

Abstract
Traumatic brain injury (TBI) is a common cause of disability in childhood, resulting in numerous physical, behavioral, and cognitive sequelae, which can influence development through the lifespan. The mechanisms by which TBI influences normal development and maturation remain largely unknown. Pediatric rodent models of TBI often do not demonstrate the spectrum of motor and cognitive deficits seen in patients. To address this problem, we developed a New Zealand white rabbit model of pediatric TBI that better mimics the neurological injury seen after TBI in children. On postnatal Day 5-7 (P5-7), rabbits were injured by a controlled cortical impact (6-mm impactor tip; 5.5 m/sec, 2-mm depth, 50-msec duration). Rabbits from the same litter served as naïve (no injury) and sham (craniotomy alone) controls. Functional abilities and activity levels were measured 1 and 5 d after injury. Maturation level was monitored daily. We performed cognitive tests during P14-24 and sacrificed the animals at 1, 3, 7, and 21 d after injury to evaluate lesion volume and microglia. TBI kits exhibited delayed achievement of normal developmental milestones. They also demonstrated significant cognitive deficits, with lower percentage of correct alternation rate in the T-maze (n=9-15/group; p<0.001) and less discrimination between novel and old objects (p<0.001). Lesion volume increased from 16% at Day 3 to 30% at Day 7 after injury, indicating ongoing secondary injury. Activated microglia were noted at the injury site and also in white matter regions of the ipsilateral and contralateral hemispheres. The neurologic and histologic changes in this model are comparable to those reported clinically. Thus, this rabbit model provides a novel platform for evaluating neuroprotective therapies in pediatric TBI.
Key words: : cognition, microglia, motor, pediatric traumatic brain injury, rabbit
Introduction
Traumatic brain injury (TBI) in childhood is a major cause of pediatric mortality and commonly leads to lasting morbidity and disability in the survivors.1 Studies have shown that infants and young children with TBI (<4 years of age) exhibit poorer cognitive outcomes than do older children and adults with TBI.2 Therefore, it is critical to understand the pathophysiologic responses of the immature and developing brain after TBI in order to optimize clinical therapeutic modalities. Unfortunately, the lack of appropriate animal models hinders our understanding of the factors that contribute to poor outcomes in the youngest population.3
Cerebral maturation is significantly different among rodents, pigs, cats, nonhuman primates, and humans.4 Rabbits and humans are perinatal brain developers, while rodents are postnatal brain developers.4 The developmental stage of the rat brain at postnatal Day 7 (P7) is considered equivalent to that of a term human infant.5 Primates and guinea pigs are considered “pecocial” with a major part of myelination occurring prenatally.4 The time-courses for cortical and white matter development is different between species and this can influence the response to injury.4 In past years, the rodent has been the major animal model for TBI owing to its relatively low cost, fast breeding cycle, and rapid maturation. However, the lissencephalic rodent brain lacks anatomical similarity to human brain. Although the pediatric rodent TBI models produce consistent cortical injury,6–8 the functional deficits have not been consistently demonstrated in all studies and outcomes have been variable between studies. For example, an immature rat model of diffuse contusive brain injury induced chronic cognitive deficits,9 whereas in immature rat models of severe focal TBI, there was significant variability in extent and persistence of neurologic deficits.10,11 Primates offer the closest anatomic similarity to humans; however, the high cost prevents them from being widely used in experimental TBI models. Large animals, such as pigs, have been used to study childhood neurotrauma because their white matter volume is similar to that of humans.12,13 However, as with primates, high costs make it difficult to screen promising therapies. Although it is very difficult to have an animal model where all developmental processes mimic that in humans, the rabbit model is unique because white matter development, microglial presence in the white matter tracts, and the pattern of brain growth parallels that in humans.14,15 In this study, we have developed a new rabbit model of pediatric TBI of focal contusion that results in neurologic responses and histologic changes similar to that seen in children, which would enable evaluation of novel therapies for treatment of TBI.
Methods
Animals
Pregnant New Zealand rabbits were purchased from Robinson Services Inc. (Mocksville, NC) and arrived at the facility one week before giving birth. All of the kits were delivered naturally and remained with their mother after birth. All animals were housed under ambient conditions (22°C, 50% relative humidity, and a 12-h light/dark cycle), and necessary precautions were taken throughout the study to minimize pain and stress associated with the experimental treatments. Experimental procedures were approved by the Johns Hopkins University Animal Care and Use Committee (ACUC).
TBI surgical procedures
New Zealand rabbits (n=98; 31 male and 67 female) were from 19 independent litters. Rabbits from the same litter were randomly distributed into three groups: naïve group (n=24; 10 male/14 female from 16 independent litters), sham group (n=24; 7 male/17 female from 14 independent litters), and TBI group (n=50; 14 male/36 female from 19 independent litters). On P5-7, rabbits were anesthetized with dexmedetomidine hydrochloride (30 μg/kg, subcutaneously) and placed in a stereotaxic frame. Anesthesia was maintained with 2% inhaled isoflurane. A computer-operated thermal blanket pad and a rectal thermometer allowed maintenance of body temperature within normal limits (∼37°C). Respirations were monitored throughout and there were no episodes of apnea or hypoventilation noted. A midline incision was made to expose the skull, then an 8-mm craniotomy was made on the left hemisphere lateral to the sagittal suture and centered between bregma and lambda. The skull at the craniotomy site was removed without disrupting the underlying dura. A controlled cortical impact (CCI) device was used to injure the exposed cortex. The CCI device used a 6-mm, flat impactor tip and was set at a velocity of 5.5 m/sec, a touching duration of 50–60 msec, and a depth of 2 mm. These injury parameters were based on previously published data by our group in postnatal rats.7,16,17 Initial pilot studies using 1.5 mm, 2 mm, and 2.5 mm depths identified that the extent of the lesion using a depth of 2 mm in rabbits was comparable to that of our immature rat model with 1.5 mm. This resulted in a moderate to severe lesion without significant mortality. After the injury, the skull was replaced over the injury site and secured with dental cement. The skin was then sutured together and the animals were placed in an animal incubator to recover. The sham group underwent craniotomy without TBI. All kits were returned to their mother and littermates after their recovery from anesthesia. The naïve group did not receive any intervention. We closely monitored all animals post-operatively with weight and health surveillance recordings, as per ACUC guidelines.
Immunohistochemistry
Cresyl violet staining and lesion volume estimation
At 1, 3, 7, and 21 d after injury, the rabbits were anesthetized and transcardially perfused with saline followed by 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS). The brains were removed, post-fixed in 4% PFA overnight, and cryoprotected in 30% sucrose. Coronal brain sections (40 μm) were cut on a frozen sliding microtome and stored in anti-freeze solution at −20°C until use. We mounted sections (1:6 series, 240 μm apart) on glass slides and stained them with cresyl violet (Sigma, St. Louis, MO) to identify degenerating neurons and confirm the core impact injury of the TBI model. Images were acquired by Intellicam software (Intellicam LLC, Ashland, VA) and analyzed with ImageJ software (NIH, Bethesda, MD). The ipsilateral and contralateral hemispheres on each section (12–15 sections, 1:6 series) were outlined, and the areas were measured and standardized with a grid spacing of 1 mm. The volume of the hemisphere was calculated as Σarea×240. The percentage of lesion volume was estimated as (1 – ipsilateral hemisphere/contralateral hemisphere)×100%.
The contralateral lateral ventricle of the contralateral hemisphere on each section (12–15 sections, 1:6 series) was outlined, and the areas were measured and standardized with a grid spacing of 1 mm. The volume of the contralateral lateral ventricle was calculated as Σarea×240. The ventricular enlargement following TBI was determined by measuring the volume of the contralateral lateral ventricle, compared with the contralateral hemisphere. This was expressed as the percentage of lateral ventricle volume.
