

Abstract
Combined kidney liver transplant is the preferred transplant option for most patients with primary hyperoxaluria type 1 (PH1) given that it removes the hepatic source of oxalate production and improves renal allograft survival. However, PH1 patients homozygous for the G170R mutation can develop normal urine oxalate levels with pyridoxine therapy and may be candidates for kidney alone transplant. We examined the efficacy of pyridoxine therapy following kidney alone transplant in five patients homozygous for G170R transplanted between 9/1999 and 7/2013. All patients were maintained on pyridoxine post-transplant. Median age at transplant was 39 years (range 33–67 years). Median follow-up post-transplant was 8.5 years (range 0.2–13.9 years). At the end of follow-up, 4 grafts were functioning. One graft failed 13.9 years post-transplant due to recurrent oxalate nephropathy following an acute medical illness. After tissue oxalate stores had cleared, post-transplant urine oxalate levels were < 0.5 mmol/24 hr the majority of times checked. Calcium oxalate crystals were noted in only 3/13 allograft biopsies. This series suggests that a subgroup of PH1 patients demonstrate sustained response to pyridoxine therapy following kidney alone transplant. Therefore, pyridoxine combined with kidney alone transplantation should be considered for PH1 patients with a homozygous G170R mutation.
INTRODUCTION
The primary hyperoxalurias are a group of rare autosomal recessive disorders characterized by aberrant metabolism of glyoxylate into oxalate. Hyperoxaluria can result in kidney stones, end-stage renal disease (ESRD) and systemic oxalosis (1). Primary hyperoxaluria type 1 (PH1) is the most common form of primary hyperoxaluria accounting for 70–80% of all cases (2). PH1 is caused by mutations in the AGXT gene which encodes the liver-specific peroxisomal enzyme alanine/glyoxylate aminotransferase (AGT). More than 170 mutations of AGXT have been described (3). The AGT enzyme is responsible for converting glyoxylate into glycine within liver peroxisomes. When the enzyme is defective or deficient, glyoxylate is aberrantly converted into oxalate (3). Since humans have no enzymes to further degrade oxalate, it must be eliminated in the urine. Excessive amounts of oxalate in fluid within the renal tubules and urine can lead to kidney stones, nephrocalcinosis, and renal failure. Once renal failure ensues, oxalate can build up in body tissues over time including bone, leading to oxalosis. After kidney transplantation, tissue oxalate stores are released and can quickly damage the allograft.
Combined liver kidney transplantation is the treatment of choice for patients with PH1 and end-stage renal disease (ESRD) (4). Concurrent liver transplantation is required in order to restore the underlying enzyme deficiency. Without liver transplantation, excessive hepatic oxalate production persists and the kidney allograft is subjected to persistent hyperoxaluria (5, 6). Historically, allograft survival following kidney alone transplant (KTx) in PH1 has been poor (7, 8).
However, not all patients with PH1 and ESRD may require liver transplantation. Pyridoxine, or vitamin B6 (VB6), is an essential cofactor for AGT enzyme function. Patients with the most common mutation of AGXT, the G170R mutation, have a unique trafficking defect. This mutation results in mistargeting of the AGT enzyme from the peroxisome to the mitochondria of the hepatocyte where catalytic activity may be retained but is ineffective in disposition of glyoxylate located in other compartments of the cell. The G170R mutation has an allelic frequency of 30% (3). Thus compound heterozygosity for the G170R mutation is common in PH1, with homozygosity less often encountered (approximately 9% of PH1 patients in the Rare Kidney Stone Consortium (RKSC) Registry). However, PH1 patients who are homozygous for the G170R mutation have a better renal prognosis (9) and can be fully responsive to treatment with pharmacologic doses of pyridoxine, or vitamin B6 (VB6)(10, 11). In the setting of the G170R mutation, pharmacologic doses of VB6 act as a chemical chaperone to increase peroxisomal targeting and expression of the mutant enzyme (12). Thus, patients with G170R homozygosity can respond to pharmacologic dose VB6 therapy with normal or near normal urine oxalate levels (10). Given these considerations, KTx may be a viable treatment option for PH1 patients homozygous for the G170R mutation, sparing them the morbidity associated with complete removal of an otherwise normal liver and the risks associated with liver transplantation (13–15). The purpose of this study was to examine the long-term clinical outcome of G170R homozygous PH1 patients following treatment with VB6 and KTx.
