

Key Points
Natural killer cell granzyme B, A, and K delivery and subsequent caspase activation is rapid after conjugation with tumor target cells.
Natural killer cells also induce caspase activation through death receptor ligation that can be monitored in real time.
Abstract
Growing interest in natural killer (NK) cell–based therapy for treating human cancer has made it imperative to develop new tools to measure early events in cell death. We recently demonstrated that protease-cleavable luciferase biosensors detect granzyme B and pro-apoptotic caspase activation within minutes of target cell recognition by murine cytotoxic lymphocytes. Here we report successful adaptation of the biosensor technology to assess perforin-dependent and -independent induction of death pathways in tumor cells recognized by human NK cell lines and primary cells. Cell-cell signaling via both Fc receptors and NK-activating receptors led to measurable luciferase signal within 15 minutes. In addition to the previously described aspartase-cleavable biosensors, we report development of granzyme A and granzyme K biosensors, for which no other functional reporters are available. The strength of signaling for granzyme biosensors was dependent on perforin expression in IL-2–activated NK effectors. Perforin-independent induction of apoptotic caspases was mediated by death receptor ligation and was detectable after 45 minutes of conjugation. Evidence of both FasL and TRAIL-mediated signaling was seen after engagement of Jurkat cells by perforin-deficient human cytotoxic lymphocytes. Although K562 cells have been reported to be insensitive to TRAIL, robust activation of pro-apoptotic caspases by NK cell–derived TRAIL was detectable in K562 cells. These studies highlight the sensitivity of protease-cleaved luciferase biosensors to measure previously undetectable events in live cells in real time. Further development of caspase and granzyme biosensors will allow interrogation of additional features of granzyme activity in live cells including localization, timing, and specificity.
Introduction
Natural killer (NK) cells are cytotoxic lymphocytes that provide the first line of defense in the human immune system by recognizing and eliminating tumor cells and virally infected cells. NK cells are reported to execute target cells by a combination of death receptor ligation and secretory granule–mediated killing involving perforin (PRF) and the granzyme family of serine proteases (Grz)1,2; however, the majority of studies support nearly exclusive usage of the PRF-mediated pathway for rapid cell death. NK cells may recognize target cells directly through a balance of inhibitory and activating receptors, or via coupling of the low-affinity IgG receptor, CD16/FcγRIIIA, and target cells.3 The latter is termed “antibody-dependent cellular cytotoxicity” (ADCC) and is one pathway used by antibody-based immunomodulators, such as rituximab, to eliminate tumor cells.
PRF and Grzs are constitutively expressed by NK cells, although expression can be augmented after cytokine stimulation.4-7 After NK cell conjugation with a target cell, granules are rapidly exocytosed within minutes,8 and PRF delivers Grzs into the target cell through Ca2+-dependent oligomerization and pore formation.1 The 5 human Grzs (A, B, H, K, and M) initiate cell death processes through proteolysis of intracellular substrates. GrzB has been shown extensively to activate pro-apoptotic, caspase-dependent pathways through 2 mechanisms dictated by tumor cell specificity: (1) direct GrzB processing and activation of caspase 3/7, 8, and 109-12; and (2) engagement of the mitochondrial pathway to achieve caspase 3/7 activation.9 Much less information is known about the remaining Grzs, largely because of a dearth of functional assays and Grz-specific inhibitors.
NK cells can also kill tumor cells by granule-independent, caspase-dependent mechanisms through death receptor (tumor necrosis factor–related apoptosis-inducing ligand [TRAIL] and Fas) ligation.13-17 After ligation of death receptors on the target cell and initiation of signaling, caspases 8 and/or 10 are reported to activate caspase 3/7 by similar pathways as described for GrzB: (1) direct cleavage of caspase 318 and, in some cells, (2) indirect activation of caspase 3 through the mitochondrial pathway.19-21 The timing and amplitude of death receptor–induced caspase activation during human NK cell conjugation is not well understood.
Using protease-activated luciferase biosensors expressed in a variety of tumor target cells, we evaluated the kinetics of Grz and pro-apoptotic caspase activation mediated by human NK cell lines and primary human NK cells in real time. We found that activated caspase and GrzB signals were rapidly detected after both Fc receptor (CD16) engagement and direct recognition of tumor target cells by cytokine-activated NK cells. Pro-apoptotic caspase activation was triggered by the secretory granule and death receptor pathways within 30 to 120 minutes of cell contact. Finally, we characterized novel tumor cell–based biosensors for GrzA and GrzK and identified nafamostat mesylate as an inhibitor of GrzK. This study highlights the utility of protease-cleavable biosensors and allows, for the first time, kinetic measurements of granzyme and caspase activity in live NK target cells using a microplate assay.
Materials and methods
Cells and cell lines
K562 and Jurkat cells were cultured in complete RPMI 1640 (supplemented with 10% fetal bovine serum [FBS] [Sigma], 100 U/mL penicillin, and 100 μg/mL streptomycin [Gibco]). P815 cells were cultured in complete Dulbecco’s modified Eagle medium. NK92 cells were cultured in α-MEM (Gibco) with 12.5% equine serum (Hyclone), 12.5% FBS, 0.02 mM folic acid, 0.2 mM inositol, and 200 U/mL rIL-2. KHYG1 (from Japanese Collection of Research Bioresources Cell Bank, JCRB0156) were maintained in complete RPMI 1640 with 100 U/mL rIL-2. The natural killer leukemia (NKL) line was cultured as described.3 Herpesvirus saimiri–transformed human cytotoxic lymphocytes (HVS-CL) were cultured as previously described.22 Human NK cells were obtained from whole blood by negative selection (Miltenyi). To expand NK cells from peripheral blood mononuclear cells (PBMCs) without previous selection, 1000 U/mL IL-2 was used for 6 days. Human blood samples were obtained according to protocols approved by our institutional review board, in accordance with the Declaration of Helsinki.
Reagents
GLS cAMP reagent (biosensor substrate) was provided by Promega. For luciferase assays, GrzB inhibitor Compound 20 (C20, Synkinase batch YAJB028-035) and pan-caspase inhibitor Q-VD-OPH (MP Biomedicals) were prepared in dimethyl sulfoxide. Mg2+-EGTA was prepared in PBS (3 mM MgCl2, 4 mM EGTA, pH 7.6). Nafamostat mesylate (Enzo Life Sciences) was prepared in water and used in substrate medium containing heat-inactivated FBS. Anti-human CD16 (3G8), anti-human TRAIL (RIK-2), anti-human FasL (NOK-1), or IgG1k isotype controls (MOPC-21) (all from Biolegend) were used in substrate medium. For immunoblots, primary antibodies recognized firefly luciferase (Luci17, Abcam); GrzB (2C5/F5, BD Biosciences); GrzK (H85, Novis Bio); GrzA (GA6, AbD Serotec); perforin (P1-8, Kamiya); and β-actin (AC-15, Sigma). Secondary antibody was goat anti-mouse IRDye 800 (Licor).