Ionized calcium-binding adapter molecule 1 (Iba1)and translocator protein (TSPO) staining
At 1, 3, 7, and 21 d after injury, the rabbits were anesthetized and transcardially perfused with saline followed by 4% PFA in PBS. The brains were removed, post-fixed in 4% PFA overnight, and cryoprotected in 30% sucrose. Coronal sections (40 μm, 1:6 series) were incubated in 0.3% hydrogen peroxide solution and blocked in 3% normal goat serum in 0.1 M PBS. Sections were then incubated overnight at 4°C with goat-anti-IBA1 antibody (1:500, Abcam, Cambridge, MA) or goat anti-TSPO antibody (1:100, Thermo Scientific, Waltham, MA). Sections were subsequently washed and incubated with biotinylated secondary antibody (1:250; Vector Laboratories, Burlingame, CA) for 4 h at room temperature. Next, the sections were incubated for 2 h in avidin–biotin substrate (ABC kit, Vector Laboratories). All sections were then incubated for 1 min in 3,3'-diaminobenzidine solution (Vector Laboratories). Sections were dehydrated in ethanol and Histo-Clear II (Fisher Scientific, Pittsburg, PA) before cover slips were applied with mounting medium. Sections were examined with a Leica DM2500 microscope (Leica Microsystems Inc., Bannockburn, IL).
Cell count
The staining of microglia was performed on 1:6 series extending from the level of the bregma. Microglia cells in three representative white matter regions, the corpus callosum, corona radiata, and internal capsule at the injured area were counted as previously described.18 On each section, four images were randomly captured at 40× magnification from each white matter region. The number of “activated” (bushy and round) and “resting” (ramified) microglia were counted as previously described.19 The ratio of the “activated cells” (bushy and round) to the total number of microglial cells (ramified+bushy and round) was calculated and compared for the naïve, sham, and TBI groups.
Real-time polymerase chain reaction (PCR)
Kits from sham and TBI groups were sacrificed at 7 d after surgery along with age-matched naïve controls. The brains were quickly harvested and stored in RNAlater solution (Life Technologies, Grand Island, NY). The tissues around the injury site (or matched region in the naïve and sham) were micro-dissected and the total RNA was extracted using TRIZOL (Life Technologies) according to the manufacturer instructions. RNA samples were quantified using the Nanodrop ND-1000 Spectrophotometer. The single-stranded complementary DNA (cDNA) was first reverse transcribed from the total RNA samples using the High Capacity cDNA Reverse Transcription Kit with RNase inhibitor (Life Technologies). The real-time PCR was performed with SYBR Green PCR Master Mix (Life Technologies) using Fast 7500 Real-time PCR systems (Life Technologies). Amplification conditions included 30 min at 48°C, 10 min at 95°C, 40 cycles at 95°C for 15 s and 60°C for 1 min. Primers were custom designed and ordered from Integrated DNA Technologies (Coralville, Iowa). GAPDH forward 5′TGACGACATCAAGAAGGTGGTG3′; GAPDH reverse 5′GAAGGTGGAGGAGTGGGTGTC3′; TSPO forward 5′ GCTCCCTCTGTAACTGCTTT3′; TSPO reverse 5′ CATAGTGGGAGGCCATGAAG 3′. Comparative Ct method was used to assess differential gene expressions. The gene expression levels for each sample were normalized to the expression level of the housekeeping gene encoding glyceraldehydes 3-phosphate dehydrogenase (GAPDH) within a given sample (ΔCt); the differences between sham, compared with the naïve group or TBI, compared with the naïve group, were used to determine the ΔΔCt. The 2-ΔΔCt gave the relative fold changes in gene expression.
Neurobehavior
Neurobehavioral tests were carried out 1 d and 5 d after TBI. The behavioral assessments were conducted blinded to the injury group. We videotaped each animal for 10 min and scored them on a scale of 0–3 (0, worst; 3, best) for posture, movements of head and limbs, and duration and intensity of movements on a flat surface as previously described for rabbits.20 We also fed the kits rabbit milk with a syringe attached to an artificial nipple and assessed suck and swallow and head turn during feeding on a scale of 0–3 (worst-best).20,21 Olfaction was tested by the intensity of aversive response to a cotton swab soaked with 100% ethanol.21,22 We assessed the righting reflex by placing the kits on their backs and recording the number of times they turned from the supine to the prone position in 10 tries. The hops and steps were scaled at 0–4 (0, no step or hop; 4, ≥10 steps or 4 hops).23 Pain sensitivity was tested by recording the time and intensity of responses to a pinch of the hind feet. Responses were scored on a 0–3 scale (from no response to a strong response, such as vigorously kicking the feet and making vocalizations).22
Neurodevelopmental tests
Developmental maturation was monitored daily from P1 until the day of sacrifice. The developmental measurements were modified from previously published studies.24 In brief, we recorded the first day the kits achieved developmental milestones such as eye opening, body elevation (simultaneous fore- and hind-limb elevation), righting reflex, mature gait (walking in a straight line with lifting up of limbs above the ground and lack of circling), cliff avoidance (backward propulsion to avoid the edge of an elevated surface), head elevation for more than 1 min, hopping (for more than half of the duration of locomotion), and locomotion without falling.
Open field
The locomotor activity of the rabbit kits was assessed with the Opto-Varimex-4 Auto Track System (Columbus Instruments, Columbus, OH). The Opto-Varimex consists of an open chamber (23.5×23.5×21 cm) with infrared optical beam sensors composed of a detector and emitter on either side of the cage. The Auto Track system senses motion in the X and Y direction with a grid of infrared beams. Vertical motion (like head movement) is detected by a second set of photocells placed above the kits. Interruptions in the X and Y beams provide the location and distance traveled by the kits. The apparatus was placed in the test room, which has the same conditions as the animal house. The open field test was performed at 8 a.m. in the morning and maintained on all test days. Naïve, sham, and TBI kits were placed in the apparatus before surgery and 1 and 5 d after TBI surgery; spontaneous activity was recorded for 10 min. The animals were not disturbed during recording. After the recording, the rabbit kits were returned to their cages. The distance traveled by the kit (cm), resting time (sec), large ambulatory movement (sec), new beam breaks, vertical sensor counts, and speed (calculated as distance traveled/large ambulatory movement) were measured and compared.
T-maze spontaneous alternation
The T-maze spontaneous alternation test was modified from previously published methods for rodents.25 Naïve, sham, and TBI rabbit kits ages P14 to P17 (corresponds to ∼4 d after eye opening) were tested for three consecutive days, four trials per animal per day. The behavioral assessments were conducted blinded to injury group. The first two trials were in the morning and the second two trials in the afternoon, with an interval of 4–5 h. Rabbits were handled daily and habituated in the test room 3 h per day for five consecutive days before the test. For each trial, the rabbits were placed in the start area in an in-house–made T-maze (goal arms, 50×10×30 cm; start arm, 50×16×30 cm) and confined for 5 sec before they were allowed to choose a goal arm. The rabbits were considered to have made a choice when they placed all four paws into one arm. Thereafter, the rabbits were allowed to explore the chosen goal arm for 20 sec. Then, they were removed from the goal arm and placed back in the cage before being given the next trial. The inter-trial interval was 5 min. The number of alternations divided by the total number of choices was calculated and averaged.
