

Abstract
Enhanced gas-phase cleavage of peptides adjacent to histidine was investigated. The peptides examined were angiotensins III (RVYIHPF) and IV (VYIHPF) as well as synthetic peptide analogs with altered key residues ((R)VYI-X-Z-F; X=F or H and Z=A, P or Sar) or a fixed charge Φ3P+CH2C(O)-VYIHPF. While all singly protonated peptide ions containing both histidine and arginine fragment non-selectively, the doubly protonated peptide ions with arginine and histidine, and the singly protonated peptides containing histidine but not arginine, cleave in a selective manner. In particular, dominant complementary b+/y+ product ions resulting from cleavage between the HP amide bond are observed. For the fixed-charge derivative, selective cleavage occurs only if a proton is added to produce a doubly charged precursor. The results are consistent with involvement of a protonated histidine in the selective cleavage. The ratio of b+/y+ is determined by the identity of the residue C-terminal to histidine and by the ability of protonated histidine to transfer a proton to the C-terminal leaving fragment. This was probed further by systematically changing the residue C-terminal to histidine and by alkylating histidine. The results indicate that while b+/y+ complementary ion pairs dominate in doubly protonated RVYIHPF, b52+ and b62+ product ions dominate the spectra of doubly protonated RVYIHAF. Also, dominant b52+ product ions are observed when the histidine side chain is alkylated (H*) in doubly protonated RVYIH*PF. Based on all of the results, a selective fragmentation mechanism for enhanced cleavage at histidine involving an atypical b ion structure is proposed.
Introduction
Proteomics and related biosciences rely on mass spectrometry and tandem mass spectrometry (MS/MS) for peptide sequencing and protein identification.1, 2 Moreover, obtaining structural information by MS/MS, especially for proteins and peptides, is of paramount importance in this “post-genome” era in which characterization of proteins is a necessary step in understanding gene function. To meet this goal, computer algorithms now exist for correlating peptide MS/MS spectra with theoretical MS/MS spectra (or predicted fragmentation spectra), generated from sequence strings in various protein and translated nucleotide databases.3–7 While such computer programs are proving highly useful for peptide sequence determination and/or protein identification following LC-MS/MS,8, 9 their utility might be improved if our basic knowledge of peptide dissociation mechanisms and energetics could be increased. Current algorithms perform this identification with little or no priori insight into what rules (either general or specific) govern peptide dissociation. Hence, false positive correlation scores can be obtained between theoretical and experimental MS/MS spectra of peptides. As a consequence, protein identification using this MS/MS approach can be wrong. Thus, incorporation of robust peptide fragmentation rules to these computer algorithms, through fundamental and applied research on peptide dissociation, may be beneficial.
In MS/MS of peptides, fragment ions result from cleavages along the peptide backbone10 and/or at the various amino acid side chains. Cleavage of the amide bond with retention of the charge on the C-terminal peptide fragment results in yn ions, whereas if the charge is retained on the N-terminal side, bn ions are generated.11–13 Ions 28 u lower in mass than bn fragment ions are designated an ions. Immonium ions produced from various amino acids present in the peptides, as well as ions that correspond formally to the loss of H2O (bn° or yn°) and/or NH3 (bn* or yn*) from y or b fragment ions, are also present in MS/MS spectra of protonated peptides.14–17 The mobile proton model8, 13–16, 18, 19 has been employed to rationalize selective and non-selective fragmentation in protonated peptides; strongly basic residues such as arginine are expected to sequester the ionizing proton,20 and have been associated with higher energy requirements for fragmentation.21
Peptides, although polymeric in nature, offer many challenges for sequencing in the gas phase because fragmentation is not always general or non-selective. Many factors can influence peptide dissociation in the gas phase including the nature of the residues present (neutral vs. acidic vs. basic),21–23 the charge state of the precursor ion,21, 24 the composition of the peptide backbone (amide vs. N-alkylated),25, 26 the size27 and the conformation of the peptide.18, 28–33 In some instances some of these factors can lead to enhanced or specific cleavages at certain peptide bonds, limiting the information obtained from the MS/MS spectrum. As an example, it has recently been shown that selective cleavages dominate the MS/MS spectra of peptides with arginine and acidic residues.23, 34–36 Selective cleavages at acidic residues dominate the MS/MS spectra of these peptides if the number of ionizing protons does not exceed the number of arginine residues. Similar selective cleavage (C-terminal to acidic residues) has also been observed on statistical analysis of a CID spectral database of doubly charged peptides with histidine, arginine and acidic residues.37 Alternatively, non-selective cleavages occur over the peptide backbone in the presence of a mobile proton (a proton in excess of the number of basic residues in the peptide). In support of the utility of this fragmentation rule, and in spite of the fact that it was developed and tested for peptides, the work of Stephenson et al.38 shows that [M+11H]11+ ions of lysozyme, a 14.3 kDa polypeptide that contains 11 arginine residues, fragment predominantly between Asp48-Gly49 and Asp18-Asn19 resulting in two complementary pairs of product ions. The [M+14H]14+ precursor ions of lysozyme, however, fragment non-selectively, consistent with the fact that the number of protons added is greater than the number of arginine residues available to sequester protons.
In this study, we attempt to gain a better understanding of the propensity of certain peptides to be ionized at a specific basic residue, histidine, and investigate the influence of this protonation on selective cleavage at histidine. Recently, data mining of a set of peptide spectra has shown that histidine is the amino acid residue most likely to show preferential cleavage at its C-terminal side in doubly-protonated tryptic peptides (when histidine is likely to be protonated at its side chain rather than N-terminal).39 In the research described below, enhanced cleavage at histidine is demonstrated for singly and doubly protonated model peptides using a variety of activation methods that deposit different internal energy distributions and access different time frames for dissociation. The dependence of selective cleavages at histidine on the nature of the residue C-terminal to histidine was tested by systematic residue substitutions from Pro to Sar to Ala. This is especially important because proline is another amino acid residue that directs selective cleavage of the peptide backbone. Either complementary b+/y+ product ions or b2+ product ions at histidine are produced depending on the nature of the residue C-terminal to histidine. Moreover, a plausible formation mechanism common to both these types of product ions is proposed. Finally, MS/MS/MS and resonance ejection experiments provide data that support a unique histidine b ion structure. A brief summary of this work was presented in an article highlighting the role of mobile protons in peptide fragmentation;40 the present paper provides the data and analysis that support the proposed mechanism and structure. A related computational study on b ion structure involving Histidine has been published.41
Experimental Section
Peptide Synthesis
Angiotensin III (RVYIHPF), and Angiotensin IV(VYIHPF) were purchased from Sigma (St. Louis, MO) as the acetate salts and used without any further purification. Iodoacetic anhydride and tris(2,4,6-trimethoxyphenyl)phosphine were also purchased from Sigma. Any synthetic peptides required were prepared by solid-phase synthesis protocols outlined by Atherton and Sheppard.42 9-Fluoroenylmethoxycarbonyl (Fmoc) derivatives of the various amino acids required (Nα-Fmoc-Ng-2,2,5,7,8-pentamethylchroman-6-sulfonyl-L-arginine; Fmoc-L-valine; Fmoc-O-t-butyl-L-tyrosine; Nα-Fmoc-Nim-trityl-L-histidine; Fmoc-L-alanine monohydrate; Fmoc-L-phenylalanine; Fmoc-L-proline) were purchased from Advanced Chemtech (Louisville, KY) with the exception of Nα-Fmoc-im-benzyl-L-histidine (H*) which was purchased from Sigma. The C-terminal residues required for peptide synthesis were purchased already attached to resins from Calbiochem/Novabiochem (San Diego, CA) as was sarcosine [denoted as Sar; Nα-Fmoc-L-sarcosine (N-methylglycine)]. All other reagents required during synthesis were from Aldrich (Milwaukee, WI). Deprotection was performed by using piperidine (50%) in dimethylformamide (DMF). Coupling reactions between successive L-amino acid residues was performed in a solution of DMF containing a 2–4 fold excess of N-hydroxybenzotriazole dihydrate (HOBt), benzotriazole-1-yl-oxy-tris-(dimethyamino)-phosphonium hexafluorophosphate (BOP; Castro’s reagent), diisopropylethylamine (DIEA), and the appropriate Fmoc-protected amino acid. The synthesized peptides were removed from the resin and deprotected using a mixture of 95% trifluoroacetic acid (TFA), 2.5% H2O and 2.5% 1,2-ethanedithiol (EDT) for 1.5 h at ambient temperature. In the case of RVYIH*PF, deprotection and cleavage from the resin was carried out for 1 h because the alkyl group is stable under these conditions.43 Once each synthetic peptide was precipitated in diethyl ether, its purity and identity were checked by mass spectrometry. For the fixed-charge derivative of peptide VYIHPF, the resin-bound peptide (0.011 mmol) with the free N-terminus was reacted with 3x molar excess iodoacetic anhydride in anhydrous DMF for 30 min, washed with DMF twice, and then reacted with 4x molar excess tris(2,4,6-trimethoxyphenyl)phosphine in toluene/DMF (1:1 v/v) for 90 min. The resin-bound fixed-charge VYIHPF derivative (i.e., Φ3P+CH2C(O)-VYIHPF), was then washed with toluene, DMF, and then finally with dicholoromethane (DCM). Cleavage from the resin and peptide isolation were carried out as described above.
