

Abstract
Background
Perioperative bronchospasm refractory to β-agonists continues to challenge anesthesiologists and intensivists. The TMEM16A calcium-activated chloride channel modulates airway smooth muscle (ASM) contraction. We hypothesized that TMEM16A antagonists would relax ASM contraction by modulating membrane potential and calcium flux.
Methods
Human ASM, guinea pig tracheal rings or mouse peripheral airways were contracted with acetylcholine (Ach) or leukotriene D4 (LTD4) and then treated with the TMEM16A antagonists: benzbromarone, T16Ainh-A01, MONNA or B25. In separate studies, guinea pig tracheal rings were contracted with Ach and then exposed to increasing concentrations of isoproterenol (0.01nM-10μM) ± benzbromarone. Plasma membrane potential and intracellular calcium concentrations were measured in human ASM cells.
Results
Benzbromarone was the most potent TMEM16A antagonist tested for relaxing an Ach-induced contraction in guinea pig tracheal rings (n=6). Further studies were done to investigate benzbromarone’s clinical utility. In human ASM, benzbromarone relaxed either an acetylcholine- or LTD4-induced contraction (n=8). Benzbromarone was also effective in relaxing peripheral airways (n=9) and potentiating relaxation by β-agonists (n=5–10). In cellular mechanistic studies, benzbromarone hyperpolarized human ASM cells (n=9–12) and attenuated intracellular calcium flux from both the plasma membrane and sarcoplasmic reticulum (n=6–12).
Conclusions
TMEM16A antagonists work synergistically with β-agonists and through a novel pathway of interrupting ion flux both at the plasma membrane and sarcoplasmic reticulum to acutely relax human airway smooth muscle.
Keywords: Benzbromarone, Eact, FLIPR, SR calcium
Introduction
Asthma is a very common respiratory disease, affecting up to 10% of adults and 30% of children in the Western world.1 These patients frequently present for surgical procedures requiring anesthesia, and instrumentation of the airway is a potent stimulus for acute bronchoconstriction. The pathogenesis of asthma involves chronic airway inflammation, increased mucous secretion, irritable airway nerves, and a hyperresponsiveness of airway smooth muscle (ASM) that can lead to acute bronchospasm that greatly impairs the ability to provide adequate ventilation. The incidence of intraoperative bronchospasm has been reported to be up to 20% in active asthmatics,2,3 and poorly controlled asthma is a major risk factor.4 While β-agonist therapy is the gold-standard for the treatment of bronchospasm, chronic β-agonist use is associated with tachyphylaxis and increased mortality.5,6 Perioperative bronchospasm refractory to β-agonists is a challenge for anesthesiologists. Since relaxation of ASM is an important goal in the treatment of bronchospasm and no new drugs have been developed in several decades to facilitate acute ASM relaxation, there is an obvious need for novel bronchodilators that work synergistically with β-agonists.
The modulation of chloride flux is a promising new approach to achieve ASM relaxation. Chloride flux is important for ASM plasma membrane depolarization following activation of calcium-activated chloride channels which enhances influx of extracellular calcium through voltage sensitive plasma membrane channels. Calcium-activated chloride channels have also been localized to the sarcoplasmic reticulum (SR) where they may play a key role in the release and re-filling of calcium by modulating charge balance across the SR membrane.7 Calcium-activated chloride current was identified in ASM as an important component of contraction several decades ago.8 However, the identity of the specific calcium-activated chloride channels remained elusive. In 2008 three separate laboratories discovered the molecular identity of this channel as the TMEM16 or anoctamin family of proteins.9–11 The discovery of this family of proteins has revolutionized the understanding of calcium regulation of chloride flux in many cell types.12 Our lab has shown that TMEM16A is expressed in ASM and others have demonstrated that blockade modulates ASM contraction.13
With the molecular discovery of TMEM16A, many groups have sought to identify potent and selective TMEM16A antagonists and agonists. Using high throughput screening methods, four different groups have identified TMEM16A antagonists: benzbromarone,14 T16Ainh-A01,15 MONNA16 and B25.17 These groups investigated antagonist activity at TMEM16A with IC50’s of less than 10 μM. Affinity for other chloride channels such as TMEM16B, CFTR, bestrophin and CLC were investigated with selected TMEM16A antagonists. Another group identified a TMEM16A agonist, Eact, and demonstrated that it increased TMEM16A activity.18 Utilizing these innovative pharmacological tools, we sought to determine whether these TMEM16A antagonists were effective at relaxing ASM and whether they would work synergistically with β-agonists. We hypothesized that relaxation was due to hyperpolarization of the ASM cell, and attenuation of a cytosolic increase in calcium from both the plasma membrane and SR. If TMEM16A antagonists work synergistically with first-line agents β-agonists and work through a novel pathway, they may be excellent therapeutics to treat acute bronchospasm.