Novel object recognition
Retention, or intact memory, was assessed by the novel object recognition test for three consecutive days on P19-21. By this age, the rabbits consistently demonstrate curiosity and exploration of the environment. The protocols were modified from previous publications for rodents.26 The behavioral assessments were conducted blinded to injury group. Rabbits were handled daily and habituated in the test room for 3 h per day for five consecutive days before the test. For habituation, the animals were placed in the open field (50×50×40 cm) box for 30 min without any objects present. The test was composed of two trials. During the first trial (training trial), rabbits were initially made to face the wall and then released to the open field to explore two identical objects for 5 min. The objects were placed at equal distances to the walls. During the second (testing) phase, one of the previously exposed “old” objects was replaced with a new “novel” object. The rabbits were allowed to explore these two objects for 5 min. The inter-trial interval was 60 min, during which the animals were placed back in their cages. On each day, we used a completely new set of novel and old objects, and there was no overlap of the objects during the 3 d of testing. The discrimination index for the second trial was used to analyze the cognitive outcomes. Discrimination index=time spent exploring the novel object/(time spent exploring the old object+time spent exploring the novel object)×100%.
Statistical analysis
All data are presented as mean values±standard deviation. One-way analysis of variance (ANOVA) or two-way ANOVA was used for group comparisons, followed by subsequent pairwise comparisons with the Bonferroni multiple comparison post hoc test. Statistical significance was set at p<0.05 for all analyses. For analysis of cliff avoidance that had a “yes” or “no” score, Fisher's exact test was used.
Results
Mortality rate
In the TBI group, six (12%) of 50 kits died post- injury (6 h [one], 24 h [two], 3 d [two], and 14 d [one]). Of those six, four kits had seizures and died at 6 h (one), 24 h (two), and 3 d (one) post-injury. None of the sham kits had seizures after surgery. No kits in the naïve group died or had any complication.
Body weight
The initial body weights of each group at the time of surgery was similar (F=1.73, p=0.19; Fig. 1A) and there was no significant difference between males and females in any of the groups (F=1.86, p=0.12). At 1 d post-injury, the kits in the TBI group had gained significantly less weight than had the naïve and sham animals (F=3.42, p=0.04); however, there was no significant difference in the weight gain at 5 d post-injury (F=1.14, p=0.33; Fig. 1B). There was no significant difference in the net weight gain between males and females at 1 or 5 d post-injury in any of the groups (p>0.05).
FIG. 1.

Weight gain, lesion volumes and lateral ventricle volume. (A) The body weight of the kits at naïve (n=15), sham (n=13) and traumatic brain injury (TBI; n=23) groups were similar before the injury. (B) Net weight gain at 1 d post injury and 5 d post-injury. (C) Lesion volumes at different time points after TBI. Rabbits were sacrificed 1, 3, 7, and 21 d post-injury, and coronal brain sections (40 μm) were stained with cresyl violet and areas of the ipsilateral and contralateral hemispheres measured with ImageJ software. The percentage of lesion volume was calculated as: % of lesion volume=(1 – ipsilateral hemisphere/contralateral hemisphere)×100%. (D) Volume of the ipsilateral and contralateral hemispheres in the naïve, sham and TBI groups 21 d post-injury. Compared with the naïve and sham, the volume of ipsilateral hemisphere was significantly decreased in TBI kits. (E) Percentage of lateral ventricle volume (contralateral hemisphere) at 21 d post-injury. Compared with that in the naïve and sham groups, the lateral ventricle in the TBI kits was significantly enlarged. The data are expressed as individual values and median to show the distribution. *p<0.05, ***p<0.001.
Lesion volume
The average weight of P5-7 rabbit brains was 2.03±0.11 g (n=4 brains). The volume of the brain was calculated as weight/density. The volume of the rabbit brain (without olfactory bulbs and cerebellum) at P5-7 was approximately 1.99±0.10 cm3 (assuming a density of 1.05 g/mL27). The volume of the piston displacement=2 mm×π×(3 mm)2=56.52 mm3=0.06 cm3. Thus, the volume of the piston displacement as a percent of the total brain volume was approximately 3.02%. Lesion volumes were calculated on Days 1 (n=8; 3 male/5 female), 3 (n=8; 2 male, 6 female), 7 (n=8; 5 male, 3 female), and 21 (n=10; 3 male/7 female) post-injury in TBI kits. Lesion volume was 11.5% at 1 d and 15.9% at 3 d post-injury. Brain tissue damage was localized mainly to the frontoparietal cortex, where the primary impact occurred, and the hippocampus was present in most of the kits at these early time-points (Day 1 and 3 post-injury). At 7 d post-injury, the lesion volume had increased to 30.5%. At 21 d post-injury, the lesion volume was 30.9%, suggesting that that injury progression had plateaued at 7 d post-injury (Fig. 1C). Moreover, we also directly compared the ipsilateral and contralateral volumes across groups at 21 d post-injury. We found that the volume of the ipsilateral hemisphere significantly decreased in the TBI group (n=10), compared with age-matched naïve (n=10) and sham (n=7) animals (Fig. 1D). A significant increase in the contralateral ventricle size at 21 d post-injury, depicted as “percent lateral ventricle volume” in the TBI kits, was noted, when compared with the naïve and sham groups (F=9.5, p<0.0001; Fig. 1E).
Microglial activation after TBI
We also evaluated the microglial response at 1 d (n=8), 3 d (n=8), 7 d (n=8), and 21 d (n=10) post-injury in TBI kits. Staining for IBA1 (a microglial marker) revealed a robust increase in microglial activation in the white matter area, including corpus callosum, angle of ventricle, corona radiate, and internal capsule at 1 d post-injury. The staining became more prominent at 3 d post-injury both ipsilateral and contralateral to the injury. In the TBI kits, the microglial morphology changed to an amoeboid shape, with hypertrophic soma and thickened, retracted processes (Fig. 2). The intense microglial activation was still present in the white matter tracts of the ipsilateral hemisphere at 7 d post-injury and was persistent at 21 d post-injury (Fig. 2). We further investigated the microglial activation at 7 d post-injury. We found that the microglial activation was visible not only at the injury site but also in white matter regions (periventricular region and internal capsule) of the ipsilateral hemisphere beyond the injury site (Fig. 3A). Moreover, the contralateral side also exhibited increased microglial activation in the white matter tracts but to a lesser extent (Fig. 3A). We also performed microglial cell count in white matter regions at the injury site in the ipsilateral hemisphere. We found that the ratio of activated microglia was significantly higher in the TBI kits (n=6), compared with naïve (n=4) and sham kits (n=4), in the corpus callosum (F=116.4, p<0.0001), corona radiata (F=47.3, p<0.0001) and internal capsule (F=117.5, p<0.0001; Fig. 3B).
FIG. 2.
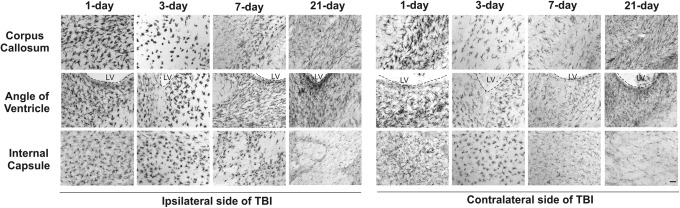
Microglial activation in white matter 1, 3, 7, and 21 d after traumatic brain injury (TBI). Ionized calcium-binding adapter molecule 1 (IBA1) staining of the ipsilateral and contralateral hemispheres revealed microglial activation at 1 d post-injury. The IBA1 staining became more prominent at 3 d post-injury in both the ipsilateral and contralateral hemispheres. The microglial activation in the ipsilateral hemisphere was still apparent at 7 and 21 d post-injury. LV, lateral ventricle. Scale bar: 50 μm.
FIG. 3.

