

Abstract
The Mdm2 oncogene is a negative regulator of the p53 tumor suppressor and recently identified inhibitor of DNA break repair. Nutlin-3 is a small molecule inhibitor of Mdm2/p53 interaction that can induce apoptosis in cancer cells through activation of p53. While this is promising therapy for those cancers with wild-type p53, half of all human cancers have inactivated p53. Here, we reveal a previously unappreciated effect of Nutlin is inhibition of DNA break repair, stemming from its ability to increase Mdm2 protein levels. The Nutlin-induced increase in Mdm2 inhibited DNA double-strand break (DSB) repair and prolonged DNA damage response signaling independent of p53. Mechanistically, this effect of Nutlin required Mdm2 and acted through Nbs1 of the Mre11/Rad50/Nbs1 DNA repair complex. In ovarian cancer cells where >90% have inactivated p53, Nutlin combined with the genotoxic agents, cisplatin or etoposide, had a cooperative lethal effect resulting in increased DNA damage and apoptosis. Therefore, these data demonstrate an unexpected consequence of pharmacologically increasing Mdm2 levels that when utilized in combination with genotoxic agents induces synthetic lethality in ovarian cancer cells, and likely other malignant cell types, that have inactivated p53.
Implications
Data reveal a therapeutically beneficial effect of pharmacologically increasing Mdm2 levels combined with chemotherapeutic agents for malignancies that have lost functional p53.
Keywords: Mdm2, p53, chemotherapy, Nutlin, ovarian cancer
Introduction
The p53 tumor suppressor can inhibit cell cycle progression and induce apoptosis following cellular stress, such as DNA damage. Consequently, p53 is mutated or deleted in approximately half of all of human cancers, with some malignancies having higher p53 mutation rates (1). In high grade serous ovarian cancer, the most common and aggressive form of the disease, >90% of the tumors have inactivated p53 (2). Typically, an initial diagnosis is made at late stages of metastatic disease, making ovarian cancer challenging to treat and one of the most deadly cancers among women (3). Primary chemotherapy for ovarian cancer involves the genotoxic platinum drugs, cisplatin or carboplatin (4). Although initial responses are good, platinum resistance occurs often, requiring the use of other genotoxic agents, such as etoposide (5).
Mdm2 is an E3 ubiquitin ligase that binds directly to p53, targeting it for proteosomal degradation (1). In recent years, the drug Nutlin-3 (Nutlin) was developed that binds in the p53-binding pocket of Mdm2, inhibiting the interaction between Mdm2 and p53. Nutlin prevents the negative regulation of p53 by Mdm2, thus, activating p53. In cancer cells with wild-type p53, Nutlin can induce p53-mediated apoptosis (6). Additionally, Nutlin has been used in combination with genotoxic agents to induce apoptosis of cancer cells retaining functional p53 (7). While this approach shows promise for tumors retaining wild-type p53, this mechanism of action is not useful for the many cancers with mutated or deleted p53. Therefore, understanding and establishing therapeutics that can act independent of p53 are needed for treating these cancers.
It has been reported that a consequence of Nutlin treatment is elevated Mdm2 protein levels, and this occurs in cells with or without p53 (6,8). While Mdm2 negatively regulates p53, Mdm2 also has p53-independent functions that negatively impact genome stability (9). We have shown elevated Mdm2 levels act through Nbs1 of the Mre11/Rad50/Nbs1 (MRN) DNA repair complex to impair DNA damage response signaling, leading to inhibition of double-strand DNA break repair and increased genome instability independent of p53 (10,11). Here, we show the Nutlin-induced increase in Mdm2 results in an inhibition of double-strand DNA break repair and DNA damage response signaling that requires both Mdm2 and Nbs1, but not p53. We also demonstrate a combined lethal effect of Nutlin and genotoxic agents in p53 inactivated ovarian cancer cells due to the inhibition of DNA repair. These studies reveal an alternative mechanism of action of Nutlin in tumor cells that have inactivated p53 and a likely therapeutic benefit of increasing Mdm2 levels pharmacologically in those tumors.
Materials and Methods
Cell culture
p53−/−, p53−/−Mdm2−/−, and Nbs1ΔB/ΔB murine embryonic fibroblasts (MEFs) were cultured as we previously described (12). 293T cells were cultured as described by the American Type Culture Collection (Manassas, VA). SKOV-3, OVCAR-5 and OVCAR-8 ovarian cancer cell lines were cultured as previously described (13).
Chemotherapeutic agents
Nutlin (Sigma) and etoposide (Sigma) were dissolved in DMSO (Sigma) at 17.2 mM and 50 mM, respectively, from which working stocks were generated. The concentration of Nutlin used refers to the entire mixture, but only half of the total concentration represents the active enantiomer A. Cisplatin (Sigma) was resuspended in 0.9% NaCl at 5 mM from which working stocks were generated.