Biosensor constructs
Biosensors derived from GloSensor pGLS-30F containing a caspase 3/7 DEVDG cleavage site (hereafter referred to as GLS.DEVD) were constructed as described23 in both the pGLS-30F plasmid and a retroviral vector pMIEG expressing GFP in cis.24 pGLS-30F plasmids containing Grz A/K–specific sites25,26 were generated using the following oligonucleotides: GLS.SGR, forward: 5′-GATCCTCGGGCCGTAGCGGA-3′, and reverse: 5′-AGCTTCCGCTACGGCCCGAG-3′; GLS.PGPR, forward: 5′-GATCCCCGGGGCCCAGGGAGGGA-3′, and reverse: 5′-AGCTTCCCTCCCTGGGCCCCGGG-3′; GLS.QGPR, forward: 5′-GATCCCAGGGACCTAGAGGA-3′, and reverse: 5′-AGCTTCCTCTAGGTCCCTGG-3′. For the sequences of control plasmids, please contact the authors.
In vitro testing of recombinant biosensors
Recombinant luciferase protein was generated and tested as described23,27 with recombinant human GrzB (ImmunoChemistry), GrzA (Millipore), or GrzK (Abcam). To inhibit GrzA and GrzK activity, 3.5 μM nafamostat mesylate was preincubated with 0.42 U/uL enzyme for 15 minutes at 37°C. Fold activation was expressed as: (Signal enzyme / Signal no enzyme). Percentage inhibition of signal was expressed as: [1 − (Signalinhibitor / Signaldiluent) × 100].
Transfection and stable transduction of cell lines
Clonal and polyclonal cell lines expressing GLS biosensors were obtained as previously described.23 KHYG1 cells were transduced with retrovirus to overexpress miR30-based shRNAs targeting the 3′UTR of PRF1 with mCherry expressed in cis.28 Transduced KHYG1 cells (KHYG1 shPRF) were sorted for mCherry expression 7 days later.
Biosensor assay
The assay was performed as described with target cells at 20 000 cells per well in quadruplicate.23 Relative light units (RLUs) were measured every 3 minutes in a GloMax-Multi+ luminometer set at 37°C (0.5-s integration time). Fold activation for each time point represents 4 replicate wells and was calculated using: (RLUE+T / RLUT alone). Statistical analysis by one-way analysis of variance (ANOVA) and Dunnett multiple comparison test was performed in GraphPad Prism v.6.0f.
51Cr-release assay
A 51Cr assay was performed as previously described.23 Target cells were incubated with 51Cr for 1 hour, then effector and target cells (5000 cells/well) were coincubated for 4 hours at 37°C. Cytotoxicity is reported as the percentage of 51Cr release into supernatant, calculated as follows: [(test − spontaneous) / (maximum lysis − spontaneous)] ×100. Maximum lysis was determined by addition of 1% Triton X-100 to 5000 target cells.
Immunoblots
Cell lysates were prepared with lysis buffer (2% NP-40; 150 mM NaCl; 50 mM TrisCl [tris(hydroxymethyl)aminomethane chloride]; 1 mM MgCl2, pH 8; 2.5 mM EDTA, 2 μg/mL leupeptin; 2 μg/mL aprotinin; 50 μg/mL phenylmethylsulfonyl fluoride; 1 μg/mL pepstatin; 10 μM Q-VD-OPH; 50 μM C20). Following standard immunoblotting procedures, nitrocellulose membranes (Millipore) were visualized with a LICOR Odyssey CLx scanner. Relative expression of perforin protein in KHYG1 shPRF to KHYG1 was calculated as follows: [(shPRF densityPRF / densityactin) / (KHYG1 densityPRF / densityactin)].
Flow cytometry
Surface staining was performed with antibodies specific to human CD3, CD8, CD56, CD16, and CD95/Fas (BD). For intracellular staining, cells were fixed and permeabilized with Cytofix/Cytoperm (BD) as per the manufacturer’s protocol, and stained with antibodies specific to perforin (clone δG9, Biolegend) and granzyme B (Invitrogen). Data were acquired on a BD FACSCanto II with BD FACSDIVA software (V8.0.1) and analyzed on FlowJo X (V10.0.7).
Results
Protease cleavable biosensors are activated by recombinant human granzymes
We have previously demonstrated murine GrzB activation of luciferase biosensors containing 3 cleavage sequences (GLS.IETD, -IEAD, and -VGPD).23 Based on cleavage prediction by GraBCas, we hypothesized that the biosensors would be activated by human GrzB (Table 1). Luciferase was generated by in vitro translation, and cleavage was tested with recombinant human GrzB (Figure 1A). Cleavage and activation of luciferase biosensors were dependent on the concentration of recombinant human GrzB (Figure 1A). Confirming previous findings,23,29 the GLS.DEVD biosensor and 3 additional control biosensors with a noncleavable alanine at P1 could not be activated by GrzB (Figure 1B).
Table 1.
Sequence and cleavage sites of granzyme and caspase biosensors
| Sensor name | Predicted cleavage site | Protease(s) predicted to cleave site |
|---|---|---|
| GLS.IEAD | SGRIEADSESL | Granzyme B; caspase 10 |
| GLS.IETD | SGIETDSGSL | Granzyme B; caspase 8 |
| GLS.VGPD | SKSVGPDFGSL | Granzyme B |
| GLS.DEVD | SDEVDGSL | Caspase 3/7 |
| GLS.SGR | GSSGRSGSL | Granzyme A; granzyme K |
| GLS.PGPR | GSPGPREGSL | Granzyme A; granzyme K |
| GLS.QGPR | GSQGPRGSL | Granzyme A; granzyme K |
Figure 1.
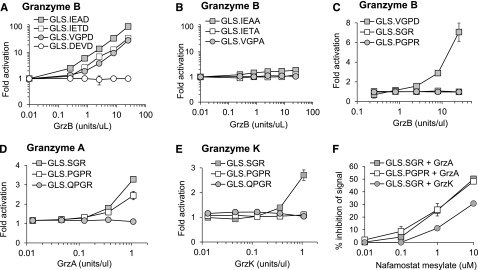
Protease-cleavable biosensors are activated by recombinant human granzymes. Luciferase was expressed by in vitro translation, and activation was tested after cleavage with recombinant human granzyme B (A-C), granzyme A (D), or granzyme K (E) over different concentrations. Recombinant granzyme A or K (0.42 U/uL) was preincubated with nafamostat mesylate before addition to GLS.SGR (granzyme A, K) or GLS.PGPR (granzyme A) (F). After 1 hour of incubation of in vitro translated luciferase with enzyme, luciferase reagent was added, RLU were measured, and fold activation or percentage inhibition was calculated as described in Methods. Error bars indicate standard error of the mean (SEM) for 3 experiments.
Because GrzA is one of the most abundant granzymes expressed by human NK cells,30 we developed 3 new luciferase biosensors containing proteolytic sequences identified to be cleaved by GrzA; GLS.SGR, GLS.PGPR, and GLS.QGPR25 (Table 1). Recombinant GrzB was unable to activate these biosensors (Figure 1C). Recombinant human GrzA activated both GLS.SGR and GLS.PGPR, with GLS.SGR demonstrating the largest fold activation (Figure 1D). Because GrzK has also been reported to recognize arginine in the P1 position,31,32 we also evaluated cleavage by this tryptase. Recombinant human GrzK activated GLS.SGR but not GLS.PGPR (Figure 1E). Neither enzyme could activate GLS.QGPR (Figure 1D-E). Based on reports of GrzA inhibition, we reasoned that the pan-tryptase inhibitor, nafamostat mesylate (NM), would inhibit both GrzA- and GrzK-induced luciferase activity.33-36 After preincubation with recombinant GrzA and GrzK, NM inhibited biosensor activation induced by both enzymes, although the magnitude of GrzA inhibition was greater than that of GrzK (Figure 1F). Although subtle, these findings represent the first report of GrzK inhibition by nafamostat mesylate.