Microglial activation 7 d after traumatic brain injury (TBI). (A) Ionized calcium-binding adapter molecule 1 staining revealed that ipsilateral microglial activation remained elevated at 7 d post-injury compared with that in naïve and sham kits. The left panel represents the brain areas that more rostral to the injury site. The right panel represents the brain area that within the injury site (anatomical areas indicated in the rabbit brain schematic). The microglial activation was present not only at the injury site, but also extended beyond the injury site. Scale bar: 20 μm. (B) The ratio of activated microglia was significantly increased in the corpus callosum (CC), corona radiate (CR), and internal capsule (IC) at the injury site of ipsilateral hemisphere. ***p<0.001.
TBI increases TSPO expression
Translocator protein (TSPO) is an 18 kDa protein and a glia marker that is expressed at low levels in healthy brain.28 It is markedly upregulated in response to injury and inflammation and is mainly expressed in activated microglia. Therefore, TSPO overexpression reflects microglial cell activation and proliferation.29 We measured the TSPO protein expression with TSPO antibody staining and TSPO messenger RNA (mRNA) expression with real-time PCR at 7 d post-injury. We found that the TSPO staining increased in the injury site and white matter, including corpus callosum, corona radiata, and internal capsule, in TBI kits (n=8), compared with that in naïve (n=8) and sham (n=3) kits (Fig. 4A). Moreover, TSPO mRNA levels were significantly increased in the ipsilateral site of the injury in the TBI kits (n=7), compared with naïve (n=5) and sham (n=5; F=4.4, p<0.01; Fig. 4B).
FIG. 4.

Translocator protein (TSPO) expression 7 d after traumatic brain injury (TBI). TSPO expression was significantly elevated in TBI kits in the ipsilateral hemisphere when compared to that of naïve and sham kits. (A) TSPO staining in the ipsilateral hemisphere was more prominent in the TBI kits. Scale bar: 20 μm. (B) TSPO messenger RNA level was significantly increased in the ipsilateral hemisphere in TBI kits. *p<0.05.
TBI causes short-term locomotion deficits and delayed maturation of milestones
We measured the overall neurobehavioral function before TBI and 1 and 5 d post-injury. We found that TBI induced significant behavioral deficits, including impaired sucking and swallowing ability and decreased head and limb movements, 1 d post-injury. Kits had fully recovered from these deficits at 5 d post-injury (Fig. 5). The results from the open field test indicated that there were no significant changes in the locomotion, including travel distance, rest time, speed, and vertical movements. Moreover, TBI kits showed a significant delay in achievement of milestones, such as hopping, head elevation, normal gait, and ability to walk without falling. Moreover, five (26%) of 19 kits lost the previously achieved milestone of cliff-avoidance post-injury (Table 1). There was no statistically significant difference in the posture, righting reflex, olfaction, and pain sensitivity tests.
FIG. 5.

Neurobehavioral function was tested before traumatic brain injury (TBI) and on Days 1 and 5 post-injury. The scores of suck and swallow, head movement, and hops were significantly lower in TBI kits than in naïve and sham kits 1 d after TBI. There was no difference between groups at 5 d post-injury. *p<0.05, ***p<0.001 vs. naïve and sham.
Table 1.
Maturation Onset (Days) in Control, Sham, and TBI Kits
| Developmental milestone | Control (n=15) | Sham (n=13) | TBI (n=19) |
|---|---|---|---|
| Eye opening | 11 (10, 12) | 11(10, 12) | 12 (11, 14)NS |
| Body elevation | 9 (7, 9) | 9 (8.5, 9.5) | 10 (9, 11) NS |
| Righting | 1 (1, 1) | 1 (1, 1) | 1 (1, 1) NS |
| Normal gait | 8 (7, 9) | 8 (8, 8.5) | 9 (8, 10)** |
| Δ Cliff avoidance | 1 | 1 | Lost after TBI in 5/19 animals * |
| Head elevation | 9 (8, 9) | 9 (8.5, 9) | 10 (9, 10) # |
| Hopping | 11 (10, 12) | 10 (10, 12.5) | 14 (12, 14)*** |
| Cessation of falling | 7 (6, 7) | 7 (7, 8) | 9 (8, 9) ## |
p<0.05, **p<0.01, ***p<0.001 control and sham vs. TBI. #p<0.05, ##p<0.01, control vs. TBI.
Data were expressed as median (interquartile range). Body elevation, simultaneous fore- and hind-limb elevation; cliff avoidance, backward propulsion to avoid the edge of an elevated surface; head elevation, head elevated for more than 1 min; hopping, hopping for more than half of the duration of locomotion; cessation of falling, locomotion without falling.
Δ: there was a significant difference between the three groups for loss of the milestone for cliff avoidance (p value for this by Fisher's exact test was 0.02). The p values for other milestones were analyzed by one-way analysis of variance (non-parametric) and Bonferroni's multiple comparison test.
TBI, traumatic brain injury; NS, not significant.
Cognitive deficits
We used spontaneous alternation in a T-maze and the novel object recognition test to assess cognitive deficits in the TBI kits. We found that the percentage of correct alternation was significantly lower in TBI kits (n=16) than in naïve (n=14) and sham (n=9) kits (p<0.001; Fig. 6A). Cognitive deficits did not differ significantly between the male (n=6) and female (n=10) kits in the TBI group (male, 33.3±10.5%; female, 36.7±13.2%; t(14)=0.5; p=0.6). In addition, TBI kits (n=12) spent significantly less time exploring the novel object than did the naïve (n=11) and sham (n=8) kits (p<0.001; Fig. 6B). There was no significant difference between male (n=4) and female (n=8) TBI kits (male, 53.9±15.7%; female, 57.8±17.2%; t(10)=0.4; p=0.7).
FIG. 6.

Traumatic brain injury (TBI) causes cognitive deficits in TBI animals. (A) Spontaneous alternation in the T-maze was tested at postnatal Days 14–17 (P14-17: after eye opening). Compared with naïve and sham kits, TBI kits showed a significant decrease in the percentage of spontaneous alternation. (B) The novel object recognition test was administered for three consecutive days at P19-21. TBI kits spent significantly less time exploring the novel object than did naïve and sham kits. ***p<0.001 vs. naïve and sham.
Discussion
Rabbit model of pediatric brain injury
In this study, we have developed a rabbit model of pediatric TBI and have characterized the neurologic deficits and pathophysiologic changes resulting from a focal contusion in immature rabbits at P5-7. New Zealand white rabbits (P5-7) were chosen because their patterns of brain development and growth parallel those of humans.30,31 Myelination in rabbits starts in the perinatal period, with rapid changes in immature oligodendrocyte density occurring between embryonic day 29 (E29) and P5. Peak myelination starts around P5, reaching 70–80% of adult levels by the third postnatal week.32,33 Microglial presence in the white matter tracts parallels that of oligodendrocyte maturation and peak myelination, extending from before birth to the postnatal period, similar to that seen in humans.14,15 In humans, myelination of sensory and motor fibers starts at birth and continues during the first year of life,34,35 with persistence of microglia in the white matter tracts up to two years of age, which corresponds to the period of peak myelination.36 Therefore, the microglial response seen after injury in relation to development would mimic that seen in humans.
Rationale for timing of injury and outcome evaluation
We have chosen P5-7 as the time of injury, which corresponds to a period of active myelination along with microglial presence.15,37 Thus, an injury at this time will enable us to evaluate the effect of the injury on microglial activation and white matter development. Moreover, at this time-point, the major cortical layers in the rabbits are already formed, but cortical development (e.g., visual and auditory) is not completed.38–41 At P5-7, the rabbits have achieved some of the major developmental milestones, such as righting reflex and cliff avoidance; however, the animals have not achieved other important developmental milestones, such as eye opening, head and body elevation, and hopping (Table 1). Therefore, we will be able to measure the impact of the injury on the cortical development and assess the achievement and/or lose of milestones after injury.