Surface-Induced Dissociation
The instrument used for the SID experiments is a custom-built tandem quadrupole mass spectrometer (Q1Q2) specifically designed for low-energy ion-surface collisions. It has been described previously in greater detail.19,21,44 The singly- and doubly protonated peptides were formed by electrospray ionization using a heated capillary electrospray design based on those of Chowdhury et al.45 and Papac et al..46 Peptide analytes were dissolved in a 70/30% (v/v) mixture of methanol/H2O containing 1% acetic acid to give the appropriate concentration (~ 30–50 pmol/µL)21, and sprayed at atmospheric pressure from a syringe needle (Syringe pump: Sage ATI Orion Model 361) held at 4.2–4.7 kV (flow rate of 2 µL/min), towards a metal capillary (120 V). The temperature of the metal capillary was maintained at 120oC to ensure proper desolvation of the ions. The desolvated ions were directed toward a skimmer cone (90 V), after which they entered into the high-vacuum region of the mass spectrometer, where they were analyzed and detected. SID was accomplished by changing the potential difference between the ion source skimmer cone and the surface; for multiply protonated peptide ions the potential difference is multiplied by the number of charges to calculate the ion kinetic energy.
The chemically modified surfaces used in the SID experiments were self-assembled monolayer films of 2-(perfluorooctyl)ethanethiol [CF3(CF2)7CH2CH2S-Au, referred to as FC10 SAM] or 2-(perfluorodecyl)ethanethiol [CF3(CF2)9CH2CH2S-Au, referred to as FC12 SAM] on gold.47 Gold surfaces (1000Å of vapor-deposited gold on silica with a 50Å adhesion layer of titanium between the silica and the gold) were obtained from Evaporated Metal Films (Ithaca, NY) and cleaned by using a UV cleaner (UV-Clean, Boekel, Philadelphia, PA) before immersion into the alkanethiol solution (1 mM in ethanol, 24 hr) as previously described.21
Sustained Off-Resonance Irradiation
A Bruker 7-tesla Fourier transform mass spectrometer (FTICR-MS)48 combined with an Analytica electrospray ionization source was also used for MS/MS studies. The peptide solutions (5 µM), were electrosprayed (4 kV) into the vacuum system at a flow rate of 0.5 µL/min using a Harvard Apparatus syringe pump. A glass capillary coated with metal on both ends transfers the ions from atmospheric pressure into the vacuum system. Desolvation of the sample was achieved by heating the capillary with counterflow N2 gas at 200°C. The CID activation method employed was sustained off-resonance irradiation (SORI),49 a slow multi-step activation process in which ion dissociation processes that occur in the several msec to sec time scale can be observed. The collision gas pulsed into the ion cyclotron resonance (ICR) cell was CO2. For the SORI-CID experiments, the radio frequency (RF) irradiation was offset by 700 Hz above or below the resonance frequency of the selected ions.49, 50 For the resonance ejection experiments, doubly protonated RVYIHAF produced in the external ESI source was isolated by using correlated sweep and then excited at its cyclotron frequency to a certain kinetic energy. The product ions of interest produced during the activation (i.e., b62+) were ejected out of the cell by applying a resonant ejection pulse (1s) in order to prevent their further dissociation. In this manner, further fragmentation of the b62+ product ions in the cell does not occur (unless the fragmentation is faster than the minimum time required for ejection) and only fragment ions (other than the ejected ions) originating from the precursor ions are observed.
Low-Energy Collision-Induced Dissociation
Low-energy CID experiments were performed with a Finnigan LCQ ion trap mass spectrometer (San Jose, CA). The peptides were dissolved in a solution of H2O:MeOH (1:1; v/v) containing 1% acetic acid to make up a concentration of 20–30 pmol/µL. The peptide solutions were then sprayed into the mass spectrometer at a rate of 3 µL/min. The applied needle voltage used was 4.8 kV and the capillary temperature was maintained at 200°C for all samples. Unit mass selection of the precursor ion was performed in order to avoid ambiguities from isotope contributions. The excitation energy (indicated as % collision energy by the manufacturer), was incremented in small steps to selectively monitor low energy fragmentation processes for the precursor ions selected. For MS/MS/MS, desired fragment ions produced from the precursor (MS/MS) were selected and isolated in the trap and then excited as described above. Energy resolved mass spectral “breakdown” curves were accomplished by selecting the product ion of interest in the ion trap and performing MS/MS/MS at incremented energy steps (% relative collision energies as referenced by the manufacturer). The percent relative abundance of the fragment ions following MS/MS/MS were plotted as a function of energy.
Molecular Modeling
The peptides RVYIHPF and RVYIHAF were built in Macromodel51 (Version 6.0) using the program’s amino acid library. They were then modified to carry two positive charges with the charges being placed on the arginine and histidine residue side chains. As either of the two nitrogens (τ- and π-) on the side chain of histidine can carry the ionizing proton, each nitrogen atom was sequentially protonated such that two versions of doubly protonated RVYIHPF and RVYIHAF were modeled. The built-in protonated peptide structures were first subjected to 500 optimization iterations in Macromodel resulting in an energetically refined starting structure. Stochastic molecular dynamics using the AMBER force field52 were then performed on each of these structures using the Batchmin program of Macromodel. AMBER force field default parameters were used for the electrostatic and hydrogen bonding treatments during the molecular dynamics. Moreover, large distance cut off values were assigned to ensure smoother convergence and the Polak-Riviere Conjugate Gradient (RPCG)53 was used for the program’s energy minimization routines. In all, 10 000 structures were sampled during the stochastic dynamics run (1 fs time step; 10 000 ps total time; 800 K initial and final temperature; 0.5 ps bath time). Our choice of temperature for the molecular dynamics was based on previous analysis of kinetic energy transfer into internal energy for singly protonated YGGFL using thermal decomposition kinetics methods.54 Those results showed that applying the thermal kinetic parameters to the dissociation rate constant for singly protonated YGGFL at the inflection point of the SID fragmentation efficiency curve yields an effective temperature of 800 K.
Results and Discussion
I. Gas-Phase Dissociation of Angiotensin III (RVYIHPF) and IV (VYIHPF)
(A) Selective Fragmentation in Doubly Protonated RVYIHPF
Figure 1 shows the MS/MS spectra of doubly protonated RVYIHPF under SORI-CID conditions. Selective fragmentation is observed for RVYIHPF with y2 and b5 as the dominant product ions. These product ions are the result of cleavage at the HP amide bond. This selective cleavage is most clearly represented in the SORI-CID spectrum of RVYIHPF (Figure 1). SID and low-energy CID also show this cleavage, although to a less striking extent. The a5, b4/a4 and b3/a3 ions produced by SID (supplementary figure 1A) are likely the result of further fragmentation of the b5 product ion, which supports an enhanced cleavage at the HP bond. It is common to see pairs of b and a ions produced by SID. The quadrupole ion trap instrument which discriminates against low mass ions of m/z below 35% of the parent mass,55 is responsible for the low abundance of the y2 product ion in the low-energy CID spectrum (supplementary figure 1B). Strong complementary b6/y2 and b6/y4 ions were also observed in the SORI-CID spectra of doubly protonated angiotensin II (DRVYIHPF) and low-energy CID spectra of doubly protonated angiotensin I (DRVYIHPFHL), respectively (supplementary figures 2 & 3).
Figure 1.

