

Abstract
In this article we propose that impaired efficiency of glutamatergic synaptic transmission and a compensatory reduction in inhibitory neurotransmission, a process called homeostatic dishinhibition, occurs in the aging brain and more dramatically in Alzheimer’s disease (AD). Homeostatic disinhibition may help understand certain features of the aging brain and AD including: 1) the increased risk for epileptic seizures, especially in the early phase of the disease; 2) the reduced ability to generate γ-oscillations and 3) the increase in neuronal activity as measured by functional MRI. Homeostatic disinhibition may be the major mechanism that activates cognitive reserve. Modulating neuronal activity may therefore be a viable therapeutic strategy in AD that can complement existing anti-amyloid strategies. Specifically, enhancing endogenous glutamatergic synaptic transmission through increased co-agonist signaling or through positive allosteric modulation of metabotropic glutamatergic receptors appears as an attractive strategy. Alternatively, further reduction of GABAergic signaling may work as well, although care has to be taken to prevent epileptic seizures.
Keywords: Disinhibition, Alzheimer′s Disease, GABA, interneuron, cognitive reserve, neuronal activity
Introduction
Alzheimer’s disease (AD) is a complex disease in which amyloid β (Aβ) plays a central role. Treatment strategies aimed at reducing Aβ load are in advanced stages of clinical trial testing and have thus far been the major focus for the development of a disease modifying therapy for AD [1]. However, results from completed clinical trials have been disappointing [2], indicating no cognitive improvement in AD patients. Consequently, it is argued that treatment should start in very early and presymptomatic phases of the disease [3; http://www.alz.org/icad/2010_release_diagnostic_071310_130pm.asp]. This would make such therapies preventive in character. Accordingly, efforts to establish biomarkers of AD are underway to identify high risk populations for intervention with preventive treatment strategies [4–6]; this strategy likely will provide useful and relevant biomarkers in the next few years [7]. However, even in a best case scenario it will take considerable time and effort to reach results like a number of patients needed to treat in order to prevent one case of AD. In view of this situation it is prudent to have a closer look at additional therapeutic options in AD. It is clear that aging and apolipoprotein E genotype are the two major risk factors for AD and that cognitive reserve is a major protective mechanism that can stave off the disease even in the presence of the most severe AD pathology [8]. A deeper insight into the underlying molecular mechansims of brain aging and cognitive reserve might thus enhance our overall understanding of AD and also lead to therapeutic strategies.
In this article we focus on the hypothesis that neuronal activity is altered in the aging brain in ways that may influence cognitive reserve and vulnerability to AD. Reviewing results from gene expression and brain imaging studies we argue that homeostatic disinhibition is an important aspect of neuronal network activity in the aging brain and in AD that can explain some important clinical findings in AD and may lead to treatment strategies that are independent of Aβ, but may nevertheless complement anti-Aβ treatment strategies in the future. For the purpose of this article we define cognitive reserve as the ability to perform normal or minimally impaired on a behavioural level despite the presence of pathological changes in the brain on a histological and macroanatomical level. We define homeostatic disinhibition as a reduction in inhibitory neurotransmission that compensates for a reduced excitatory drive.
Does AD = Aβ + aging?
Aβ and tau pathology are defining features of AD [9, 10] but it is important to realize that AD pathology displays additional features. It also includes a marked increase in astrogliosis [11] and reactive microglia [12, 13], a loss of cholinergic projection neurons in the basal forebrain [14] a reduction in aminergic projections from midbrain regions [15, 16], a reduction in somatostatin levels [17] and most notably a loss of synapses and neurons [18]. The loss of synapses is believed to precede the loss of neurons in AD and also to be accompanied by a (compensatory) increase in synapse size [18]. Basal forebrain neurons are severely affected by cell death early on during AD [14], and glutamatergic neurons in the transenthorhinal cortex are affected by tau pathology before these changes spread to the enthorhinal regions, the hippocampus and ultimately to neocortical areas with layer II/III and V neurons most significantly affected [9]. GABAergic interneurons are traditionally considered to be not significantly affected by cell loss in AD [19]. Loss of aminergic neurotransmitters from midbrain projection neurons has come into focus more recently, but may be an early event in AD pathology [15, 16]. Although AD is characterized by a net loss of neurons and synapses, axonal sprouting has also been observed, especially around plaques [20].
Importantly, a significant number of features of AD pathology is also present when old and young control brains are compared, albeit to a lesser degree. In mouse models of AD, amyloid pathology, astrogliosis and microglia activation are well replicated but the other aspects of AD pathology are less evident [21]. Accordingly, patients with AD suffer from severe dementia, whereas the cognitive deficits of mouse models are comparatively modest, typically involving a slightly shortened retention span in the Morris water maze test. This is also true for the so-called triple transgenic model of AD which, in addition to plaques, also displays neurofibrillary tangle-like tau pathology [22]. A potential explanation for these differences between mouse models and patients is that the full picture of AD pathology requires both aging and Aβ pathology and in mouse models the former aspect is not well replicated. Indeed, a systematic and extensive comparison of aging-associated changes in gene expression in brains of humans, monkeys and mice revealed no significant correlation between expression patterns of aging mice and humans, whereas a modest degree of correlation was observed between humans and monkeys [23]. This suggests differences in the aging process of humans and mice.
The case for homeostatic disinhibition in the aging brain
The two most prominent changes in gene expression of the aged human brain are reduced expression of transcripts associated with GABAergic transcription and cAMP-dependent signaling [23]. Many transcripts related to GABAergic neurotransmission are activity-dependently transcribed, e.g. glutamate decarboxylase (GAD) 1 and 2 or somatostatin (Sst), [24–27] and have binding sites for cAMP response element binding protein (CREB) in their promoter regions [28]. Apart from cAMP, (nuclear) calcium is the major activator of CREB and CREB is the prototypical activity-dependent transcription factor in neurons. Also, GABAergic interneurons express high levels of NMDA-receptors and are highly sensitive to NMDA-receptor blockade [29]. Thus, the aging human brain transcriptome can theoretically be explained by reduced activity-dependent transcription resulting in (compensatory) reduced expression of transcripts related to inhibitory neurotransmission (Fig. 1). In a situation of reduced excitatory (glutamatergic) drive, a homeostatic response results in reduced inhibitory tone [24]. Such homeostatic disinhibition keeps excitatory and inhibitory neurotransmission in balance. Maintaining this balance is a basic property of neuronal networks. It can be replicated in cultured neurons in which neuronal network activity is pharmacologically blocked, e.g. by the sodium channel inhibitor tetrodotoxin (TTX) or the NMDA receptor antagonist MK 801 [30]. A gene expression analysis in neurons treated with these compounds demonstrated that transcripts related to inhibitory neurotransmission, together with transcripts related to cholesterol synthesis and transcripts belonging to the calpacitin family, were most consistently and robustly down-regulated [30]. Pathway analysis of gene expression changes showed that TTX or MK 801-induced gene expression changes significantly overlap with gene expression changes observed in the aging human brain [23, 31] and in the hippocampus of AD patients [32]. Accordingly, both studies found transcripts related to GABAergic transmission down-regulated, including GAD1 and 2 and Sst. These findings were replicated in a recent smaller study studying AD-related gene expression changes in the neocortex [33]. In line with similar gene expression changes in hippocampus [32] and neocortex [33] in AD, aging-related changes in gene expression in hippocampus overlap with those of cortical regions but not of the cerebellum [34].