We have chosen P14-21 as the time of cognitive evaluation, when development of the auditory cortex,41 visual cortex,39 primary somatosensory cortex,38 and cerebellum42 are closer to that of adult levels. In addition, the rabbits have achieved all the developmental milestones at this age; thus, we will be able to evaluate the impact of the injury on cognition and learning.
Neurobehavioral and cognitive tests
In this model the most robust neurobehavioral changes were observed in the delayed achievement of milestones related to gait and locomotion, and in the cognitive tests (T-maze and novel object recognition). Normal neonatal rabbits have a typical circular movement, walk with their body very close to the ground and have a tendency to fall when walking in a straight line. This locomotion pattern changes to a more mature gait, with predominantly hops, cessation of falls, and body elevation, by about 8 d of age. We observed significant delays in the achievement of these normal developmental milestones. Moreover, five of 19 TBI kits lost cliff-avoidance, an important milestone that is present in majority of the rabbits by Day 1 of life gestation day 32 (G32). We observed significant decreases in suck and swallow, head movement, and number of hops at 1 d post- injury. However, these short-term changes may be related to cerebral edema from the injury, which may explain them returning to normal by 5 d post-injury. The TBI kits also exhibited significant cognitive deficits, with poor performance in the T-maze and novel object recognition tests (p<0.001). Short-term neurobehavioral tests that are specific for juvenile rabbits such as suck and swallow, head movement, and number of hops, may be reliable for assessing an early response to treatment in the immediate post-injury period. The tests for cognition, such as the T-maze and novel object recognition, will be helpful for assessing delayed efficacies. Delay in achievement of normal motor milestones which is an important parameter to be assessed in injuries to the developing brain, also will be an appropriate outcome measure for therapeutic interventions.
Lesion volume and microglial activation
In this model, the volume of the piston displacement as a percent of the total brain volume was approximately 3.02% in P5-7 rabbits. The controlled cortical impact (6-mm metal impactor tip, a velocity of 5.5±0.3 m/sec, and a duration of 50 msec) with a depth of 2.0 mm caused a lesion volume of ∼30% at 7 d post-injury. To determine if the extent of injury induced by an impact is greater in this rabbit model, we compared the volume of piston displacement and lesion volume with a standard, pediatric rat TBI model that has been previously characterized by members of our group using the same CCI instrument and operator (M.S.).43 In the P16-17 immature rats, volume of piston displacement as a percent of total brain volume was approximately 4.3% (brain volume at this age ∼0.93 cm3; volume of the piston displacement=1.5 mm×π×(3 mm)2=42.39 mm3=0.04 cm3). This is 1.4 times greater than in the rabbit model but resulted in a lesion volume of only ∼10% at 7 d post-injury in the rat, which is a third of the lesion volume seen in the rabbit.43 Compared with rodents, the white matter development of rabbits and microglial presence and distribution in the brain is closer to that in humans,14 which may increase the vulnerability of the developing rabbit brain, resulting in a more severe secondary injury as seen clinically.
In this study, we found that lesion volume increased from 16% at 3 d post-injury to 30% at 7 d. In addition, we also observed enlarged ventricles in the contralateral hemisphere. These results are consistent with the clinical neuroimaging findings from patients injured at very young ages.44–46 The progressive, diffuse tissue loss and brain atrophy indicate ongoing secondary injury that may be related to ongoing microglia activation. We found that TBI causes intensive microglial activation and TSPO expression not only at the injury site but also in white matter regions (periventricular region and internal capsule) far beyond the injury site in both ipsilateral and contralateral hemispheres. This diffuse injury pattern is similar to the neuropathologic findings seen clinically.47 Moreover, the intense microglial activation lasts for up to 21 d post-injury. This exaggerated microglial activation in the white matter may have a key role in the pathogenesis of neuronal damage and developmental deficits. Inflammation in TBI also may have an important function in recovery and repair, as both pro- and anti-inflammatory cytokines such as interleukin (IL)-1, tumor necrosis factor-α, IL-10, IL-8, and MCP1 are altered in the presence of TBI.48,49 Although a microglial response is necessary for removal of necrotic tissues and neuronal repair, prolonged microglial activation can potentially cause detrimental and neurotoxic effects.50 Microglial activation has been imaged in vivo using TSPO ligands as PET imaging biomarkers both clinically51 and in animal models52 to determine the extent of microglial activation after injury and to predict severity of injury. 53We have previously shown in our rabbit model of cerebral palsy that TSPO binding by PET imaging increases with worsening motor deficits.53,54 A rabbit pediatric TBI model provides the additional advantage of enabling high resolution imaging due to the larger size of the brain.
In the developing human brain, amoeboid microglia are present in the white matter tracts from the late second trimester onward. They decrease in density in the postnatal period but they remain in the white matter tracts up to two years of age and eventually migrating to the cortex by adulthood.36 During development, activated microglia render a supportive role in myelinogenesis and axonogenesis by stimulating synthesis of myelin basic protein and the induction of laminin, an extracellular matrix molecule that enhances neurite outgrowth.36,55 Microglia can exist in two activation states, M1 (classical activation, or pro-inflammatory state) and M2 (alternative activation, or anti-inflammatory form responsible for clearance of debris, repair, and regeneration).56,57 In the immature brain, microglial cells have a more amoeboid structure and express surface markers for the M2 phenotype.58 However, the increased presence of microglia during development may not only increase the vulnerability of the immature brain to diverse brain insults but may also interfere with repair and regeneration. This may partially explain the extensive secondary injury leading to larger lesion volumes seen in this model.
Comparison with other models and translational relevance
Although some acute studies have been carried out with CCI in young piglets,59 the majority of the pediatric studies of CCI have used immature rodents,60–62 which often do not consistently demonstrate the spectrum of cognitive deficits seen in pediatric TBI patients. Deficits in working memory and new learning have been shown in children after TBI 63 and in rodents.64 Our data indicate that TBI at P5-7 causes significant cognitive deficits at P22-26 in the rabbit, suggesting that pediatric TBI impaired cognitive function. Moreover, the site of injury is critical for the outcomes of TBI. In an immature rodent CCI model, a unilateral frontal cortex injury induced significant motor deficits but no significant impairment of social behavior or spatial memory performance.65 Midline brain injury in immature rats led to diffuse neurodegeneration in both hemispheres that was felt to be responsible for the sustained cognitive deficits observed in these animals.66 In our model, we used unilateral injury to the parietal cortex, which led to significant cognitive deficits and delay in developmental maturation. These findings are consistent with other rodent models with parietal cortex injury.67,68 It is also possible that the diffuse bilateral neuroinflammatory response seen in the immature rabbit following a focal contusion leads to a more diffuse injury, which could explain the robust cognitive deficits seen in the rabbit model. In our future studies, we will alter the injury site and monitor the change in motor function, social behavior and cognition.