MS/MS spectrum of doubly protonated RVYIHPF obtained by sustained off-resonance irradiation at an excitation energy of 6 eV; Electrospray ionization.
Selective cleavage C-terminal to histidine as seen here (Figure 1), has been previously observed under CID conditions with an ESI double focusing sector instrument by Hu and Loo for Ni2+, Co2+, and Zn2+ complexes of peptides.56 Moreover, dominant b+/y+ product ion pairs (b6/y2 in DRVYIHPF and b5/y2 in RVYIHPF) have also been documented by Adams et al. and Sullards et al.57, 58 who measured the kinetic energy release of the b5 and y2 ions and concluded that these were complementary ions resulting from cleavage at the HP amide bond in doubly protonated RVYIHPF. These authors however, stated that it is not clear whether the fragmentation is charge-directed or charge-remote. The work of Tang and Boyd also supports the direct dissociation to complementary b+/y+ product ions from doubly protonated ions of tryptic peptides.59
(B) Selective Cleavage in Singly Protonated VYIHPF
Figure 2 shows the MS/MS spectra of singly protonated VYIHPF under SORI-CID and SID conditions (see supplementary figure 4 for ion-trap CID spectrum). Once again, selective cleavage at the HP amide bond is seen leading to either a b4 or, in SID, also a y2 product ion. Differences in the time frame of the dissociation event and the energy imparted to the ion during the collision contribute to the differences seen in the spectra (compare Figures 2A and 2B). Because singly protonated VYIHPF does not contain a highly basic arginine to sequester the ionizing proton, these results suggest that the single ionizing proton, likely located on the basic side chain of His60, 61 is promoting the selective cleavage at the HP amide bond, consistent with the selective cleavage that occurs for peptides containing arginine, histidine and two ionizing protons (Figure 1). Upon dissociation, if the charge is retained on the N-terminal fragment, a b-type ion results (i.e., b4), while if the charge is retained on the C-terminal fragment, a y-type ion (i.e., y2) is formed. Singly protonated AAHAA and AAAAAHAA were also dissociated by SID and low-energy CID (supplementary figures 5 & 6). The MS/MS results reveal that abundant product ions are produced that correspond to cleavage C-terminal to histidine (b3 and b6, respectively), although other product ions, including a strong bm-1 (m= total number of residues), are also detected.
Figure 2.

MS/MS spectrum of singly protonated VYIHPF obtained by (A) sustained off-resonance irradiation at an excitation energy of 19 eV and (B) SID at a collision energy of 35 eV on a 2-(perfluorooctyl)ethanethiolate [CF3(CF2)7CH2CH2S-Au] monolayer surface; Electrospray ionization.
(C) Non-selective Fragmentation in Singly Protonated RVYIHPF
Figure 3 shows the SORI-CID MS/MS spectrum of singly protonated RVYIHPF (see supplementary figure 7 for SID and ion-trap CID spectra). It is apparent from a comparison of this spectrum (Figure 3; 34eV excitation energy) with that for the doubly protonated peptide (Figure 1; 6eV excitation energy), that singly protonated RVYIHPF ions require higher energies for the fragmentation process (as predicted by the mobile proton model for a peptide with one arginine and one proton21) and fragment to produce a number of sequence ions across the mass range. Fragment ions that differ from b and a ions by the formal loss of ammonia15 are also abundant in the spectrum (designated by *). Previous work by Starrett and DiDonato62 using collision induced dissociation in a reversed-geometry double-focusing mass spectrometer has also demonstrated non-selective fragmentation seen here for singly protonated angiotensin II (DRVYIHPF) (supplementary figure 8 and 9).
Figure 3.

MS/MS spectrum of singly protonated RVYIHPF obtained by sustained off-resonance irradiation at an excitation energy of 34 eV; Electrospray ionization.
(D) Non-selective Fragmentation in Φ3P+CH2C(O)-VYIHPF and Selective Fragmentation in [Φ3P+CH2C(O)-VYIHPF +H+]2+
As described above, the addition of two protons to RVYIHPF with subsequent activated dissociation results in selective cleavage of the HP amide bond while addition of only one proton gives non-selective fragmentation. A fixed charge [i.e., tris(2,4,6-trimethoxy phenyl)phosphine)] was placed at the N-terminus of VYIHPF to mimic the protonated arginine in RVYIHPF (i.e., to provide charge that is not mobile, that doesn’t readily intramolecularly transfer to another basic site). The fragmentation of this fixed-charge derivative was evaluated in the absence and presence of an ionizing proton. Figure 4A shows the low-energy (ion trap) CID spectrum of Φ3P+CH2C(O)-VYIHPF and as expected, the fragmentation is non-selective. Selective fragmentation at the HP amide bond, which mirrors that seen with doubly protonated RVYIHPF, is observed when a proton is added to this fixed-charge peptide derivative (Φ3P+CH2C(O)-VYIHPF+H+; Figure 4B). The data suggest that protonation at the histidine side chain by an available proton directs this selective cleavage.
Figure 4.

Low-energy CID spectrum of (A) Φ3P+CH2C(O)-VYIHPF at 30% relative collision energy, and (B) [Φ3P+CH2C(O)-VYIHPF+H]2+ at 22% relative collision energy; Electrospray ionization.
II. Histidine b ion Formation and Structure
In general, the combined mass spectral results presented above indicate that the addition of a second ionizing proton to angiotensin III (RVYIHPF) has a significant effect on the fragmentation pattern with the doubly protonated peptides dissociating in a selective manner. This is in contrast to peptides containing a single Arg and no His, whose doubly protonated precursor ions exhibit non-selective cleavage at the various amide bonds.11, 15, 19, 21, 22, 34, 63 Because it is well established that a highly basic arginine side chain will sequester one of the ionizing protons in the doubly protonated peptide ions formed by ESI,21, 22 it follows that the location of the second ionizing proton is important in directing the selective cleavages C-terminal to histidine in the peptides under investigation here.
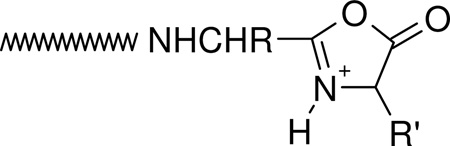 |
(1) |
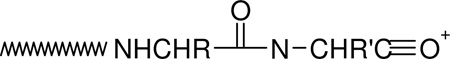 |
(2) |
The nature (including the formation and structure) of typical charge-directed b ions in protonated peptides has been studied extensively in the past.26, 63–69 Both protonated oxazolone (1)26, 63–67 and acylium (2)68, 69 structures have been proposed as the structures of b ions, with compelling evidence given for both. Alternatively, an equilibrium might exist in the gas-phase between these two different b ion structures which would bring harmony to all the different types of experimental evidence. The experimental MS/MS data in Figures 1 and 2 prompts the question of whether the dominant b ions at histidine might be oxazolone, acylium or “other” in structure. The side chain of histidine is known to be basic and a potential site of protonation in peptides.60, 61 With this in mind, we propose the mechanism depicted in Scheme 1 to account for histidine b ion formation in singly protonated VYIHPF. As shown in Scheme 1, following protonation at the side chain of histidine, one proton is transferred intramolecularly from the histidine imidazole to the histidine carbonyl oxygen. This results in an electropositive histidine carbonyl carbon which can be attacked by the nucleophilic imino nitrogen of histidine resulting in a fused bicyclic 5-membered ring structure. Next, a 1–3 hydrogen shift or a more complicated intramolecular atom shuttling induces cleavage.59, 68–72 The result is a unique protonated histidine b ion and a neutral C-terminal leaving piece (Scheme 1). Alternatively, the proton could initially be transferred from the histidine imidazole to the amide-N of the His-Pro bond; the question of involvement of protonated amide carbonyl oxygen vs nitrogen has not been definitively answered.40, 73, 74 To produce the b+/y+ pair from a doubly protonated peptide, a proton transfer to the C-terminal leaving piece is required as discussed below. For a peptide containing arginine to which a single proton is added, (e.g., RVYIHPF) arginine is expected to sequester the single ionizing proton.21, 22 Following activation of singly protonated RVYIHPF, the proton is immobile15, 16, 19, 21, 22, 63, 75–77 to a great extent, leading to initial loss of ammonia. Further fragmentation occurs by charge-remote fragmentation or fragmentation initiated by a high energy proton transfer that does not favor transfer to any one particular site. As a result the fragmentation is non-selective (Figure 3). However, upon addition of the second ionizing proton which is expected to reside (at least initially) at the side chain of histidine, the fragmentation becomes selective. In this case, a b+/y+ complementary ion pair results (Figure 1).
Scheme 1.