Figure 1. Homeostatic disinhibition in AD.
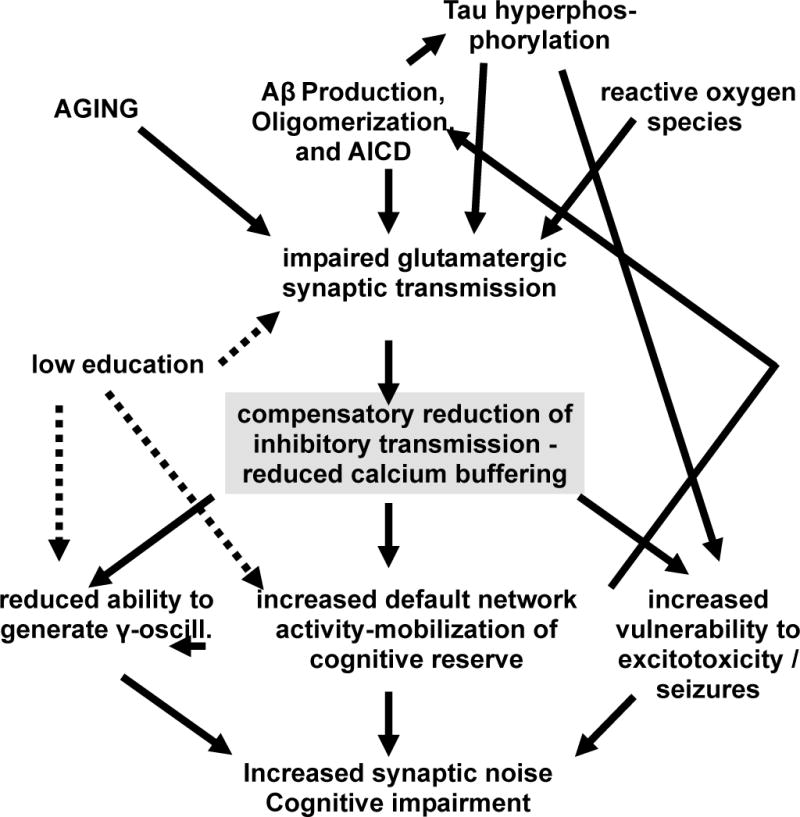
The diagrams depicts how Aβ and possibly AICD as well as aging-associated changes impair efficiency of glutamatergic synaptic transmission, leading to homeostatic disinhibition as a compensatory mechanism. Such changes in excitatory balance may mobilize cognitive reserve, but may nevertheless lead to negative consequences for information processing efficiency, seizure vulnerability and possibly excitotoxicity.
Homeostatic disinhibition does not involve cell death, but affects gene expression regulation. This is consistent with the old finding that GABAergic neurons are relatively resistant in AD [19]. Thus, cell death is an unlikely explanation for the observed gene expression changes in AD and the aging brain. In line with this hypothesis is the result from an in silico study using the dataset from Blalock et al [31]. An upstream analysis found that altered CREB-mediated transcription is the single most significant change taking place in AD and incipient AD [35]. Reduced expression of transcripts related to GABAergic transmission is seen both in aging (i.e. in old vs young brains) and in AD (i.e. in AD vs. age matched controls). Thus, at the gene expression level AD appears to be in part an extension or aggravation of the regular aging process. Similarly, certain histological features are present in AD and brain aging (astrogliosis, microglia activation, loss of cholinergic neurons in the basal forebrain). The close association between aging and AD is also exemplified by the doubling of dementia incidence every 5.5 years in old people [36].
Causes for reduced excitatory drive in the aging brain and in AD
Homeostatic dishinhibition describes a reduction in inhibitory neurotransmission that compensates for a reduced excitatory drive. Aβ has been repeatedly associated with impaired glutamatergic synaptic transmission [37–39]. The exact form of Aβ that mediates this effect as well as the molecular targets of Aβ and the sequence of events remain fairly controversial. E.g., Aβ dimers as well as oligomers comprised of 100 or more peptides or so called globulomers of 60 kDa were associated with impaired LTP through binding to NMDA receptors, calcium channels, the prion protein and other molecules [37–40]. Also, it has been reported that rather than being an antagonist for NMDA receptors, Aβ in fact activates NMDA receptors, which will then trigger an acute desensitization of receptors, the net effect being impaired NMDA receptor-dependent synaptic transmission [41]. A decline in NMDA receptor-dependent long term potentiation (LTP) has also been observed in aged rats [42]. However, the aging associated decline in glutamatergic drive could theoretically also be caused by other products of APP processing, e.g. by the APP intracellular domain (AICD). Moreover, accumulation of oxidatively modified proteins and lipids together with reduced metabolic capacity of mitochondria likely also contribute to an aging-associated decline in LTP.
It should be mentioned that excitatory effects of Aβ peptides have been reported as well [43]. Although this is seemingly in contrast with our hypothesis and findings of other groups [37–39], it should be kept mind that experiments with synthetic Aβ peptides have some caveats. Specifically, Minkeveciene et al. [43] used Aβ peptide 1–42 with mouse sequence and 25–35 with rat sequence at 1 μM for 60 min. Therefore the findings allow for multiple interpretations. One possible explanation would be that Aβ exerts dose dependent effects, e.g. inhibitory at lower doses and excitatory at higher doses [44].