Patient outcomes are variable following severe TBI.69 Studies show that factors including mitochondrial polymorphisms,70 aromatase genetic variation71 and glucose variability72 may impact patient outcomes after TBI. Investigators have reported an interaction of sex and age on lesion volume in the piglet CCI model,73 as well as gender differences in learning and memory after pediatric traumatic brain injury in humans.74 Some of the variability in our model may be related to individual differences between different litters. Although we did not find a significant difference between male and female kits in lesion volume or in the T-maze or novel object recognition test, it is possible that these differences may become more obvious with greater animal numbers. It has been shown in the rodent pediatric TBI model that certain deficits, such as deficit in social recognition or memory, are only fully manifested after the associated skills reach maturity,64 whereas changes in certain behavioral phenotypes, such as locomotor activity and social investigation, are age-dependent but also injury-independent.65 Because rabbits are larger and have more complex behavior and social interactions, this model will allow us to use more advanced physiologic and neurologic monitoring in future studies to measure injury-related and/or age-related changes in the behavioral phenotype. The ability to use these techniques will help in better evaluation of responses to therapy increasing the translational relevance of this model. Clarification of the time-dependent emergence of abnormal behavioral patterns will provide valuable information for screening potential therapeutics in pediatric TBI. Future work will focus on sex differences after injury and evaluation of delayed, long-term neurobehavioral outcomes.
Study limitations
The P5-7 rabbit is closest to infants and toddlers in humans. TBI in approximately 30% of children <2 years old is secondary to non-accidental trauma.75 This rabbit model mimics focal contusion but not necessarily the diffuse axonal injury component of inflicted TBI. This model potentially mimics severe brain injury, such as those caused by road traffic accidents, which has a mortality rate of ∼40% in children <2 years of age.76 In our future studies, we plan to study the effect of CCI in rabbits of older ages. We also plan to study the weight-drop technique in P5-7 rabbits to assess the diffuse axonal injury aspects in the rabbit.
In conclusion, this pediatric TBI model produces neurologic and neuroinflammatory responses that have similarities to those observed clinically in children. Therefore, this model should provide a valuable tool for preclinical screening of therapeutic interventions for pediatric TBI.
Acknowledgments
This work was funded in part by R01HD069562, NICHD, NIH; by the Department of Anesthesiology and Critical Care Medicine, and the Brain Science Institute, Johns Hopkins University School of Medicine.
Author Disclosure Statement
No competing financial interests exist.
Reference
- 1.Morrison G., Fraser D.D., and Cepinskas G. (2013). Mechanisms and consequences of acquired brain injury during development. Pathophysiology 20, 49–57 [DOI] [PubMed] [Google Scholar]
- 2.Di Battista A., Soo C., Catroppa C., and Anderson V. (2012). Quality of life in children and adolescents post-TBI: a systematic review and meta-analysis. J. Neurotrauma 29, 1717–1727 [DOI] [PubMed] [Google Scholar]
- 3.Prins M.L. and Hovda D.A. (2003). Developing experimental models to address traumatic brain injury in children. J. Neurotrauma 20, 123–137 [DOI] [PubMed] [Google Scholar]
- 4.Clancy B., Finlay B.L., Darlington R.B., and Anand K.J. (2007). Extrapolating brain development from experimental species to humans. Neurotoxicology 28, 931–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dobbing J. and Sands J. (1973). Quantitative growth and development of human brain. Arch. Dis. Child 48, 757–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fan P., Yamauchi T., Noble L.J., and Ferriero D.M. (2003). Age-dependent differences in glutathione peroxidase activity after traumatic brain injury. J. Neurotrauma 20, 437–445 [DOI] [PubMed] [Google Scholar]
- 7.Robertson C.L., Saraswati M., Scafidi S., Fiskum G., Casey P., and McKenna M.C. (2013). Cerebral glucose metabolism in an immature rat model of pediatric traumatic brain injury. J. Neurotrauma 30, 2066–2072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dapul H.R., Park J., Zhang J., Lee C., DanEshmand A., Lok J., Ayata C., Gray T., Scalzo A., Qiu J., Lo E.H., and Whalen M.J. (2013). Concussive injury before or after controlled cortical impact exacerbates histopathology and functional outcome in a mixed traumatic brain injury model in mice. J. Neurotrauma 30, 382–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huh J.W. and Raghupathi R. (2007). Chronic cognitive deficits and long-term histopathological alterations following contusive brain injury in the immature rat. J. Neurotrauma 24, 1460–1474 [DOI] [PubMed] [Google Scholar]
- 10.Prins M.L. and Hovda D.A. (1998). Traumatic brain injury in the developing rat: effects of maturation on Morris water maze acquisition. J. Neurotrauma 15, 799–811 [DOI] [PubMed] [Google Scholar]
- 11.Adelson P.D., Fellows-Mayle W., Kochanek P.M., and Dixon C.E. (2013). Morris water maze function and histologic characterization of two age-at-injury experimental models of controlled cortical impact in the immature rat. Childs Nerv. Syst. 29, 43–53 [DOI] [PubMed] [Google Scholar]
- 12.Raghupathi R. and Margulies S.S. (2002). Traumatic axonal injury after closed head injury in the neonatal pig. J. Neurotrauma 19, 843–853 [DOI] [PubMed] [Google Scholar]
- 13.Armstead W.M. and Kurth C.D. (1994). Different cerebral hemodynamic responses following fluid percussion brain injury in the newborn and juvenile pig. J. Neurotrauma 11, 487–497 [DOI] [PubMed] [Google Scholar]
- 14.Saadani-Makki F., Kannan S., Lu X., Janisse J., Dawe E., Edwin S., Romero R., and Chugani D. (2008). Intrauterine administration of endotoxin leads to motor deficits in a rabbit model: a link between prenatal infection and cerebral palsy. Am. J. Obstet. Gynecol. 199, 651 e651–e657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bass W.T., Singer G.A., and Liuzzi F.J. (1998). Transient lectin binding by white matter tract border zone microglia in the foetal rabbit brain. Histochem. J. 30, 657–666 [DOI] [PubMed] [Google Scholar]
- 16.Casey P.A., McKenna M.C., Fiskum G., Saraswati M., and Robertson C.L. (2008). Early and sustained alterations in cerebral metabolism after traumatic brain injury in immature rats. J. Neurotrauma 25, 603–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kilbaugh T.J., Bhandare S., Lorom D.H., Saraswati M., Robertson C.L., and Margulies S.S. (2011). Cyclosporin A preserves mitochondrial function after traumatic brain injury in the immature rat and piglet. J. Neurotrauma 28, 76–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saadani-Makki F., Kannan S., Makki M., Muzik O., Janisse J., Romero R., and Chugani D. (2009). Intrauterine endotoxin administration leads to white matter diffusivity changes in newborn rabbits. J. Child Neurol. 24, 1179–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kannan S., Saadani-Makki F., Muzik O., Chakraborty P., Mangner T.J., Janisse J., Romero R., and Chugani D.C. (2007). Microglial activation in perinatal rabbit brain induced by intrauterine inflammation: detection with 11C-(R)-PK11195 and small-animal PET. J. Nucl. Med. 48, 946–954 [DOI] [PubMed] [Google Scholar]