Proposed mechanism for the formation of the histidine b ion. Transfer of the ionizing proton by protonated histidine to the peptide oxygen renders the carbonyl carbon electropositive. Nucleophilic attack by the imino nitrogen of histidine on the electropositive carbonyl carbon along with N-protonation leads to breaking of the peptide bond and the fused bicyclic ring b ion structure. The C-terminal leaving piece is neutral.
III. Enhanced Cleavage at Histidine For Different His-Xxx Bonds
Proline, due to its cyclic nature and alkylated nitrogen, has long been recognized to direct cleavage N-terminal to this residue resulting in dominant y-type fragment ion formation.78–80 Indeed, the MS/MS spectra of doubly protonated peptides with no His, RVYIFPF and singly protonated VYIFPF, demonstrate dominant b5/y2 and b4 product ions, respectively, while those of doubly protonated RVYIFAF and singly protonated VYIFAF ions show non-selective fragmentation (supplementary figures 10–13). This suggests that it is not the histidine alone that is responsible for the enhanced HP cleavage in doubly protonated RVYIHPF. To ascertain if enhanced cleavage at histidine persists irrespective of its C-terminal residue, proline was substituted by sarcosine (Sar; N-methylglycine) and then by alanine. Sar contains an alkylated nitrogen mimicking the alkylation of Pro while Ala does not contain an alkylated nitrogen. The MS/MS spectra of doubly protonated RVYIHPF, RVYIHSarF and RVYIHAF ions obtained under SORI-CID conditions are shown in Figure 5A,B,C. It is evident from the mass spectral data that while complementary b5/y2 product ions dominate the spectrum of RVYIHPF (Figure 5A), b52+ and b62+ product ions become more significant on substitution of Pro by Ala (Figure 5C). The spectrum of doubly protonated RVYIHSarF (Figure 5B) is intermediate to those of the other two peptides, with complementary b5+/y2+ product ions in comparable abundance to b62+ product ions. The ratio of complementary b+/y+ product ions to doubly charged b product ions in RVYIHPF and RVYIHSarF (Figure 5) suggests that the cyclic nature of Pro and the conformation it induces in RVYIHPF are more important in aiding complementary b+/y+ product ion formation than an alkylated nitrogen because alternate low-energy fragmentation pathways begin to compete following the Pro → Sar substitution. A commonality in the MS/MS spectra of the current peptides (Figure 5) is dominant cleavage C-terminal to the histidine residue resulting in sequence-dependent selective product ions (i.e., dominant complementary b5+/y2+ product ions in RVYIHPF and dominant b52+and b62+ product ions in RVYIHAF). Recently, it has been shown by statistical analysis of doubly charged tryptic peptides that histidine leads to preferential cleavage on its C-terminal side in b ion formation.39 A mechanism which accounts for the formation of doubly charged b-type product ions C-terminal to histidine in RVYIHAF is discussed below.
Figure 5.

SORI-CID spectra of doubly protonated (A) RVYIHPF, (B) RVYIHSarF and (C) RVYIHAF at an excitation energy of 9 eV; Electrospray ionization. Fragments of interested are circled as squares (singly charged fragments) and as ovals (doubly charged fragments).
IV. A Common Intermediate in the Production of Selective b2+ or b+/y+ Complementary Product Ions at Histidine Residues
The mass spectral data presented above (Figures 1, 2 and 5) as well as previous work by Adams et al. and Sullards et al.57, 58 suggest that histidine is a very likely protonation site in the peptides under investigation. A means must exist by which the doubly protonated precursor ions of RVYIHPF, protonated at both the side chains of Arg and histidine, dissociate to give complementary b+/y+ product ions. Moreover, while dominant complementary b+/y+ product ions are produced for His-Pro cleavage, b52+ product ions dominate in RVYIHAF. Following the postulate that the ionizing proton resides on the side chain of histidine in singly protonated VYIHPF (see Section II; b4 product ion formation), we propose a common mechanism, presented in Scheme 2, which demonstrates the formation of b52+ or b5+/y2+ product ions. Following initial protonation at the two most basic sites in the peptide (arginine and histidine side chains),57, 80, 81 one proton is transferred intramolecularly from the histidine side chain π-nitrogen to the histidine carbonyl oxygen (see Scheme 1). This starting peptide ion structure is supported by our molecular dynamics calculations which reveal that this particular proton bridge (within 2.5 Å) occurs in ~ 30% and ~21% of the 10000 structures of RVYIHPF and RVYIHAF sampled, respectively. It is of interest that a similar proton bridge from an ionizing proton on the histidine side chain τ-nitrogen, to the histidine carbonyl oxygen, is not supported by the modeling results clearly implying that initial proton transfer must occur from the ionized π-nitrogen of histidine. Nucleophilic attack by the imino (i.e., π-) nitrogen of histidine on the now electropositive carbonyl carbon results in the fused bicyclic 5-membered ring structure involving the histidine side chain (Scheme 2). The molecular modeling results showed that in this closed-ring doubly protonated RVYIHPF peptide structure (top structure, Scheme 2), the transferred proton is now within H-bonding distance to the Pro backbone nitrogen in one third (i.e. ~ 29%) of 10000 structures sampled. A similar hydrogen bonding interaction is seen in ~80% of 10000 sampled structures of RVYIHAF. During this proton transfer step, the amide bond between HP or HA is weakened and is cleaved. This results in a complex intermediate where the formally separated product species might be in close proximity to each other, either via hydrogen bonds or electrostatic interactions in an ion-molecule complex. At this step, a branching point in the fragmentation can occur. Namely, in the case of RVYIHPF, it is proposed that a proton transfer takes place from the protonated histidine side chain to the basic amine nitrogen of Pro within the complex intermediate, leading to simultaneous formation of the b5+/y2+ product ions. In contrast to RVYIHPF where a second hydrogen is also transferred to the C-terminal fragment (PF) from the τ-nitrogen of histidine to result in y2 product ion formation (Figures 1A and 5A), no final proton transfer step takes place from [RVYIH + 2H]2+ to the departing AF (Figure 5C). This leaves both protons on the N-terminal fragment resulting in the b52+ product ion and a neutral C-terminal fragment.
Scheme 2.