Effects of homeostatic disinhibition
While homeostatic disinhibition keeps an overall balance between glutamatergic and GABAergic neurotransmission, not all aspects of neuronal network activity remain identical. GABAergic interneurons are the main firewall against epileptic seizures as well as the principle generators of γ-oscillations in the brain (repeated synchronized depolarizations of groups of neurons in the 30–80 Hz range). A reduction in GABAergic innervation is thus expected to result in increased vulnerability to epileptic seizures and a reduced capacity to generate γ-oscillations. Notably, AD is characterized by both features. Epidemiological studies show an increased risk for epileptic seizures in AD [45]. Interestingly, the risk for seizures is most pronounced in the early symptomatic period of the disease. However, in sporadic AD antiepileptic medication is rarely required and seizures do not tend to occur in high frequency. Homeostatic disinhibition provides a good explanation for these findings. Homeostatic disinhibition only poses a risk for seizures in situations with high excitatory input. As outlined above, this is generally not the case. However, in situations of emotional agitation, sensory overstimulation or metabolic challenges (e.g. low blood glucose levels) excitatory input may reach critical levels that then can not be adequately met by inhibitory neurotransmission. It can be speculated that this risk is higher early in the disease when glutamatergic drive is less severely impaired and homeostatic disinhibition may in fact result in some form of over-compensation. In later stages glutamatergic transmission is further impaired, making critical levels of excitatory input less likely.
It has been noted before that AD is associated with two seemingly paradoxical features, i.e. Aβ-mediated impairment of excitatory synaptic transmission and increased network excitability (examplified by increased seizure frequency) [46, 47]. So far few explanations for this seeming paradox have been proposed [44]. We think that homeostatic disinhibition offers a straight forward explanation for this well recognized paradox.
While seizure frequency in sporadic AD is increased in comparison to age matched control populations, it is not a very common event and rarely requires medication. In familial AD this can be quite different and many cases have been published that were associated where intractable seizures posed a serious therapeutic challenge [45]. However, in the majority of these cases mutations in the Presenilin 1 gene were involved. It is now well established that the Presenilin 1 protein has a direct function in maintaining intraneuronal calcium homeostasis, which, in addition to γ-secretase function, is also affected by mutation associated with familial AD [48–50]
γ-Oscillations are believed to be critical for encoding information and higher power of the γ-spectrum is believed to be associated with more efficient information processing [51]. The ability to generate γ-oscillations declines with age and is further reduced in AD patients and is correlated with cognitive decline in these groups [51]. These findings are in line with a theory of homeostatic disinhibition in the aging brain and in AD.
Findings in brain imaging studies
EEG and MEG, to study brain activity, have high temporal resolution but comparatively low spatial resolution. Glucose uptake is a metabolic marker that can be studied with positron emission tomography (PET). It is believed to be closely connected to neuronal acitivity. Similarly, the blood oxygenation level difference (BOLD) signal that is measured by functional MRI (fMRI) is often used as a marker for neuronal activity. fMRI is attractive because it has higher spatial and temporal resolution than PET and is non-invasive, requiring no radioactive substances. PET studies have shown a decline of glucose uptake in the aging and in AD [52]. This has been interpreted as a result of reduced neuronal activity as well as a loss of synaptic connections and in AD also a loss of neurons. Therefore, it was initially surprising that fMRI studies found greater areas of the brain activated (as determined by BOLD signal) during cognitive tasks in older subjects and subjects in the early stages of AD [53, 54]. These findings have been interpreted to reflect superior efficiency of the young brain. For the same task fewer brain regions are activated in the young brain, whereas in older brains more brain regions become or stay active. At the same time cognitive performance is better in younger subjects. This has been well documented for a region coined the default network that shows reduced activity in memory recall tasks in brains of young subjects. In old subjects this reduction in activity weakens and in (pre)MCI and AD patients this regions remains active throughout the task [55]. Interestingly, this region also shows intense Aβ deposition that correlates with BOLD signal intensity. Homeostatic disinhibition offers an attractive hypothesis to reconcile electrophysiological and imaging findings in the aging brain. Old age and Aβ cause impaired glutamatergic synaptic transmission and thus homeostatic disinhibition. As a result the capacity to generate γ-oscillations declines and information processing becomes less efficient. At the same time reduced inhibitory tone will faciliate the activation of other brain areas, facilitating recruitment of the cognitive reserve. Recent studies combining MRI spectroscopy and fMRI [56, 57] and MEG [58] found that resting state GABA levels are indeed negatively correlated with BOLD signal intensity and positively correlated with γ-oscillation power. It should be noted, however, that these studies were performed in young healthy control subjects and not in all studies was the default network area part of the analysis. Interestingly, a longitudinal study found that in the early phase of transition to AD BOLD signal intensity in the hippocampus can exceed levels of non demented control subjects [59]. It may be that homeostatic inhibition initially causes an overshoot-reaction. This would fit with the observation of maximal risk for epileptic seizures in the early phase of AD [45].
Homeostatic Disinhibition and Reduction in Neurotrophic Support
Activation of neural circuits, as occurs during performance of cognitive tasks and during exercise, results in increased expression of neurotrophic factors that support neuronal survival and synaptic plasticity [60]. Among the neurotrophic factors produced in response to neuronal activity, brain-derived neurotrophic factor (BDNF) plays a particularly important role in learning and memory. BDNF is critical for long-term potentiation of synaptic transmission and neurogenesis, and mediates at least some of the beneficial effects of exercise and cognitive stimulation on neuronal resiliency [61, 62]. Our homeostatic disinhibition model predicts that an increase in GABAergic signaling in brain aging and AD would result in decreased BDNF expression. Indeed, it has been reported that levels of BDNF in the cerebrospinal fluid of elderly subjects are positively correlated with performance on cognitive tests [63], and the expression of BDNF by hippocampal cells is significantly reduced in AD patients compared to age-matched control subjects [64]. Studies of transgenic AD mouse models have provided evidence that Aβ reduces BDNF production [65], possibly by impairment of synaptic transmission and homeostatic disinhibition. That a reduction in BDNF levels plays a role in cognitive impairment in aging and AD is suggested by studies showing that BDNF delivery to the brain can improve cognitive performance in rodent and monkey models of AD [66, 67]. With regards to its possible effects on homeostatic disinhibition, it was shown that BDNF can counteract the adverse effects of chronic silencing of neuronal network activity in an experimental model [30]. On the other hand, because of its ability to enhance synaptic plasticity it is possible that BDNF could lower seizure threshold; however, this seems unlikely because BDNF levels are decreased in the brain during aging and in AD.