MATERIALS AND METHODS
Patient population
The case series included all G170R homozygous patients enrolled in the RKSC PH Registry. Detailed analysis was completed on the subset of patients who developed ESRD and underwent KTx alone with follow-up at our center post-transplant. The RKSC PH Registry is a voluntary, observational registry that obtains clinical information from medical records provided by each participant’s healthcare providers following patient consent and under approval from the Institutional Review Board at the Mayo Clinic. Thus the subset of patients cared for at Mayo Clinic post-transplant (n=5), the major focus of the current report, were all maintained on pharmacologic doses of VB6 (approximately 5–8 mg/kg/d) and citrate therapy, in addition to immunosuppressive medications. VB6 was initiated prior to transplant in all patients as soon as the diagnosis of PH1 was recognized, and was continued without interruption after transplantation. Doses were given orally, unless patients were fasting for more than 24 hours, in which case intravenous pyridoxine was administered.
Laboratory and histologic analysis
DNA was extracted from peripheral blood lymphocytes and PCR was used to identify homozygosity for the G170R mutation of the AGXT gene. Allograft function was assessed by frequent determinations of serum creatinine, from which the eGFR was obtained using the 4-variable Modification of Diet in Renal Disease equation (MDRD) (16). Iothalamate clearance was measured annually per our post-transplant protocol. In 1998 our center also began performing protocol allograft biopsies at the time of transplant, 4 months, 12 months, 2 years and 5 years post-transplant. For-cause biopsies are performed at the discretion of a treating nephrologist. All biopsies were scored using Banff criteria (17, 18). Plasma and 24-hour urine oxalate levels were monitored at frequent intervals during the early months after transplant, and at least annually thereafter. Pyridoxal 5-phosphate levels were measured at multiple time points following KTx, typically every year.
Data analysis
Data were expressed as means and standard deviation or median and range as appropriate. Patient follow-up was censored at the time of death, graft failure or date of analysis. Graft failure was defined as return to dialysis.
RESULTS
Patient characteristics
33 of 380 (8.9%) patients in the RKSC PH Registry were identified as homozygous for the G170R mutation. Fifteen of those patients had developed ESRD at time of last registry follow-up. Five homozygous patients received KTx and were followed at our institution post-transplant. These five patients received transplants between 9/1999 and 7/2013 and were included in our series. Baseline clinical and laboratory characteristics of the five recipients are shown in Table 1. Median age at transplant was 39 years (range 33–67 years), 4 were female and 4 received a living donor KTx. Three patients were transplanted at our center and two patients were transplanted elsewhere but followed by our program. PH patients followed at our center routinely undergo physical examination for skin manifestations of oxalosis including livedo reticularis, retinal examinations by ophthalmology for oxalate deposits, bone density assessments, electrocardiograms, and echocardiograms both before and after transplantation to assess systemic oxalosis. Only Patient 1 exhibited evidence of systemic oxalosis detectable by these measures; retinal oxalate crystals were seen on eye examination prior to transplant. Patient 2 underwent bilateral native nephrectomy one week following KTx (patient 2) and one patient underwent bilateral native nephrectomy six weeks prior to KTx (patient 5) in order to remove significant oxalate stores accumulated in the native kidneys. Patient 1 had received a living donor kidney transplant prior to a diagnosis of PH1 which failed due to recurrent oxalate nephropathy. Due to late diagnosis of PH1 in four of the patients, only patient 4 had taken VB6 prior to ESRD. Median post-transplant dose of VB6 was 7.7 mg/kg/d (range 5.4–9.4 mg/kg/d). Median post-transplant follow-up was 8.5 years (range 0.2–13.9 years).