- 20.Derrick M., Luo N.L., Bregman J.C., Jilling T., Ji X., Fisher K., Gladson C.L., Beardsley D.J., Murdoch G., Back S.A., and Tan S. (2004). Preterm fetal hypoxia-ischemia causes hypertonia and motor deficits in the neonatal rabbit: a model for human cerebral palsy? J. Neurosci. 24, 24–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eixarch E., Batalle D., Illa M., Munoz-Moreno E., Arbat-Plana A., Amat-Roldan I., Figueras F., and Gratacos E. (2012). Neonatal neurobehavior and diffusion MRI changes in brain reorganization due to intrauterine growth restriction in a rabbit model. PLoS One 7, e31497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Georgiadis P., Xu H., Chua C., Hu F., Collins L., Huynh C., Lagamma E.F., and Ballabh P. (2008). Characterization of acute brain injuries and neurobehavioral profiles in a rabbit model of germinal matrix hemorrhage. Stroke 39, 3378–3388 [DOI] [PubMed] [Google Scholar]
- 23.Kannan S., Dai H., Navath R.S., Balakrishnan B., Jyoti A., Janisse J., Romero R., and Kannan R.M. (2012). Dendrimer-based postnatal therapy for neuroinflammation and cerebral palsy in a rabbit model. Sci. Transl. Med. 4, 130ra146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mitchell R.L., Barbano T.E., Losken H.W., Siegel M.I., and Mooney M.P. (2003). Early neuromotor behavior in craniosynostotic rabbits. Cleft Palate Craniofac. J 40, 486–492 [DOI] [PubMed] [Google Scholar]
- 25.Deacon R.M., and Rawlins J.N. (2006). T-maze alternation in the rodent. Nat. Protoc. 1, 7–12 [DOI] [PubMed] [Google Scholar]
- 26.Bevins R.A. and Besheer J. (2006). Object recognition in rats and mice: a one-trial non-matching-to-sample learning task to study ‘recognition memory’. Nat. Protoc. 1, 1306–1311 [DOI] [PubMed] [Google Scholar]
- 27.DiResta G.R., Lee J., Lau N., Ali F., Galicich J.H., and Arbit E. (1990). Measurement of brain tissue density using pycnometry. Acta Neurochir. Suppl. (Wien) 51, 34–36 [DOI] [PubMed] [Google Scholar]
- 28.Chen M.K. and Guilarte T.R. (2008). Translocator protein 18 kDa (TSPO): molecular sensor of brain injury and repair. Pharmacol. Ther. 118, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Venneti S., Lopresti B.J., and Wiley C.A. (2006). The peripheral benzodiazepine receptor (Translocator protein 18kDa) in microglia: from pathology to imaging. Prog. Neurobiol. 80, 308–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Derrick M., Drobyshevsky A., Ji X., and Tan S. (2007). A model of cerebral palsy from fetal hypoxia-ischemia. Stroke 38, 731–735 [DOI] [PubMed] [Google Scholar]
- 31.Harel S., Watanabe K., Linke I., and Schain R.J. (1972). Growth and development of the rabbit brain. Biol. Neonate 21, 381–399 [DOI] [PubMed] [Google Scholar]
- 32.Drobyshevsky A., Song S.K., Gamkrelidze G., Wyrwicz A.M., Derrick M., Meng F., Li L., Ji X., Trommer B., Beardsley D.J., Luo N.L., Back S.A., and Tan S. (2005). Developmental changes in diffusion anisotropy coincide with immature oligodendrocyte progression and maturation of compound action potential. J. Neurosci. 25, 5988–5997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Drobyshevsky A., Jiang R., Derrick M., Luo K., and Tan S. (2014). Functional correlates of central white matter maturation in perinatal period in rabbits. Exp. Neurol. 261, 76–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Graaf-Peters V.B. and Hadders-Algra M. (2006). Ontogeny of the human central nervous system: what is happening when? Early Hum. Dev. 82, 257–266 [DOI] [PubMed] [Google Scholar]
- 35.Brody B.A., Kinney H.C., Kloman A.S., and Gilles F.H. (1987). Sequence of central nervous system myelination in human infancy. I. An autopsy study of myelination. J. Neuropathol. Exp. Neurol. 46, 283–301 [DOI] [PubMed] [Google Scholar]
- 36.Billiards S.S., Haynes R.L., Folkerth R.D., Trachtenberg F.L., Liu L.G., Volpe J.J., and Kinney H.C. (2006). Development of microglia in the cerebral white matter of the human fetus and infant. J. Comp. Neurol. 497, 199–208 [DOI] [PubMed] [Google Scholar]
- 37.Tan S., Drobyshevsky A., Jilling T., Ji X., Ullman L.M., Englof I., and Derrick M. (2005). Model of cerebral palsy in the perinatal rabbit. J. Child Neurol. 20, 972–979 [DOI] [PubMed] [Google Scholar]
- 38.Rice F.L., Gomez C., Barstow C., Burnet A., and Sands P. (1985). A comparative analysis of the development of the primary somatosensory cortex: interspecies similarities during barrel and laminar development. J. Comp. Neurol. 236, 477–495 [DOI] [PubMed] [Google Scholar]
- 39.Mathers L.H., Jr (1979). Postnatal dendritic development in the rabbit visual cortex. Brain Res. 168, 21–29 [DOI] [PubMed] [Google Scholar]
- 40.De Groot D., and Vrensen G. (1978). Postnatal development of synaptic contact zones in the visual cortex of rabbits. Brain Res. 147, 362–369 [DOI] [PubMed] [Google Scholar]
- 41.McMullen N.T., Goldberger B., and Glaser E.M. (1988). Postnatal development of lamina III/IV nonpyramidal neurons in rabbit auditory cortex: quantitative and spatial analyses of Golgi-impregnated material. J. Comp. Neurol. 278, 139–155 [DOI] [PubMed] [Google Scholar]
- 42.Delhaye-Bouchaud N. (1971). Activity of Purkinje cells, parallel fibers, and climbing fibers in the developing rabbit cerebellum. Dev. Psychobiol. 4, 375–390 [DOI] [PubMed] [Google Scholar]
- 43.Robertson C.L., Saraswati M., and Fiskum G. (2007). Mitochondrial dysfunction early after traumatic brain injury in immature rats. J. Neurochem. 101, 1248–1257 [DOI] [PubMed] [Google Scholar]
- 44.Wilde E.A., Hunter J.V., Newsome M.R., Scheibel R.S., Bigler E.D., Johnson J.L., Fearing M.A., Cleavinger H.B., Li X., Swank P.R., Pedroza C., Roberson G.S., Bachevalier J., and Levin H.S. (2005). Frontal and temporal morphometric findings on MRI in children after moderate to severe traumatic brain injury. J. Neurotrauma 22, 333–344 [DOI] [PubMed] [Google Scholar]
- 45.Levin H.S., Benavidez D.A., Verger-Maestre K., Perachio N., Song J., Mendelsohn D.B., and Fletcher J.M. (2000). Reduction of corpus callosum growth after severe traumatic brain injury in children. Neurology 54, 647–653 [DOI] [PubMed] [Google Scholar]
- 46.Pinto P.S., Poretti A., Meoded A., Tekes A., and Huisman T.A. (2012). The unique features of traumatic brain injury in children. Review of the characteristics of the pediatric skull and brain, mechanisms of trauma, patterns of injury, complications and their imaging findings–part 1. J. Neuroimaging 22, e1–e17 [DOI] [PubMed] [Google Scholar]
- 47.Graham D.I., Ford I., Adams J.H., Doyle D., Lawrence A.E., McLellan D.R., and Ng H.K. (1989). Fatal head injury in children. J. Clin. Pathol. 42, 18–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Buttram S.D., Wisniewski S.R., Jackson E.K., Adelson P.D., Feldman K., Bayir H., Berger R.P., Clark R.S., and Kochanek P.M. (2007). Multiplex assessment of cytokine and chemokine levels in cerebrospinal fluid following severe pediatric traumatic brain injury: effects of moderate hypothermia. J. Neurotrauma 24, 1707–1717 [DOI] [PubMed] [Google Scholar]