NK stimulation by CD16 delivers granzyme B and activates pro-apoptotic caspases in target cells
We previously reported that anti-CD3 triggered degranulation of HVS-CL.23 Degranulation led to activation of the caspase 3/7 biosensor (GLS.DEVD) expressed by P815 target cells, a murine tumor line that binds murine-derived antibodies. Unfortunately, HVS-CL do not reliably express the NK Fc receptor CD16. Therefore, to evaluate Fc receptor–mediated NK function, we evaluated CD16/ FcγRIIIA expression by flow cytometry on NK92, KHYG1, and NKL human cell lines and, consistent with previous publications,3,37,38 determined that only NKL cells expressed CD16 (supplemental Figure 1A). We also confirmed that the NK effectors expressed PRF and GrzB by flow cytometry (supplemental Figure 1B). Using a redirected killing assay as a surrogate for ADCC, we conjugated NK effector cells with anti-CD16 and P815 target cells expressing GLS.DEVD or GLS.VGPD. Luminescence was measured in live cells every 3 minutes for 240 minutes on a plate reader warmed to 37°C. Figure 2A shows that NKL cells were capable of robustly activating the GLS.DEVD biosensors in P815 cells, similar to our previous findings with anti-CD3 stimulation.23 The caspase-mediated luciferase signal was rapid, peaking at 60 minutes, followed by a slow decline over the 240-minute time course. We also measured CD16-mediated GrzB delivery by NKL cells using P815 cells expressing GLS.VGPD and noted a similar pattern of rapid activation (Figure 2A). In contrast, KHYG1 cells that lacked surface CD16 exhibited no biosensor activation (Figure 2B), despite expressing PRF and GrzB (supplemental Figure 1B).
Figure 2.
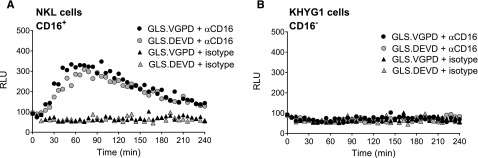
Granzyme B delivery and caspase 3/7 activation are detected during redirected NK killing. Human NK cell lines activate caspase 3/7 and granzyme B biosensors in P815 tumor target cells during redirected killing, as a model for ADCC. P815 cells expressing GLS.DEVD and GLS.VGPD were preincubated with anti-CD16 antibody or isotype control for 30 minutes at 37°C before incubation with NK effector cells at 1:1 (20 000 target cells/well). Cells were spun into contact and RLU was measured in a luminometer set at 37°C every 3 minutes for 240 minutes; every 6 minutes is plotted for clarity. Each datum point represents the mean of 4 replicate wells. Shown is a representative figure from 3 independent experiments using 2 effector cells lines: (A) NKL cells, and (B) KHYG1 cells.
Granzyme and pro-apoptotic caspase biosensors are activated through direct NK cell recognition of tumor target cells
Using primary murine NK cells cultured in IL2, we previously demonstrated modest activation of GLS.DEVD and GLS.VGPD in YAC cells, a murine NK target.23 To expand our studies to human NK cells, we transduced Jurkat and K562 cells with retroviruses expressing the GLS.DEVD, -VGPD, -IETD, or -IEAD biosensors and sorted for GFP+ cells (supplemental Figure 2A). We next evaluated direct NK-mediated killing using NK92, KHYG1, and NKL cells as effectors. The most robust signal in both target cells was mediated by NK92 cells, the cell line that expressed the highest levels of PRF and GrzB39,40 (supplemental Figure 1B). NK92 cells rapidly activated all 4 biosensors within minutes in Jurkat and K562 (Figure 3A-B). The maximal fold induction differed between the target cells and was not predicted by the relative amount of retroviral expression (supplemental Figure 2A). Jurkat cells demonstrated preferential activation of the GLS.IETD sensor. In K562 cells, the GLS.VGPD generated the highest fold activation in signal. Variation in peak biosensor signal was also observed between experiments; for example, GLS.VGPD ranged from 16- to 80-fold in K562, and from 4- to 10-fold in Jurkat cells. For this reason, one representative experiment of several is graphed. Because of the optimal, rapid signals obtained using K562 vs Jurkat target cells, we focused the majority of NK experiments using K562 target cells.
Figure 3.

Biosensors are activated after direct NK cell recognition of tumor target cells. Caspase and granzyme biosensors are activated in tumor target cells after NK effector conjugation. NK92 cells were conjugated at 1:1, with each target cell line expressing biosensors, and RLU was measured every 3 minutes for 240 minutes. The mean datum point at every 6 minutes is plotted. Fold activation in signal is shown for (A) Jurkat cells and (B) K562 cells. For inhibitor studies, K562 cells were preincubated with Q-VD-OPH (10 μM) or diluent, and NK92 cells were preincubated with C20 (50 μM); then fold activation in signal was measured for: (C) K562-GLS.VGPD and (D) K562-GLS.DEVD. (E) NK92 cells were conjugated at 3:1 with a K562 clonal line (GLS.SGR.5) expressing the GrzA/K sensor with diluent (phosphate-buffered saline [PBS]) or Mg2+-EGTA in PBS (4 mM). (F) To test specificity of GLS.SGR, K562 cells were preincubated with Q-VD-OPH (10 μM) or diluent, and NK92 cells were preincubated with C20 (50 μM), nafamostat mesylate (NM, 3.5 μM) or diluent, before conjugation at 3:1. Only NM inhibited GLS.SGR activation (P < .0001). The fold change in signal from 3 independent reads at 96 minutes is indicated. Data are represented as mean ± standard deviation (SD). ***P < .001; ns, not significant by one-way ANOVA. Shown is a representative figure from at least 3 independent experiments.
We assessed the dependence of the luciferase signal on GrzB and/or caspase activity by pretreating cells with a pan-caspase inhibitor (Q-VD-OPH)41 or a selective human GrzB inhibitor (C20).42 Based on cleavage prediction by GraBCas that the VGPD site would be restricted to GrzB proteolysis, we expected the GLS.VGPD signal to be suppressed by the inhibitor of GrzB, but not the pan-caspase inhibitor. This is clearly illustrated in Figure 3C, where there is no apparent impact of Q-VD-OPH on the NK92-mediated signal, and near complete suppression with C20 using K562 target cells expressing GLS.VGPD. Using this same concentration of C20, we measured caspase 3/7 activation in K562 cells. We anticipated that C20 would also fully inhibit the GLS.DEVD biosensor. As expected, Q-VD-OPH ablated the GLS.DEVD signal (Figure 3D). Surprisingly, C20 only partially blocked caspase 3/7 activation; a late signal was consistently detected in K562 expressing GLS.DEVD (Figure 3D). The most likely explanation for this finding is that the GLS.DEVD signal resulted from residual GrzB activity; however, we could not exclude the alternate hypothesis that GLS.DEVD activation was mediated via death receptor activation.
Novel granzyme A/K biosensor is activated through direct NK cell recognition of tumor target cells
We next tested the capacity of the newly developed GrzA/K biosensors to be triggered in the same kinetic live cell assay. Because GLS.SGR gave the highest signal after in vitro cleavage with both GrzA and GrzK, K562 cells were transfected with GLS.SGR, and a clonal line (GLS.SGR.5) that expressed the highest levels of luciferase was selected to further maximize the GLS.SGR signal (supplemental Figure 2B). NK92 cells were conjugated at an E:T of 3:1 with K562 cells to maximize the modest GLS.SGR signal (Figure 3E). Activation of the GLS.SGR biosensor was secretory granule–dependent, because Mg2+-EGTA eliminated the signal. We tested the specificity of activation of GLS.SGR by performing assays in the presence of NM, C20, or Q-VD-OPH. The GLS.SGR signal was inhibited only by NM, indicating that GrzB and caspases do not contribute to GLS.SGR activation (Figure 3F).