Table 1.
Baseline clinical and laboratory characteristics of G170R homozygous PH1 patients who received kidney alone transplant and were followed post-transplant at Mayo*
| Patient | Age at symptom onset (yrs) | Pre-KTx Uox (mmol/24 hrs) off VB6 | Age at PH diagnosis and start of pyridoxine therapy (yrs) | Age at ESRD (yrs) | Age at KTx (yrs) | Donor type | HLA mismatch | Induction immuno-suppression | Maintenance immuno-suppression |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 33 | n/a | 38 | 33 | 33/39 | LURD | n/a | n/a | Tac/MMF/pred |
| 2 | 23 | 1.71 | 33 | 32 | 33 | DD | 5 | Alemtuzumab | Tac/MMF |
| 3 | 6 | 1.36 | 67 | 67 | 67 | LRD | 2 | Thymoglobulin | Tac/MMF/pred |
| 4 | 12 | 2.27 | 12 | 33 | 33 | LRD | 0 | n/a | MMF/pred/CSA→SRL |
| 5 | 37 | n/a | 38 | 38 | 38 | LRD | 0 | Basiliximab | Tac/MMF |
Symptoms = nephrolithiasis or development of chronic kidney disease; normal Uox < 0.46 mmol/24 hrs; n/a = not available; LURD = living unrelated donor; DD = deceased donor; LRD = living related donor; Tac = tacrolimus; MMF = mycophenolate mofetil; Pred = prednisone; CSA = cyclosporine; SRL = sirolimus.
Oxalate levels, allograft function and histology following transplant
Post-transplant urine oxalate levels are depicted in Figure 1. Following transplant, mobilization and excretion of oxalate tissue stores accumulated during ESRD took approximately 1.5 months. After that time period, urine oxalate decreased into the normal range and remained normal or near normal at the majority of visits throughout follow-up (< 0.5 mmol/24 hr at 33/50 times checked). The urine oxalate of patient 2 did not normalize until 9 months post-KTx. She had previously developed significant tissue oxalate stores while receiving standard 3 times per week hemodialysis without VB6 for over one year prior to transplantation. Post-transplant plasma oxalate levels among all patients are displayed in Figure 2. After falling quickly post-KTx, most were less than the reference range in a healthy non-PH population (< 1.8 mcmol/L) and 43/45 readings obtained after the first 1.5 months post-transplant were < 5 mcmol/L.
Figure 1.
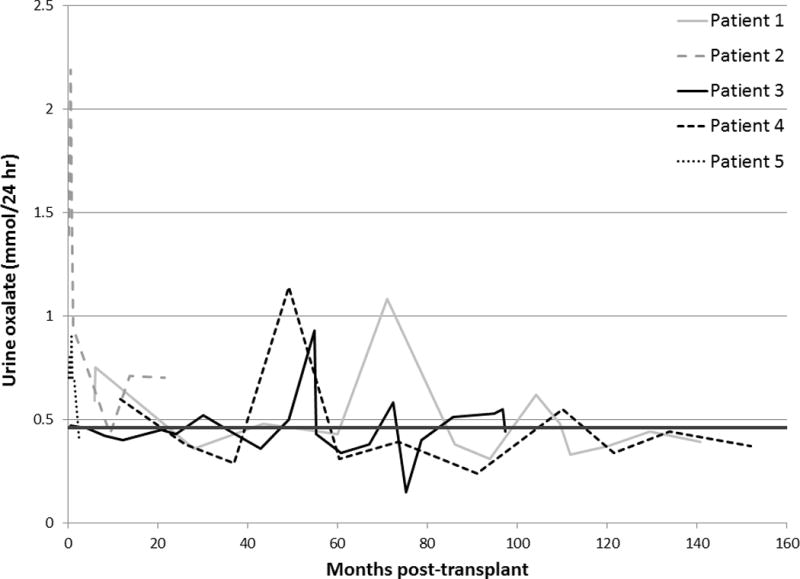
Urine oxalate levels following KTx alone in PH1 patients homozygous for the G170R mutation. The dark line is the upper limit of urine oxalate excretion in normal subjects (<0.46 mmol/24 hours).