Western blotting and Protein half-life analysis
Following addition of Nutlin (10 µM) or vehicle control (DMSO) for 1 hour, cycloheximide (20 µg/ml; Sigma) was added to cultures of p53−/− MEFs. At intervals, cells were placed on ice and harvested for western blot analysis. Whole cell lysates were prepared, subjected to SDS-PAGE, transferred to nitrocellulose, and Western blotted as we previously described (11). Antibodies specific for Mdm2 (2A10 for mouse, Calbiochem; 3G9 for human, Millipore), cleaved Caspase 3 (ASP175, Cell Signaling), and β-actin (Sigma) were used. Densitometry for quantification of Mdm2 protein bands was performed using Image J software (National Institutes of Health) and were relative to band intensities of β-actin.
Comet assay
Cells were left untreated or were treated with Nutlin (10 µM) or vehicle control (DMSO) 24 hours prior to γ-radiation (137Cs source), where indicated. As a control, cells were either transfected or infected with a bicistronic retroviral vector encoding Mdm2 and GFP (10). Neutral comet assays were performed at intervals following γ-radiation, as previously described (10,11). A minimum of three independent experiments were performed for all analyses. Statistical significance was determined by student’s t-tests.
Immunofluorescence
Cells were cultured on coverslips and, where indicated, treated for 24 hours with the specified drug. Following γ-radiation, cells were fixed and γH2AX and pS/TQ foci were detected by immunofluorescence and analyzed as previously described (11,14). Antibodies against pSer139-H2AX (Millipore), pSer1981-ATM (Rockland), and secondary antibodies Alexa Fluor 594 and Alexa Fluor 488 (Invitrogen) were used. The number of γH2AX and pS/TQ foci per cell for at least 40 cells per individual condition was quantified. A minimum of 2–3 independent experiments were performed for these analyses. Statistical significance was determined using a confidence interval of 95%. The assays determining the percentage of γH2AX positive cells 24 hours following removal of cisplatin or etoposide were performed two times as previously published (15), except Nutlin (10 µM) was added to the samples indicated. Cells that had 5 or more foci were considered positive; statistical significance was determined using a student’s t-test.
Proliferation, viability, and apoptosis analysis
For MTT assays, cells (5×103–1×104) were cultured in 96-well plates and treated in quadruplicate with chemotherapeutic agents (Nutlin, etoposide, cisplatin, or a combination) at varying concentrations for 72 hours. At intervals, MTT assays (Sigma) were performed according to manufacturer’s protocol. To evaluate apoptosis, cells were treated with chemotherapeutic agents for 48 hours, photographed and harvested. Whole cell protein lysates were prepared as described above. Cleaved Caspase 3 was evaluated by Western blot (see above).
Results
Nutlin increases Mdm2 protein stability independent of p53
It was previously reported that cell lines treated with Nutlin can lead to increased levels of Mdm2 as detected by Western blot, and this can occur in the absence of p53 (6,8). To determine whether Nutlin influences Mdm2 protein stability, we evaluated Mdm2 protein levels following addition of the protein synthesis inhibitor, cycloheximide. To exclude effects on Mdm2 from p53 activation due to Nutlin treatment, we utilized p53−/− MEFs. We also only exposed the MEFs to Nutlin for one hour to evaluate direct effects of Nutlin and eliminate any secondary effects that may occur with prolonged treatment. Mdm2 protein remained present for longer in cells with Nutlin compared to vehicle control treated cells (Figure 1A). Specifically, there were high levels of Mdm2 protein remaining after 20 minutes with cycloheximide in the Nutlin treated cells, whereas in the vehicle control treated cells, Mdm2 was barely detectable. These data indicate increased stability of Mdm2 protein in the presence of Nutlin, resulting in elevated levels of Mdm2.
Figure 1. Nutlin inhibits DNA break repair independent of p53 and through Mdm2 and Nbs1.

(A) Cycloheximide (CHX) was added to p53−/− MEFs pretreated with Nutlin (10 µM) or vehicle control (DMSO) for 1 hour. At the intervals indicated cells were harvested and whole cell protein lysates were subjected to Western blot analysis for the protein indicated. Quantification of Mdm2 relative to β-actin was performed by densitometry. (B–E) 293T cells (B), p53−/− MEFs (C), p53−/−Mdm2−/− MEFs (D), and Nbs1ΔB/ΔB MEFs (E) were cultured with Nutlin (10 µM) or vehicle control (DMSO). Ectopic Mdm2 overexpression in each cell line served as a positive control. Whole cell lysates were Western blotted for the indicated proteins. Following 5 Gy of γ-radiation, neutral comet assays were used to evaluate the percentage of cells with a tail moment ≥4 (damaged DNA). All graphs represent of a minimum of three independent experiments and each circle indicates one experimental sample consisting of a minimum of 40 cells analyzed. For D, 120 minutes following γ-radiation is shown. B, *p=0.0004, **p=0.011; C, *p=0.01, **p=0.007; D, *p=0.001; **p=0.0093; E, *p=0.031, all as compared to DMSO, t-test.