Human primary NK cells require IL-2 activation to activate granzyme biosensors
Like NK cell lines, ex vivo human NK cells are cytotoxic,43,44 constitutively express perforin and granzymes,45 and exhibit enhanced function upon IL-2 stimulation.46 We hypothesized that primary NK could induce biosensor activation and that IL-2 stimulation would further enhance biosensor signal. After isolation of primary human NK cells (>60% CD3– CD56+) from PBMCs, we measured NK cytolysis of K562 cells by Cr51 release assay. Function was nearly comparable with NK92 cells and significantly enhanced after culture in IL-2 for 24 hours (Figure 4A).
Figure 4.

Primary human NK cells elicit limited biosensor activation without IL-2 stimulation. Primary and IL-2–activated human NK cells were assessed for biosensor activation with K562 biosensors at an E:T of 3:1. Human NK cells were negatively selected from healthy donor PBMCs by magnetic bead isolation and confirmed to be >60% CD3– CD56+ by flow cytometry. NK92 was used as a positive control. (A) NK and 24-hour IL-2–stimulated NK are cytotoxic against K562 targets. Error bars represent SD from quadruplicate wells. One representative experiment of 2 is shown. (B) NK and 24-hour IL-2–stimulated NK are unable to activate the GLS.VGPD sensor in K562 clonal lines at an E:T of 0.75:1 (left), although GLS.DEVD activation (right) was observed by 90 minutes after stimulation with IL-2. One representative experiment of 3 with and without IL-2 stimulation is shown. (C) To maximize the biosensor signal, isolated NK were conjugated at a higher E:T (3:1) and the read extended to 240 minutes. NK cells were unable to activate GLS.VGPD (left) and GLS.SGR (middle), although consistent late activation of GLS.DEVD (>90 min) was observed (right). (D) After 6 days’ culture in 1000 U/mL IL-2, NK expanded from PBMCs readily activated GLS.VGPD (left), GLS.SGR (middle), and GLS.DEVD (right) expressed in K562 clonal lines at 3:1. The fold activation for 1 representative experiment of 3 is shown.
Next, we assessed the ability of primary NK to activate GrzB and caspase 3/7 biosensors, using either retrovirally-transduced K562 cells, or K562 clonal cell lines derived to express the highest level of biosensor signal. As shown in Figure 4B, primary human NK and IL-2–stimulated NK were unable to activate the GrzB biosensor, GLS.VGPD, expressed in K562 clone 1 (Figure 4B, left), suggesting that 24-hour IL-2 stimulation is insufficient to upregulate adequate GrzB. However, primary NK elicited minimal activation of the caspase 3/7 biosensor (GLS.DEVD) in K562 clone 6 by 90 minutes, and this signal was amplified after IL-2 stimulation (Figure 4B, right). To further enhance the biosensor signal, we increased the E:T of the conjugation assay to 3:1 and extended the assay to 240 minutes. However, we were unable to detect activation of GLS.VGPD or GLS.SGR. Minimal detection of late GLS.DEVD activation after 90 minutes was again observed, similar to that shown in Figure 4B. The data obtained using retrovirally transduced, polyclonal K562 cells were identical: no GLS.VGPD or GLS.SGR biosensor signal was noted in the absence of IL-2 stimulation, and minimal activation of GLS.DEVD (data not shown).
NK cell function was also tested using >20 individual donor PBMCs at an E:T of 20:1 (equivalent of NK E:T of 0.25-2:1, depending on donor). In the absence of IL-2 stimulation, a measurable signal was never obtained using K562-expressing GLS.VGPD or GLS.SGR. Samples sometimes showed a minimal, late GLS.DEVD signal. We cultured NK from PBMCs using the same donors shown in Figure 4C in 1000 units IL-2 for 6 days, and conjugated effectors at 3:1 with K562 clonal lines. Figure 4D indicates that this alternate expansion NK cell protocol led to a robust biosensor signal for GLS.VGPD and GLS.DEVD. The magnitude of increase of GLS.SGR signal was less dramatic, yet also approached the modest signal induced by NK92 cells.
Granzyme B and caspase biosensor activity correlates with NK cell PRF content
To evaluate PRF dependence of the NK kinetic biosensor assay, we silenced PRF protein expression in the KHYG1 NK cell line using a shRNA expressed by a retroviral vector.28 KHYG1 was chosen because this line gave reproducibly higher retroviral transduction levels compared with other NK effectors. PRF protein was reduced by >80% as confirmed by immunoblot (Figure 5A), and PRF-deficient KHYG1 cells demonstrated reduced cytolytic ability (Figure 5B). Analogous to the results obtained by pharmacologic inhibition of GrzB, inhibition of PRF expression in KHYG1 cells suppressed activation of all 4 biosensors in K562 cells. Figure 5 shows that the K562 lines with the highest signal were the GrzB sensor, GLS.VGPD (Figure 5C), and GrzB/caspase 8 sensor, GLS.IETD (Figure 5D). Because the PRF knockdown in KHYG1 shPRF was not 100%, it is possible that some of the GrzB biosensor activation could be caused by residual GrzB delivery.
Figure 5.
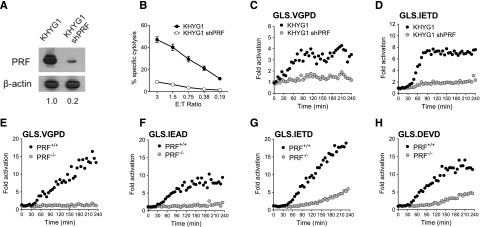
Caspase biosensors are activated by perforin-dependent and -independent mechanisms. The PRF dependence of the biosensor signal was tested with shRNA silencing of PRF1 in KHYG1 cells (A-D) and a patient-derived line deficient in PRF (E-H). (A) Expression of PRF in parental KHYG1 line and in KHYG1 shPRF transduced with shRNA specific to PRF1 3′UTR. PRF expression relative to KHYG1 is shown. Relative PRF expression to KHYG1 is shown below. (B) Cytotoxicity of KHYG1 shPRF cells against K562 targets is decreased. Error bars represent SEM from >10 independent experiments. Luciferase biosensors were sensitive to PRF knockdown in KHYG1 shPRF cells after 1:1 conjugation with K562 cells expressing (C) GLS.VGPD or (D) GLS.IETD. (E-H) HVS-CL line from PRF−/− patient or PRF+/+ donor reveals late caspase activation after 1:1 conjugation with K562 expressing: (E) GLS.VGPD, (F) GLS.IEAD, (G) GLS.IETD, or (H) GLS.DEVD. For luciferase assays, each datum point represents the mean of 4 replicate wells. One representative graph is shown from 3 independent experiments with similar results.