Proposed mechanism for the formation of complementary b+/y+ and b2+ product ions at histidine from doubly protonated peptides. Following formation of an ion neutral complex as in Scheme 1, a proton transfer step to the C-terminal leaving piece results in the complementary b5+/y2+ ions in RVYIHPF (left side). No proton transfer step leaves both charges on the N-terminal piece resulting in a b52+ product ion in RVYIHAF (right side).
It has recently been reported by Nold et al.82 that during dissociation in protonated peptides, competition exists for the added proton between the N– and C-terminal fragments. This competition is based on the proton affinities of the two segments. Typically, N-terminal fragments with cyclic (or oxazolone like)63–65, 83, 84 structures and C-terminal fragments with linear structures83, 85 are produced on peptide dissociation. From this it follows that the relative basicity of the two penultimate amino acid segments (PF and AF), must also play a role in the transfer of the added proton from histidine that leads to a charged (i.e., y2) and/or neutral linear C-terminal fragment with concomitant b5+ or b52+ product ion formation, respectively. Indeed the side chain of alanine is less basic than that of proline and therefore less likely to promote proton transfer, consistent with the observed intensities of the b5+ and b52+ product ions (b52+ > b5+ for RVYIHAF and b52+ < b5+ for RVYIHPF; Figures 5C and A).
V. Controlling the Formation of Selective b2+ Product Ions at Histidine Residues
In support of the above model for complementary b+/y+ or doubly charged b2+ product ion formation in doubly protonated histidine-containing peptides, MS/MS experiments were carried out on an angiotensin III analog where the histidine side chain τ-nitrogen was alkylated. This was done to test the mechanism in Scheme 2 by blocking one of the two hydrogen transfers from the protonated histidine side chain to the departing C-terminal product (PF). Figure 6 shows the low-energy (ion trap) CID and SID of doubly protonated RVYIH*PF, where H* stands for alkylated histidine. Clearly, the b52+ product ion is the dominant sequence ion peak. Formation of the H* immonium ion is a favorable process by SID (Figure 6B), although b52+ is the most abundant sequence ion. No b5+ or y2+ product ions were observed at any of the activation energies investigated in contrast to the fragmentation behavior of doubly protonated RVYIHPF (compare Figures 1 with 6). This is presumably because there is no available proton for transfer from the side chain of the alkylated histidine in doubly protonated RVYIH*PF. In contrast, if the b ion formed were a typical protonated oxazolone intermediate, alkylation of the His-nitrogen should not influence subsequent H-transfer. These SID and low-energy CID results also underscore the notion that protonated histidine is directly involved in formation of the b5+/y2+ product ions in doubly protonated RVYIHPF and that histidine is a favorable site of protonation in this set of angiotensin peptides. The involvement of protonated histidine in the cleavage is also supported by the selective cleavage of Φ3P+CH2C(O)-VYIHPF to form [Φ3P+CH2C(O)-VYIH + H]2+ only when an added proton is present (a mimic of doubly protonated RVYIHPF, Figure 4B).
Figure 6.

Dominant b52+ product ion formation in RVYIH*PF by (A) low-energy CID at a 18.2 % relative collision energy and (B) SID at a collision energy of 30 eV on a 2-(perfluorodecyl)ethanethiolate [CF3(CF2)9CH2CH2S-Au] monolayer surface; Electrospray ionization.

VI. Enhanced Cleavage One-Residue Away from Histidine
It is of interest that in addition to the dominant b52+ product ions seen for doubly protonated RVYIHAF, b62+ product ions are also abundant in the MS/MS spectrum of this peptide (Figure 5C). This is also true for doubly protonated RVYIHSarF (Figure 5B). Molecular modeling indicates that a very favorable interaction between protonated histidine and the carbonyl group of the AF peptide bond occurs in ~ 40% of 10000 structures of doubly protonated RVYIHAF. This dominant H-bond from the side chain of histidine to the XX peptide bond in HXX-containing peptides along with a subsequent proton transfer could lead to enhanced cleavage of the XX bond. It is of interest that while b62+ product ions are of minor abundance from doubly protonated RVYIHPF, the molecular modeling indicates that the proton on the side chain of histidine comes within H-bonding distance (2.5 Å) to the PF peptide carbonyl oxygen in 78% of structures sampled. Thus, although thermodynamically favorable, O-protonation at the PF peptide carbonyl oxygen does not lead to fragmentation in RVYIHPF because of the same stereo chemical factors which typically disfavor formation of an oxazolone structure C-terminal to a Pro residue.
VII. Evidence for the Unique Histidine b Ion Structure
In Sections II and IV and in Schemes 1 and 2, we propose that in the presence of a proton, selective cleavage C-terminal to histidine leads to a unique bicyclic b ion. Recent ab initio calculations, which followed our initial unpublished data of the bicyclic His b ion structure, have shown that this unique b ion structure40 is stabilized by the aromaticity of the imidazole ring.41 In order to probe further the structure of the histidine b ion relative to other typical b ions (e.g., oxazolone or acylium ions) in protonated peptides, the doubly and singly charged product ions (b52+ and b5+) were isolated in the ion trap and subjected to further fragmentation. In addition, resonance ejection experiments (Figure 7) were performed in the FTICR-MS to see if the b52+ and b62+ product ions of RVYIHAF are formed by independent mechanisms or if the b52+ ions are further fragments of the b62+ product ions. If the latter situation holds true, ejection of b62+ product ions should result in no detectable b52+ fragment ions assuming that multi step dissociation is not faster than ejection. It is clearly seen that upon ejection of the b62+ product ions from doubly protonated RVYIHAF, b52+ product ions still persist in the spectrum. Thus, within the limits of the timescale of the resonance ejection experiment, the b52+ ions are not further fragments of the b62+ product ions. Independent formation of these two doubly charged b ions is consistent with our proposed model of how bn2+ ions (n=position of histidine) are formed immediately C-terminal to histidine residues (Scheme 2).
Figure 7.

On-resonance CID spectrum of doubly protonated RVYIHAF with (A) no resonance ejection and (B) with resonance ejection of the b62+ product ion; Electrospray ionization.
To probe further the structures of the b52+ and b62+ from RVYIHAF, MS/MS/MS experiments were carried out. The trapping capabilities of the FTICR-MS and LCQ were exploited to isolate the b52+ ions and then dissociate them further. The SORI-CID spectrum of the b52+ product ions is shown in Figure 8A. The results demonstrate that the major fragments are a52+ ions, and formation of the (possibly complementary) b4 and histidine immonium ions. Presuming b52+ product ion generation as shown in Scheme 2, the loss of CO to give mainly a52+ fragments is illustrated in Scheme 3.
Figure 8.

(A) 9-eV MS/MS/MS SORI-CID spectrum of the selected b52+ product ion of doubly protonated RVYIHAF at 26 eV collision and (B) 19-eV MS/MS/MS SORI-CID spectrum of the selected b62+ product ion of doubly protonated RVYIHAF at 17 eV collision; Electrospray ionization.
Scheme 3.

Proposed mechanism for the fragmentation of b52+ product ions from doubly protonated RVYIHAF precursor ions.
Unlike MS/MS/MS of the b52+ product ions of RVYIHAF which leads to formation of a52+ fragment ions, the b62+ product ions of RVYIHAF fragment further to form exclusively b52+ fragment ions (Figure 8B). The lack of CO loss from b62+ product ions suggests that these product ions do not have the same structure as the b52+ product ions shown in Schemes 2 and 3. Recently, cyclic oxazolone-like structures for generic b ions produced in MS/MS of protonated peptides have been proposed.26, 63–67 With this in mind and considering the MS/MS/MS results for the b62+ products ions of RVYIHAF, Scheme 4 shows how a protonated oxazolone b62+ product ion could be formed and subsequently dissociate to give b52+ fragment ions. Following protonation on the alanyl peptide carbonyl oxygen (e.g. through a proton transfer step from the protonated side chain of histidine), nucleophilic attack by the carbonyl oxygen C-terminal to histidine occurs on the alanyl carbonyl carbon along with concomitant cleavage of the AF bond. This leads to formation of the protonated 5-membered ring oxazolone-like b62+ product ion and a neutral C-terminal fragment. The b52+ fragment ion is formed from the b62+ product ion by opening of the oxazolone 5-membered ring structure through a nucleophilic attack by the histidine imino nitrogen on the electropositive oxazolone carbon. This produces a b52+ fragment ion whose local structure is identical to that shown in Schemes 2 and 3. Fourth stage MS experiments by low-energy CID of the b52+ fragment ions produced from the b62+ product ions indicated favorable loss of CO to give a52+ fragment ions (supplementary figure 14). This is consistent with Scheme 3 above.
Scheme 4.