Nutlin inhibits DNA break repair through Mdm2 and Nbs1, independent of p53
We have previously shown that increased levels of Mdm2 protein inhibit double-strand DNA break repair independent of p53 (10,11,14). To determine whether the increase in Mdm2 levels caused by Nutlin would result in an inhibition in double-strand DNA break repair, we first tested this by neutral comet assay in 293T cells, which have inactivated p53 due to expression of SV40 large T antigen. We detected 60.1% (+/−1.57%) of the cells exposed to Nutlin had DNA damage remaining 60 minutes post γ-radiation, compared to only 29.7% (+/−2.35%) of DMSO treated cells (Figure 1B). The reduction in repaired DNA caused by Nutlin was comparable to cells that ectopically overexpressed Mdm2. To more fully assess whether this effect of Nutlin is independent of p53, we evaluated DNA repair in p53−/− MEFs in the presence of Nutlin. Sixty minutes after γ-radiation, p53−/− MEFs with Nutlin had reduced repair of double-strand DNA breaks resulting in significantly more cells harboring damaged DNA compared to cells treated with vehicle control (Figure 1C). Again, the inhibition of DNA repair by Nutlin was comparable to cells overexpressing Mdm2 from a vector.
To determine the requirement of Mdm2 for this effect on DNA repair of Nutlin, we exposed p53−/−Mdm2−/− MEFs to Nutlin or DMSO vehicle control. We observed at both 60 and 90 minutes following γ-radiation, Nutlin treated cells repaired their DNA to a similar extent as DMSO control (Figure 1D), whereas MEFs overexpressing Mdm2 had significantly reduced DNA repair. These results demonstrate the Nutlin-induced inhibition of double-strand DNA break repair is mediated by Mdm2.
The inhibition in DNA break repair induced by Mdm2 is mediated through its interactions with Nbs1 of the MRN complex (11). To evaluate the requirement of Nbs1 for the effects of Nutlin on DNA break repair, we utilized MEFs where Nbs1 has been mutated resulting in instability and severely reduced Nbs1 expression (Nbs1ΔB/ΔB (16)). DNA break repair is already delayed in the Nbs1ΔB/ΔB MEFs and addition of Nutlin did not further inhibit DNA repair (Figure 1E). As we reported previously (11), expression of wild-type Nbs1 rescues the DNA repair kinetics in the Nbs1ΔB/ΔB MEFs. However, when a mutant of Nbs1 that cannot bind Mdm2 (mut 8; (11)) was expressed, Nutlin treatment did not result in a delay in DNA repair (Figure 1E). Therefore, Nutlin-induced inhibition of DNA break repair is dependent on Mdm2 and Nbs1 interaction.
Nutlin inhibits the DNA damage signal through Mdm2
To determine whether the reduction in DNA break repair caused by Nutlin is due to a delay in DNA damage signaling that occurs when Mdm2 is overexpressed (10,11), we evaluated γH2AX foci formation and resolution. H2AX is rapidly phosphorylated (γH2AX) upon DNA damage and returns to an unphosphorylated form upon repair (17). Assessment of γH2AX foci formation showed that 10 minutes following γ-radiation, p53−/− MEFs in the presence of Nutlin formed 31% fewer foci than vehicle treated cells (Figure 2A and Supplemental Figure S1A). These results were comparable to cells overexpressing Mdm2 from a retroviral vector. At 150 minutes post γ-radiation, there were significantly increased numbers of γH2AX foci in the Nutlin exposed MEFs compared to DMSO treated controls, indicating a delay in foci resolution. To determine the role of Mdm2, we measured foci in p53−/−Mdm2−/− MEFs. When Mdm2 was not present, Nutlin no longer inhibited the formation or the resolution of γH2AX foci compared to DMSO treated cells (Figure 2B and Supplemental Figure S1B). These results indicate Nutlin treatment has a comparable inhibitory effect on γH2AX foci formation and resolution as Mdm2 overexpression, and this effect is independent of p53, but requires Mdm2.
Figure 2. DNA damage signaling is inhibited by Nutlin and requires Mdm2.

(A, C) p53−/− MEFs and (B, D) p53−/−Mdm2−/− MEFs were treated with Nutlin (10 µM) or vehicle control (DMSO). MEFs with ectopic Mdm2 overexpression served as a positive control. Following exposure to 5 Gy of γ-radiation, MEFs were fixed at intervals and immunofluorescence for γH2AX (A, B) and pS/TQ (C, D) was performed. The number of foci per cell was quantified. The mean of 2–3 independent experiments is graphed. Error bars represent SEM, and significance (*) determined using a confidence interval of 95%.