Figure 2.
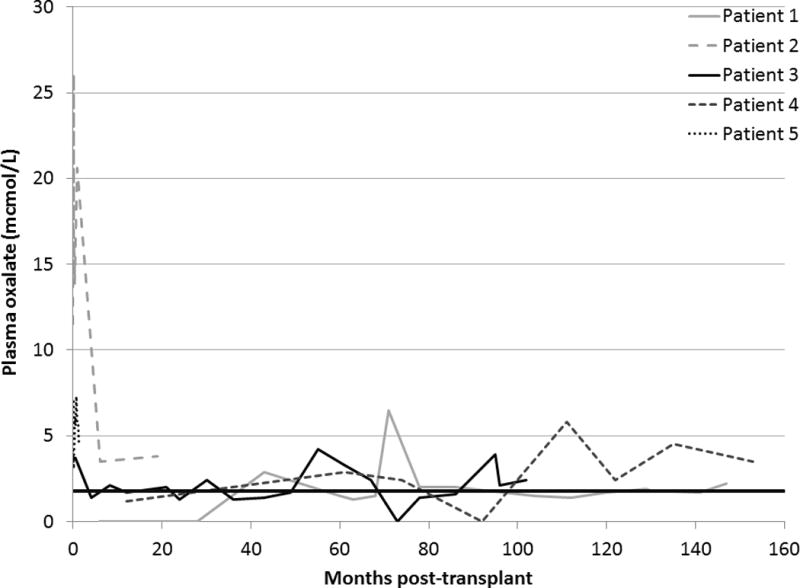
Plasma oxalate levels following KTx alone in PH1 patients homozygous for the G170R mutation. The dark line is the upper limit of plasma oxalate in normal subjects (<1.8 mcmol/L).
Four of the five allografts functioned throughout the study. At last follow-up, serum creatinine ranged from 1.2 to 1.8 mg/dL and eGFR ranged from 34 to 57 ml/min/1.73 m2 (Table 2 and Figure 3). In the four patients with functioning allografts, the corrected iothalamate clearance obtained closest to last follow-up ranged from 39 to 80 ml/min/1.73 m2. One allograft failed during follow-up. Patient 4 had stable allograft function and no evidence of oxalate deposition within the allograft until 13.9 years post-transplant when she developed allograft failure in the setting of an acute medical illness. After an episode of severe cholecystitis requiring cholecystectomy, the patient experienced nausea, vomiting, diarrhea and profound volume contraction followed by sudden allograft failure requiring initiation of hemodialysis. Whether VB6 was absorbed during her illness and hospitalizations is uncertain.
Table 2.
Renal allograft function following kidney alone transplant in G170R homozygous PH1 patients
| Patient | Vitamin B6 dose at last follow-up (mg/kg/d) | Follow-up post-KTx (yrs) | Creatinine at last follow-up (mg/dL) | Estimated GFR at last follow-up (ml/min/1.73m2) | Allograft status at last follow-up |
|---|---|---|---|---|---|
| 1 | 7.7 | 12.4 | 1.3 | 45 | Functioning |
| 2 | 9.4 | 1.8 | 1.8 | 34 | Functioning |
| 3 | 7.5 | 8.5 | 1.3 | 57 | Functioning |
| 4 | 5.4 | 13.9 | Failed | Failed | Failed due to oxalate nephropathy |
| 5 | 9.4 | 0.2 | 1.2 | 50 | Functioning |
Figure 3.