Because previous reports indicated that the majority of HVS-CL exhibited NK-like activity against K562 and Jurkat tumor target cells,22,47 we tested the capacity for PRF-sufficient (PRF+/+) and PRF-deficient (PRF−/−) HVS-CL to activate the GrzB and caspase biosensors in K562 cells. The PRF−/− cells were derived from a patient with hemophagocytic lymphohistiocytosis with no PRF protein (supplemental Figure 3A-B) as a result of biallelic 50delT truncations in PRF1.23 After 1:1 conjugation with biosensor-expressing K562 cells, PRF−/− HVS-CL did not activate GLS.VGPD or GLS.IEAD compared with PRF+/+ HVS-CL (Figure 5E-F). Despite being unable to kill K562 target cells in a cytolytic assay (supplemental Figure 3C), PRF−/− cells activated the GLS.IETD (GrzB/caspase 8) and GLS.DEVD (caspase 3/7) biosensors at a slightly delayed onset and a significantly lower level (Figure 5G-H). We previously showed that PRF−/− HVS-CL activate the caspase 3/7 biosensor in P815 cells through Fas activation,23 but K562 cells do not express Fas (supplemental Figure 2C).48,49 Taken together, these data suggested that the caspase 3/7 (GLS.DEVD) and caspase 8 biosensors (GLS.IETD) were activated by PRF and Fas-independent mechanisms during receptor-mediated direct recognition of K562 by HVS-CL.
Perforin-deficient human cytotoxic lymphocytes activate pro-apoptotic caspases in tumor target cells by death receptor ligation
PRF−/− murine NK cells can kill target cells by activating death receptor pathways,23 but similar findings using PRF−/− human NK cells have not been described. K562 and Jurkat cells both express TRAIL receptors, DR4/DR5, but only Jurkat cells are reported to be sensitive to rapid onset, TRAIL-mediated death.50,51 Likewise, Fas is also expressed by Jurkat,48,49 and reports have indicated killing by NK92.52 To measure the effect of death receptor function on caspase biosensor activation in K562 cells, we incubated PRF−/− HVS-CL with blocking antibodies against TRAIL before and during conjugation with target cells. Surprisingly, TRAIL blockade completely inhibited caspase 3/7, 8, and 10 biosensor activation in K562 cells (Figure 6A-B). To our knowledge, this is the first report of early caspase activation in K562 cells through NK cell–mediated TRAIL ligation. We repeated these experiments with PRF−/− HVS-CL and Jurkat cells, focusing on both TRAIL and Fas. Figure 6C indicates that blockade of TRAIL ligation led to incomplete diminution of caspase 8 biosensor (GLS.IETD) activation in Jurkat cells. Blocking FasL did not reliably inhibit the GLS.IETD signal unless TRAIL was concurrently blocked, demonstrating that both FasL and TRAIL death receptors were activated on Jurkat target cells by PRF−/− NK cells (Figure 6C-D). Taken together, these results indicate that caspase activation in the context of PRF−/− NK cells is dependent on death receptor ligation, and highlights differing sensitivities of tumor cell lines to caspase activation after death receptor signaling.
Figure 6.
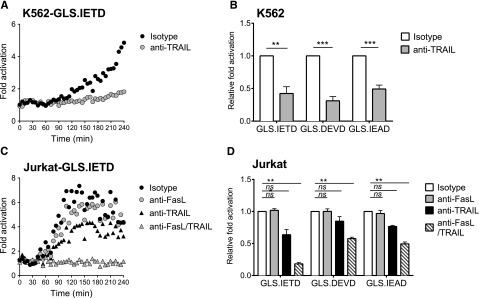
Perforin-deficient NK cells activate caspase biosensors by Fas and TRAIL death receptor ligation. HVS-CL cells from a PRF−/− patient were preincubated with blocking antibodies or with isotype control antibodies, and then incubated with tumor target cells in a 4-hour luciferase assay at 1:1. Fold signal activation over target cells alone was calculated. (A,C) Representative time course of signal activation in target cells expressing GLS.IETD caspase 8 biosensor. Each datum point represents the mean of 4 replicate wells. (B,D) The fold change in signal from 3 independent reads at 240 minutes was normalized to the isotype control. Data are represented as mean ± SEM from 3 experiments. (A-B) K562 target cells. (C-D) Jurkat target cells. **P < .01; ***P < .001; ****P < .0001; ns, not significant by one-way ANOVA.
Discussion
Although the 5 human Grz activate distinct methods of caspase-dependent and -independent cell death in tumor cells,1,2 and cytotoxicity is initiated rapidly by human NK cells,8,53 techniques to study kinetic Grz and caspase activation during target cell death are limited. Recent developments in murine systems now permit examination of the kinetics of Grz and caspase activation during CTL-mediated apoptosis,23 and visualization of tumoricidal caspase 3/7 activity in animals.29,54 Here, we have used luciferase biosensors to measure GrzB, GrzA, GrzK, and pro-apoptotic caspase activation in real time after human NK cell cytotoxicity induced by CD16 ligation and NK receptor–mediated recognition.
Our study highlights the differences in biosensor magnitude between 2 different tumor target cell lines, K562 and Jurkat, despite similar vector expression levels (supplemental Figure 2). For example, K562-expressed caspase 3/7 biosensors exhibited far superior magnitude compared with P815 (Figure 2) and Jurkat biosensors (Figure 3). In addition, the activity of GLS.IETD was robust in Jurkat cells compared with the other Jurkat biosensors (Figure 3A). Whether the differences in biosensor signal among P815, K562, and Jurkat cells are ascribable to cell-specific Grz inhibitor expression, death receptor activity, luciferase stability, adenosine triphosphate content, or another mechanism, is currently unknown.
The flexibility of the luciferase technology allowed the development of biosensors to measure the kinetics of delivery of GrzA and GrzK by human NK cells, and subsequent characterization of GrzA and GrzK inhibitors. Using the GLS.SGR biosensor at an E:T of 3:1, we detected delivery and activation of GrzA/K within 30 minutes, analogous to GrzB activation. The signal generated by GLS.SGR was modest compared with the other biosensors, which was surprising despite the higher E:T used when evaluating its activity in K562 cells, and the abundance of GrzA, GrzK, and PRF in NK92 cells (supplemental Figure 1C). The low signal could be explained by differences in accessibility of the enzyme active site55,56 or proteolytic sequence as a result of steric hindrance.25 It has also been suggested that GrzA accumulates in the target cell nucleus, rather than the cytoplasm, where the biosensors are expressed.57
The identification of nafamostat mesylate (NM) as an inhibitor of NK cell–derived GrzA and GrzK is an important finding. NM was previously shown to be a GrzA inhibitor in vitro and to partially inhibit T cell–mediated cytolysis.34 Several reports indicate that NM reduces cytotoxic lymphocyte-mediated target cell lysis without identifying the mechanism of inhibition.34,58 Despite evidence from murine models59,60 and a study demonstrating decreased NK92 cytotoxicity after GrzK knockdown,40 the importance of GrzA and GrzK to the cytotoxic capacity of human NK is unclear. Studies with combinations of NM and other protease inhibitors may begin to address these questions.
The magnitude of GrzB and pro-apoptotic caspase biosensor activation in K562 cells was largely dependent on GrzB and PRF delivery, because we found that IL-2–activated NK cell lines expressing high levels of PRF and GrzB induced rapid, robust biosensor activation, compared with NK cells expressing lower concentrations of cytotoxic proteins. Because PRF and GrzB are constitutively expressed in resting, primary human NK cells,45,46 we were surprised by the absence of GrzA and GrzB biosensors activation at 3:1 E:T using K562 clones with high-level luciferase expression (Figure 4B-C). Although it is possible that the biosensors lack sensitivity to detect the cytoplasmic granzyme signal, an alternate possibility is that primary NK cells do not deliver sufficient granzymes to the cytoplasm to cleave the protease-activated luciferase. In addition, it was surprising that GLS.DEVD biosensor activation was minimal and delayed in response to primary NK (Figure 4C). Whether the late caspase 3/7 activation elicited by primary NK is caused by GrzB activity below the level of biosensor detection, or by death receptor ligation, is under further investigation.