γ-Radiation induces DNA breaks, which activates ATM, the DNA damage-induced kinase, that then phosphorylates motifs with a serine or threonine followed by a glutamine (S/TQ) (18). Another means to measure DNA damage signaling is to evaluate these phosphorylated S/TQ (pS/TQ) sites. Following γ-radiation, pS/TQ foci were assessed in p53−/− MEFs cultured with Nutlin or vehicle control. Cells with Nutlin had 28% fewer foci immediately after γ-radiation, and 62% more foci at 240 minutes post γ-radiation compared to controls (Figure 2C and Supplemental Figure S2A). When p53−/−Mdm2−/− MEFs were used, Nutlin had neither an impact on the formation nor the resolution of the pS/TQ foci (Figure 2D and Supplemental Figure S2B). In both cell types, pS/TQ foci formation and resolution was impaired when Mdm2 was ectopically overexpressed, as we previously reported (11). Therefore, Nutlin inhibits the initial DNA damage response signal leading to prolonged signaling, and this requires Mdm2 but is independent of p53.
Nutlin inhibits DNA break repair in ovarian cancer cells
Ovarian cancer is a difficult to treat malignancy with the aggressive high grade serous ovarian carcinoma type harboring mutated or deleted p53 in >90% of the tumors (2). To determine whether Nutlin could increase levels of Mdm2 and inhibit DNA break repair in malignant cells that have inactivated p53, we assessed ovarian cancer cells that had either deleted or mutated p53. SKOV-3 ovarian cancer cells have deleted p53 and showed increased levels of Mdm2 following exposure to Nutlin (Figure 3A). We then assessed the effect of Nutlin on their ability to repair damaged DNA by exposing the SKOV-3 cells to Nutlin or vehicle control, and performing comet assays following γ-radiation. Only 57.6% of the cells treated with Nutlin were able to repair their DNA damage within 90 minutes, compared to 74.5% of cells exposed to vehicle control (Figure 3A).
Figure 3. Delayed DNA repair by Nutlin in ovarian cancer cells with inactivated p53.

SKOV-3 (A) OVCAR-5 (B), and OVCAR-8 (C) ovarian cancer cell lines were cultured with Nutlin (10 µM) or vehicle control (DMSO). Whole cell protein lysates were evaluated by Western blot. Following 5 Gy of γ-radiation, neutral comet assays were used to measure DNA breaks. Each bar represents the average of 2–4 independent experiments with a minimum of 40 cells analyzed for each sample. A, *p=0.0073, **p=0.035; B, *p=0.0047, **p=0.01; C, *p=0.004, **p=0.0073, student’s t-test.
We also evaluated ovarian cancer cells that harbor mutant p53, OVCAR-5 (insertion at codon 224) and OVCAR-8 (deletion of codons 126–132) (19). Nutlin treatment led to increased Mdm2 protein levels in both cell lines (Figure 3B and 3C). In addition, both lines exposed to Nutlin had significantly reduced DNA break repair at 60 and 90 minutes post γ-radiation (Figure 3B and 3C). These results indicate Nutlin-induced increase in Mdm2 results in a delay in double-strand DNA break repair in ovarian cancer cells that contain deleted or mutated p53.
Delay of DNA damage signal in ovarian cancer cells caused by Nutlin
To assess the DNA damage signal in ovarian cancer cells, we evaluated γH2AX foci formation and resolution. Ten minutes after γ-radiation, there were 29% fewer γH2AX foci in the Nutlin treated p53 deleted SKOV-3 cells compared to vehicle control (Figure 4A and Supplemental Figure S3A). At 240 minutes post γ-radiation, the Nutlin treated SKOV-3 cells had 46% more foci remaining compared to DMSO treated cells. Similar results were obtained with the p53 mutant ovarian cancer cell lines, OVCAR-5 and OVCAR-8 (Figure 4B and 4C, respectively and Supplemental Figure S3B and S3C). Therefore, Nutlin inhibits DNA repair and DNA damage response signals in ovarian cancer cells with inactivated p53, analogous to what was observed in MEFs.
Figure 4. Nutlin inhibits DNA damage signaling in ovarian cancer cells.