Therapeutic implications
If homeostatic dishinhibition is considered valid and relevant for the aging brain and AD, at least two therapeutic strategies can be adopted. Excitatory drive can be enhanced or inhibitory neurotransmisson further reduced. Since the glutamatergic deficit is considered the primary cause for homeostatic disinhibition, enhancing glutamatergic synaptic transmission may be regarded as the more upstream strategy. In this case it is expected that GABAergic transmission will adapt to the higher levels of excitatory drive, ultimately improving the ability for gamma oscillation generation and for efficient information processing. However, further reducing inhibitory neurotransmission may just as well be a valid therapeutic strategy. Here the rationale is to shift the glutamate/GABA balance more towards glutamate. The first approach may be limited by the metabolic capacity of old neurons. Both strategies may carry a risk for inducing seizures, especially the second, as the balancing capacity of the GABAergic system will be hampered. The second approach also will come without an increase in gamma oscillations. One can speculate that the second approach is more effective in later stages of the disease, where the mechanism of homeostatic disinhibition is close to being exhausted. Further pharmacological disinhibition would then extend the potential benefit of homeostatic disinhibition.
Pharmacological intervention
If the first approach is chosen, many pharmacological targets that are investigated in schizophrenia research may also have potential in AD. A very similar hypothesis of GABAergic disinhibition in response to a glutamatergic deficit has been formulated in schizophrenia research [68]. However, in schizophrenia the glutamatergic deficit is considered to be a developmental deficit, rather than a progressively worsening aging phenomenon. The glutamatergic deficit is the primary reason for the cognitive deficit associated with schizophrenia. Current medications for schizophrenia do not improve the cognitive deficit, that is, however, more relevant for long term prognosis. Therefore, drugs that can enhance glutamatergic drive are investigated in schizophrenia research. They may also be used for AD therapy. Strategies include increasing activation of the NMDA coagonist binding sites (cyclo-D-serine) or inhibitors of glycine re-uptake, or targeting receptors of modulating neurotransmitters, such as metabotropic glutamate receptors (mGluRs), cannabinoid receptors, noradrenergic receptors. In principle, allosteric modulation offers the most attractive mechanism to modulate synaptic transmission, as it will only enhance (positive allosteric modulators) or weaken (negative allosteric modulators) endogenous neurotransmission. In general, neurotransmitter receptors expression levels are high in the CNS and low outside the CNS. This, too, makes them good drug targets. However, the pharmacology for specific allosteric modulators is challenging and few compounds have made it into clinical trials.
Viewing information processing in the brain merely as the sum of excitatory and inhibitory drive oversimplifies things and it is unlikely that all brain regions will have a similar extend of homeostatic disinhibition. A global increase in excitatory drive may thus have the unintended effect of increasing noise in information processing; for example, when enhanced excitatory drive results in increased activity in the hippocampus but also in the default network it is easy to anticipate an unfavourable result. Therefore, strategies aimed at increasing excitatory drive will benefit from a detailed understanding of cortical and hippocampal microcircuits and their activity patterns in aging and AD. Mining existing databases such as the Allen brain atlas may also help to identify targets with a favourable expression pattern. Another option to increase specificity of the therapeutic approach may lie in designing co-agonists that bind specifically and reversibly to the closed resting state of ion channels. Such properties would target the drug preferentially to inactive neuron populations, while sparing active regions, thereby reducing the risk of generating seizures.
Further reducing inhibitory tone might be accomplished through the same classes of drug targets, depending on which subtype of recptor is targeted and whether positive or negative allosteric modulators are used. Using antagonists for GABA-A receptors would seem like a dangerous strategy, carrying a high risk of inducing epileptic seizures. However, antagonist (or negative allosteric modulators) for GABA-B receptors (which are G-protein coupled receptors, not ion channels) is an interesting alternative. In fact, this strategy was tested with a compound called SGS742 [69]. This compound gave positive results in a phase II study in subjects with MCI but most likely failed in a phase II study with AD patients (http://clinicaltrials.gov/ct2/show/NCT00093951?term=sgs742&rank=1). Interestingly, the first effect of NMDA inhibition usually is GABAergic disinhibition, due to the fact that GABAergic interneurons display a high density of NMDA receptors and are thus most sensitive to NMDA inhibition. In this respect it is conceivable that the beneficial effect of the weak NMDA antagonist memantine may in fact be based on disinhibition and activation of cognitive reserve. In line with this theory are some of the observed side effects of memantine, such as hallucinations that are most apparent in cases of Lewy body dementia [70], but anecdotal cases in AD patients have also been reported [71; MM personal observation]. Also, the fact that memantine is more effective in moderate to severe stages of AD, as opposed to mild stages, would be in line with a mechanism based on disinhibiton. However, it should be noted that no consensus exists over memantine’s mode of action. Other hypotheses include prevention of excitotoxicity and prevention of NMDA receptor desensitization [72].
This list of pharmacological intervention does not claim to be exhaustive. Other therapeutic are conceivable as well. Altered neuronal activity and learning are likely to affect histone modification, i.e. epigenetic modulation. Consequently, drugs that target posttranslational modifications of proteins, including methylation and acetylation, may also have potential in AD therapy [73].
Behavioral intervention
Apart from pharmacological intervention, excitatory drive may also be modulated through behavior. Homeostatic disinhibition would therefore also provide a clear rationale for behavioral cognitive stimulation. Similarly, the beneficial effect of education on cognitive reserve may be based on increased excitatory drive and compensatory increased inhibitory tone in younger years. Later in life, the degree to which inhibitory tone can be reduced will then be higher in individuals with more education.
Computer programs are used to improve cognitive function in a variety of conditions. A recent large clinical study, however, found no evidence that computer programs improve cognitive performance in a fashion relevant to activities of daily living [74]. However, performance in the trained tasks did increase significantly, demonstrating that cognitive stimulation results in some degree of neuronal plasticity even in an aged population. Also, smaller studies with more intensive training have reported some modest improvement of cognitive function not directly related to the learned task [75]. In the aged canine model of AD, which is closer to sporadic AD than transgenic mouse models, environmental enrichment caused a significant improvement in cognitive performance, but Aβ-vaccination did not [76–78]. Interestingly, both strategies reduced Aβ load, but this was not strongly correlated with cognitive performance.
Thus, currently no known behavioral intervention in AD patients has been proven effective but future improvements in such intervention protocols, possibly also including computer programs, may change this.
