblast differentiation. Cell 1997;89:747–54. [DOI] [PubMed] [Google Scholar]
- 10.Wang XP, Aberg T, James MJ, et al. RUNX2 (Cbfa1) inhibits SHH signalling in the lower but not upper molars of mouse embryos and prevents hebud. J Dental Res 2005;84:138–43. [DOI] [PubMed] [Google Scholar]
- 11.Thirunavukkarasu K, Magajaan M, McLarren KW, et al. Two domains unique to osteoblast specific transcription factor Osf2/Cbfa1contribute to its transactivation function and its inability to heterodimerize with Cbfb. Mol Cell Biol 1998;18:4197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thesleff I. The genetic basis of tooth development and dental defects. Am J Med Genet 2006;140:2530–5. [DOI] [PubMed] [Google Scholar]
- 13.D'Alessandro G, Tagariello T, Piana G. Craniofacial change and treatment of the stomatognathic system in subjects with cleidocranial dysplasia. Orth Craniofac Res 2010;13:197–202. [PubMed] [Google Scholar]