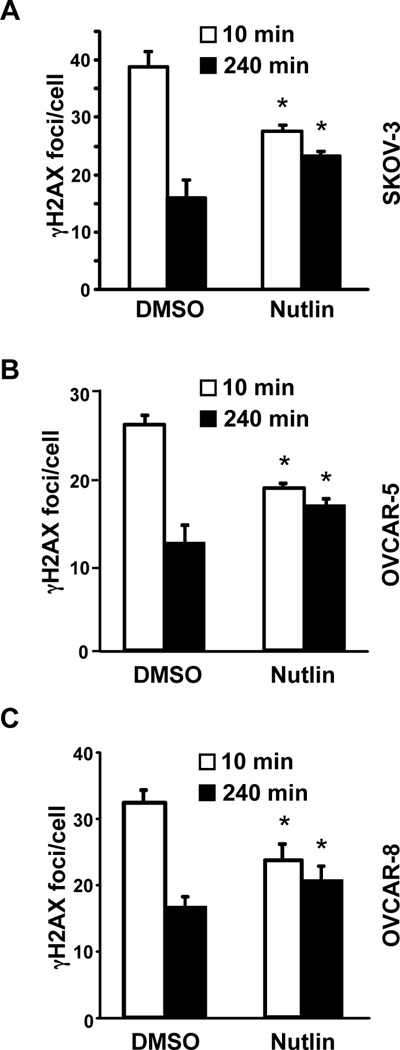
SKOV-3 (A) OVCAR-5 (B), and OVCAR-8 (C) ovarian cancer cell lines were cultured with Nutlin (10 µM) or vehicle control (DMSO), and following γ-radiation, γH2AX foci were detected by immunofluorescence. The number of foci per cell was quantified. The mean of 2–3 independent experiments is graphed. Error bars represent SEM, and significance (*) determined using a confidence interval of 95%.
Inhibition of ovarian cancer cell expansion with combined treatment of Nutlin and genotoxic agents
Cisplatin, used in first-line therapy for ovarian cancer, is a genotoxic agent that causes DNA double-strand breaks (20). To determine whether inhibition in DNA repair by Nutlin would cooperate with genotoxic chemotherapeutic agents, we evaluated the combination effect of Nutlin and cisplatin. SKOV-3 cells were treated with Nutlin alone, cisplatin alone, or Nutlin and cisplatin in combination. SKOV-3 cells treated with 1 µM of cisplatin alone or 10 µM of Nutlin alone resulted in a maximum of 12.3% +/− 0.43% reduction in cell growth at 72 hours compared to vehicle treated control (Figure 5A and Supplemental Figure S4A). However, treatment with both 1 µM of cisplatin and 10 µM of Nutlin resulted in a 26.9% +/− 0.62% reduction in cell growth at 72 hours compared to vehicle treated cells (Supplemental Figure S4A). This cooperative effect was more apparent between Nutlin and 5 µM cisplatin, which combined inhibited growth by 51.5% +/− 0.49%, while 5 µM cisplatin alone only inhibited 35.0% +/− 1.08% (Figure 5A). Nutlin enhanced the negative effect of 5 µM cisplatin on cell expansion over that of the 10 µM cisplatin alone. Similar results were obtained with the OVCAR-5 and OVCAR-8 mutant p53 ovarian cancer cells (Figure 5A and Supplemental Figure S4A). These results indicate pharmacologically increasing Mdm2 with Nutlin enhanced the lethality of cisplatin in ovarian cancer cells with inactivated p53.
Figure 5. Nutlin cooperates with genotoxic agents to reduce cell viability independent of p53 in ovarian cancer cells.

The indicated ovarian cancer cell lines were treated with the indicated drug(s) or vehicle control (DMSO). Nutlin was always at 10 µM. Cisplatin (A) or etoposide (B) was at 5 or 10 µM. MTT assays were performed at intervals. Representative graphs of 3 independent experiments are shown. Error bars are SEM.
Etoposide also results in double-strand DNA breaks and is used to treat relapsed ovarian cancer, following platinum-resistance (5). We evaluated the combination effect of Nutlin with etoposide in SKOV-3, OVCAR-5, and OVCAR-8 ovarian cancer cell lines. For all three cell lines, treatment with either Nutlin or 1 µM etoposide had a very slight effect on the expansion of the cells, but when combined, cell growth was significantly inhibited (35.9–56.2%) (Supplemental Figure S4B). Similarly, when treated with Nutlin and 5 µM etoposide, cells were inhibited by 60.2–75.3% compared to only 49–53% when 5 µM etoposide alone was used (Figure 5B). The effects of Nutlin with 5 µM etoposide was similar to treating the cells with 2–4 times the amount of etoposide alone (Figure 5B). Therefore, ovarian cancer cells treated with both Nutlin and etoposide were significantly impaired in their ability to expand, and this was more than the effect of either drug alone, indicating cooperation.