Electrical or magnetic stimulation
A third option to enhance excitatory drive is electrophysiologically through transcranial magnetic stimulation (TMS) or transcranial direct current stimulation (tDCS). tDCS is applied more easily at home. In vitro models and computer simulations demonstrate that the effect of tDCS is not outright depolarization of neurons. It is more a facilitation of depolarization in the presence of an incoming stimulus, and thus bears some resemblance to allosteric modulation, although it is more likely mediated throug very minor alterations of the resting membrane potential in the 1 to 2 mV range [79]. Recently tDCS has been shown to facilitate a cortical form of long term potentiation in acute slice cultures [80]. Proof of principle studies have consistently shown that tDCS improves learning of motor skills in control subjects but data are less clear in patients recovering from stroke [81, 82]. Nor are effects on every day motor performance and on cognition well established. A technical limitation of the method is that it affects mostly brain regions close to the skull but not in deeper regions. In AD tDCS might activate prefrontal and parietal regions, which may be useful, although medial temporal region would be most desirable.
Combining different modalities to increase exctitatory drive is an attractive idea. Specifically, combining behavioral interventions with pharmacological or electrophysiological interventions makes sense [83].
Conclusion
In this article we argue that homeostatic dishinhibition is a key feature of neuronal network activity in the aging brain and in AD. In Figure 1 we have summarized how homeostatic disinhibition and modulation of network activity are an essential part of Alzheimer’s disease. The additive (or synergistic) effect of Aβ and aging on impaired glutamatergic drive may ultimately exhaust compensation by homeostatic disinhibition, resulting in more rapid cognitive deterioration. This model provides a rationale for a treatment strategy that involves enhancing efficiency of glutamatergic neurotransmission. Alternatively, a strategy of reducing GABAergic inhibitory tone might be pursued, although we believe this option will be less efficient and more prone to unwanted side effects. We think that homeostatic disinhibition is closely linked to mobilisation of cognitive reserve. Boosting glutamatergic drive might thus increase an individual’s cognitive reserve. This would be a symptomatic, rather than disease modifying treatment strategy. However, symptomatic treatment does not necessarily preclude strong therapeutic effects. Higher education is associated with increased cognitive reserve and a roughly 30% lower prevalence and incidence of AD [84, 85]. Any treatment that would reduce AD incidence by 30% would be considered a major therapeutic breakthrough by today’s standards. Moreover, we believe that such a symptomatic treatment strategy might act synergistally with anti-Aβ therapies and increase efficacy of anti-Aβ therapies in later stages of the disease.
Homeostatic disinhibition involves a compensatory adjustment of the balance between excitatory and inhibitory synaptic input. This likely oversimplifies the anatomy of the brain, but is nevertheless useful to generate testable hypotheses. Microcircuits in hippocampus and cortex obviously are more complex and involve many more neuromodulatory transmitters. A more thorough understanding of such microcircuits in critical anatomical areas may therefore reveal additional therapeutic options.
References
- 1.Citron M. Alzheimer’s disease: strategies for disease modification. Nat Rev Drug Discov. 2010;9:387–398. doi: 10.1038/nrd2896. [DOI] [PubMed] [Google Scholar]
- 2.Creed MC, Milgram NW. Amyloid-modifying therapies for Alzheimer’s disease: therapeutic progress and its implications. Age (Dordr) 2008;32:365–384. doi: 10.1007/s11357-010-9142-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reiman EM, Langbaum JB, Tariot PN. Alzheimer’s prevention initiative: a proposal to evaluate presymptomatic treatments as quickly as possible. Biomark Med. 2010;4:3–14. doi: 10.2217/bmm.09.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khachaturian ZS. Perspective on the Alzheimer’s disease neuroimaging initiative: progress report and future plans. Alzheimers Dement. 2010;6:199–201. doi: 10.1016/j.jalz.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 5.Beckett LA, Harvey DJ, Gamst A, Donohue M, Kornak J, Zhang H, Kuo JH, Alzheimer’s Disease Neuroimaging Initiative The Alzheimer’s Disease Neuroimaging Initiative: Annual change in biomarkers and clinical outcomes. Alzheimers Dement. 2010;6:257–264. doi: 10.1016/j.jalz.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weiner MW, Aisen PS, Jack CR, Jr, Jagust WJ, Trojanowski JQ, Shaw L, Saykin AJ, Morris JC, Cairns N, Beckett LA, Toga A, Green R, Walter S, Soares H, Snyder P, Siemers E, Potter W, Cole PE, Schmidt M, Alzheimer’s Disease Neuroimaging Initiative The Alzheimer’s disease neuroimaging initiative: progress report and future plans. Alzheimers Dement. 2010;6:202–211. doi: 10.1016/j.jalz.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fagan AM, Holtzman DM. Cerebrospinal fluid biomarkers of Alzheimer’s disease. Biomarker Med. 2010;4:51–63. doi: 10.2217/BMM.09.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Snowdon DA, Nun Study Healthy aging and dementia: findings from the Nun Study. Ann Intern Med. 2003;139:450–454. doi: 10.7326/0003-4819-139-5_part_2-200309021-00014. [DOI] [PubMed] [Google Scholar]
- 9.Braak H, Braak E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging. 1995;16:271–278. doi: 10.1016/0197-4580(95)00021-6. [DOI] [PubMed] [Google Scholar]
- 10.Nelson PT, Braak H, Markesbery WR. Neuropathology and cognitive impairment in Alzheimer disease: a complex but coherent relationship. J Neuropathol Exp Neurol. 2009;68:1–14. doi: 10.1097/NEN.0b013e3181919a48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kashon ML, Ross GW, O’Callaghan JP, Miller DB, Petrovitch H, Burchfiel CM, Sharp DS, Markesbery WR, Davis DG, Hardman J, Nelson J, White LR. Associations of cortical astrogliosis with cognitive performance and dementia status. J Alzheimers Dis. 2004;6:595–604. doi: 10.3233/jad-2004-6604. [DOI] [PubMed] [Google Scholar]