We evaluated primary NK cell function using K562 cells rather than Jurkat or P815 because of the enhanced signal-to-noise found with K562 cells, and the reported absence of death receptor activity.50,51 Future studies will focus on evaluating whether primary NK cells may activate pro-apoptotic caspases by death receptor activation in more susceptible cell lines, such as Jurkat or P815.
We previously identified that PRF–/– HVS-CL use FasL to trigger-activate caspase 3/7 in P815 cells.23 In the current study, we found that caspase activation in Jurkat was also partially dependent on Fas ligation (Figure 6), confirming previous reports.61,62 The highest signal induced by PRF–/– cells was with the caspase 8 sensor, GLS.IETD (Figure 6), in both K562 and Jurkat, confirming that caspase 8 is the first initiator caspase activated by DR4/DR5 and Fas receptor ligation in these tumor lines.18 In contrast, the signal from the caspase 10 biosensor, GLS.IEAD, was lower in magnitude. This may relate to suboptimal luciferase function or reflect the limited role reported for caspase 10 in death receptor signaling in these cells.21
The finding of significant caspase 3/7 activation mediated by TRAIL in K562 cells was quite surprising because previous reports noted the resistance of this cell line to TRAIL-induced cell death at 24 hours.63,64 Our studies indicate that early TRAIL signaling may lead to low level of caspase 3/7 activation in K562 (Figure 6B), with an insufficient signal to bring about cell death. Future studies will be required to determine whether the caspase signal induced by PRF–/– cytotoxic lymphocytes is limited to robust activation of a small fraction of the tumor cells whose death is not detectable by cytolytic assays, or is caused by activation of pro-apoptotic caspases at an insufficient magnitude to induce cell death.
In summary, the current study presents target cell–expressed biosensors as powerful tools to interrogate differences in cytotoxic function of human NK cells, and to allow further delineation of PRF and death receptor–mediated pathways. The ability to create and define novel GrzA and GrzK luciferase biosensors demonstrates the possibility of adaptation of the technology to understanding the kinetics of other granzymes involved in NK cell killing. In addition, using this novel methodology, we have revealed previously undetectable events in TRAIL-mediated early caspase activation in K562 cells, opening the door for future studies exploring the resistance to death in this myeloid leukemia cell line.
Acknowledgments
The authors gratefully acknowledge the gifts of the shRNA targeting PRF1 from Ilia Voskoboinik, GrzK from C. Alexander Valencia, and NKL cells from Jerome Ritz. IL-2 was obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH. Promega Corporation generated the GrzB/caspase 3 biosensor vectors and tested in vitro translated products at no cost.
The project described was supported by the National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant 8 UL1 TR000077-05 (K.A.R) and supported by a National Institutes of Health, National Institute of Allergy and Infectious Diseases training award (T32 AI-060515) (J.L.); and the HLH Center of Excellence at Cincinnati Children’s Hospital Medical Center.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: A.C.V. designed and performed research, analyzed data, and wrote the paper; A.E.H., S.K.F., J.M.F., B.F.B., B.L.B., and J.L. designed and performed research, analyzed data, and revised the paper; K.A.R. designed research, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: B.F.B. and B.L.B. are employees of Promega. The remaining authors declare no competing financial interests.
Correspondence: Kimberly Risma, Cincinnati Children's Hospital Medical Center, 3333 Burnet Ave, MLC 2000, Cincinnati, OH 45229; e-mail: kimberly.risma@cchmc.org.
References
- 1.Chowdhury D, Lieberman J. Death by a thousand cuts: granzyme pathways of programmed cell death. Annu Rev Immunol. 2008;26:389–420. doi: 10.1146/annurev.immunol.26.021607.090404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Susanto O, Trapani JA, Brasacchio D. Controversies in granzyme biology. Tissue Antigens. 2012;80(6):477–487. doi: 10.1111/tan.12014. [DOI] [PubMed] [Google Scholar]
- 3.Robertson MJ, Cochran KJ, Cameron C, Le JM, Tantravahi R, Ritz J. Characterization of a cell line, NKL, derived from an aggressive human natural killer cell leukemia. Exp Hematol. 1996;24(3):406–415. [PubMed] [Google Scholar]
- 4.Yamamoto K, Shibata F, Miyasaka N, Miura O. The human perforin gene is a direct target of STAT4 activated by IL-12 in NK cells. Biochem Biophys Res Commun. 2002;297(5):1245–1252. doi: 10.1016/s0006-291x(02)02378-1. [DOI] [PubMed] [Google Scholar]
- 5.Carayol G, Robin C, Bourhis JH, et al. NK cells differentiated from bone marrow, cord blood and peripheral blood stem cells exhibit similar phenotype and functions. Eur J Immunol. 1998;28(6):1991–2002. doi: 10.1002/(SICI)1521-4141(199806)28:06<1991::AID-IMMU1991>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 6.Liu CC, Rafii S, Granelli-Piperno A, Trapani JA, Young JD. Perforin and serine esterase gene expression in stimulated human T cells. Kinetics, mitogen requirements, and effects of cyclosporin A. J Exp Med. 1989;170(6):2105–2118. doi: 10.1084/jem.170.6.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fehniger TA, Cai SF, Cao X, et al. Acquisition of murine NK cell cytotoxicity requires the translation of a pre-existing pool of granzyme B and perforin mRNAs. Immunity. 2007;26(6):798–811. doi: 10.1016/j.immuni.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 8.Lopez JA, Susanto O, Jenkins MR, et al. Perforin forms transient pores on the target cell plasma membrane to facilitate rapid access of granzymes during killer cell attack. Blood. 2013;121(14):2659–2668. doi: 10.1182/blood-2012-07-446146. [DOI] [PubMed] [Google Scholar]
- 9.Metkar SS, Wang B, Ebbs ML, et al. Granzyme B activates procaspase-3 which signals a mitochondrial amplification loop for maximal apoptosis. J Cell Biol. 2003;160(6):875–885. doi: 10.1083/jcb.200210158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sutton VR, Wowk ME, Cancilla M, Trapani JA. Caspase activation by granzyme B is indirect, and caspase autoprocessing requires the release of proapoptotic mitochondrial factors. Immunity. 2003;18(3):319–329. doi: 10.1016/s1074-7613(03)00050-5. [DOI] [PubMed] [Google Scholar]
- 11.Andrade F, Roy S, Nicholson D, Thornberry N, Rosen A, Casciola-Rosen L. Granzyme B directly and efficiently cleaves several downstream caspase substrates: implications for CTL-induced apoptosis. Immunity. 1998;8(4):451–460. doi: 10.1016/s1074-7613(00)80550-6. [DOI] [PubMed] [Google Scholar]