Lethal combination for ovarian cancer cells of Nutlin and genotoxic agents
To determine if cell death caused the observed reduction in cell growth, SKOV-3, OVCAR-5 and OVCAR-8 ovarian cancer cell lines were first visually evaluated. We observed fewer cells in addition to an increase in dead and dying cells in the cultures exposed to both Nutlin and cisplatin (Figure 6A and Supplemental Figure S5) as well as Nutlin and etoposide (Figure 6B and Supplemental Figure S5) in comparison to either drug alone. These results suggest that the ovarian cancer cells are dying, but to test whether apoptosis was occurring, we assessed cleaved Caspase 3. Very little or no cleaved Caspase 3 was detected in Nutlin or vehicle control treated cells (Figure 6C and 6D). However, cleaved Caspase 3 was readily apparent in ovarian cancer cells treated with the combination of Nutlin and 5 µM cisplatin, and was similar to the amount of cleaved Caspase 3 observed in cells treated with 10 µM cisplatin alone (Figure 6C). Moreover, Nutlin combined with 5 µM of etoposide resulted in increased cleaved Caspase 3 to levels similar to 10 µM of etoposide alone (Figure 6D). Comparable results were obtained in all three ovarian cancer cell lines with both genotoxic drugs (Figure 6C and 6D). These results indicate Nutlin has a cooperative effect with cisplatin and etoposide in ovarian cancer cells with inactivated p53, resulting in apoptosis.
Figure 6. Nutlin and genotoxic agents are a lethal combination for ovarian cancer cells that have inactivated p53.

The indicated ovarian cancer cell line was treated with the indicated drug(s) or vehicle control (DMSO). Nutlin was always at 10 µM and cisplatin (A, C, E) or etoposide (B, D, F) was at 5 µM. For C and D, 10 µM cisplatin or etoposide, respectively, was also used. After 48 hours, representative pictures (A and B) were taken and whole cell protein lysates were evaluated by Western blot (C and D) for cleaved Caspase 3 and β-actin. (E) Immunofluorescence for γH2AX was performed 24 hours following removal of cisplatin or etoposide from SKOV-3 cells incubated with Nutlin or DMSO. Error bars represent SEM, *p<0.04, t-test.
One established method to measure effects of drugs on ovarian cancer cells at the DNA level is to evaluate γH2AX foci after a prolonged time (i.e. 24 hours) after removal of the genotoxic drug; this is a measure of damage remaining and indicative of cells that are likely to die (15). Using this method, we observed an increased percentage of ovarian cancer cells that were γH2AX positive after incubation with Nutlin or cisplatin alone, but significantly more when cells were treated with both Nutlin and cisplatin (Figure 6E). Analogous results were obtained when etoposide was used (Figure 6F). Combined, the data indicate that inhibition of double-strand DNA break repair resulting from Nutlin-induced increased Mdm2 levels cooperates with genotoxic agents, such as cisplatin or etoposide, to cause apoptosis of ovarian cancer cells lacking functional p53.
Discussion
Since the development of Nutlin, scientists have been using it as a research tool to investigate the involvement of the p53 pathway in various settings and for determining the chemotherapeutic potential of activating p53 to treat cancer (6,7). Although studies have extensively demonstrated p53-dependent properties of Nutlin, p53-independent effects of Nutlin have also emerged. One observed p53-independent effect of Nutlin is the increase in Mdm2 protein levels (6,21), which our data indicate is due to Mdm2 protein stabilization. We had previously shown Mdm2 overexpression inhibits DNA break repair and promotes genome instability independent of p53 (10,11). Here our data reveal that stabilization of Mdm2 by Nutlin resulted in an inhibition in the DNA damage signal and delayed DNA break repair. This effect of Nutlin was dependent on Mdm2 and its interactions with Nbs1. When Nutlin was combined with genotoxic agents in ovarian cancer cells, a significant increase in DNA damage and apoptosis independent of p53 was induced. These results provide insight into a novel p53-independent, Mdm2-dependent mechanism of Nutlin. Importantly, our data show the therapeutic benefit of pharmacologically increasing Mdm2 in conjunction with genotoxic agents as a means of inducing synthetic lethality in cancers with inactivated p53, such as ovarian cancer.
Numerous studies have described a cooperative effect between Nutlin and genotoxic agents in the context of wild-type p53. Our data suggest that in addition to p53 activation, Nutlin is also stabilizing Mdm2 causing an increase in the levels of Mdm2, which dampens the p53 response to DNA damage, resulting in increased sensitivity to genotoxic drugs (22,23). A small number of studies have also explored the effect Nutlin in combination with genotoxic drugs in cancer cells with mutated or deleted p53, but the mechanism for the observed cooperation between the two in this context was poorly understood. For example, a cooperative effect between Nutlin and topoisomerase II inhibitors in pancreatic cancer cells with inactivated p53 has been described (24). They reported Nutlin caused an increase in DNA damage response signals in response to genotoxic insults in three hours. Consistent with this, we observed an increase in DNA damage response signals four hours after γ-radiation, but we also detected a reduction in the early DNA damage response signal. Our results indicate Nutlin delays the DNA damage signal leading to more DNA damage signaling at later times after the genotoxic drug. Another study proposes Nutlin cooperates with cisplatin and doxorubicin by activating a p53 family member, p73, leading to E2F1 activation to induce apoptosis (25–28). Mdm2 can bind and regulate p73, and both p73 and E2F1 can be activated following DNA damage (29). Our data established that in the absence of functional Nbs1, Nutlin was unable to inhibit double-strand DNA break repair. Thus, if p73 and E2F1 are involved it appears that they do not contribute to the delay in DNA break repair induced by Nutlin.