- 12.McGeer PL, Kawamata T, Walker DG, Akiyama H, Tooyama I, McGeer EG. Microglia in degenerative neurological disease. Glia. 1993;7:84–92. doi: 10.1002/glia.440070114. [DOI] [PubMed] [Google Scholar]
- 13.McGeer EG, McGeer PL. Neuroinflammation in Alzheimer’s disease and mild cognitive impairment: a field in its infancy. J Alzheimers Dis. 2010;19:355–361. doi: 10.3233/JAD-2010-1219. [DOI] [PubMed] [Google Scholar]
- 14.Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delon MR. Alzheimer’s disease and senile dementia: loss of neurons in the basal forebrain. Science. 1982;215:1237–1239. doi: 10.1126/science.7058341. [DOI] [PubMed] [Google Scholar]
- 15.Samuels ER, Szabadi E. Functional neuroanatomy of the noradrenergic locus coeruleus: its roles in the regulation of arousal and autonomic function part II: physiological and pharmacological manipulations and pathological alterations of locus coeruleus activity in humans. Curr Neuropharmacol. 2010;6:254–285. doi: 10.2174/157015908785777193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simic G, Stanic G, Mladinov M, Jovanov-Milosevic N, Kostovic I, Hof PR. Does Alzheimer’s disease begin in the brainstem? Neuropathol Appl Neurobiol. 2009;35:532–554. doi: 10.1111/j.1365-2990.2009.01038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davies P, Katzman R, Terry RD. Reduced somatostatin-like immunoreactivity in cerebral cortex from cases of Alzheimer disease and Alzheimer senile dementa. Nature. 1980;288:279–280. doi: 10.1038/288279a0. [DOI] [PubMed] [Google Scholar]
- 18.Davies CA, Mann DM, Sumpter PQ, Yates PO. A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer’s disease. J Neurol Sci. 1987;78:151–164. doi: 10.1016/0022-510x(87)90057-8. [DOI] [PubMed] [Google Scholar]
- 19.Davies P, Anderton B, Kirsch J, Konnerth A, Nitsch R, Sheetz M. First one in, last one out: the role of gabaergic transmission in generation and degeneration. Prog Neurobiol. 1998;55:651–658. doi: 10.1016/s0301-0082(98)00024-0. [DOI] [PubMed] [Google Scholar]
- 20.Masliah E, Honer WG, Mallory M, Voigt M, Kushner P, Hansen L, Terry R. Topographical distribution of synaptic-associated proteins in the neuritic plaques of Alzheimer’s disease hippocampus. Acta Neuropathol. 1994;87:135–142. doi: 10.1007/BF00296182. [DOI] [PubMed] [Google Scholar]
- 21.Howlett DR, Richardson JC. The pathology of APP transgenic mice: a model of Alzheimer’s disease or simply overexpression of APP? Histol Histopathol. 2009;24:83–100. doi: 10.14670/HH-24.83. [DOI] [PubMed] [Google Scholar]
- 22.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 23.Loerch PM, Lu T, Dakin KA, Vann JM, Isaacs A, Geula C, Wang J, Pan Y, Gabuzda DH, Li C, Prolla TA, Yankner BA. Evolution of the aging brain transcriptome and synaptic regulation. PLoS One. 2008;3:e3329. doi: 10.1371/journal.pone.0003329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hendry SH, Jones EG. Activity-dependent regulation of GABA expression in the visual cortex of adult monkeys. Neuron. 1988;1:701–712. doi: 10.1016/0896-6273(88)90169-9. [DOI] [PubMed] [Google Scholar]
- 25.Benson DL, Huntsman MM, Jones EG. Activity-dependent changes in GAD and preprotachykinin mRNAs in visual cortex of adult monkeys. Cereb Cortex. 1994;4:40–51. doi: 10.1093/cercor/4.1.40. [DOI] [PubMed] [Google Scholar]
- 26.Tallent MK. Somatostatin in the dentate gyrus. Prog Brain Res. 2007;163:265–284. doi: 10.1016/S0079-6123(07)63016-7. [DOI] [PubMed] [Google Scholar]
- 27.de Lima AD, Opitz T, Voigt T. Irreversible loss of a subpopulation of cortical interneurons in the absence of glutamatergic network activity. Eur J Neurosci. 2004;19:2931–2943. doi: 10.1111/j.0953-816X.2004.03403.x. [DOI] [PubMed] [Google Scholar]
- 28.Zhang X, Odom DT, Koo SH, Conkright MD, Canettieri G, Best J, Chen H, Jenner R, Herbolsheimer E, Jacobsen E, Kadam S, Ecker JR, Emerson B, Hogenesch JB, Unterman T, Young RA, Montminy M. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci U S A. 2005;102:4459–4464. doi: 10.1073/pnas.0501076102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Braun I, Genius J, Grunze H, Bender A, Möller HJ, Rujescu D. Alterations of hippocampal and prefrontal GABAergic interneurons in an animal model of psychosis induced by NMDA receptor antagonism. Schizophr Res. 2007;97:254–263. doi: 10.1016/j.schres.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 30.Gleichmann M, Zhang Y, Wood WH, Becker KG, Mughal MR, Pazin MJ, van Praag H, Kobilo T, Zonderman AB, Troncoso JC, Markesbery WR, Mattson MP. Molecular changes in brain aging and Alzheimer′s disease are mirrored in experimentally silenced cortical neuron networks. Neurobiol Aging. 2010 Oct 12; doi: 10.1016/j.neurobiolaging.2010.08.012. 2010. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–891. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- 32.Blalock EM, Geddes JW, Chen KC, Porter NM, Markesbery WR, Landfield PW. Incipient Alzheimer’s disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc Natl Acad Sci U S A. 2004;101:2173–2178. doi: 10.1073/pnas.0308512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tan MG, Chua WT, Esiri MM, Smith AD, Vinters HV, Lai MK. Genome wide profiling of altered gene expression in the neocortex of Alzheimer’s disease. J Neurosci Res. 2010;88:1157–1169. doi: 10.1002/jnr.22290. [DOI] [PubMed] [Google Scholar]