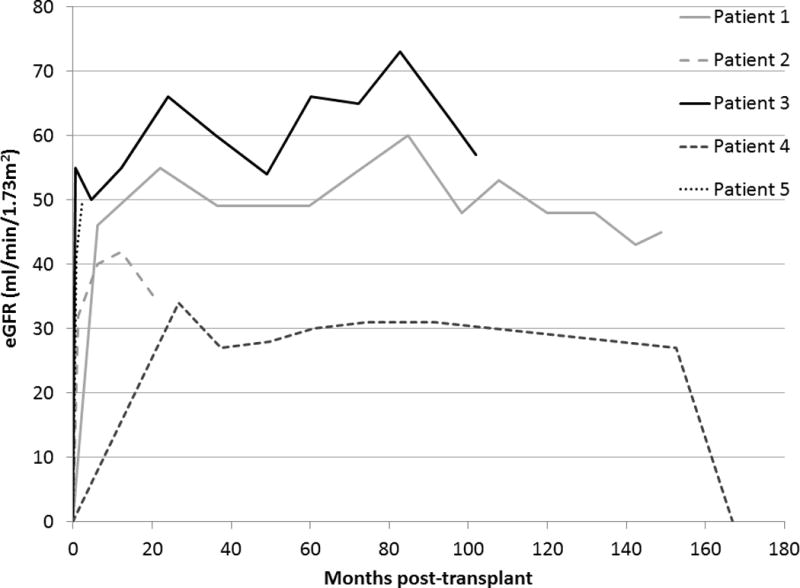
Estimated glomerular filtration rate (eGFR) following KTx alone in PH1 patients homozygous for the G170R mutation.
Allograft histology is outlined in Table 3. Calcium oxalate crystals were noted in only 3/13 allograft biopsies. The crystals were rare in 2 biopsies and present in moderate degree only in the patient who lost allograft function following an episode of dehydration. Most recent allograft biopsies in all 4 patients with functioning KTx did not reveal calcium oxalate deposition. Despite the early post-transplant elevations in plasma and urine oxalate levels in patient 2, the allograft biopsy 1.6 years post-KTx did not reveal calcium oxalate crystals and contained only mild fibrosis.
Table 3.
Allograft histology following KTx in G170R homozygous PH1 patients
| Patient | Time post-KTx (months) | Protocol biopsy |
Banff scores
|
Calcium oxalate | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| g | t | i | v | Ah | cg | ct | ci | cv | ||||
|
| ||||||||||||
| 1 | 61 | Yes | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | None |
|
| ||||||||||||
| 2 | 0 | Yes | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | None |
| 1 | Yes | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | Rare | |
| 6 | Yes | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | Rare | |
| 19 | Yes | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | None | |
|
| ||||||||||||
| 3 | 0 | Yes | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | None |
| 4 | Yes | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | None | |
| 12 | Yes | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | None | |
| 24 | Yes | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | None | |
| 61 | Yes | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | None | |
|
| ||||||||||||
| 4 | 37 | No | 0 | 0 | 0 | 0 | 3 | 0 | 2 | 3 | 1 | None |
| 165 | No | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Moderate | |
|
| ||||||||||||
| 5 | 0 | Yes | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | None |
n/a = not available; g = glomerulitis, t = tubulitis, i = interstitial inflammation, v = intimal arteritis, ah = arteriolar hyalinosis, cg = glomerulopathy, ct = tubular atrophy, ci = interstitial fibrosis, cv = vascular fibrous intimal thickening.
DISCUSSION
This case series is the first to show that KTx combined with VB6 therapy can be a successful long-term treatment option for PH1 patients homozygous for the G170R mutation. Over a median 8.5 years post-KTx, urine oxalate was < 0.5 mmol/24 hr the majority of the times it was monitored after initial normalization. Allograft function remained excellent throughout follow-up in four of five patients, and the fifth patient maintained good graft function for 13.9 years before the graft was lost. Plasma oxalate concentrations fell rapidly following transplantation. The significance of the mild elevations in plasma oxalate (1.8–5 mcmol/L) observed over time after transplantation and VB6 treatment remains to be established. Although patients with a significant burden of systemic oxalosis prior to transplant may experience persistent hyperoxaluria in the early post-transplant period, this problem can be avoided by earlier diagnosis of PH1 and prompt initiation of treatment with VB6. Even with systemic oxalosis, urinary oxalate levels improve over time and with careful management, oxalate nephropathy can be avoided in most patients. Allograft failure in one patient was precipitated by an acute medical illness resulting in severe volume depletion and uncertain absorption of VB6 therapy.