- 49.Amick J.E., Yandora K.A., Bell M.J., Wisniewski S.R., Adelson P.D., Carcillo J.A., Janesko K.L., DeKosky S.T., Carlos T.M., Clark R.S., and Kochanek P.M. (2001). The Th1 versus Th2 cytokine profile in cerebrospinal fluid after severe traumatic brain injury in infants and children. Pediatr. Crit. Care Med. 2, 260–264 [DOI] [PubMed] [Google Scholar]
- 50.Block M.L., Zecca L., and Hong J.S. (2007). Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci. 8, 57–69 [DOI] [PubMed] [Google Scholar]
- 51.Kenk M., Selvanathan T., Rao N., Suridjan I., Rusjan P., Remington G., Meyer J.H., Wilson A.A., Houle S., and Mizrahi R. (2014). Imaging neuroinflammation in gray and white matter in schizophrenia: an in-vivo PET study with [18F]-FEPPA. Schizophr. Bull. 41, 85–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang Y., Yue X., Kiesewetter D.O., Niu G., Teng G., and Chen X. (2014). PET imaging of neuroinflammation in a rat traumatic brain injury model with radiolabeled TSPO ligand DPA-714. Eur. J. Nucl. Med. Mol. Imaging 41, 1440–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kannan S., Balakrishnan B., Muzik O., Romero R., and Chugani D. (2009). Positron emission tomography imaging of neuroinflammation. J. Child Neurol. 24, 1190–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kannan S., Saadani-Makki F., Balakrishnan B., Chakraborty P., Janisse J., Lu X., Muzik O., Romero R., and Chugani D.C. (2011). Magnitude of [(11)C]PK11195 binding is related to severity of motor deficits in a rabbit model of cerebral palsy induced by intrauterine endotoxin exposure. Dev. Neurosci. 33, 231–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hagberg H., Gressens P., and Mallard C. (2012). Inflammation during fetal and neonatal life: implications for neurologic and neuropsychiatric disease in children and adults. Ann. Neurol. 71, 444–457 [DOI] [PubMed] [Google Scholar]
- 56.Kigerl K.A., Gensel J.C., Ankeny D.P., Alexander J.K., Donnelly D.J., and Popovich P.G. (2009). Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J. Neurosci. 29, 13435–13444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Durafourt B.A., Moore C.S., Zammit D.A., Johnson T.A., Zaguia F., Guiot M.C., Bar-Or A., and Antel J.P. (2012). Comparison of polarization properties of human adult microglia and blood-derived macrophages. Glia 60, 717–727 [DOI] [PubMed] [Google Scholar]
- 58.Prinz M. and Priller J. (2014). Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat. Rev. Neurosci. 15, 300–312 [DOI] [PubMed] [Google Scholar]
- 59.Duhaime A.C., Margulies S.S., Durham S.R., O'Rourke M.M., Golden J.A., Marwaha S., and Raghupathi R. (2000). Maturation-dependent response of the piglet brain to scaled cortical impact. J. Neurosurg. 93, 455–462 [DOI] [PubMed] [Google Scholar]
- 60.Grundl P.D., Biagas K.V., Kochanek P.M., Schiding J.K., Barmada M.A., and Nemoto E.M. (1994). Early cerebrovascular response to head injury in immature and mature rats. J. Neurotrauma 11, 135–148 [DOI] [PubMed] [Google Scholar]
- 61.Hutchison J.S., Derrane R.E., Johnston D.L., Gendron N., Barnes D., Fliss H., King W.J., Rasquinha I., MacManus J., Robertson G.S., and MacKenzie A.E. (2001). Neuronal apoptosis inhibitory protein expression after traumatic brain injury in the mouse. J. Neurotrauma 18, 1333–1347 [DOI] [PubMed] [Google Scholar]
- 62.Mendez D.R., Cherian L., Moore N., Arora T., Liu P.K., and Robertson C.S. (2004). Oxidative DNA lesions in a rodent model of traumatic brain injury. J. Trauma 56, 1235–1240 [DOI] [PubMed] [Google Scholar]
- 63.Bauman Johnson W.L., Maricle D.E., Miller D.C., Allen D.N., and Mayfield J. (2010). Utilization of the comprehensive trail making test as a measure of executive functioning in children and adolescents with traumatic brain injuries. Arch. Clin. Neuropsychol. 25, 601–609 [DOI] [PubMed] [Google Scholar]
- 64.Semple B.D., Canchola S.A., and Noble-Haeusslein L.J. (2012). Deficits in social behavior emerge during development after pediatric traumatic brain injury in mice. J. Neurotrauma 29, 2672–2683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen C.Y., Noble-Haeusslein L.J., Ferriero D., and Semple B.D. (2013). Traumatic injury to the immature frontal lobe: a new murine model of long-term motor impairment in the absence of psychosocial or cognitive deficits. Dev. Neurosci. 35, 474–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huh J.W., Widing A.G., and Raghupathi R. (2008). Midline brain injury in the immature rat induces sustained cognitive deficits, bihemispheric axonal injury and neurodegeneration. Exp. Neurol. 213, 84–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tong W., Igarashi T., Ferriero D.M., and Noble L.J. (2002). Traumatic brain injury in the immature mouse brain: characterization of regional vulnerability. Exp Neurol 176, 105–116 [DOI] [PubMed] [Google Scholar]
- 68.Pullela R., Raber J., Pfankuch T., Ferriero D.M., Claus C.P., Koh S.E., Yamauchi T., Rola R., Fike J.R., and Noble-Haeusslein L.J. (2006). Traumatic injury to the immature brain results in progressive neuronal loss, hyperactivity and delayed cognitive impairments. Dev. Neurosci. 28, 396–409 [DOI] [PubMed] [Google Scholar]
- 69.Bigler E.D., Abildskov T.J., Petrie J., Farrer T.J., Dennis M., Simic N., Taylor H.G., Rubin K.H., Vannatta K., Gerhardt C.A., Stancin T., and Owen Yeates K. (2013). Heterogeneity of brain lesions in pediatric traumatic brain injury. Neuropsychology 27, 438–451 [DOI] [PubMed] [Google Scholar]
- 70.Conley Y.P., Okonkwo D.O., Deslouches S., Alexander S., Puccio A.M., Beers S.R., and Ren D. (2014). Mitochondrial polymorphisms impact outcomes after severe traumatic brain injury. J. Neurotrauma 31, 34–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Garringer J.A., Niyonkuru C., McCullough E.H., Loucks T., Dixon C.E., Conley Y.P., Berga S., and Wagner A.K. (2013). Impact of aromatase genetic variation on hormone levels and global outcome after severe TBI. J. Neurotrauma 30, 1415–1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Matsushima K., Peng M., Velasco C., Schaefer E., Diaz-Arrastia R., and Frankel H. (2012). Glucose variability negatively impacts long-term functional outcome in patients with traumatic brain injury. J. Crit. Care 27, 125–131 [DOI] [PubMed] [Google Scholar]
- 73.Missios S., Harris B.T., Dodge C.P., Simoni M.K., Costine B.A., Lee Y.L., Quebada P.B., Hillier S.C., Adams L.B., and Duhaime A.C. (2009). Scaled cortical impact in immature swine: effect of age and gender on lesion volume. J. Neurotrauma 26, 1943–1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Donders J. and Hoffman N.M. (2002). Gender differences in learning and memory after pediatric traumatic brain injury. Neuropsychology 16, 491–499 [DOI] [PubMed] [Google Scholar]
- 75.Duhaime A.C., Alario A.J., Lewander W.J., Schut L., Sutton L.N., Seidl T.S., Nudelman S., Budenz D., Hertle R., Tsiaras W., and et al. (1992). Head injury in very young children: mechanisms, injury types, and ophthalmologic findings in 100 hospitalized patients younger than 2 years of age. Pediatrics 90, 179–185 [PubMed] [Google Scholar]
- 76.Tude Melo J.R., Di Rocco F., Blanot S., Oliveira-Filho J., Roujeau T., Sainte-Rose C., Duracher C., Vecchione A., Meyer P., and Zerah M. (2010). Mortality in children with severe head trauma: predictive factors and proposal for a new predictive scale. Neurosurgery 67, 1542–1547 [DOI] [PubMed] [Google Scholar]