- 34.Fraser HB, Khaitovich P, Plotkin JB, Pääbo S, Eisen MB. Aging and gene expression in the primate brain. PLoS Biol. 2005;3:e274. doi: 10.1371/journal.pbio.0030274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Satoh J, Tabunoki H, Arima K. Molecular network analysis suggests aberrant CREB-mediated gene regulation in the Alzheimer disease hippocampus. Dis Markers. 2009;27:239–252. doi: 10.3233/DMA-2009-0670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Corrada MM, Brookmeyer R, Paganini-Hill A, Berlau D, Kawas CH. Dementia incidence continues to increase with age in the oldest old: the 90+ study. Ann Neurol. 2010;67:114–121. doi: 10.1002/ana.21915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nimmrich V, Grimm C, Draguhn A, Barghorn S, Lehmann A, Schoemaker H, Hillen H, Gross G, Ebert U, Bruehl C. Amyloid beta oligomers (A beta(1–42) globulomer) suppress spontaneous synaptic activity by inhibition of P/Q-type calcium currents. J Neurosci. 2008;28:788–797. doi: 10.1523/JNEUROSCI.4771-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hillen H, Barghorn S, Striebinger A, Labkovsky B, Müller R, Nimmrich V, Nolte MW, Perez-Cruz C, van der Auwera I, van Leuven F, van Gaalen M, Bespalov AY, Schoemaker H, Sullivan JP, Ebert U. Generation and therapeutic efficacy of highly oligomer-specific beta-amyloid antibodies. J Neurosci. 2010;30:10369–10379. doi: 10.1523/JNEUROSCI.5721-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ondrejcak T, Klyubin I, Hu NW, Barry AE, Cullen WK, Rowan MJ. Alzheimer’s disease amyloid beta-protein and synaptic function. Neuromolecular Med. 2010;12:13–26. doi: 10.1007/s12017-009-8091-0. [DOI] [PubMed] [Google Scholar]
- 42.Boric K, Muñoz P, Gallagher M, Kirkwood A. Potential adaptive function for altered long-term potentiation mechanisms in aging hippocampus. J Neurosci. 2008;28:8034–8039. doi: 10.1523/JNEUROSCI.2036-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Minkeviciene R, Rheims S, Dobszay MB, Zilberter M, Hartikainen J, Fülöp L, Penke B, Zilberter Y, Harkany T, Pitkänen A, Tanila H. Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci. 2009;29:3453–362. doi: 10.1523/JNEUROSCI.5215-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Palop JJ, Mucke L. Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat Neurosci. 2010;13:812–818. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Larner AJ. Epileptic seizures in AD patients. Neuromolecular Med. 2010;12:71–77. doi: 10.1007/s12017-009-8076-z. [DOI] [PubMed] [Google Scholar]
- 46.Palop JJ, Mucke L. Synaptic depression and aberrant excitatory network activity in Alzheimer’s disease: two faces of the same coin? Neuromolecular Med. 2010;12:48–55. doi: 10.1007/s12017-009-8097-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bishop NA, Lu T, Yankner BA. Neural mechanisms of ageing and cognitive decline. Nature. 2010;464:529–535. doi: 10.1038/nature08983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheung KH, Mei L, Mak DO, Hayashi I, Iwatsubo T, Kang DE, Foskett JK. Gain-of-function enhancement of IP3 receptor modal gating by familial Alzheimer’s disease-linked presenilin mutants in human cells and mouse neurons. Sci Signal. 2010;3:ra22. doi: 10.1126/scisignal.2000818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gleichmann M, Mattson M. Neuronal Calcium Homeostasis and Dysregulation. Antioxid Redox Signal. 2010 doi: 10.1089/ars.2010.3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008;31:454–463. doi: 10.1016/j.tins.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Herrmann CS, Demiralp T. Human EEG gamma oscillations in neuropsychiatric disorders. Clin Neurophysiol. 2005;116:2719–2733. doi: 10.1016/j.clinph.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 52.Jagust WJ, Bandy D, Chen K, Foster NL, Landau SM, Mathis CA, Price JC, Reiman EM, Skovronsky D, Koeppe RA, Alzheimer’s Disease Neuroimaging Initiative The Alzheimer’s Disease Neuroimaging Initiative positron emission tomography core. Alzheimers Dement. 2010;6:221–229. doi: 10.1016/j.jalz.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sperling RA, Dickerson BC, Pihlajamaki M, Vannini P, LaViolette PS, Vitolo OV, Hedden T, Becker JA, Rentz DM, Selkoe DJ, Johnson KA. Functional alterations in memory networks in early Alzheimer’s disease. Neuromolecular Med. 2010;12:27–43. doi: 10.1007/s12017-009-8109-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Park DC, Reuter-Lorenz P. The adaptive brain: aging and neurocognitive scaffolding. Annu Rev Psychol. 2009;60:173–196. doi: 10.1146/annurev.psych.59.103006.093656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sperling RA, Laviolette PS, O’Keefe K, O’Brien J, Rentz DM, Pihlajamaki M, Marshall G, Hyman BT, Selkoe DJ, Hedden T, Buckner RL, Becker JA, Johnson KA. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron. 2009;63:178–188. doi: 10.1016/j.neuron.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Northoff G, Walter M, Schulte RF, Beck J, Dydak U, Henning A, Boeker H, Grimm S, Boesiger P. GABA concentrations in the human anterior cingulate cortex predict negative BOLD responses in fMRI. Nat Neurosci. 2007;10:1515–1517. doi: 10.1038/nn2001. [DOI] [PubMed] [Google Scholar]
- 57.Donahue MJ, Near J, Blicher JU, Jezzard P. Baseline GABA concentration and fMRI response. Neuroimage. 2010;53:392–398. doi: 10.1016/j.neuroimage.2010.07.017. [DOI] [PubMed] [Google Scholar]
- 58.Muthukumaraswamy SD, Edden RA, Jones DK, Swettenham JB, Singh KD. Resting GABA concentration predicts peak gamma frequency and fMRI amplitude in response to visual stimulation in humans. Proc Natl Acad Sci U S A. 2009;106:8356–8361. doi: 10.1073/pnas.0900728106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.O’Brien JL, O’Keefe KM, LaViolette PS, DeLuca AN, Blacker D, Dickerson BC, Sperling RA. Longitudinal fMRI in elderly reveals loss of hippocampal activation with clinical decline. Neurology. 2010;74:1969–1976. doi: 10.1212/WNL.0b013e3181e3966e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mattson MP. Glutamate and neurotrophic factors in neuronal plasticity and disease. Ann N Y Acad Sci. 2008;1144:97–112. doi: 10.1196/annals.1418.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rex CS, Lauterborn JC, Lin CY, Kramár EA, Rogers GA, Gall CM, Lynch G. Restoration of long-term potentiation in middle-aged hippocampus after induction of brain-derived neurotrophic factor. J Neurophysiol. 2006;96:677–685. doi: 10.1152/jn.00336.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stranahan AM, Lee K, Martin B, Maudsley S, Golden E, Cutler RG, Mattson MP. Voluntary exercise and caloric restriction enhance hippocampal dendritic spine density and BDNF levels in diabetic mice. Hippocampus. 2009;19:951–961. doi: 10.1002/hipo.20577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li G, Peskind ER, Millard SP, Chi P, Sokal I, Yu CE, Bekris LM, Raskind MA, Galasko DR, Montine TJ. Cerebrospinal fluid concentration of brain-derived neurotrophic factor and cognitive function in non-demented subjects. PLoS One. 2009;4(5):e5424. doi: 10.1371/journal.pone.0005424. 2009. Epub 2009 May 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Phillips HS, Hains JM, Armanini M, Laramee GR, Johnson SA, Winslow JW. BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer’s disease. Neuron. 1991;7:695–702. doi: 10.1016/0896-6273(91)90273-3. [DOI] [PubMed] [Google Scholar]