KTx alone in G170R homozygotes was first described by van Woerden et al. in 2004 and is a potentially attractive option because it spares patients the risks and morbidity associated with complete removal of the native liver and liver transplantation (11). Liver transplant also requires a scare resource (donated liver) and waiting times can be long. Combined liver kidney transplant can be associated with significant morbidity and early patient and graft survival rates are lower compared to kidney alone transplant (19, 20). Furthermore, with recent advances in molecular genetic testing VB6 responsiveness can be more easily assessed, even in patients with reduced kidney function since G170R homozygosity identifies this subgroup of PH1 (10). Testing for the G170R mutation determined by DNA sequencing is highly accurate. Patients with available living donors or older individuals with cardiovascular disease who may be intolerant of the stress of liver transplantation may particularly benefit from more tailored therapy with KTx alone.
While the impact of VB6 on urine oxalate excretion has been recognized for over fifty years (21), the mechanism by which VB6 increases AGT enzyme activity is still being elucidated, and likely involves more than one mechanism (12). The G170R mutation is a missense mutation. In cases of G170R homozygosity, AGT is mistargeted to the mitochondria instead of the peroxisome. Although the mistargeted enzyme remains catalytically active, it cannot metabolize oxalate within the peroxisome. Recent studies suggest that VB6 has many effects on AGT within the hepatocyte, including increasing production of the enzyme, increasing its metabolic activity and increasing targeting of the enzyme to the peroxisome (12). Although our current patient cohort demonstrated sustained response to VB6 therapy, and our previous experience has supported consistent association of G170R homozygosity and VB6 responsiveness, not all centers have reported uniform VB6 response based upon genotype (22). Therefore, assessment of urine oxalate excretion before and after initiation of pharmacologic VB6 dosing remains clinically helpful. Ideally, patients homozygous for the G170R mutation should be tested for responsiveness to VB6 prior to transplant. However, VB6 responsiveness can be particularly hard to assess among patients with advanced renal insufficiency, and in such cases relying upon genotyping results alone may be necessary.
While KTx alone appears to be an excellent option for the majority of G170R homozygotes, the procedure requires strict compliance with VB6 therapy post-transplant, plus avoidance of volume depletion. While the effect of VB6 was sustained over time, transient rises in both plasma and urine oxalate were noted, possibly related to noncompliance with VB6 therapy or other unexplained factors related to the effect of the medication. In addition, some authors have observed that urine oxalate levels improve in G170R homozygous patients but do not always remain consistently normal (22). Patients with a history of nonadherence or difficulty tolerating VB6 prior to transplant may not be good candidates for KTx alone. Patients who develop nausea or vomiting post-transplant or who are unable to tolerate oral medications for a period of time may require bridging therapy with intravenous VB6 and intravenous fluids to maintain urine output.
Close follow-up of PH1 patients receiving KTx alone is paramount. We recommend that patients undergo close monitoring of their urinary oxalate levels throughout the life of their transplant, including frequent follow-up with a nephrologist during the first post-transplant year and at least annual visits thereafter. In addition to VB6 therapy, other strategies to decrease the risk of calcium oxalate precipitation within the allograft include maintenance of an adequate urine volume and initiation of oral citrate therapy to reduce the risk for oxalate crystallization within the urine. The dose of VB6 should be regularly assessed. Most patients will receive maximum benefit from a dose of approximately 5–8 mg/kg/d (10). Pyridoxal 5-phosphate levels can also be monitored to ensure compliance with the drug. Although side effects have occasionally been reported at very high doses (> 1000 mgm per day), VB6 is generally well tolerated over many years. To date, we have observed no side effects of concern in any of our PH patients treated with long-term VB6 therapy. Lastly, protocol biopsies should be considered throughout the life of the allograft. Even early post-transplant biopsies help determine adequacy of treatment when the clearance of systemic oxalate stores is highest.