- 12.McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol. 2013;5(4):a008656. doi: 10.1101/cshperspect.a008656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zamai L, Ahmad M, Bennett IM, Azzoni L, Alnemri ES, Perussia B. Natural killer (NK) cell-mediated cytotoxicity: differential use of TRAIL and Fas ligand by immature and mature primary human NK cells. J Exp Med. 1998;188(12):2375–2380. doi: 10.1084/jem.188.12.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Screpanti V, Wallin RP, Grandien A, Ljunggren HG. Impact of FASL-induced apoptosis in the elimination of tumor cells by NK cells. Mol Immunol. 2005;42(4):495–499. doi: 10.1016/j.molimm.2004.07.033. [DOI] [PubMed] [Google Scholar]
- 15.Eischen CM, Schilling JD, Lynch DH, Krammer PH, Leibson PJ. Fc receptor-induced expression of Fas ligand on activated NK cells facilitates cell-mediated cytotoxicity and subsequent autocrine NK cell apoptosis. J Immunol. 1996;156(8):2693–2699. [PubMed] [Google Scholar]
- 16.Sheard MA, Asgharzadeh S, Liu Y, et al. Membrane-bound TRAIL supplements natural killer cell cytotoxicity against neuroblastoma cells. J Immunother. 2013;36(5):319–329. doi: 10.1097/CJI.0b013e31829b4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Montel AH, Bochan MR, Hobbs JA, Lynch DH, Brahmi Z. Fas involvement in cytotoxicity mediated by human NK cells. Cell Immunol. 1995;166(2):236–246. doi: 10.1006/cimm.1995.9974. [DOI] [PubMed] [Google Scholar]
- 18.Crowder RN, El-Deiry WS. Caspase-8 regulation of TRAIL-mediated cell death. Exp Oncol. 2012;34(3):160–164. [PubMed] [Google Scholar]
- 19.Ashkenazi A, Holland P, Eckhardt SG. Ligand-based targeting of apoptosis in cancer: the potential of recombinant human apoptosis ligand 2/Tumor necrosis factor-related apoptosis-inducing ligand (rhApo2L/TRAIL). J Clin Oncol. 2008;26(21):3621–3630. doi: 10.1200/JCO.2007.15.7198. [DOI] [PubMed] [Google Scholar]
- 20.Walczak H, Miller RE, Ariail K, et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat Med. 1999;5(2):157–163. doi: 10.1038/5517. [DOI] [PubMed] [Google Scholar]
- 21.Sprick MR, Rieser E, Stahl H, Grosse-Wilde A, Weigand MA, Walczak H. Caspase-10 is recruited to and activated at the native TRAIL and CD95 death-inducing signalling complexes in a FADD-dependent manner but can not functionally substitute caspase-8. EMBO J. 2002;21(17):4520–4530. doi: 10.1093/emboj/cdf441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Biesinger B, Müller-Fleckenstein I, Simmer B, et al. Stable growth transformation of human T lymphocytes by herpesvirus saimiri. Proc Natl Acad Sci USA. 1992;89(7):3116–3119. doi: 10.1073/pnas.89.7.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li J, Figueira SK, Vrazo AC, et al. Real-time detection of CTL function reveals distinct patterns of caspase activation mediated by Fas versus granzyme B. J Immunol. 2014;193(2):519–528. doi: 10.4049/jimmunol.1301668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williams DA, Tao W, Yang F, et al. Dominant negative mutation of the hematopoietic-specific Rho GTPase, Rac2, is associated with a human phagocyte immunodeficiency. Blood. 2000;96(5):1646–1654. [PubMed] [Google Scholar]
- 25.Van Damme P, Maurer-Stroh S, Hao H, et al. The substrate specificity profile of human granzyme A. Biol Chem. 2010;391(8):983–997. doi: 10.1515/BC.2010.096. [DOI] [PubMed] [Google Scholar]
- 26.Hirata Y, Inagaki H, Shimizu T, et al. Expression of enzymatically active human granzyme 3 in Escherichia coli for analysis of its substrate specificity. Arch Biochem Biophys. 2006;446(1):35–43. doi: 10.1016/j.abb.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 27.Fan F, Binkowski BF, Butler BL, Stecha PF, Lewis MK, Wood KV. Novel genetically encoded biosensors using firefly luciferase. ACS Chem Biol. 2008;3(6):346–351. doi: 10.1021/cb8000414. [DOI] [PubMed] [Google Scholar]
- 28.Brennan AJ, Chia J, Browne KA, et al. Protection from endogenous perforin: glycans and the C terminus regulate exocytic trafficking in cytotoxic lymphocytes. Immunity. 2011;34(6):879–892. doi: 10.1016/j.immuni.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 29.Galbán S, Jeon YH, Bowman BM, et al. Imaging proteolytic activity in live cells and animal models. PLoS One. 2013;8(6):e66248. doi: 10.1371/journal.pone.0066248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bade B, Boettcher HE, Lohrmann J, et al. Differential expression of the granzymes A, K and M and perforin in human peripheral blood lymphocytes. Int Immunol. 2005;17(11):1419–1428. doi: 10.1093/intimm/dxh320. [DOI] [PubMed] [Google Scholar]
- 31.Bovenschen N, Quadir R, van den Berg AL, et al. Granzyme K displays highly restricted substrate specificity that only partially overlaps with granzyme A. J Biol Chem. 2009;284(6):3504–3512. doi: 10.1074/jbc.M806716200. [DOI] [PubMed] [Google Scholar]
- 32.Shi L, Kam CM, Powers JC, Aebersold R, Greenberg AH. Purification of three cytotoxic lymphocyte granule serine proteases that induce apoptosis through distinct substrate and target cell interactions. J Exp Med. 1992;176(6):1521–1529. doi: 10.1084/jem.176.6.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mori S, Itoh Y, Shinohata R, Sendo T, Oishi R, Nishibori M. Nafamostat mesilate is an extremely potent inhibitor of human tryptase. J Pharmacol Sci. 2003;92(4):420–423. doi: 10.1254/jphs.92.420. [DOI] [PubMed] [Google Scholar]
- 34.Poe M, Wu JK, Blake JT, Zweerink HJ, Sigal NH. The enzymatic activity of human cytotoxic T-lymphocyte granzyme A and cytolysis mediated by cytotoxic T-lymphocytes are potently inhibited by a synthetic antiprotease, FUT-175. Arch Biochem Biophys. 1991;284(1):215–218. doi: 10.1016/0003-9861(91)90286-r. [DOI] [PubMed] [Google Scholar]
- 35.Shivakumar P, Mourya R, Bezerra JA. Perforin and granzymes work in synergy to mediate cholangiocyte injury in experimental biliary atresia. J Hepatol. 2014;60(2):370–376. doi: 10.1016/j.jhep.2013.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Legrand F, Driss V, Delbeke M, et al. Human eosinophils exert TNF-α and granzyme A-mediated tumoricidal activity toward colon carcinoma cells. J Immunol. 2010;185(12):7443–7451. doi: 10.4049/jimmunol.1000446. [DOI] [PubMed] [Google Scholar]
- 37.Yagita M, Huang CL, Umehara H, et al. A novel natural killer cell line (KHYG-1) from a patient with aggressive natural killer cell leukemia carrying a p53 point mutation. Leukemia. 2000;14(5):922–930. doi: 10.1038/sj.leu.2401769. [DOI] [PubMed] [Google Scholar]
- 38.Gong JH, Maki G, Klingemann HG. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia. 1994;8(4):652–658. [PubMed] [Google Scholar]