- 65.Peng S, Garzon DJ, Marchese M, Klein W, Ginsberg SD, Francis BM, Mount HT, Mufson EJ, Salehi A, Fahnestock M. Decreased brain-derived neurotrophic factor depends on amyloid aggregation state in transgenic mouse models of Alzheimer’s disease. J Neurosci. 2009;29:9321–9329. doi: 10.1523/JNEUROSCI.4736-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Blurton-Jones M, Kitazawa M, Martinez-Coria H, Castello NA, Müller FJ, Loring JF, Yamasaki TR, Poon WW, Green KN, LaFerla FM. Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc Natl Acad Sci U S A. 2009;106:13594–13599. doi: 10.1073/pnas.0901402106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, Wang L, Blesch A, Kim A, Conner JM, Rockenstein E, Chao MV, Koo EH, Geschwind D, Masliah E, Chiba AA, Tuszynski MH. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat Med. 2009;15:331–337. doi: 10.1038/nm.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lisman JE, Coyle JT, Green RW, Javitt DC, Benes FM, Heckers S, Grace AA. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci. 2008;31:234–242. doi: 10.1016/j.tins.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Froestl W, Gallagher M, Jenkins H, Madrid A, Melcher T, Teichman S, Mondadori CG, Pearlman R. SGS742: the first GABA(B) receptor antagonist in clinical trials. Biochem Pharmacol. 2004;68:1479–1487. doi: 10.1016/j.bcp.2004.07.030. [DOI] [PubMed] [Google Scholar]
- 70.Ridha BH, Josephs KA, Rossor MN. Delusions and hallucinations in dementia with Lewy bodies: worsening with memantine. Neurology. 2005:65481–6482. doi: 10.1212/01.wnl.0000172351.95783.8e. [DOI] [PubMed] [Google Scholar]
- 71.Monastero R, Camarda C, Pipia C, Camarda R. Visual hallucinations and agitation in Alzheimer’s disease due to memantine: report of three cases. J Neurol Neurosurg Psychiatry. 2007;78:546. doi: 10.1136/jnnp.2006.096420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lipton SA. Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nat Rev Drug Discov. 2006;5:5160–5170. doi: 10.1038/nrd1958. [DOI] [PubMed] [Google Scholar]
- 73.Penner MR, Roth TL, Barnes CA, Sweatt JD. An epigenetic hypothesis of aging-related cognitive dysfunction. Front Aging Neurosci. 2010;2:9. doi: 10.3389/fnagi.2010.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Owen AM, Hampshire A, Grahn JA, Stenton R, Dajani S, Burns AS, Howard RJ, Ballard CG. Putting brain training to the test. Nature. 2010;465:775–778. doi: 10.1038/nature09042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Basak C, Boot WR, Voss MW, Kramer AF. Can training in a real-time strategy video game attenuate cognitive decline in older adults? Psychol Aging. 2008;23:765–777. doi: 10.1037/a0013494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Milgram NW, Head E, Zicker SC, Ikeda-Douglas CJ, Murphey H, Muggenburg B, Siwak C, Tapp D, Cotman CW. Learning ability in aged beagle dogs is preserved by behavioral enrichment and dietary fortification: a two-year longitudinal study. Neurobiol Aging. 2005;26:77–90. doi: 10.1016/j.neurobiolaging.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 77.Cotman CW, Head E. The canine (dog) model of human aging and disease: dietary, environmental and immunotherapy approaches. J Alzheimers Dis. 2008;15:685–707. doi: 10.3233/jad-2008-15413. [DOI] [PubMed] [Google Scholar]
- 78.Pop V, Head E, Hill MA, Gillen D, Berchtold NC, Muggenburg BA, Milgram NW, Murphy MP, Cotman CW. Synergistic effects of long-term antioxidant diet and behavioral enrichment on beta-amyloid load and non-amyloidogenic processing in aged canines. J Neurosci. 2010;30:9831–9830. doi: 10.1523/JNEUROSCI.6194-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Radman T, Parra L, Bikson M. Amplification of small electric fields by neurons; implications for spike timing. Conf Proc IEEE Eng Med Biol Soc. 2006;2006:9831–9839. doi: 10.1109/IEMBS.2006.259636. [DOI] [PubMed] [Google Scholar]
- 80.Fritsch B, Reis J, Martinowich K, Schambra HM, Ji Y, Cohen LG, Lu B. Direct current stimulation promotes BDNF-dependent synaptic plasticity: potential implications for motor learning. Neuron. 2010;6:198–204. doi: 10.1016/j.neuron.2010.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Reis J, Robertson E, Krakauer JW, Rothwell J, Marshall L, Gerloff C, Wassermann E, Pascual-Leone A, Hummel F, Celnik PA, Classen J, Floel A, Ziemann U, Paulus W, Siebner HR, Born J, Cohen LG. Consensus: “Can tDCS and TMS enhance motor learning and memory formation? Brain Stimul. 2008;1:363–369. doi: 10.1016/j.brs.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hummel FC, Celnik P, Pascual-Leone A, Fregni F, Byblow WD, Buetefisch CM, Rothwell J, Cohen LG, Gerloff C. Controversy: Noninvasive and invasive cortical stimulation show efficacy in treating stroke patients. Brain Stimul. 2008;1:370–382. doi: 10.1016/j.brs.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 83.Rozzini L, Costardi D, Chilovi BV, Franzoni S, Trabucchi M, Padovani A. Efficacy of cognitive rehabilitation in patients with mild cognitive impairment treated with cholinesterase inhibitors. Int J Geriatr Psychiatry. 2007;22:356–360. doi: 10.1002/gps.1681. [DOI] [PubMed] [Google Scholar]
- 84.Kukull WA, Higdon R, Bowen JD, McCormick WC, Teri L, Schellenberg GD, van Belle G, Jolley L, Larson EB. Dementia and Alzheimer disease incidence: a prospective cohort study. Arch Neurol. 2002;59:1737–1746. doi: 10.1001/archneur.59.11.1737. [DOI] [PubMed] [Google Scholar]
- 85.Plassman BL, Langa KM, Fisher GG, Heeringa SG, Weir DR, Ofstedal MB, Burke JR, Hurd MD, Potter GG, Rodgers WL, Steffens DC, Willis RJ, Wallace RB. Prevalence of dementia in the United States: the aging, demographics, and memory study. Neuroepidemiology. 2007;29:125–1232. doi: 10.1159/000109998. [DOI] [PMC free article] [PubMed] [Google Scholar]