Our series demonstrates that PH1 patients homozygous for the G170R mutation may experience a sustained response to VB6 following KTx alone. Although the number of patients in our series was small and follow-up is ongoing, the majority of allograft biopsies in our cohort showed no evidence of recurrent oxalosis and long-term graft outcomes were excellent. KTx alone combined with long-term therapy with VB6 is an appropriate consideration in PH1 patients homozygous for the G170R mutation.
Acknowledgments
Funding for this work was provided by the National Institute of Diabetes and Digestive and Kidney Diseases, the National Center for Advancing Translational Sciences Rare Disease Clinical Research Network (U54KD083908) and the Oxalosis and Hyperoxaluria Foundation. The Rare Kidney Stone Consortium is a member of the Rare Diseases Clinical Research Network of the NIH. We also want to thank the staff of the Mayo Clinic Hyperoxaluria Center, referring physicians Dr. A. Abualfa, Dr. G. Posen and Dr. R Mallavarpu, and the many physicians and patients who have contributed to the RKSC PH Registry.
References
- 1.Milliner DS, Wilson DM, Smith LH. Phenotypic expression of primary hyperoxaluria: comparative features of types I and II. Kidney Int. 2001;59(1):31–36. doi: 10.1046/j.1523-1755.2001.00462.x. [DOI] [PubMed] [Google Scholar]
- 2.Hoppe B, Beck BB, Milliner DS. The primary hyperoxalurias. Kidney Int. 2009;75(12):1264–1271. doi: 10.1038/ki.2009.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rumsby G, Cochat P. Primary hyperoxaluria. N Engl J Med. 2013;369(22):2163. doi: 10.1056/NEJMc1311606. [DOI] [PubMed] [Google Scholar]
- 4.Cochat P, Hulton SA, Acquaviva C, Danpure CJ, Daudon M, De Marchi M, et al. Primary hyperoxaluria Type 1: indications for screening and guidance for diagnosis and treatment. Nephrol Dial Transplant. 2012;27(5):1729–1736. doi: 10.1093/ndt/gfs078. [DOI] [PubMed] [Google Scholar]
- 5.Katz A, Kim Y, Scheinman J, Najarian JS, Mauer SM. Long-term outcome of kidney transplantation in children with oxalosis. Transplant Proc. 1989;21(1 Pt 2):2033–2035. [PubMed] [Google Scholar]
- 6.Ruder H, Otto G, Schutgens RB, Querfeld U, Wanders RJ, Herzog KH, et al. Excessive urinary oxalate excretion after combined renal and hepatic transplantation for correction of hyperoxaluria type 1. Eur J Pediatr. 1990;150(1):56–58. doi: 10.1007/BF01959482. [DOI] [PubMed] [Google Scholar]
- 7.Bergstralh EJ, Monico CG, Lieske JC, Herges RM, Langman CB, Hoppe B, et al. Transplantation outcomes in primary hyperoxaluria. Am J Transplant. 2010;10(11):2493–2501. doi: 10.1111/j.1600-6143.2010.03271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Broyer M, Brunner FP, Brynger H, Dykes SR, Ehrich JH, Fassbinder W, et al. Kidney transplantation in primary oxalosis: data from the EDTA Registry. Nephrol Dial Transplant. 1990;5(5):332–336. doi: 10.1093/ndt/5.5.332. [DOI] [PubMed] [Google Scholar]
- 9.Harambat J, Fargue S, Acquaviva C, Gagnadoux MF, Janssen F, Liutkus A, et al. Genotype-phenotype correlation in primary hyperoxaluria type 1: the p.Gly170Arg AGXT mutation is associated with a better outcome. Kidney Int. 2010;77(5):443–449. doi: 10.1038/ki.2009.435. [DOI] [PubMed] [Google Scholar]