Materials and Methods
All materials were obtained from Sigma-Aldrich® (St. Louis, MO) unless otherwise specified. Tetrodotoxin was purchased from Calbiochem, EMD Biosciences (La Jolla, CA). MK571, T16Ainh-A01, Eact and albuterol were purchased from Tocris (Minneapolis, MN). Membrane potential dye [fluorescent imaging plate reader (FLIPR) blue reagent] was obtained from Molecular Devices (Sunnyvale, CA). Fura-2, AM and mag-fluo-4, AM were obtained from Molecular Probes. B25 (5-[(2,6-difluorobenzyl)oxy]-2-(2-naphthyl)benzofuran-3-carboxylic acid) was synthesized as in Kumar et al.17 MONNA (N-((4-methoxy)-2-naphthyl)-5-nitroanthranilic acid) was a gift from Dr. C. Justin Lee from Korea Institute of Science and Technology.
Guinea Pig Tracheal Rings
Animal protocols were approved by the Columbia University Animal Care and Use Committee. Male Hartley guinea pigs weighing approximately 400 g were anesthetized with an intraperitoneal injection of sodium pentobarbital (100 mg/kg). Trachea were removed and transferred to ice-cold Krebs-Henseleit (KH) buffer of the following composition (in mM): NaCl 118, KCl 5.6, CaCl2 0.5, MgSO4 0.24, NaH2PO4 1.3, NaHCO3 25, glucose 5.6, pH 7.4. Epithelial-denuded tracheal rings were prepared using a dissecting microscope. Epithelium was removed by gently passing cotton fibers through the lumen of the tracheal rings. The rings were suspended in 4mL water jacketed organ baths (37°C: Radnoti Glass Technology, Monrovia, CA) and connected to Grass FT03 force transducers (Grass Telefactor, West Warwick, RI) using silk sutures. The transducers were coupled to the computer with BioPac hardware and data was collected using Acqknowledge 7.3.3 software (BioPac Systems, Goleta, CA). KH buffer was bubbled with a gas mixture of 95% oxygen and 5% carbon dioxide and buffer was exchanged every 15 minutes for 1 hour during equilibration of tracheal rings at 1 g resting tension. All baths received 10 μM indomethacin to block the synthesis of endogenous prostanoids, 10 μM N-vanillylnonanamide (capsaicin analogue) to activate and subsequently block the effects of non-adrenergic, non-cholinergic nerves, 1 μM tetrodotoxin to block neuronal effects, and 10 μM of pyrilamine to block effects of endogenous histamine release. Histamine and neuronal effects can affect intrinsic tone and confound results when studying an Ach contraction. Preliminary contractile challenges of each ring consisted of 2 cycles of acetylcholine (Ach) dose responses (100 nM – 100 μM) to define the EC50 of Ach for each tracheal ring. Rings were contracted with an EC50 concentration of Ach and force was allowed to plateau. For all organ bath experiments, tissues were randomized to receive vehicle or TMEM16A antagonists, but the investigator was not blinded. Vehicle (DMSO) or TMEM16A blockers were then added to organ bath in cumulatively increasing concentrations at 30 minute intervals (10 μM, 50 μM and 100 μM). The amount of contractile force remaining at 30 minutes after each concentration was expressed as the percent of initial force immediately before antagonist addition.
Separate studies were performed to determine if the treatment with benzbromarone could potentiate a β-agonist induced relaxation of an Ach contraction. Rings were contracted with an EC50 concentration of Ach and relaxation was induced with ½ log increments of isoproterenol (0.01 nM-10 μM) in the presence of vehicle (0.1% DMSO), 10 μM benzbromarone or 25 μM benzbromarone added simultaneously with the 0.5 nM concentration of isoproterenol.
Human Tracheal Strips
Studies using de-identified human tissue were reviewed by the Columbia University Institutional Review Board and deemed not human subjects research. Tissues were obtained from healthy, normal organ donor discarded surgical waste subsequent to lung transplant surgery. Tissues were collected and placed in DMEM media (GIBCO) and bubbled overnight in 5% carbon dioxide and 95% oxygen at 4°C. The following morning, the smooth muscle tissue was carefully dissected under a dissecting microscope and the epithelium removed. The strips were anchored in the organ bath as described above (in Guinea Pig Tracheal Rings section of Methods) using KH buffer of the same composition. The strips were allowed to equilibrate for 1 hour with buffer exchanges every 15 minutes and were treated with 1 μM tetrodotoxin and 10 μM pyrilamine. MK571 (10 μM) was added to baths contracted with Ach but not leukotriene D4 (LTD4) to block endogenous leukotriene release. Endogenous leukotriene release contributes to the intrinsic tone of human airway smooth muscle, and may confound results when studying an Ach contraction.19 Preliminary contractile challenges of each human airway smooth muscle strip consisted of 2 cycles of Ach dose responses (100 nM – 1 mM) to define the EC50 of Ach for each strip. If tissue was not stored properly, stored too long, cut in a way that is not optimal for our contraction studies or did not contract properly, the experiment was terminated prior to treatment with drugs or vehicle. Tissue was contracted with an EC50 dose of Ach or 20 nM LTD4. When a plateau in contractile force was achieved, TMEM16A antagonists in cumulatively increasing concentrations or vehicle were added to organ baths and the remaining contractile force was calculated as in guinea pig studies above.