Half of all human cancers have directly inactivated p53 and many cancers that progress or relapse from therapies have inactivated p53 (1). Therefore, there is much need for identifying new chemotherapeutic approaches for those malignancies that have inactivated p53, such as ovarian cancer. Current standard chemotherapy treatment for ovarian cancer involves the platinum-based genotoxic agents cisplatin or carboplatin (4). Our results reveal Nutlin used in combination with cisplatin increases the efficacy of cisplatin, showing that in the presence of Nutlin, 2-fold lower concentrations of cisplatin are effective. Relapse in ovarian cancer often results from platinum-resistance and requires the need to use combination therapy with other genotoxic agents, such as etoposide (5). Our data demonstrate that Nutlin also cooperates with etoposide to increase its efficacy, resulting in 2–4-fold lower concentrations of etoposide being equally effective. Our results have significant therapeutic implications, as they reveal that pharmacologically modulating Mdm2 levels could be used to more effectively treat malignancies that have inactivated p53, as well as allow lower concentrations of toxic chemotherapeutic drugs to be used. This study also emphasizes the importance of assessing compounds that lead to increased Mdm2 levels for therapeutic purposes. Moreover, since increasing Mdmx levels also leads to an inhibition of DNA break repair (14), compounds that stabilize Mdmx or both Mdm2 and Mdmx are also likely to have similar results.
Supplementary Material
Acknowledgements
We thank Pia Arrate, Robert Duszynski, Kristin Peterson, Jeanette Saskowski, and Andrew Wilson for technical assistance. This work was supported by F31CA150546 (AMC), R01CA181204 (CME), T32CA093240 (MH), and K08CA148887 (DK).
Financial Support: This work was supported by F31CA150546 (AMC), R01CA181204 (CME), T32CA093240 (MH), and K08CA148887 (DK).
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
References
- 1.Eischen CM, Lozano G. The Mdm network and its regulation of p53 activities: a rheostat of cancer risk. Human mutation. 2014;35(6):728–737. doi: 10.1002/humu.22524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.TCGA. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 4.Kyrgiou M, Salanti G, Pavlidis N, Paraskevaidis E, Ioannidis JP. Survival benefits with diverse chemotherapy regimens for ovarian cancer: meta-analysis of multiple treatments. Journal of the National Cancer Institute. 2006;98(22):1655–1663. doi: 10.1093/jnci/djj443. [DOI] [PubMed] [Google Scholar]
- 5.Rose PG, Blessing JA, Mayer AR, Homesley HD. Prolonged oral etoposide as second-line therapy for platinum-resistant and platinum-sensitive ovarian carcinoma: a Gynecologic Oncology Group study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1998;16(2):405–410. doi: 10.1200/JCO.1998.16.2.405. [DOI] [PubMed] [Google Scholar]
- 6.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In Vivo Activation of the p53 Pathway by Small-Molecule Antagonists of MDM2. Science. 2004;303(5659):844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 7.Khoo KH, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nature reviews Drug discovery. 2014;13(3):217–236. doi: 10.1038/nrd4236. [DOI] [PubMed] [Google Scholar]
- 8.Li X, Gilkes D, Li B, Cheng Q, Pernazza D, Lawrence H, et al. Abnormal MDMX degradation in tumor cells due to ARF deficiency. Oncogene. 2012;31(32):3721–3732. doi: 10.1038/onc.2011.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Melo AN, Eischen CM. Protecting the genome from mdm2 and mdmx. Genes Cancer. 2012;3(3–4):283–290. doi: 10.1177/1947601912454139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alt JR, Bouska A, Fernandez MR, Cerny RL, Xiao H, Eischen CM. Mdm2 binds to Nbs1 at sites of DNA damage and regulates double strand break repair. J Biol Chem. 2005;280(19):18771–18781. doi: 10.1074/jbc.M413387200. [DOI] [PubMed] [Google Scholar]
- 11.Bouska A, Lushnikova T, Plaza S, Eischen CM. Mdm2 promotes genetic instability and transformation independent of p53. Mol Cell Biol. 2008;28(15):4862–4874. doi: 10.1128/MCB.01584-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, et al. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 1998;12(15):2424–2433. doi: 10.1101/gad.12.15.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khabele D, Son DS, Parl AK, Goldberg GL, Augenlicht LH, Mariadason JM, et al. Drug-induced inactivation or gene silencing of class I histone deacetylases suppresses ovarian cancer cell growth: implications for therapy. Cancer Biol Ther. 2007;6(5):795–801. doi: 10.4161/cbt.6.5.4007. [DOI] [PubMed] [Google Scholar]