- 39.Suck G, Branch DR, Smyth MJ, et al. KHYG-1, a model for the study of enhanced natural killer cell cytotoxicity. Exp Hematol. 2005;33(10):1160–1171. doi: 10.1016/j.exphem.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 40.Jiang W, Chai NR, Maric D, Bielekova B. Unexpected role for granzyme K in CD56bright NK cell-mediated immunoregulation of multiple sclerosis. J Immunol. 2011;187(2):781–790. doi: 10.4049/jimmunol.1100789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Caserta TM, Smith AN, Gultice AD, Reedy MA, Brown TL. Q-VD-OPh, a broad spectrum caspase inhibitor with potent antiapoptotic properties. Apoptosis. 2003;8(4):345–352. doi: 10.1023/a:1024116916932. [DOI] [PubMed] [Google Scholar]
- 42.Willoughby CA, Bull HG, Garcia-Calvo M, Jiang J, Chapman KT, Thornberry NA. Discovery of potent, selective human granzyme B inhibitors that inhibit CTL mediated apoptosis. Bioorg Med Chem Lett. 2002;12(16):2197–2200. doi: 10.1016/s0960-894x(02)00363-3. [DOI] [PubMed] [Google Scholar]
- 43.Caligiuri MA, Murray C, Robertson MJ, et al. Selective modulation of human natural killer cells in vivo after prolonged infusion of low dose recombinant interleukin 2. J Clin Invest. 1993;91(1):123–132. doi: 10.1172/JCI116161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Molleran Lee S, Villanueva J, Sumegi J, et al. Characterisation of diverse PRF1 mutations leading to decreased natural killer cell activity in North American families with haemophagocytic lymphohistiocytosis. J Med Genet. 2004;41(2):137–144. doi: 10.1136/jmg.2003.011528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mellor-Heineke S, Villanueva J, Jordan MB, et al. Elevated Granzyme B in Cytotoxic Lymphocytes is a Signature of Immune Activation in Hemophagocytic Lymphohistiocytosis. Front Immunol. 2013;4(72):72. doi: 10.3389/fimmu.2013.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dybkaer K, Iqbal J, Zhou G, et al. Genome wide transcriptional analysis of resting and IL2 activated human natural killer cells: gene expression signatures indicative of novel molecular signaling pathways. BMC Genomics. 2007;8(230):230. doi: 10.1186/1471-2164-8-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pon RA, Freedman MS. Study of Herpesvirus saimiri immortalization of gammadelta T cells derived from peripheral blood and CSF of multiple sclerosis patients. J Neuroimmunol. 2003;139(1-2):119–132. doi: 10.1016/s0165-5728(03)00157-7. [DOI] [PubMed] [Google Scholar]
- 48.Owen-Schaub LB, Yonehara S, Crump WL, III, Grimm EA. DNA fragmentation and cell death is selectively triggered in activated human lymphocytes by Fas antigen engagement. Cell Immunol. 1992;140(1):197–205. doi: 10.1016/0008-8749(92)90187-t. [DOI] [PubMed] [Google Scholar]
- 49.Owen-Schaub LB, Zhang W, Cusack JC, et al. Wild-type human p53 and a temperature-sensitive mutant induce Fas/APO-1 expression. Mol Cell Biol. 1995;15(6):3032–3040. doi: 10.1128/mcb.15.6.3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Simon AK, Williams O, Mongkolsapaya J, et al. Tumor necrosis factor-related apoptosis-inducing ligand in T cell development: sensitivity of human thymocytes. Proc Natl Acad Sci USA. 2001;98(9):5158–5163. doi: 10.1073/pnas.091100398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hietakangas V, Poukkula M, Heiskanen KM, Karvinen JT, Sistonen L, Eriksson JE. Erythroid differentiation sensitizes K562 leukemia cells to TRAIL-induced apoptosis by downregulation of c-FLIP. Mol Cell Biol. 2003;23(4):1278–1291. doi: 10.1128/MCB.23.4.1278-1291.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reid GSD, Bharya S, Klingemann HG, Schultz KR. Differential killing of pre-B acute lymphoblastic leukaemia cells by activated NK cells and the NK-92 ci cell line. Clin Exp Immunol. 2002;129(2):265–271. doi: 10.1046/j.1365-2249.2002.01919.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jenkins MR, Rudd-Schmidt JA, Lopez JA, et al. Failed CTL/NK cell killing and cytokine hypersecretion are directly linked through prolonged synapse time. J Exp Med. 2015;212(3):307–317. doi: 10.1084/jem.20140964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hoff BA, Chughtai K, Jeon YH, et al. Multimodality imaging of tumor and bone response in a mouse model of bony metastasis. Transl Oncol. 2012;5(6):415–421. doi: 10.1593/tlo.12298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hink-Schauer C, Estébanez-Perpiñá E, Kurschus FC, Bode W, Jenne DE. Crystal structure of the apoptosis-inducing human granzyme A dimer. Nat Struct Biol. 2003;10(7):535–540. doi: 10.1038/nsb945. [DOI] [PubMed] [Google Scholar]
- 56.Hink-Schauer C, Estébanez-Perpiñá E, Wilharm E, et al. The 2.2-A crystal structure of human pro-granzyme K reveals a rigid zymogen with unusual features. J Biol Chem. 2002;277(52):50923–50933. doi: 10.1074/jbc.M207962200. [DOI] [PubMed] [Google Scholar]
- 57.Lieberman J. Granzyme A activates another way to die. Immunol Rev. 2010;235(1):93–104. doi: 10.1111/j.0105-2896.2010.00902.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yamamoto M, Saigo K, Koshiba M, et al. Inhibition of natural killer cytotoxicity in vitro by clinical grade serine protease inhibitors. Haematologia (Budap) 2002;32(2):103–111. doi: 10.1163/156855902320387934. [DOI] [PubMed] [Google Scholar]
- 59.Susanto O, Stewart SE, Voskoboinik I, et al. Mouse granzyme A induces a novel death with writhing morphology that is mechanistically distinct from granzyme B-induced apoptosis. Cell Death Differ. 2013;20(9):1183–1193. doi: 10.1038/cdd.2013.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pardo J, Bosque A, Brehm R, et al. Apoptotic pathways are selectively activated by granzyme A and/or granzyme B in CTL-mediated target cell lysis. J Cell Biol. 2004;167(3):457–468. doi: 10.1083/jcb.200406115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Munker R, Marini F, Jiang S, Savary C, Owen-Schaub L, Andreeff M. Expression of CD95(FAS) by gene transfer does not sensitize K562 to Fas-killing. Hematol Cell Ther. 1997;39(2):75–78. doi: 10.1007/s00282-997-0075-7. [DOI] [PubMed] [Google Scholar]
- 62.McGahon AJ, Nishioka WK, Martin SJ, Mahboubi A, Cotter TG, Green DR. Regulation of the Fas apoptotic cell death pathway by Abl. J Biol Chem. 1995;270(38):22625–22631. doi: 10.1074/jbc.270.38.22625. [DOI] [PubMed] [Google Scholar]
- 63.Hao J-H, Yu M, Liu F-T, Newland AC, Jia L. Bcl-2 inhibitors sensitize tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by uncoupling of mitochondrial respiration in human leukemic CEM cells. Cancer Res. 2004;64(10):3607–3616. doi: 10.1158/0008-5472.CAN-03-3648. [DOI] [PubMed] [Google Scholar]
- 64.Jia L, Patwari Y, Srinivasula SM, et al. Bax translocation is crucial for the sensitivity of leukaemic cells to etoposide-induced apoptosis. Oncogene. 2001;20(35):4817–4826. doi: 10.1038/sj.onc.1204628. [DOI] [PubMed] [Google Scholar]