Additional studies in human airway smooth muscle were performed to determine if the treatment with benzbromarone could augment an albuterol (β2 selective agonist)-induced relaxation of an Ach contraction. Strips were contracted with an EC50 concentration of Ach and relaxation was induced with ½ log increments of albuterol (0.1 nM-10 μM) in the presence of vehicle (0.1% DMSO) or 25 μM benzbromarone added simultaneously with the 0.5 nM concentration of albuterol.
Mouse Lung Slices
Animal protocols were approved by Texas Tech University Health Sciences Center Institutional Animal Care and Use Committee. Mouse lung slices were prepared as described previously.20 Briefly, male C57BL/6 mice, 8–12 wk old, were euthanized with an intraperitoneal injection of sodium pentobarbital (40 mg/kg). The chest cavity was opened, and the trachea was exposed and cannulated with an intravenous catheter (20-gauge Intima; BD Biosciences). The lungs were inflated with 1.4 ± 0.1 ml of 2% agarose (low-melting temperature agarose; USB Corporation) in sterile HBSS followed by 0.2 ml of air to flush the agarose out of the airways and into the distal alveolar space. The agarose was gelled by cooling the animal at 4°C for 20 min. Lung lobes were separated and trimmed near to the main bronchus to create a base. Each lobe was transferred to the specimen syringe tube of a tissue slicer (Compresstome VF-300; Precisionary Instruments), embedded in agarose, and prepared for sectioning. Sections (140 μm) were prepared starting at the peripheral edge of each lung lobe, and 15–20 sections containing small terminal airways with a diameter of 100–300 μm were collected in sterile HBSS. The sections were incubated overnight in low-glucose DMEM (Invitrogen) supplemented with antibiotics at 37°C and 10% CO2.
Lung slices were mounted at the center of a 22 × 40 mm cover glass in a perfusion chamber. The chamber was placed on the stage of an inverted phase-contrast microscope and lung slices were imaged with a 10X objective. Lungs were contracted with 0.3 μM methacholine, then exposed to 10 μM benzbromarone and washed. The area of the airway lumen was calculated from each image using a custom-written script in Video Savant that distinguishes the lumen from the surrounding tissue. The lumen area was normalized to the area before stimulation, and the changes in lumen area were plotted against time.
Cultured Human Airway Smooth Muscle Cells
Immortalized human airway smooth muscle cell lines modified to stably express human telomerase reverse transcriptase were a kind gift from Dr. William Gerthoffer (University of South Alabama, Mobile, AL) prepared as described previously21 and were grown in Dulbecco’s Modified Eagle’s Medium/F12 media (GIBCO, Grand Island, NY), with 10% FBS and antibiotics.
For calcium studies with acetylcholine, human airway smooth muscle cells stably transfected to express the human M3 muscarinic receptor were used as this receptor is not highly expressed in our cell line. Production of the lentiviral vector and transduction of cells was described previously.22
FLIPR membrane potentiometric dye assay
Cells were prepared as described previously.23 Briefly, immortalized human airway smooth muscle cells were cultured on black-walled 96-well plates to 100 % confluence and were washed four times with warmed (37°C) assay buffer. A stock solution (100% dye) of FLIPR blue dye was prepared by reconstitution of 1 vial (125 mg) with 100 mL of assay buffer. Cells loaded with a 50% working stock over 45 min at 37°C in a humidified cell culture incubator (95% air/5% CO2). Fluorescence was measured with a FlexStation 3 microplate reader (Molecular Devices) using an excitation wavelength of 530 and emission wavelength of 565. Baseline fluorescence was measured every 2 seconds for 1 min, then for 2 min after injection of the vehicle or drug of interest as indicated and the maximal change in fluorescence was recorded.
Fura-2 Calcium Assay
Immortalized human airway smooth muscle cells were cultured on black-walled 96-well plates to 100 % confluence and washed four times with warmed modified (37°C) Hank’s Balanced Salt Solution buffer (concentration in mM: NaCl 137.93, KCl 5.33, CaCl2 2, MgSO4 1, Hepes 2.38, glucose 5.5, pH to 7.4). Cells were loaded with 5 μM fura-2 AM for 45 minutes and washed an additional three times with buffer. Cells were then pretreated with vehicle (0.1% DMSO) or benzbromarone (10 μM, 50 μM and 100 μM) for 10 minutes after which they were placed in a FlexStation 3 microplate reader. Baseline fluorescence was measured, and histamine or bradykinin (10 μM final concentration) was pipetted into the wells of the plate. Fluorescence was read every 4 s for 400 s using excitation wavelengths of 340 and 380, an emission wavelength of 510, and a cutoff filter of 495. