

Abstract
5′,8-cyclopurine-2′-deoxynucleosides including 5′,8-cyclo-dA (cdA) and 5′,8-cyclo-dG (cdG) are induced by hydroxyl radicals resulting from oxidative stress such as ionizing radiation. 5′,8-cyclopurine-2′-deoxynucleoside lesions are repaired by nucleotide excision repair with low efficiency, thereby leading to their accumulation in the human genome and lesion bypass by DNA polymerases during DNA replication and base excision repair (BER). In this study, for the first time, we discovered that DNA polymerase β (pol β) efficiently bypassed a 5′R-cdA, but inefficiently bypassed a 5′S-cdA during DNA replication and BER. We found that cell extracts from pol β wild-type mouse embryonic fibroblasts exhibited significant DNA synthesis activity in bypassing a cdA lesion located in replication and BER intermediates. However, pol β knock-out cell extracts exhibited little DNA synthesis to bypass the lesion. This indicates that pol β plays an important role in bypassing a cdA lesion during DNA replication and BER. Furthermore, we demonstrated that pol β inserted both a correct and incorrect nucleotide to bypass a cdA at a low concentration. Nucleotide misinsertion was significantly stimulated by a high concentration of pol β, indicating a mutagenic effect induced by pol β lesion bypass synthesis of a 5′,8-cyclopurine-2′-deoxynucleoside. Moreover, we found that bypass of a 5′S-cdA by pol β generated an intermediate that failed to be extended by pol β, resulting in accumulation of single-strand DNA breaks. Our study provides the first evidence that pol β plays an important role in bypassing a 5′,8-cyclo-dA during DNA replication and repair, as well as new insight into mutagenic effects and genome instability resulting from pol β bypassing of a cdA lesion.
Keywords: 5′8-cyclopurine-2′-deoxynucleoside, DNA polymerase β, Lesion bypass synthesis, Base excision repair, DNA replication, Mutagenesis
1. Introduction
Reactive oxygen species (ROS) induced by endogenous oxidative stress and environmental factors such as ionizing radiation can attack genomic DNA and result in various types of DNA damage such as oxidized DNA base lesions, strand breaks and DNA-protein cross-links in the human genome (1,2). Because of its high reactivity with DNA bases, the hydroxyl radical (•OH) leads to the formation of the majority of oxidized base lesions (3). One type of oxidized DNA base lesion induced by •OH is the 5′,8-cyclopurine-2′-deoxynucleosides (cdPu), which includes 5′,8-cyclo-2′-deoxyadenosine (cdA) and 5′,8-cyclo-2′-deoxyguanosine (cdG). These lesions can exist in a 5′R or 5′S configuration. It has been found that both configurations of cdPu lesions including 5′R cdA and 5′S cdA lesions can form naturally in the genomic DNA of mammals (4–10). Because cdPu lesions caused by the highly reactive free radical, •OH, which can be generated in cells by both endogenous and exogenous sources such as ionizing radiation and carcinogens, cdPu lesions have been also proposed to be used as an oxidative DNA damage marker to evaluate the level of oxidative stress in mammals (5,6,11). Previous studies have shown that 180–320 cdPus/cell can be produced in fetal and postnatal rat liver per day (4). In addition, it has been estimated that cdA lesions occur at a frequency of one lesion/107 DNA bases (12). In vitro studies have shown that the ratio of the amount of 5′R cdA to 5′S cdA in DNA is about 3:1 (6), suggesting a different efficiency in production of various forms of cyclopurine lesions. cdPus contain an extra covalent bond that links the C5′ of the 2′-deoxyribose and C8 of the purine in addition to the glycosidic bond. The extra covalent bond can alter DNA structure (13), thereby stabilizing the glycosidic bond of the lesions (14–16) and preventing the cleavage of the bond by a DNA glycosylase (14–16). Because 5′,8-cyclopurine-2′-deoxynucleoside lesions can induce distortions to double-stranded DNA that can be recognized by nucleotide excision repair (NER) proteins, NER is the only repair pathway that can remove this type of DNA damage (17–19). However, the efficiency of removal of a 5′R- and 5′S-cdA lesion by NER is 2- to 4-fold less than that for other bulky DNA adducts such as a cis-B[α]P-N2-dG adduct (18). This results in the accumulation of a high level of cdPus in the genome. This is supported by previous studies showing that a significant amount of cdPu lesions were detected in genomic DNA of cells and tissues of mammals and other organisms (5,6,12). cdPus can severely impair normal cellular functions such as DNA replication and transcription (20). It has been shown that both 5′R-cdA and 5′S-cdA can directly block RNA polymerase II synthesis by inhibiting recruitment of RNA polymerase II to a promoter region (17,21). cdPus can also completely block DNA synthesis of human replication polymerases such as pol δ, causing replication fork stalling (19). To resolve the stalled replication fork and restart DNA replication, Y family DNA translesion polymerases are recruited to perform translesion synthesis to bypass the base lesions (22). However, a template 5′S-cdA can also block lesion bypass synthesis by Y family polymerases such as pol η and pol ι, resulting in accumulation of DNA strand breaks and apoptosis (23–25). Furthermore, the lesion bypass of a cdA by Y family polymerases is highly mutagenic (25). Thus, unrepaired cdPus in the genome may lead to adverse biological effects, resulting in the development of human diseases and pathological conditions such as cancer, neurodegeneration and aging (5,26–29).
DNA polymerase β (pol β) is the smallest DNA polymerase identified in cells thus far (30). It belongs to the X family of DNA polymerases (31). Pol β is composed of two subdomains, a 31 kDa polymerase domain and an 8 kDa deoxyribose phosphate (dRP) lyase domain (32). It plays an essential role in filling gaps and removing a 5′-dRP group during BER (30,32). Similar to the Y family translesion synthesis polymerases, pol β can also perform translesion synthesis to bypass a DNA base lesion during replication and BER (33–43). Previous studies have shown that pol β can bypass small base lesions including 8-oxoG and apurinic/apyrimidinic sites (AP sites), as well as bulky DNA lesions including a cisplatin adduct, propano-2′-deoxyguanosine (PdG), benzo(a)pyrene (BaP) diolepoxide (BPDE) DNA adduct, BaP-dG adduct, pyrimidine(6-4)pyrimidone photoproduct (6,4-PP) and cyclobutane pyrimidine dimer (CPD) (33–43). Because pol β lacks 3′–5′ exonuclease activity for proofreading, it can insert an incorrect nucleotide to bypass a DNA base lesion, leading to a high frequency of mutation (30,33–39,41,42). For example, during its bypass of an AP site, pol β can insert an adenosine opposite to the lesion that may ultimately result in C to T transition if the abasic site is originally derived from an 8-oxoG (35). Pol β bypass of bulky DNA adducts such as Pt-adducts can also induce mutations in the genome with a high frequency (33,34,36,38,39,41,42). Thus, pol β lesion bypass DNA synthesis can lead to mutations that may further result in the development of human cancer.
Previous studies have shown that several Y family DNA translesion synthesis polymerases can bypass a cdPu lesion during DNA replication with moderate efficiency (25). It remains to be elucidated whether other DNA repair polymerases that can bypass DNA base lesions, such as pol β, may also bypass a cdPu during DNA replication and repair and whether the lesion bypass of a cdPu by these DNA polymerases may affect genome integrity and stability. We hypothesized that pol β can bypass a 5′R-cdA and a 5′S-cdA during DNA replication and BER, and this may subsequently cause genome instability. This is supported by our recent study showing that pol β can bypass both a 5′R-cdA and 5′S-cdA located in a CAG repeat tract by skipping over the lesions, thereby resulting in CTG repeat deletion (44). To test this hypothesis, in this study, we initially examined DNA synthesis for bypassing a cdA with pol β deficient and proficient cell extracts. We then characterized pol β DNA synthesis in bypassing a cdA during DNA replication and BER and determined nucleotide insertions during pol β lesion bypass of a template cdA. For the first time, we found that pol β efficiently bypassed a 5′R-cdA during DNA replication and BER. However, the enzyme bypassed a 5′S-cdA with a low efficiency. Moreover, we further demonstrated that pol β bypass of a cdA also led to nucleotide misinsertions and single-strand DNA breaks, indicating that pol β lesion bypass of a cdA lesion during DNA replication and repair can induce mutations and strand breaks in the genome that ultimately results in genome instability.
2. Materials and methods
2.1 Materials
DNA oligonucleotides containing a 5′R-cdA or 5′S-cdA were synthesized and purified by HPLC as described previously (45). The structures of 5′R-cdA and 5′S-cdA were illustrated in Figure 1. All other oligonucleotides were synthesized by Integrated DNA Technologies (IDT, Coralville, IA, USA). Deoxynucleoside 5′-triphosphates (dNTPs) were purchased from Fermentas (Glen Burnie, MD, USA). Radionucleotides, [γ-32P] ATP (6000 mCi/mmol) and cordycepin 5′-triphosphate 3′-[α-32P] (5000 mCi/mmol) were purchased from Perkin Elmer Inc. (Boston, MA, USA). Micro Bio-Spin 6 chromatography columns were from Bio-Rad (Hercules, CA, USA). All other standard chemical reagents were from Sigma-Aldrich (St. Louis, MO, USA) and Thermo Fisher Scientific (Pittsburgh, PA, USA). Purified pol β, flap endonuclease 1 (FEN1) and DNA ligase I (LIG I) were generous gifts from Dr. Samuel H. Wilson at the National Institute of Environmental Health Sciences (NIEHS)/National Institutes of Health, Research Triangle Park, North Carolina.
Figure 1.
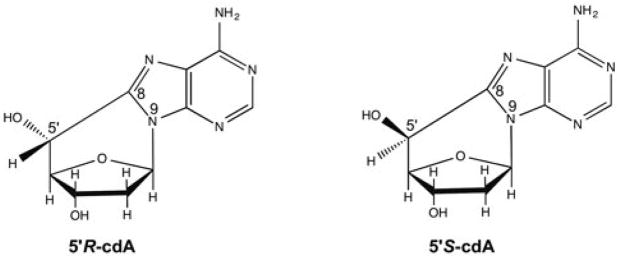
The structures of 5′,8-cyclo-2′-deoxyadenosines.
2.2 Oligonucleotide substrates
Substrates containing a 31-nt template strand with a 5′R-cdA or 5′S-cdA located at the 19th nucleotide counted from the 3′-end were designed to mimic DNA replication or BER intermediates with a cdA lesion on the template strand. The upstream primer of the substrates is an 18-nt strand without or with a 3′-terminus dT that base paired with the template cdA. The downstream primer of the substrates is a 13-nt strand with a 5′-phosphate or 5′-phosphorylated tetrahydrofuran (THF) residue, an abasic site analog. Substrates that mimic the lesion bypass intermediates formed during DNA leading strand synthesis were constructed by annealing an 18-nt upstream primer containing a 3′-terminus dT with the cdA-containing template strand at a molar ratio of 1:2. Substrates representing the intermediates with a 1-nt gap opposite a template cdA formed during DNA lagging strand synthesis and BER were constructed by annealing the 18-nt upstream primer and a 13-nt downstream primer with a 5′-phosphate or 5′-THF residue with the template strand containing a 5′R-cdA or 5′S-cdA at a molar ratio of 1:1:2. Substrates were radiolabeled at the 5′-end of the upstream primers or the 3′-end of downstream primers for measuring the activities of different enzymes. The sequences of the oligonucleotide substrates are listed in the Supplementary Table S1.
2.3 Translesion DNA synthesis in bypassing a cdA in mouse embryonic fibroblast (MEF) cell extracts
MEF cell extracts were prepared according to the procedure described previously (46). Briefly, pol β null (pol β−/−) and wild-type pol β (pol β+/+) MEF cells were grown to near confluence. Cells were then harvested and lysed with rotation at 4 °C in lysis buffer. Cell lysates were then subject to centrifugation at 12,000 rpm for 30 min to obtain whole-cell extracts. The whole-cell extracts were subsequently dialyzed into BER reaction buffer containing 50 mM Tris-HCl, pH 7.5, 50 mM KCl, 0.1 mM EDTA, 0.1 mg/ml bovine serum albumin and 0.01% Nonidet P-40. Levels of pol β protein in different cell extracts were determined by immunoblotting using rabbit anti-mouse pol β antibody (a generous gift from Samuel H. Wilson at NIEHS/NIH) (Data not shown). DNA synthesis activity was measured by incubating 60 μg of cell extracts with open template substrates with an upstream primer, that contained a 5′R-cdA or 5′S-cdA on the template strand, or with gapped substrates with a 1-nt gap opposite a template 5′R-cdA or 5′S-cdA with a 5′-phosphate or 5′-phosphorylated THF residue. Reaction mixtures were assembled on ice and incubated at 37 °C for 30 min. Reactions were terminated by addition of 15 μl of stopping buffer containing 95 % formamide and 10 mM EDTA and subsequent incubation at 95 °C for 5 min. Substrates and DNA synthesis products were separated by 15% or 18% urea-denaturing PAGE and detected by Pharos FX Plus PhosphorImager from Bio-Rad (Hercules, CA, USA).
2.4 In vitro BER reconstituted with purified enzymes
In vitro BER of an abasic lesion that is opposite a cdA was performed by incubating purified pol β, FEN1, LIG I with the substrates containing a 1-nt gap opposite a 5′R-cdA or a 5′S-cdA on the template with a downstream 5′-THF residue. The 10-μl reactions were reconstituted with the indicated concentrations of BER enzymes and substrates in BER reaction buffer with 50 μM dNTPs, 5 mM MgCl2 and 2 mM ATP. Reaction mixtures were assembled on ice and incubated at 37°C for 30 min. Reactions were terminated by addition of stopping buffer containing 95% formamide and 10 mM EDTA and incubation at 95 °C for 5 min. Substrates and products were separated by 15% or 18% urea-denaturing PAGE and detected by Pharos FX Plus PhosphorImager. All substrates were 32P-labeled at the 5′-end of the upstream primer.
2.5 Enzymatic activity assays
Lesion bypass synthesis activity of pol β was determined with the open template and gapped substrates with a 5′R-cdA or 5′S-cdA in the template. Pol β lesion bypass DNA synthesis activity on the open template was determined with the substrates without or with a 3′-terminus dT that is opposite a template 5′R-cdA or 5′S-cdA. The lesion bypass DNA synthesis activity was determined at 37 °C for 30 min in a 10-μl reaction mixture in BER reaction buffer with 50 μM dNTPs and 5 mM MgCl2. Reaction mixtures containing various concentrations of pol β and 25 nM substrates and BER buffer with 5 mM MgCl2 and 50 μM dNTPs, were assembled on ice and incubated at 37 °C for 30 min. FEN1 cleavage activity was measured by incubating the substrates containing a 1-nt gap without or with a downstream 5′-phosphorylated THF with the enzyme in the absence or presence of pol β in BER buffer with 5 mM MgCl2 and 50 μM dNTP at 37 °C for 30 min. Reactions were terminated by addition of stopping buffer containing 95% formamide and 10 mM EDTA and incubation at 95°C for 5 min. Substrates and products were separated by 15% or 18% urea-denaturing PAGE and detected by a Pharos FX Plus PhosphorImager.
3. Results
3.1 Pol β plays an important role in bypassing a cdA lesion
Pol β is a key enzyme that plays an essential role in mediating efficient BER in mammalian cells by removing a 5′-dRP group as well as filling in a single- or multi-nucleotide gap (47). It also plays a critical role in lesion bypass synthesis during DNA leading and lagging strand synthesis (40,43) to facilitate Okazaki fragment maturation (40). Therefore, pol β may also play an important role in bypassing a cdA lesion during DNA replication and BER. To determine whether pol β can play a role in bypassing a cdA, we initially determined DNA synthesis in bypassing a 5′R-cdA and 5′S-cdA in cell extracts with pol β deficiency (pol β−/−) and pol β proficiency (pol β+/+) with the open template substrates containing a 5′R-cdA or 5′S-cdA on the template strand (Figure 2A) as well as the 1 nt-gapped substrates containing a template 5′R- or 5′S-cdA with a 5′-phosphate (Figure 2B) and 5′-phosphorylated THF residue (Figure 2C). The results showed that a significant amount of lesion bypass products were generated with the substrates containing a template 5′R-cdA in pol β+/+ cell extracts (lane 3 of Figure 2A, 2B and 2C). However, pol β−/− cell extracts exhibited very weak lesion bypass synthesis with the substrates (lane 2 of Figure 2A, 2B, and 2C). On the other hand, pol β+/+ cell extracts performed weak DNA synthesis on the substrates containing a template 5′S-cdA (lane 6 of Figure 2A, 2B and 2C). However, little lesion bypass synthesis products was generated by pol β−/− cell extract with the substrates (lane 5 of Figure 2A, 2B and 2C). The results showed that in the presence of pol β, the amount of lesion bypass products was increased by about 2 to 5-fold (the bottom panel of Figure 2A, 2B and 2C). This indicated that pol β played an important role in bypassing a cdA lesion during DNA replication and BER.
Figure 2. Pol β plays an important role in bypassing a cdA lesion during DNA replication and BER.



To test whether pol β is involved in bypassing a cdA lesion during DNA replication (the leading strand synthesis and Okazaki fragment maturation) and BER, DNA synthesis was examined by incubating cell extracts of pol β−/− and pol β+/+ MEFs with open template substrates that contained a template 5′R-cdA or 5′S-cdA (A) and 1-nt gapped substrates containing a template 5′R-cdA or 5′S-cdA (B) under the conditions described in Materials and Methods. (C) Pol β DNA synthesis in bypassing a template cdA lesion during BER was measured by incubating substrates that contained a 1-nt gap opposite a template 5′S-cdA or 5′R-cdA and the downstream primer containing a 5′-phosphorylated THF residue. In all panels, Lanes 1 and 4 correspond to substrates only. Lanes 2 and 5 correspond to reaction mixtures with 60 μg pol β−/− MEFs extracts. Lanes 3 and 6 correspond to reaction mixtures with 60 μg pol β+/+ MEFs extracts. The bottom panels illustrate the quantitative analysis of pol β DNA synthesis in bypassing a cdA lesion of the substrates. Substrates were 32P-labeled at the 5′-end of the upstream primer as indicated. Substrates are illustrated schematically above the gel.
3.2 Pol β bypasses a 5′R-cdA efficiently and stalls at a 5′S-cdA
Previous studies have shown that some Y family translesion synthesis polymerases such as pol η can bypass a 5′R-cdA and 5′S-cdA with different efficiency (19,23). To further determine if pol β can bypass a 5′R-cdA and 5′S-cdA during DNA replication and BER, we examined pol β DNA synthesis in bypassing cdA lesions by incubating increasing concentrations of purified pol β with open template substrates, as well as 1 nt-gapped substrates containing a template cdA. The results showed that with the open template substrates, 10 nM of pol β efficiently bypassed a 5′R-cdA and performed its multi-nucleotide DNA synthesis (Figure 3A, lane 2). With increasing concentrations of pol β (25 nM and 50 nM), the amount of pol β DNA synthesis products from bypass of a 5′R-cdA was significantly increased (Figure 3A, lanes 3–4). However, pol β exhibited weak DNA synthesis activity during its bypass of a 5′S-cdA (Figure 3A, lane 6). With increasing concentrations of pol β, the amount of its 1-nt insertion product was increased along with a slight increase in its multi-nucleotide DNA synthesis (Figure 3A, lanes 7–8). The results indicated that pol β efficiently bypassed a 5′R-cdA, but stalled after it inserted a dT for bypassing a 5′S-cdA. The stalled pol β DNA synthesis suggests that the base lesion conferred a strong inhibitory effect on polymerase lesion bypass synthesis during DNA leading strand synthesis. To further determine whether pol β can continue to perform its lesion bypass synthesis after it inserts one nucleotide that base pairs with a template cdA, we examined pol β synthesis activity with the open template substrates containing a 3′-terminus dT that base paired with a template 5′R-cdA or 5′S-cdA (Figure 3B). These substrates mimic the intermediates resulting from pol β 1-nt insertion that are generated during leading strand synthesis. We found that pol β at 10–50 nM performed efficient DNA synthesis to extend a dT, which base paired with a 5′R-cdA (Figure 3B, lanes 2–4). However, the polymerase failed to extend a dT that base paired with a 5′S-cdA (Figure 3B, lanes 6–8), indicating that the 5′S-cdA lesion completely inhibited pol β DNA synthesis. Similar to its lesion bypass DNA synthesis with the open template substrates, pol β exhibited efficient DNA synthesis with the 1-nt gapped substrates containing a 5′R-cdA without or with a 5′-phosphorylated THF residue that mimic a lagging strand maturation and BER intermediate, respectively (Figure 3C, lanes 2–4 and lanes 10–12). However, with the gapped substrates containing a 5′S-cdA, the enzyme only inefficiently inserted one nucleotide to base pair with the lesion (Figure 3C, lanes 6–8 and lanes 14–16). For the nicked substrates containing a 3′-terminus T that base paired with a 5′R-cdA without or with a 5′-THF residue, pol β efficiently extended the dT (Figure 3D, lanes 2–4 and lanes 10–12), but failed to extend a 3′-terminus dT that paired with a 5′S-cdA (Figure 3D, lanes 6–8 and lanes 14–16). The results indicated that pol β effectively bypassed a 5′R-cdA, but stalled at a 5′S-cdA after it inserted a dT that base paired with the lesion. This further suggests that a 5′S-cdA on the template distorted the DNA double-helix structure. This subsequently disrupted pol β binding to the substrates and its nucleotide insertion and further inhibited pol β multi-nucleotide DNA synthesis during its lesion bypass.
Figure 3. Pol β DNA synthesis with a template cdA lesion during DNA replication and BER.




(A) Pol β bypass of a cdA at an open template during DNA replication was examined with the substrates containing a 5′R-cdA or 5′S-cdA with an upstream primer alone annealed with the template strand as described in Materials and Methods. (B) Pol β DNA synthesis in extending a 3′-terminus dT that base paired with the template cdA lesion during DNA replication was measured with the open template substrates under the experimental conditions as described in Materials and Methods. For panels (A) and (B), lanes 1 and 5 represent substrates only. Lanes 2–4 and lanes 6–8 correspond to reaction mixtures with 10 nM, 25 nM and 50 nM pol β, respectively. (C) Pol β bypass of a cdA during Okazaki fragment maturation and BER was examined with the substrates containing a 1-nt gap opposite a template 5′R-cdA or 5′S-cdA and the downstream primer with a 5′-phosphate or a 5′-phosphorylated THF residue as described in Materials and Methods. (D) Pol β DNA synthesis after a template cdA was bypassed during Okazaki fragment maturation and BER was examined with nick substrates in which the cdA was base paired with a dT. Pol β DNA synthesis to extend a dT that was base paired with a template cdA during BER was measured by using nick substrates that contained a downstream primer with a 5′-phosphorylated THF residue. For both panels (C) and (D), lanes 1, 5, 9 and 13 correspond to substrates only. Lanes 2–4, 6–8, 10–12 and 14–16 correspond to reaction mixtures with 5, 10 and 25 nM pol β, respectively. Substrates were 32P-labeled at the 5′-end of the upstream primer as indicated. Substrates are illustrated schematically above the gel.
3.3 FEN1 cleavage activity during pol β lesion bypass of a 5′R-cdA and 5′S-cdA
FEN1 plays an essential role in both Okazaki fragment maturation and long-patch BER by removing a 5′-flap created by strand displacement synthesis of replicative DNA polymerases, pol δ/ε, or repair polymerases such as pol β (48,49). Removal of an oxidized or reduced deoxyribose phosphate during long-patch BER is dependent on FEN1 flap cleavage activity (47,50). Because pol β DNA synthesis can result in the formation of a flap, thus, it may affect FEN1 cleavage. To further determine if FEN1 cleavage during Okazaki fragment maturation and BER can be affected by pol β lesion bypass of a cdA, we examined FEN1 cleavage activity on the 1 nt-gapped substrates containing a template cdA without or with a 5′-THF residue in the absence and presence of pol β (Figure 4). The results showed that for the substrates with a template 5′R-cdA, in the absence of pol β, FEN1 mainly removed 1 or 2 nucleotides from the downstream strand (Figure 4, lanes 2 and 8). In the presence of pol β, FEN1 cleavage was significantly stimulated, and it predominantly removed a flap containing 5 or 6 nucleotides (Figure 4, lanes 3 and 9). This indicated that after pol β efficiently bypassed a 5′R-cdA, it kept inserting nucleotides to perform strand displacement synthesis, creating a 5-nt or 6-nt downstream flap for FEN1 cleavage. In contrast, for the 5′S-cdA containing substrates, in the absence of pol β, FEN1 removed 2 to 4 nucleotides (Figure 4, lanes 5 and 11). The presence of pol β did not significantly alter FEN1 cleavage activity (Figure 4, lanes 6 and 12), indicating that pol β failed to efficiently bypass a 5′S-cdA. This allowed FEN1 to remove more nucleotides than were inserted by pol β, leaving a gapped intermediate that failed to be sealed by LIG I. This subsequently led to accumulation of a single-strand break intermediate.
Figure 4. FEN1 flap cleavage during pol β bypass of a cdA.
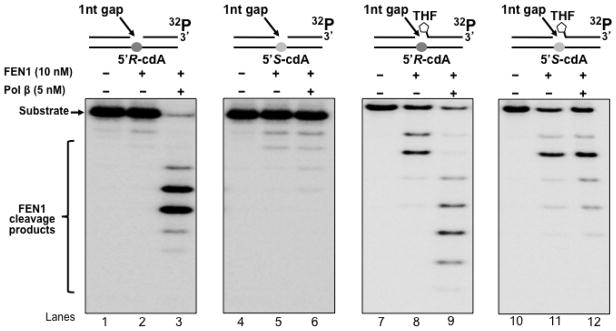
FEN1 flap cleavage on the substrates containing a 5′-phosphate or 5′-THF residue with a 1-nt gap opposite a template 5′R-cdA or 5′S-cdA was examined as described in Materials and Methods. Lanes 1, 4, 7 and 10 represent substrates only. Lanes 2, 5, 8 and 11 correspond to reaction mixtures with 10 nM FEN1. Lanes 3, 6, 9 and 12 correspond to reaction mixtures with 10 nM FEN1 in the presence of 5 nM pol β. Substrates were 32P-labeled at the 3′-end of the downstream primer as indicated. Substrates are illustrated schematically above the gel.
3.4 A 5′S-cdA lesion disrupts the completion of Okazaki fragment maturation and BER by inhibiting pol β lesion bypass synthesis
When genomic DNA is under the challenge of oxidative stress, oxidative DNA damage including 8-oxoGs and cdPus can be generated simultaneously in DNA in vitro and in vivo at a significant percentage (51,52). Because cdPus including cdA lesions can only be repaired by nucleotide excision repair with low efficiency, this allows the accumulation of the lesions in the genome readily. Thus, 8-oxoGs and cdA lesions may occur simultaneously in the complementary strands of DNA with high frequency, and it is likely that BER of an 8-oxoG and an abasic lesion in one strand of the genome may encounter a cdA base lesion accumulated in the template strand. This could allow pol β to perform DNA synthesis to bypass a cdPu lesion, thereby subsequently altering the efficiency of lagging strand maturation and BER. It has also been suggested that BER of a uracil opposite a 5′S-cdA is compromised because the lesion can alter the geometry of duplex DNA (53). To test this possibility, we reconstituted DNA lagging strand maturation and BER with the substrates containing a 1-nt gap opposite a 5′R-cdA or 5′S-cdA without or with a 5′-THF residue (Figure 5). With the substrates containing a 5′R-cdA, pol β exhibited efficient lesion bypass synthesis leading to the production of a significant amount of replication and repair products (Figure 5, lanes 2–4 and 10–12), indicating that pol β bypass of a 5′R-cdA led to the completion of lagging strand maturation and BER. However, with the substrates containing a 5′S-cdA, pol β only inserted a dT to bypass a template 5′S-cdA and failed to further extend the nucleotide by inserting additional nucleotides (Figure 5, lanes 6–8 and 14–16), resulting in the accumulation of a single-strand DNA break intermediate with a template 5′S-cdA. These results demonstrated that inefficient bypass of a 5′S-cdA by pol β prevented the completion of lagging strand maturation and BER. This further suggests that a 5′S-cdA induced DNA strand distortion more severely than a 5′R-cdA, thereby inhibiting pol β lesion bypass and preventing the completion of Okazaki fragment maturation and BER.
Figure 5. Completion of DNA lagging strand maturation and BER through pol β bypass of a cdA lesion.
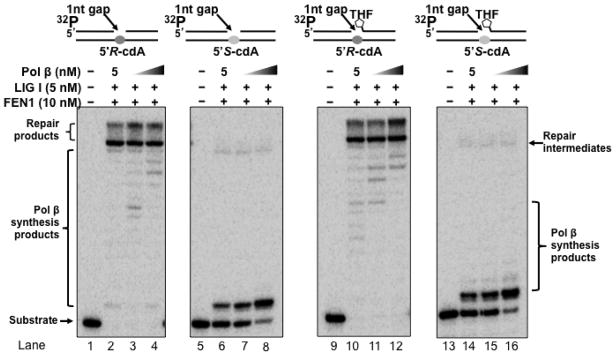
Reconstituted DNA lagging strand maturation and BER were performed to determine the effects of pol β bypass of a cdA on completion of DNA lagging strand maturation and BER. Substrates containing a 5′-phosphate or 5′-THF residue with a 1-nt gap opposite a template 5′R-cdA or 5′S-cdA were incubated with indicated BER enzymes at the experimental conditions described in Materials and Methods. Lanes 1, 5, 9 and 13 represent substrates only. Lanes 2–4, 6–8, 10–12 and 14–16 correspond to reconstitution reaction mixtures with 10 nM FEN1 and 5 nM LIG I in the presence of 5, 10 and 25 nM pol β, respectively. Substrates were 32P-labeled at the 5′-end of the upstream primer as indicated. Substrates are illustrated schematically above the gel.
3.5 Pol β can bypass a 5′,8-cdA through nucleotide misinsertion during DNA replication and BER
Previous studies have shown that cdA lesions can completely block DNA synthesis of human replication polymerases such as pol δ, causing replication fork stalling through inducing the distortion of DNA backbone (19). To resolve the stalled replication fork and restart DNA replication, Y family DNA translesion polymerases are recruited to perform translesion synthesis for bypassing the base lesions (22). However, a template 5′S-cdA can also block the lesion bypass synthesis by Y family polymerases such as pol η and pol ι, resulting in the accumulation of DNA strand breaks leading to apoptosis (23–25). Because pol β has also been shown to exhibit translesion synthesis to bypass a DNA base lesion during DNA replication and BER (33,36,38–43) Thus, it is possible that pol β can be recruited to perform translesion synthesis to bypass a cdA lesion. However, several studies have shown that pol β lesion bypass synthesis can induce a high frequency of mutations in the genome (30,33–39,41,42). Furthermore, because of lacking a proofreading 3′–5′ exonuclease activity, high levels of pol β are associated with high mutation rates in a variety of cancer cells (54–56). To investigate whether pol β bypass of a cdA can induce mutations in the genome, pol β nucleotide insertion during its bypass of a cdA was determined by incubating the open template and gapped substrates that contained a template 5′R-cdA or 5′S-cdA with 5 nM and 50 nM pol β. The results showed that at a low concentration (5 nM), pol β mainly inserted a dT to base pair with a 5′R-cdA with the open template substrates (Figure 6A, lane 3). However, the enzyme also inefficiently inserted a dA, dC or dG to basepair with the 5′R-cdA (Figure 6A, lanes 2, 4 and 5). On the other hand, the same concentration of pol β only inserted dT to base pair with a template 5′S-cdA (Figure 6A, lane 8) and failed to insert a dA, dC or dG to bypass the lesion (Figure 6A, lane 7 and lanes 9–10). Surprisingly, we found that with the 1-nt gapped substrates without a 5′-THF residue, 5 nM pol β efficiently inserted both dT and dC to bypass a template 5′R-cdA (Figure 6A, lanes 13–14) and inefficiently inserted a dA and dG to bypass the lesion (Figure 6A, lanes 12 and 15). For the 1-nt gap substrate containing a 5′S-cdA, 5 nM pol β mainly inserted a dT to bypass the lesion (Figure 6A, lane 18). It also inserted a dA with a very low efficiency (Figure 6A, lane 17), but it failed to insert dC and dG (Figure 6A, lanes 19–20). For the 1-nt gap substrates with a 5′-THF residue, 5 nM pol β predominantly inserted a dT and dC to bypass a 5′R-cdA (Figure 6A, lanes 23–24), although the enzyme also managed to insert a dA and dG inefficiently to bypass the lesion (Figure 6A, lanes 22 and 25). The same concentration of pol β inefficiently inserted a dT to bypass a 5′S-cdA (Figure 6A, lane 28) and failed to insert other types of nucleotides to bypass the lesion (Figure 6A, lanes 27, 29 and 30). At a high concentration of 50 nM, pol β misinsertion in bypassing a cdA lesion with all the substrates was significantly increased (Figure 6B, lanes 2, 4–5, lanes 12, 14–15, lanes 17, 19–20, and lanes 22, 24–25, 27). The high concentrations also significantly increased pol β correct nucleotide insertion in bypassing a 5′S-cdA (Figure 6B, lanes 8, 18 and 28). However, the high concentrations of pol β failed to increase the pol β misinsertion of a dA to pair with a 5′S-cdA with the open template substrate (Figure 6B, lane 7). The results indicated that at both a low and high concentration, pol β efficiently misinserted dC to bypass a 5′R-cdA during DNA lagging strand maturation and BER. However, a high concentration of pol β also significantly promoted its misinsertation of dA and dG during its lesion bypass. The results further suggest that pol β bypass of a 5′R-cdA or 5′S-cdA lesion during replication and BER may be mutagenic, especially in the presence of a high level of pol β.
Figure 6. Pol β nucleotide insertions in bypassing a cdA lesion during DNA replication and BER.


Pol β nucleotide insertion in bypassing a cdA during DNA lagging strand maturation and BER was examined by incubating the open-template substrates and 1-nt gap substrates that contained a downstream 5′-phosphate or 5′-THF residue and a template 5′R-cdA or 5′S-cdA with 5 nM (A) or 50 nM (B) pol β in the presence of 50 μM dATP or dTTP or dCTP or dGTP respectively under the experimental conditions described in Materials and Methods. In both panels, lanes 1, 6, 11, 16, 21 and 26 represent substrates only. Lanes 2–5, 7–10, 12–15, 17–20, 22–25 and 27–30 correspond to reaction mixtures with 5 nM or 50 nM pol β in the presence of 50 μM dATP, dTTP, dCTP or dGTP. Substrates were 32P-labeled at the 5′-end of the upstream primer as indicated. Substrates are illustrated schematically above the gel.
4. Discussion
In this study, for the first time we discovered that pol β played an important role in bypassing a cdA lesion during DNA replication and BER (Figure 2). We provided the first evidence that pol β efficiently bypassed a 5′R-cdA via multi-nucleotide insertions, whereas the enzyme mainly inserted 1 nt to bypass a 5′S-cdA lesion and failed to further perform its multi-nucleotide lesion bypass DNA synthesis (Figures 2–3). This resulted in disruption of its DNA synthesis in bypassing a 5′S-cdA during DNA replication and BER. Furthermore, we discovered that a 5′S-cdA on the template strand also slightly facilitated FEN1 removal of a long flap (Figure 4), suggesting that the lesion induced a distortion on the DNA structure, thereby promoting dissociation of the downstream DNA strand from its template. This further resulted in the formation of a flap that can be efficiently cleaved by FEN1 leaving gapped DNA thereby inhibiting the completion of DNA replication and repair and leading to accumulation of single-strand intermediates (Figure 5). Moreover, we found that during DNA replication and BER, a low concentration of pol β inserted both a correct and incorrect nucleotide to bypass a cdA, whereas a high concentration of pol β stimulated its nucleotide misincorporation in bypassing a cdA (Figure 6). Our results support a model in which a 5′R-cdA induced by ROS during DNA replication or BER can be efficiently bypassed by pol β. In the presence of a low concentration of pol β, a correct nucleotide is inserted to base pair with the lesion. Pol β continues to perform strand displacement synthesis, creating a long flap which is subsequently cleaved by FEN1. This leads to the production of a nicked DNA that is sealed by LIG I, resulting in no mutation in the replication and repair products (Figure 7, sub-pathway 1, left). However, both a low and high concentration of pol β can also insert a dC or dG or dA to bypass a 5′R-cdA, resulting in mutations (Figure 7, sub-pathway 1, right). On the other hand, a template 5′S-cdA induces the DNA distortion promoting dissociation of the downstream strand from its template strand resulting in the formation of a small flap. Pol β inefficiently inserts a dT to base pair with a template 5′S-cdA and stalls (Figure 7, sub-pathway 2). Subsequently, this allows FEN1 to remove more nucleotides than pol β synthesizes, resulting in accumulation of single-strand DNA break intermediates (Figure 7, sub-pathway 2).
Figure 7. Pol β bypass of a cdA lesion leads to nucleotide misinsertion and DNA strand breaks during DNA replication and BER.

Reactive oxygen species (ROS) can result in oxidized DNA lesion, including cdAs with two different configurations, 5′R-cdA or 5′S-cdA., during DNA replication and BER. A 5′R-cdA can be efficiently bypassed by pol β. In the presence of low concentration of pol β, a correct nucleotide can be inserted to base pair with a template cdA lesion. Pol β can continue to perform strand displacement synthesis, resulting in the formation of a long flap which is subsequently cleaved by FEN1. This leads to a nicked DNA for LIG I to seal, resulting in no mutation in the replication and repaired product (Sub-pathway 1, left). However, a low level of pol β can also insert a dC to base pair a cdA, and the nucleotide misinsertion is promoted at a high level of pol β (Sub-pathway 1, right). This subsequently results in mutation during DNA replication and BER. On the other hand, a template 5′S-cdA can induce DNA distortion that promotes dissociation of the downstream strand from its template strand, thereby leading to the formation of an intermediate with a gap and short flap (Sub-pathway 2). The lesion also strongly inhibits pol β lesion bypass synthesis as well as extension of the lesion bypass intermediate (Sub-pathway 2). Subsequently, FEN1 cleaves the flap removing more nucleotides than pol β synthesizes, causing accumulation of gapped intermediates that can further lead to DNA strand break intermediates (Sub-pathway 2).
Employing the cell extract-based in vitro approach, we have demonstrated the involvement of pol β in cdA lesion bypass during DNA replication and repair explicitly. And our results obtained from pol β+/+ and pol β−/− MEFs indicate that pol β plays an important role in bypassing cdA lesions (Figure 2). This is also supported by a study from the Wilson group showing that pol β−/− MEFs exhibited hypersensitivity to the oxidative DNA damaging agent, hydrogen peroxide than pol β+/+ MEFs (57). Because hydrogen peroxide is the precursor for generating hydroxyl radicals that can subsequently lead to cdA lesions, this further indicates that the hypersensitivity of pol β−/− MEFs to hydrogen peroxide results from the accumulation of oxidized DNA lesions including cdA lesions and DNA strand breaks caused by the deficiency of lesion bypass in the absence of pol β.
Our results indicate that pol β efficiently bypassed a 5′R-cdA (Figure 1–2). However, it also inserted a significant amount of incorrect nucleotides to bypass the lesion at a low concentration (5 nM), and this was significantly stimulated at a high concentration (50 nM) (Figure 6). Given the fact that in mammalian cells, during BER of oxidative DNA damage, a large number of pol β molecules can be recruited to damaged sites resulting in a high concentration of the enzyme that is accumulated at oxidized base lesions (58), it is likely that pol β nucleotide misinsertions can frequently occur during its bypass of a cdA in cells. This can then lead to a high frequency of mutations in genomic DNA. Our results are also consistent with previous findings showing that in human cancer cells, pol β protein level has been increased significantly compared with normal cells, thereby inducing a high frequency of mutations (54–56). Moreover, because of the lack of an intrinsic 3′–5′ exonuclease, pol β lesion bypass can induce more mutations in the genome than replication polymerases through its nucleotide misinsertion during its lesion bypass synthesis (59). It is conceivable that an extrinsic 3′–5′ exonuclease that can remove a mismatched base may improve pol β fidelity during its bypass of a template cdA. It has been reported that in addition to its cleavage of an abasic site during BER, APE1 also has 3′–5′ exonuclease activity that can remove mismatched bases at the 3′-end in nicked or gapped DNA (60,61). Previous studies have also found that p53 can efficiently remove a misinserted base prior to the extension of the mismatched 3′-terminus by a DNA polymerase (62,63). Because APE1 can form a complex with pol β (64), and p53 can interact with pol β (65), it is possible that these proteins may cooperate with pol β to remove a misinserted nucleotide incorporated by pol β during its bypass of a cdA lesion. It is of interest to determine if APE1 and p53 can remove a mismatched base that pairs with a template 5′R-cdA, improving pol β fidelity during its lesion bypass synthesis of a 5′,8-cyclopurine-2′-deoxynucleoside.
Our results also demonstrated that bypass of a 5′S-cdA is a challenge for pol β (Figures 2–3). We found that pol β failed to extend the 3′-terminus after it inserted one nucleotide to base pair with a 5′S-cdA. This subsequently results in the accumulation of single-strand break intermediates that may subsequently lead to double-strand breaks and cell death. Previous studies have shown that pol β DNA synthesis in bypassing an AP site during Okazaki fragment maturation can be promoted by proliferating cell nuclear antigen (PCNA) and replication protein A (RPA) (40). It has been also shown that through coordination with FEN1 and RPA, pol β strand displacement synthesis activity is also facilitated after it bypasses an abasic site (40). It is conceivable that the replication and repair cofactors that can facilitate pol β lesion bypass activity and primer extension may facilitate pol β bypass of a 5′S-cdA as well as its extension of the lesion bypass intermediates. Therefore, it is of interest to investigate how BER cofactors, PCNA and RPA may also facilitate pol β bypass of a 5′S-cdA, preventing the formation of DNA strand break intermediates and cell death.
In summary, in this study, we identified that pol β played an important role in bypassing a template cdA lesion during DNA replication and BER. We provided the first evidence that pol β can efficiently bypass a 5′R-cdA, but inefficiently bypass a 5′S-cdA, resulting in the incompletion of DNA replication and repair and accumulation of strand break intermediates. Moreover, we found that pol β inserted an incorrect nucleotide to bypass a cdA, and this was promoted by a high concentration of pol β, indicating that the fidelity of pol β bypass of a cdA lesion during DNA replication and BER can be modulated by the concentrations of pol β. This may further result in mutations in the genome. In addition, we demonstrated that a template 5′S-cdA severely inhibited pol β lesion bypass synthesis and induced dissociation of the downstream strand from the template strand resulting in the formation of a short flap (Figure 4 and Figure 7 sub-pathway 2). This subsequently led to poor pol β DNA synthesis along with relatively efficient FEN1 flap cleavage on the downstream strand, resulting in the accumulation of strand break intermediates (Figure 5, lanes 6–8 and lanes 14–16). Our results reveal that pol β bypass of a cdA can result in nucleotide misinsertions and DNA strand breaks during DNA replication and BER, thereby resulting in genome instability.
Supplementary Material
Highlights.
Pol β plays an important role in cdA lesion bypass during DNA replication and BER.
Pol β can efficiently bypass a 5′R-cdA lesion via nucleotide misinsertions.
Pol β stalls during its bypass of a 5′S-cdA lesion.
Pol β bypass of cdPus leads to genome instability.
Acknowledgments
Funding
National Institutes of Health [ES023569 to Y.L.]; Ministero dell’Istruzione, dell’Università e della Ricerca [PRIN 2009K3RH7N_002, in part]; Marie Curie Intra-European Fellowship [CYCLOGUO-298555]
We thank Samuel H. Wilson, Laboratory of Structural Biology, National Institute of Environmental Health Sciences (NIEHS), National Institutes of Health (NIH), for generously providing purified BER enzymes and plasmids for expressing BER enzymes. The support and sponsorship of the COST Action CM1201 on Biomimetic Radical Chemistry is gratefully acknowledged. We thank Jill Beaver for critical readings and insightful comments.
Footnotes
Conflict of Interest Statement
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cadet J, Delatour T, Douki T, Gasparutto D, Pouget JP, Ravanat JL, Sauvaigo S. Hydroxyl radicals and DNA base damage. Mutat Res. 1999;424:9–21. doi: 10.1016/s0027-5107(99)00004-4. [DOI] [PubMed] [Google Scholar]
- 2.Dizdaroglu M, Jaruga P, Rodriguez H. Measurement of 8-hydroxy-2′-deoxyguanosine in DNA by high-performance liquid chromatography-mass spectrometry: comparison with measurement by gas chromatography-mass spectrometry. Nucleic Acids Res. 2001;29:E12. doi: 10.1093/nar/29.3.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cadet J, Douki T, Gasparutto D, Ravanat JL. Oxidative damage to DNA: formation, measurement and biochemical features. Mutat Res. 2003;531:5–23. doi: 10.1016/j.mrfmmm.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 4.Randerath K, Zhou GD, Somers RL, Robbins JH, Brooks PJ. A 32P-postlabeling assay for the oxidative DNA lesion 8,5′-cyclo-2′-deoxyadenosine in mammalian tissues: evidence that four type II I-compounds are dinucleotides containing the lesion in the 3′ nucleotide. J Biol Chem. 2001;276:36051–36057. doi: 10.1074/jbc.M105472200. [DOI] [PubMed] [Google Scholar]
- 5.Jaruga P, Dizdaroglu M. 8,5′-Cyclopurine-2′-deoxynucleosides in DNA: mechanisms of formation, measurement, repair and biological effects. DNA Repair. 2008;7:1413–1425. doi: 10.1016/j.dnarep.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 6.Chatgilialoglu C, Ferreri C, Terzidis MA. Purine 5′,8-cyclonucleoside lesions: chemistry and biology. Chem Soc Rev. 2011;40:1368–1382. doi: 10.1039/c0cs00061b. [DOI] [PubMed] [Google Scholar]
- 7.Shaked H, Hofseth LJ, Chumanevich A, Chumanevich AA, Wang J, Wang Y, Taniguchi K, Guma M, Shenouda S, Clevers H, et al. Chronic epithelial NF-kappaB activation accelerates APC loss and intestinal tumor initiation through iNOS up-regulation. P Natl Acad Sci USA. 2012;109:14007–14012. doi: 10.1073/pnas.1211509109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mitra D, Luo X, Morgan A, Wang J, Hoang MP, Lo J, Guerrero CR, Lennerz JK, Mihm MC, Wargo JA, et al. An ultraviolet-radiation-independent pathway to melanoma carcinogenesis in the red hair/fair skin background. Nature. 2012;491:449–453. doi: 10.1038/nature11624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tilstra JS, Robinson AR, Wang J, Gregg SQ, Clauson CL, Reay DP, Nasto LA, St Croix CM, Usas A, Vo N, et al. NF-kappaB inhibition delays DNA damage-induced senescence and aging in mice. J Clin Invest. 2012;122:2601–2612. doi: 10.1172/JCI45785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang F, Fu L, Wang Y. 6-thioguanine induces mitochondrial dysfunction and oxidative DNA damage in acute lymphoblastic leukemia cells. Mol Cell Proteomics. 2013;12:3803–3811. doi: 10.1074/mcp.M113.029595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y. Bulky DNA lesions induced by reactive oxygen species. Chem Res Toxicol. 2008;21:276–281. doi: 10.1021/tx700411g. [DOI] [PubMed] [Google Scholar]
- 12.Dizdaroglu M, Jaruga P, Rodriguez H. Identification and quantification of 8,5′-cyclo-2′-deoxy-adenosine in DNA by liquid chromatography/mass spectrometry. Free Radical Bio Med. 2001;30:774–784. doi: 10.1016/s0891-5849(01)00464-6. [DOI] [PubMed] [Google Scholar]
- 13.Chatgilialoglu C, D’Angelantonio M, Kciuk G, Bobrowski K. New insights into the reaction paths of hydroxyl radicals with 2′-deoxyguanosine. Chem Res Toxicol. 2011;24:2200–2206. doi: 10.1021/tx2003245. [DOI] [PubMed] [Google Scholar]
- 14.Das RS, Samaraweera M, Morton M, Gascon JA, Basu AK. Stability of N-glycosidic bond of (5′S)-8,5′-cyclo-2′-deoxyguanosine. Chem Res Toxicol. 2012;25:2451–2461. doi: 10.1021/tx300302a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kamakura N, Yamamoto J, Brooks PJ, Iwai S, Kuraoka I. Effects of 5′,8-cyclodeoxyadenosine triphosphates on DNA synthesis. Chem Res Toxicol. 2012;25:2718–2724. doi: 10.1021/tx300351p. [DOI] [PubMed] [Google Scholar]
- 16.Lindahl T, Wood RD. Quality control by DNA repair. Science. 1999;286:1897–1905. doi: 10.1126/science.286.5446.1897. [DOI] [PubMed] [Google Scholar]
- 17.Brooks PJ, Wise DS, Berry DA, Kosmoski JV, Smerdon MJ, Somers RL, Mackie H, Spoonde AY, Ackerman EJ, Coleman K, et al. The oxidative DNA lesion 8,5′-(S)-cyclo-2′-deoxyadenosine is repaired by the nucleotide excision repair pathway and blocks gene expression in mammalian cells. J Biol Chem. 2000;275:22355–22362. doi: 10.1074/jbc.M002259200. [DOI] [PubMed] [Google Scholar]
- 18.Kropachev K, Ding S, Terzidis MA, Masi A, Liu Z, Cai Y, Kolbanovskiy M, Chatgilialoglu C, Broyde S, Geacintov NE, et al. Structural basis for the recognition of diastereomeric 5′,8-cyclo-2′-deoxypurine lesions by the human nucleotide excision repair system. Nucleic Acids Res. 2014;42:5020–5032. doi: 10.1093/nar/gku162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuraoka I, Bender C, Romieu A, Cadet J, Wood RD, Lindahl T. Removal of oxygen free-radical-induced 5′,8-purine cyclodeoxynucleosides from DNA by the nucleotide excision-repair pathway in human cells. P Natl Acad Sci USA. 2000;97:3832–3837. doi: 10.1073/pnas.070471597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yuan B, Wang J, Cao H, Sun R, Wang Y. High-throughput analysis of the mutagenic and cytotoxic properties of DNA lesions by next-generation sequencing. Nucleic Acids Res. 2011;39:5945–5954. doi: 10.1093/nar/gkr159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marietta C, Gulam H, Brooks PJ. A single 8,5′-cyclo-2′-deoxyadenosine lesion in a TATA box prevents binding of the TATA binding protein and strongly reduces transcription in vivo. DNA repair. 2002;1:967–975. doi: 10.1016/s1568-7864(02)00148-9. [DOI] [PubMed] [Google Scholar]
- 22.Friedberg EC, Lehmann AR, Fuchs RP. Trading places: how do DNA polymerases switch during translesion DNA synthesis? Mol Cell. 2005;18:499–505. doi: 10.1016/j.molcel.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 23.Kuraoka I, Robins P, Masutani C, Hanaoka F, Gasparutto D, Cadet J, Wood RD, Lindahl T. Oxygen free radical damage to DNA. Translesion synthesis by human DNA polymerase eta and resistance to exonuclease action at cyclopurine deoxynucleoside residues. J Biol Chem. 2001;276:49283–49288. doi: 10.1074/jbc.M107779200. [DOI] [PubMed] [Google Scholar]
- 24.Pednekar V, Weerasooriya S, Jasti VP, Basu AK. Mutagenicity and genotoxicity of (5′S)-8,5′-cyclo-2′-deoxyadenosine in Escherichia coli and replication of (5′S)-8,5′-cyclopurine-2′-deoxynucleosides in vitro by DNA polymerase IV, exo-free Klenow fragment, and Dpo4. Chem Res Toxicol. 2014;27:200–210. doi: 10.1021/tx4002786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.You C, Swanson AL, Dai X, Yuan B, Wang J, Wang Y. Translesion synthesis of 8,5′-cyclopurine-2′-deoxynucleosides by DNA polymerases eta, iota, and zeta. J Biol Chem. 2013;288:28548–28556. doi: 10.1074/jbc.M113.480459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brooks PJ. The 8,5′-cyclopurine-2′-deoxynucleosides: candidate neurodegenerative DNA lesions in xeroderma pigmentosum, and unique probes of transcription and nucleotide excision repair. DNA Repair. 2008;7:1168–1179. doi: 10.1016/j.dnarep.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kirkali G, de Souza-Pinto NC, Jaruga P, Bohr VA, Dizdaroglu M. Accumulation of (5′S)-8,5′-cyclo-2′-deoxyadenosine in organs of Cockayne syndrome complementation group B gene knockout mice. DNA Repair. 2009;8:274–278. doi: 10.1016/j.dnarep.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J, Clauson CL, Robbins PD, Niedernhofer LJ, Wang Y. The oxidative DNA lesions 8,5′-cyclopurines accumulate with aging in a tissue-specific manner. Aging Cell. 2012;11:714–716. doi: 10.1111/j.1474-9726.2012.00828.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J, Yuan B, Guerrero C, Bahde R, Gupta S, Wang Y. Quantification of oxidative DNA lesions in tissues of Long-Evans Cinnamon rats by capillary high-performance liquid chromatography-tandem mass spectrometry coupled with stable isotope-dilution method. Anal Chem. 2011;83:2201–2209. doi: 10.1021/ac103099s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beard WA, Wilson SH. Structure and mechanism of DNA polymerase beta. Biochemistry. 2014;53:2768–2780. doi: 10.1021/bi500139h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Braithwaite DK, Ito J. Compilation, alignment, and phylogenetic relationships of DNA polymerases. Nucleic Acids Res. 1993;21:787–802. doi: 10.1093/nar/21.4.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beard WA, Wilson SH. Structure and mechanism of DNA polymerase Beta. Chem Rev. 2006;106:361–382. doi: 10.1021/cr0404904. [DOI] [PubMed] [Google Scholar]
- 33.Bassett E, Vaisman A, Tropea KA, McCall CM, Masutani C, Hanaoka F, Chaney SG. Frameshifts and deletions during in vitro translesion synthesis past Pt-DNA adducts by DNA polymerases beta and eta. DNA Repair. 2002;1:1003–1016. doi: 10.1016/s1568-7864(02)00150-7. [DOI] [PubMed] [Google Scholar]
- 34.Batra VK, Shock DD, Prasad R, Beard WA, Hou EW, Pedersen LC, Sayer JM, Yagi H, Kumar S, Jerina DM, et al. Structure of DNA polymerase beta with a benzo[c]phenanthrene diol epoxide-adducted template exhibits mutagenic features. P Natl Acad Sci USA. 2006;103:17231–17236. doi: 10.1073/pnas.0605069103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beard WA, Shock DD, Batra VK, Pedersen LC, Wilson SH. DNA polymerase beta substrate specificity: side chain modulation of the “A-rule”. J Biol Chem. 2009;284:31680–31689. doi: 10.1074/jbc.M109.029843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chary P, Beard WA, Wilson SH, Lloyd RS. DNA polymerase beta gap-filling translesion DNA synthesis. Chem Res Toxicol. 2012;25:2744–2754. doi: 10.1021/tx300368f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Efrati E, Tocco G, Eritja R, Wilson SH, Goodman MF. Abasic translesion synthesis by DNA polymerase beta violates the “A-rule”. Novel types of nucleotide incorporation by human DNA polymerase beta at an abasic lesion in different sequence contexts. J Biol Chem. 1997;272:2559–2569. doi: 10.1074/jbc.272.4.2559. [DOI] [PubMed] [Google Scholar]