- 10.Monico CG, Rossetti S, Olson JB, Milliner DS. Pyridoxine effect in type I primary hyperoxaluria is associated with the most common mutant allele. Kidney Int. 2005;67(5):1704–1709. doi: 10.1111/j.1523-1755.2005.00267.x. [DOI] [PubMed] [Google Scholar]
- 11.van Woerden CS, Groothoff JW, Wijburg FA, Annink C, Wanders RJ, Waterham HR. Clinical implications of mutation analysis in primary hyperoxaluria type 1. Kidney Int. 2004;66(2):746–752. doi: 10.1111/j.1523-1755.2004.00796.x. [DOI] [PubMed] [Google Scholar]
- 12.Fargue S, Rumsby G, Danpure CJ. Multiple mechanisms of action of pyridoxine in primary hyperoxaluria type 1. Biochim Biophys Acta. 2013;1832(10):1776–1783. doi: 10.1016/j.bbadis.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 13.Gibbs DA, Watts RW. The action of pyridoxine in primary hyperoxaluria. Clin Sci. 1970;38(2):277–286. doi: 10.1042/cs0380277. [DOI] [PubMed] [Google Scholar]
- 14.Monico CG, Milliner DS. Combined liver-kidney and kidney-alone transplantation in primary hyperoxaluria. Liver Transpl. 2001;7(11):954–963. doi: 10.1053/jlts.2001.28741. [DOI] [PubMed] [Google Scholar]
- 15.Watts RW, Veall N, Purkiss P, Mansell MA, Haywood EF. The effect of pyridoxine on oxalate dynamics in three cases of primary hyperoxaluria (with glycollic aciduria) Clin Sci. 1985;69(1):87–90. doi: 10.1042/cs0690087. [DOI] [PubMed] [Google Scholar]
- 16.Levey AS, Bosch JP, Lewis JB, Greene T, Rogers N, Roth D. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of Diet in Renal Disease Study Group. Ann Intern Med. 1999;130(6):461–470. doi: 10.7326/0003-4819-130-6-199903160-00002. [DOI] [PubMed] [Google Scholar]
- 17.Racusen LC, Solez K, Colvin RB, Bonsib SM, Castro MC, Cavallo T, et al. The Banff 97 working classification of renal allograft pathology. Kidney Int. 1999;55(2):713–723. doi: 10.1046/j.1523-1755.1999.00299.x. [DOI] [PubMed] [Google Scholar]
- 18.Solez K, Colvin RB, Racusen LC, Haas M, Sis B, Mengel M, et al. Banff 07 classification of renal allograft pathology: updates and future directions. Am J Transplant. 2008;8(4):753–760. doi: 10.1111/j.1600-6143.2008.02159.x. [DOI] [PubMed] [Google Scholar]
- 19.Chava SP, Singh B, Zaman MB, Rela M, Heaton ND. Current indications for combined liver and kidney transplantation in adults. Transplant Rev (Orlando) 2009;23(2):111–119. doi: 10.1016/j.trre.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 20.Saborio P, Scheinman JI. Transplantation for primary hyperoxaluria in the United States. Kidney Int. 1999;56(3):1094–1100. doi: 10.1046/j.1523-1755.1999.00619.x. [DOI] [PubMed] [Google Scholar]
- 21.McLaurin AW, Beisel WR, McCormick GJ, Scalettar R, Herman RH. Primary hyperoxaluria. Ann Intern Med. 1961;55:70–80. doi: 10.7326/0003-4819-55-1-70. [DOI] [PubMed] [Google Scholar]
- 22.Hoyer-Kuhn H, Kohbrok S, Volland R, Franklin J, Hero B, Beck BB, et al. Vitamin B6 in Primary Hyperoxaluria I: First Prospective Trial after 40 Years of Practice. Clin J Am Soc Nephrol. 2014 doi: 10.2215/CJN.06820613. [DOI] [PMC free article] [PubMed] [Google Scholar]