Proposed mechanism for the fragmentation of b62+ product ions of doubly protonated RVYIHAF. Following cleavage of the Ala-Phe bond to produce an oxazolone b62+ product ion, nucleophilic attack by the histidine imino nitrogen on the electropositive carbon of the oxazolone occurs. This results in a b52+ fragment ion with same structure as the b52+ ions shown in Scheme 2.
Additional evidence for a unique histidine b ion structure vis a vis a generic b ion was obtained by MS/MS/MS experiments of the b5+ product ions produced from RVYIHPF and RVYIFPF. Figure 9 shows the energy resolved fragmentation curves of the b5+ product ions from doubly protonated RVYIFPF (Figure 9A) and RVYIHPF (Figure 9B). It is clearly evident that the two types of b5+ product ions (FP cleavage and HP cleavage) further dissociate by accessing different low-energy fragmentation pathways resulting in different fractional abundances of a5+, b5+* and a5+* fragment ions. Whereas the b5+ product ions from RVYIFPF lose CO readily to give a5+ ions, b5+* and a5+* are also favorable products from the b5+ of RVYIHPF. This indicates that the stability of the b5+ product ions from RVYIFPF and RVYIHPF are different, likely as a consequence of different structures (i.e., typical oxazolone or acylium b ions from RVYIFPF versus a unique histidine b ion from RVYIHPF; Scheme 1).
Figure 9.