- 38.Hashim MF, Schnetz-Boutaud N, Marnett LJ. Replication of template-primers containing propanodeoxyguanosine by DNA polymerase beta. Induction of base pair substitution and frameshift mutations by template slippage and deoxynucleoside triphosphate stabilization. J Biol Chem. 1997;272:20205–20212. doi: 10.1074/jbc.272.32.20205. [DOI] [PubMed] [Google Scholar]
- 39.Hoffmann JS, Pillaire MJ, Garcia-Estefania D, Lapalu S, Villani G. In vitro bypass replication of the cisplatin-d(GpG) lesion by calf thymus DNA polymerase beta and human immunodeficiency virus type I reverse transcriptase is highly mutagenic. J Biol Chem. 1996;271:15386–15392. doi: 10.1074/jbc.271.26.15386. [DOI] [PubMed] [Google Scholar]
- 40.Maga G, van Loon B, Crespan E, Villani G, Hubscher U. The block of DNA polymerase delta strand displacement activity by an abasic site can be rescued by the concerted action of DNA polymerase beta and Flap endonuclease 1. J Biol Chem. 2009;284:14267–14275. doi: 10.1074/jbc.M900759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Servant L, Cazaux C, Bieth A, Iwai S, Hanaoka F, Hoffmann JS. A role for DNA polymerase beta in mutagenic UV lesion bypass. J Biol Chem. 2002;277:50046–50053. doi: 10.1074/jbc.M207101200. [DOI] [PubMed] [Google Scholar]
- 42.Vaisman A, Chaney SG. The efficiency and fidelity of translesion synthesis past cisplatin and oxaliplatin GpG adducts by human DNA polymerase beta. J Biol Chem. 2000;275:13017–13025. doi: 10.1074/jbc.275.17.13017. [DOI] [PubMed] [Google Scholar]
- 43.Villani G, Hubscher U, Gironis N, Parkkinen S, Pospiech H, Shevelev I, di Cicco G, Markkanen E, Syvaoja JE, Tanguy Le Gac N. In vitro gap-directed translesion DNA synthesis of an abasic site involving human DNA polymerases epsilon, lambda, and beta. J Biol Chem. 2011;286:32094–32104. doi: 10.1074/jbc.M111.246611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu M, Lai Y, Jiang Z, Terzidis MA, Masi A, Chatgilialoglu C, Liu Y. A 5′, 8-cyclo-2′-deoxypurine lesion induces trinucleotide repeat deletion via a unique lesion bypass by DNA polymerase beta. Nucleic Acids Res. 2014;42:13749–13763. doi: 10.1093/nar/gku1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chatgilialoglu C, Ferreri C, Masi A, Sansone A, Terzidis MA, Tsakos M. A problem solving approach for the diastereoselective synthesis of (5′S)- and (5′R)-5′,8-cyclopurine lesions. Org Chem Front. 2014;1:698–702. [Google Scholar]
- 46.Biade S, Sobol RW, Wilson SH, Matsumoto Y. Impairment of proliferating cell nuclear antigen-dependent apurinic/apyrimidinic site repair on linear DNA. J Biol Chem. 1998;273:898–902. doi: 10.1074/jbc.273.2.898. [DOI] [PubMed] [Google Scholar]
- 47.Liu Y, Wilson SH. DNA base excision repair: a mechanism of trinucleotide repeat expansion. Trends Biochem Sci. 2012;37:162–172. doi: 10.1016/j.tibs.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Balakrishnan L, Bambara RA. Flap endonuclease 1. Annu Rev Biochem. 2013;82:119–138. doi: 10.1146/annurev-biochem-072511-122603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu Y, Kao HI, Bambara RA. Flap endonuclease 1: a central component of DNA metabolism. Annu Rev Biochem. 2004;73:589–615. doi: 10.1146/annurev.biochem.73.012803.092453. [DOI] [PubMed] [Google Scholar]
- 50.Wilson SH, Beard WA, Shock DD, Batra VK, Cavanaugh NA, Prasad R, Hou EW, Liu Y, Asagoshi K, Horton JK, et al. Base excision repair and design of small molecule inhibitors of human DNA polymerase beta. Cell Mol Life Sci. 2010;67:3633–3647. doi: 10.1007/s00018-010-0489-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Belmadoui N, Boussicault F, Guerra M, Ravanat JL, Chatgilialoglu C, Cadet J. Radiation-induced formation of purine 5′,8-cyclonucleosides in isolated and cellular DNA: high stereospecificity and modulating effect of oxygen. Org Biomol Chem. 2010;8:3211–3219. doi: 10.1039/c004531d. [DOI] [PubMed] [Google Scholar]
- 52.Kirkali G, Tunca M, Genc S, Jaruga P, Dizdaroglu M. Oxidative DNA damage in polymorphonuclear leukocytes of patients with familial Mediterranean fever. Free Radical Bio Med. 2008;44:386–393. doi: 10.1016/j.freeradbiomed.2007.09.020. [DOI] [PubMed] [Google Scholar]
- 53.Karwowski BT, Bellon S, O’Neill P, Lomax ME, Cadet J. Effects of (5′S)-5′,8-cyclo-2′-deoxyadenosine on the base excision repair of oxidatively generated clustered DNA damage. A biochemical and theoretical study. Org Biomol Chem. 2014;12:8671–8682. doi: 10.1039/c4ob01089b. [DOI] [PubMed] [Google Scholar]
- 54.Canitrot Y, Hoffmann JS, Calsou P, Hayakawa H, Salles B, Cazaux C. Nucleotide excision repair DNA synthesis by excess DNA polymerase beta: a potential source of genetic instability in cancer cells. FASEB J. 2000;14:1765–1774. doi: 10.1096/fj.99-1063com. [DOI] [PubMed] [Google Scholar]
- 55.Scanlon KJ, Kashani-Sabet M, Miyachi H. Differential gene expression in human cancer cells resistant to cisplatin. Cancer Invest. 1989;7:581–587. doi: 10.3109/07357908909017533. [DOI] [PubMed] [Google Scholar]
- 56.Srivastava DK, Husain I, Arteaga CL, Wilson SH. DNA polymerase beta expression differences in selected human tumors and cell lines. Carcinogenesis. 1999;20:1049–1054. doi: 10.1093/carcin/20.6.1049. [DOI] [PubMed] [Google Scholar]
- 57.Horton JK, Baker A, Berg BJ, Sobol RW, Wilson SH. Involvement of DNA polymerase beta in protection against the cytotoxicity of oxidative DNA damage. DNA Repair. 2002;1:317–333. doi: 10.1016/s1568-7864(02)00008-3. [DOI] [PubMed] [Google Scholar]
- 58.Lan L, Nakajima S, Oohata Y, Takao M, Okano S, Masutani M, Wilson SH, Yasui A. In situ analysis of repair processes for oxidative DNA damage in mammalian cells. P Natl Acad Sci USA. 2004;101:13738–13743. doi: 10.1073/pnas.0406048101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Beard WA, Shock DD, Batra VK, Prasad R, Wilson SH. Substrate-induced DNA Polymerase beta Activation. J Biol Chem. 2014;289:31411–31422. doi: 10.1074/jbc.M114.607432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chou KM, Cheng YC. An exonucleolytic activity of human apurinic/apyrimidinic endonuclease on 3′ mispaired DNA. Nature. 2002;415:655–659. doi: 10.1038/415655a. [DOI] [PubMed] [Google Scholar]
- 61.Parsons JL, Dianova II, Dianov GL. APE1-dependent repair of DNA single-strand breaks containing 3′-end 8-oxoguanine. Nucleic Acids Res. 2005;33:2204–2209. doi: 10.1093/nar/gki518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bakhanashvili M. Exonucleolytic proofreading by p53 protein. Eur J Biochem. 2001;268:2047–2054. doi: 10.1046/j.1432-1327.2001.02075.x. [DOI] [PubMed] [Google Scholar]
- 63.Rahav GaB. In: New Research Directions in DNA Repair. Chen C, editor. InTech; 2013. pp. 281–308. [Google Scholar]
- 64.Liu Y, Prasad R, Beard WA, Kedar PS, Hou EW, Shock DD, Wilson SH. Coordination of steps in single-nucleotide base excision repair mediated by apurinic/apyrimidinic endonuclease 1 and DNA polymerase beta. J Biol Chem. 2007;282:13532–13541. doi: 10.1074/jbc.M611295200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhou J, Ahn J, Wilson SH, Prives C. A role for p53 in base excision repair. EMBO J. 2001;20:914–923. doi: 10.1093/emboj/20.4.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.