- 14.Carrillo AM, Bouska A, Arrate MP, Eischen CM. Mdmx promotes genomic instability independent of p53 and Mdm2. Oncogene. 2015;34(7):846–856. doi: 10.1038/onc.2014.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Konstantinopoulos PA, Wilson AJ, Saskowski J, Wass E, Khabele D. Suberoylanilide hydroxamic acid (SAHA) enhances olaparib activity by targeting homologous recombination DNA repair in ovarian cancer. Gynecologic oncology. 2014;133(3):599–606. doi: 10.1016/j.ygyno.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Williams BR, Mirzoeva OK, Morgan WF, Lin J, Dunnick W, Petrini JH. A murine model of Nijmegen breakage syndrome. Curr Biol. 2002;12(8):648–653. doi: 10.1016/s0960-9822(02)00763-7. [DOI] [PubMed] [Google Scholar]
- 17.Bekker-Jensen S, Mailand N. Assembly and function of DNA double-strand break repair foci in mammalian cells. DNA Repair (Amst) 2010;9(12):1219–1228. doi: 10.1016/j.dnarep.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 18.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, 3rd, Hurov KE, Luo J, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316(5828):1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 19.O'Connor PM, Jackman J, Bae I, Myers TG, Fan S, Mutoh M, et al. Characterization of the p53 tumor suppressor pathway in cell lines of the National Cancer Institute anticancer drug screen and correlations with the growth-inhibitory potency of 123 anticancer agents. Cancer Res. 1997;57(19):4285–4300. [PubMed] [Google Scholar]
- 20.Muggia F. Platinum compounds 30 years after the introduction of cisplatin: implications for the treatment of ovarian cancer. Gynecologic oncology. 2009;112(1):275–281. doi: 10.1016/j.ygyno.2008.09.034. [DOI] [PubMed] [Google Scholar]
- 21.Li X, Gilkes D, Li B, Cheng Q, Pernazza D, Lawrence H, et al. Abnormal MDMX degradation in tumor cells due to ARF deficiency. Oncogene. 2011 doi: 10.1038/onc.2011.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang P, Lushnikova T, Odvody J, Greiner TC, Jones SN, Eischen CM. Elevated Mdm2 expression induces chromosomal instability and confers a survival and growth advantage to B cells. Oncogene. 2008;27(11):1590–1598. doi: 10.1038/sj.onc.1210788. [DOI] [PubMed] [Google Scholar]
- 23.Li Q, Zhang Y, El-Naggar AK, Xiong S, Yang P, Jackson JG, et al. Therapeutic efficacy of p53 restoration in Mdm2-overexpressing tumors. Molecular Cancer Res. 2014;12(6):901–911. doi: 10.1158/1541-7786.MCR-14-0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Conradt L, Henrich A, Wirth M, Reichert M, Lesina M, Algul H, et al. Mdm2 inhibitors synergize with topoisomerase II inhibitors to induce p53-independent pancreatic cancer cell death. Int J Cancer. 2013;132(10):2248–2257. doi: 10.1002/ijc.27916. [DOI] [PubMed] [Google Scholar]
- 25.Ambrosini G, Sambol EB, Carvajal D, Vassilev LT, Singer S, Schwartz GK. Mouse double minute antagonist Nutlin-3a enhances chemotherapy-induced apoptosis in cancer cells with mutant p53 by activating E2F1. Oncogene. 2007;26(24):3473–3481. doi: 10.1038/sj.onc.1210136. [DOI] [PubMed] [Google Scholar]
- 26.Lau LM, Nugent JK, Zhao X, Irwin MS. HDM2 antagonist Nutlin-3 disrupts p73-HDM2 binding and enhances p73 function. Oncogene. 2008;27(7):997–1003. doi: 10.1038/sj.onc.1210707. [DOI] [PubMed] [Google Scholar]
- 27.Peirce SK, Findley HW. The MDM2 antagonist nutlin-3 sensitizes p53-null neuroblastoma cells to doxorubicin via E2F1 and TAp73. International journal of oncology. 2009;34(5):1395–1402. [PubMed] [Google Scholar]
- 28.Ray RM, Bhattacharya S, Johnson LR. Mdm2 inhibition induces apoptosis in p53 deficient human colon cancer cells by activating p73- and E2F1-mediated expression of PUMA and Siva-1. Apoptosis. 2011;16(1):35–44. doi: 10.1007/s10495-010-0538-0. [DOI] [PubMed] [Google Scholar]
- 29.Urist M, Tanaka T, Poyurovsky MV, Prives C. p73 induction after DNA damage is regulated by checkpoint kinases Chk1 and Chk2. Genes Dev. 2004;18(24):3041–3054. doi: 10.1101/gad.1221004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.