Low-energy CID resolved fragmentation curves upon further dissociation of the b5+ product ions (■) from (A) doubly protonated RVYIFPF and (B) doubly protonated RVYIHPF. The fragment ions plotted are the a5+ ions (●), b5* (▲), and a5* (⋆) ions. The inserts show the structures of a generic b ion (oxazolone; part A) and the proposed b ion at histidine.
Conclusions
In this work, the occurrence of selective cleavage at protonated histidine in the spectra of a systematically varied set of model peptides is supported by data obtained with three different activation methods that deposit different distributions of energy and that are associated with different time frames for dissociation. Strongly preferred cleavages at His-Zzz have been observed. The strong cleavage at His-Pro is in agreement with previous findings of similar dominant cleavages.56, 57 A mechanism explaining the selective cleavage at histidine is proposed. These findings are of general interest and of importance because if high throughput MS-based peptide sequencing based on correlation analysis between experimental and theoretical spectra is to be reliable, predictive rules for peptide dissociation such as those presented here must be developed, tested and incorporated into algorithms.
Our proposed mechanism for selective cleavage at histidine is based on solvation of the ionizing proton on histidine by the carbonyl oxygen of the His-Xxx and His-Xxx-Xxx bonds along with nucleophilic attack of the imino nitrogen on the electropositive peptide carbonyl carbon, to result in a unique histidine b product ion. This b ion incorporates the imidazole ring of histidine and C-terminal end of the His-terminated peptide in a bicyclic structure. Evidence for this different b ion structure at histidine (i.e., not acylium or oxazolone) is based on MS/MS/MS data for both singly and doubly charged b ions. In this model for selective cleavage at histidine, a critical proton transfer step from the N-terminal ionic species to the neutral C-terminal fragment during dissociation is important in giving rise to complementary b+/y+ product ions (Scheme 2). Alternatively, lack of such a proton transfer step leads to formation of a doubly charged histidine b ion. Evidence for this comes from the MS/MS/MS spectra of doubly protonated precursor ions of RVYIH*PF, and RVYIHAF. Thus, although selective cleavage at histidine can be said to be independent of the residue C-terminal to histidine, the charge of the developing b product ion is determined by the identity of the residue C-terminal to histidine and by the ability of protonated histidine to transfer a proton to the C-terminal leaving fragment. This implies that secondary interactions (e.g., hydrogen bonds or electrostatic interactions) that hold the complex together in the final proton transfer step must be important and that involvement of a proton in the vicinity of histidine to initiate selective cleavage is also clearly required.
Supplementary Material
Acknowledgment
This work was financially supported by a grant from the National Institutes of Health (GM RO15387 to V.H.W.), the National Science Foundation (CHE-9616711 to J.H.F), and a postdoctoral fellowship from the Fonds pour la Formation de Chercheurs et l’Aide à la Recherche (to G.T.). We wish to thank Chungang Gu for providing the AAHAA SID spectra and Dr. Árpad Somogyi for helpful discussions.
Footnotes
- MS/MS spectrum of doubly protonated RVYIHPF obtained by (A) SID at a collision energy of 45 eV on a 2-(perfluorooctyl)ethanethiolate [CF3(CF2)7CH2CH2S-Au] monolayer surface and (B) low-energy CID at a 13.0 % relative collision energy; Electrospray ionization.
- MS/MS spectrum of doubly protonated DRVYIHPF obtained by (A) sustained off-resonance irradiation at an excitation energy of 9 eV, (B) SID at a collision energy of 55 eV on a 2-(perfluooctyll)ethanethiolate [CF3(CF2)7CH2CH2S-Au] monolayer surface and (C) low-energy CID at a 13.5 % relative collision energy; Electrospray ionization.
- ESI-LCQ spectrum of [M+2H]2+ ions of DRVYIHPFHL (16.0 % relative collision energy).
- MS/MS spectrum of singly protonated VYIHPF obtained by low-energy CID at a 22.5 % relative collision energy; Electrospray ionization.
- Electrospray ionization Spectrum of the [M+H]+ ions of AAHAA obtained by SID (30 eV).
- ESI-LCQ spectrum of [M+H]+ ions of AAAAAHAA (22.8 % relative collision energy).
- MS/MS spectrum of singly protonated RVYIHPF obtained by (A) SID at a collision energy of 60 eV on a 2-(perfluorooctyl)ethanethiolate [CF3(CF2)7CH2CH2S-Au] monolayer surface and (B) low-energy CID at a 28.0 % relative collision energy; Electrospray ionization.
- MS/MS spectrum of singly protonated DRVYIHPF obtained by (A) sustained off-resonance irradiation at an excitation energy of 24 eV, (B) SID at a collision energy of 65 eV on a 2-(perfluorooctyl)ethanethiol [CF3(CF2)7CH2CH2S-Au] monolayer surface and (C) low-energy CID at a 31.0 % relative collision energy.
- ESI-LCQ spectrum of [M+H]+ ions of DRVYIHPFHL (37.0 % relative collision energy).
- MS/MS spectrum of [M+2H]2+ ions of RVYIFPF obtained by SID (20 eV), SORI-CID (10 eV) and low-energy CID (8.0 % relative collision energy).
- MS/MS spectrum of [M+H]+ ions of VYIFPF obtained by SID (17.5 eV), SORI-CID (11 eV) and low-energy CID (19.0 % relative collision energy).
- MS/MS spectrum of [M+2H]2+ ions of RVYIFAF obtained by SID (20 eV) and low-energy CID (8.5 % relative collision energy).
- MS/MS spectrum of [M+H]+ ions of VYIFAF obtained by SID (20 eV) and low-energy CID (18.5 % relative collision energy).
- MS4 by low-energy CID of the b52+ fragments (RVYIH) produced on MS3 of the b62+ product (RVYIHA) from the [M+2H]2+ ions of RVYIHAF.
References
- 1.Anderson NL, Anderson NG. Electrophoresis. 1998;19:1853–1861. doi: 10.1002/elps.1150191103. [DOI] [PubMed] [Google Scholar]
- 2.Yates JR., III J. Mass Spectrom. 1998;33:1–19. doi: 10.1002/(SICI)1096-9888(199801)33:1<1::AID-JMS624>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 3.Eng JK, McCormack AL, Yates JR., III J. Am. Soc. Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 4.Yates JR, III, Eng JK, McCormack AL, Schieltz D. Anal. Chem. 1995;67:1426–1436. doi: 10.1021/ac00104a020. [DOI] [PubMed] [Google Scholar]
- 5.Mann M, Wilm M. Anal. Chem. 1994;66:4390–4399. doi: 10.1021/ac00096a002. [DOI] [PubMed] [Google Scholar]
- 6.Clauser KR, Baker P, Burlingame AL. Portland, OR: 1996. p. 365. [Google Scholar]
- 7.Perkins DN, Pappin DJC, Creasy DM, Cottrell JS. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 8.Stahl DC, Swiderek KM, Davis MT, Lee TD. J. Am. Soc. Mass Spectrom. 1996;7:532–540. doi: 10.1016/1044-0305(96)00057-8. [DOI] [PubMed] [Google Scholar]
- 9.McCormack AL, Schieltz DM, Goode B, Yang S, Barnes, Drubin D, Yates JR., III Anal. Chem. 1997;69:767–776. doi: 10.1021/ac960799q. [DOI] [PubMed] [Google Scholar]
- 10.Biemann K, McCloskey JA, editors. New York: Academic Press; 1990. pp. 455–479. [Google Scholar]
- 11.Biemann K. Methods Enzymol. 1990;193:455–479. doi: 10.1016/0076-6879(90)93433-l. [DOI] [PubMed] [Google Scholar]
- 12.Roepstorff P, Fohlman J. Biomed. Mass Spectrom. 1984;11:601–603. doi: 10.1002/bms.1200111109. [DOI] [PubMed] [Google Scholar]
- 13.Papayannopoulos IA. Mass Spectrom. Rev. 1995;14:49–73. [Google Scholar]
- 14.Alexander AJ, Thibault P, Boyd RK, Curtis JM, Rinehart KL. Int. J. Mass. Spectrom. Ion Processes. 1990;98:107–134. [Google Scholar]
- 15.McCormack AL, Somogyi Á, Dongré AR, Wysocki VH. Anal. Chem. 1993;65:2859–2872. doi: 10.1021/ac00068a024. [DOI] [PubMed] [Google Scholar]
- 16.Poulter L, Taylor LCE. Int. J. Mass. Spectrom. Ion Processes. 1989;91:183–197. [Google Scholar]
- 17.Hunt DF, Henderson RA, Shabanowitz J, Sakaguchi K, Michel H, Sevilir N, Cox AL, Appella E, Engelhard VH. Science. 1992;255:1261–1263. doi: 10.1126/science.1546328. [DOI] [PubMed] [Google Scholar]
- 18.Ballard KD, Gaskell SJ. J. Am. Chem. Soc. 1992;114:64–71. [Google Scholar]
- 19.Dongré AR, Somogyi Á, Wysocki VH. J. Mass Spectrom. 1996;31:339–350. doi: 10.1002/(SICI)1096-9888(199604)31:4<339::AID-JMS322>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 20.Carr SR, Cassady CJ. J. Mass Spectrom. 1997;32:959–967. doi: 10.1002/(SICI)1096-9888(199709)32:9<959::AID-JMS552>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 21.Dongré AR, Jones JL, Somogyi Á, Wysocki VH. J. Am. Chem. Soc. 1996;118:8365–8374. [Google Scholar]
- 22.Summerfield SG, Whiting A, Gaskell SJ. Int. J. Mass. Spectrom. Ion Process. 1997;162:149–161. [Google Scholar]
- 23.Qin J, Chait BT. J. Am. Chem. Soc. 1995;117:5411–5412. [Google Scholar]
- 24.Tang X-J, Thibault P, Boyd RK. Anal. Chem. 1993;65:2824–2834. doi: 10.1021/ac00068a020. [DOI] [PubMed] [Google Scholar]
- 25.Nair H, Somogyi Á, Wysocki VH. J. Mass Spectrom. 1996;31:1141–1148. doi: 10.1002/(SICI)1096-9888(199610)31:10<1141::AID-JMS402>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 26.Vaisar T, Urban J. J. Mass Spectrom. 1998;33:505–524. [Google Scholar]
- 27.Downard KM, Biemann K. Int. J. Mass. Spectrom. Ion Processes. 1995;148:191–202. [Google Scholar]
- 28.Dongré AR, Hayward MJ, Wysocki VH. Atlanta, GA; 1995. p. 628. [Google Scholar]
- 29.Wu QY, van Orden S, Cheng XH, Bakhtiar R, Smith RD. Anal. Chem. 1995;67:2498–2509. doi: 10.1021/ac00110a027. [DOI] [PubMed] [Google Scholar]
- 30.Schnier PD, Price WD, Jockusch RA, Williams ER. J. Am. Chem. Soc. 1996;118:7178–7189. doi: 10.1021/ja9609157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wyttenbach T, von Helden GV, Bowers MT. J. Am. Chem. Soc. 1996;118:8355–8364. [Google Scholar]
- 32.Vachet RW, Asam MR, Glish GL. J. Am. Chem. Soc. 1996;118:6252–6256. [Google Scholar]
- 33.Thorne GC, Ballard KD, Gaskell SJ. J. Am. Soc. Mass Spectrom. 1990;1:249–257. [Google Scholar]
- 34.Tsaprailis G, Nair H, Somogyi Á, Wysocki VH, Zhong W, Futrell JH, Summerfield SG, Gaskell SJ. J. Am. Chem. Soc. 1999;121:5142–5154. [Google Scholar]
- 35.Tsaprailis G, Somogyi A, Nikolaev E, Wysocki V. Int. J. Mass Spectrom. 2000;195/196:467–479. [Google Scholar]
- 36.Yu W, Vath JE, Huberty MC, Martin SA. Anal. Chem. 1993;65:3015–3023. doi: 10.1021/ac00069a014. [DOI] [PubMed] [Google Scholar]
- 37.Huang YY, Wysocki VH, Tabb DL, Yates JR. Int. J. Mass Spectrom. 2002;219:233–244. [Google Scholar]
- 38.Stephenson J, Jr., Cargile BJ, McLuckey SA. Rapid. Commun. Mass Spectrom. 1999;13:2040–2048. doi: 10.1002/(SICI)1097-0231(19991030)13:20<2040::AID-RCM754>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 39.Tabb DL, Smith LL, Breci LA, Wysocki VH, Lin D, Yates JRI. Anal. Chem. 2003;75:1155–1163. doi: 10.1021/ac026122m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wysocki VH, Tsaprailis G, Smith LL, Breci LA. J. Mass Spectrom. 2000;35:1399–1406. doi: 10.1002/1096-9888(200012)35:12<1399::AID-JMS86>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 41.Farrugia JM, Taverner T, O’Hair RAJ. Int. J. Mass Spectrom. 2001;209:99–112. [Google Scholar]
- 42.Atherton E, Sheppard RC. In: Solid Phase Peptide Synthesis: A Practical Approach. Rickwood D, Hames BD, editors. Oxford, UK: Oxford University Press; 1989. [Google Scholar]
- 43.Fletcher A, Jones J, Ramage W, Stachulski A. J. Chem. Soc. Perkin Trans. I. 1979:2261–2267. [Google Scholar]
- 44.Wysocki VH, Ding J-M, Jones JL, Callahan JH, King FL. J. Am. Soc. Mass Spectrom. 1992;3:27–32. doi: 10.1016/1044-0305(92)85015-C. [DOI] [PubMed] [Google Scholar]
- 45.Chowdhury SK, Katta V, Chait BF. Rapid Commun. Mass Spectrom. 1990;4:81–87. doi: 10.1002/rcm.1290040305. [DOI] [PubMed] [Google Scholar]
- 46.Papac DI, Schey KL, Knapp DR. Anal. Chem. 1991;63:1658–1660. doi: 10.1021/ac00015a029. [DOI] [PubMed] [Google Scholar]
- 47.Ulman A. Chem. Rev. 1996;96:1533–1554. doi: 10.1021/cr9502357. [DOI] [PubMed] [Google Scholar]
- 48.Marshall AG, Grosshans PB. Anal. Chem. 1991;63:215A–229A. [Google Scholar]
- 49.Gauthier JW, Trautman TR, Jacobson DB. Anal. Chim. Acta. 1991;246:211–225. [Google Scholar]
- 50.Senko MW, Speir JP, McLafferty FW. Anal. Chem. 1994;66:2801–2808. doi: 10.1021/ac00090a003. [DOI] [PubMed] [Google Scholar]
- 51.Mohamadi F, Richards NGJ, Guida WC, Liskamp R, Lipton M, Caufield C, Chang G, Henderickson T, Still WC. J. Comput. Chem. 1990;11:440–467. [Google Scholar]
- 52.Weiner S, Kollman P, Case D, Singh U, Ghio C, Alagona G, Profeta S, Weiner P. J. Am. Chem. Soc. 1984;106:765–784. [Google Scholar]
- 53.Polak E, Ribiere G. Revue Française Inf. Rech. Oper. 1969;16-R1:35–42. [Google Scholar]
- 54.Meot-Ner M, Dongré AR, Somogyi Á, Wysocki VH. Rapid Commun. Mass Spectrom. 1995;9:829–836. doi: 10.1002/rcm.1290090922. [DOI] [PubMed] [Google Scholar]
- 55.Gordon D, Woods M. Rapid Commun. Mass Spectrom. 1993;7:215–218. [Google Scholar]
- 56.Hu P, Loo JA. J. Am. Chem. Soc. 1995;117:11314–11319. [Google Scholar]
- 57.Adams J, Strobel FH, Reiter A, Sullards MC. J. Am. Soc. Mass Spectrom. 1996;7:30–41. doi: 10.1016/1044-0305(95)00604-4. [DOI] [PubMed] [Google Scholar]
- 58.Sullards M, Reiter J. J. Am. Soc. Mass Spectrom. 2000;11:40–53. doi: 10.1016/S1044-0305(99)00115-4. [DOI] [PubMed] [Google Scholar]
- 59.Tang X-J, Boyd RK. Rapid Commun. Mass Spectrom. 1992;6:651–657. doi: 10.1002/rcm.1290061105. [DOI] [PubMed] [Google Scholar]
- 60.Campbell S, Beauchamp JL, Rempe M, Lichtenberger DL. Int. J. Mass. Spec. Ion Processes. 1992;117:83–99. [Google Scholar]
- 61.Wu Z, Fenselau C. Rapid Commun. Mass Spectrom. 1992;6:403–405. doi: 10.1002/rcm.1290080926. [DOI] [PubMed] [Google Scholar]
- 62.Starrett AM, DiDonato GC. Rapid Commun. Mass Spectrom. 1993;7:12–15. [Google Scholar]
- 63.Nold MJ, Wesdemiotis C, Yalcin T, Harrison AG. Int. J. Mass. Spectrom. Ion Processes. 1997;164:137–153. [Google Scholar]
- 64.Yalcin T, Khouw C, Csizmadia IG, Peterson MR, Harrison AG. J. Am. Soc. Mass Spectrom. 1995;6:1165–1174. doi: 10.1016/1044-0305(95)00569-2. [DOI] [PubMed] [Google Scholar]
- 65.Yalcin T, Csizmadia IG, Peterson MR, Harrison AG. J. Am. Soc. Mass Spectrom. 1996;7:233–242. doi: 10.1016/1044-0305(95)00677-X. [DOI] [PubMed] [Google Scholar]
- 66.Arnott D, Kottmeir D, Yates JR, III, Shabanowitz J, Hunt DF. Chicago, IL: 1994. p. 470. [Google Scholar]
- 67.Summerfield SG, Bolgar MS, Gaskell SJ. J. Mass Spectrom. 1997;32:225–231. [Google Scholar]
- 68.Johnson RS, Martin SA, Biemann K. Int. J. Mass. Spectrom. Ion Processes. 1988;86:137–154. [Google Scholar]
- 69.Biemann K, Martin SA. Mass. Spectrom. Rev. 1987;6:1–76. [Google Scholar]
- 70.Downard KM, Biemenn K. J. Am. Soc. Mass Spectrom. 1994;5:966–975. doi: 10.1016/1044-0305(94)80015-4. [DOI] [PubMed] [Google Scholar]
- 71.Ishikawa K, Nishimura T, Koga Y, Niwa Y. Rapid Commun. Mass Spectrom. 1994;8:933–938. doi: 10.1002/rcm.1290081205. [DOI] [PubMed] [Google Scholar]
- 72.Kiryushkin AA, Fales HM, Axenrod T, Gilbert EJ, Milne GW. Org. Mass Spectrom. 1971;5:19–31. [Google Scholar]
- 73.Schlosser A, Lehmann WD. J. Mass Spectrom. 2000;35:1382–1390. doi: 10.1002/1096-9888(200012)35:12<1382::AID-JMS84>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 74.Csonka IP, Paizs B, Lendvay G, Suhai S. Rapid Commun. Mass Spectrom. 2000;14:417–431. doi: 10.1002/(SICI)1097-0231(20000331)14:6<417::AID-RCM885>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 75.Burlet O, Orkiszewski RS, Ballard KD, Gaskell SJ. Rapid Commun. Mass Spectrom. 1992;6:658–662. doi: 10.1002/rcm.1290061106. [DOI] [PubMed] [Google Scholar]
- 76.Burlingame AL, Boyd RK, Gaskell SJ. Anal. Chem. 1996;68:599R–651R. doi: 10.1021/a1960021u. [DOI] [PubMed] [Google Scholar]
- 77.Summerfield SG, Cox KA, Gaskell SJ. J. Am. Soc. Mass Spectrom. 1997;8:25–31. [Google Scholar]
- 78.Loo JA, Edmonds CG, Smith RD. Anal. Chem. 1993;65:425–438. doi: 10.1021/ac00052a020. [DOI] [PubMed] [Google Scholar]
- 79.Vaisar T, Urban J. J. Mass Spectrom. 1996;31:1185–1187. doi: 10.1002/(SICI)1096-9888(199610)31:10<1185::AID-JMS396>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 80.Chowdhury SK, Katta V, Chait BT. J. Am. Chem. Soc. 1990;112:9012–9013. [Google Scholar]
- 81.Guevremont R, Siu KWM, LeBlanc JCY, Berman SS. J. Am. Soc. Mass Spectrom. 1992;3:216–224. doi: 10.1016/1044-0305(92)87005-J. [DOI] [PubMed] [Google Scholar]
- 82.Nold MJ, Cerda B, Wesdemiotis C. J. Am. Soc. Mass Spectrom. 1999;10:1–8. doi: 10.1016/S1044-0305(98)00120-2. [DOI] [PubMed] [Google Scholar]
- 83.Cordero MM, Houser JJ, Wesdemiotis C. Anal. Chem. 1993;65:1594–1601. doi: 10.1021/ac00059a019. [DOI] [PubMed] [Google Scholar]
- 84.Cordero M, Wesdemiotis C. Org. Mass Spectrom. 1994;29:382–390. [Google Scholar]
- 85.Mueller DR, Eckersley M, Richter WJ. Org. Mass Spectrom. 1988:217–222. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.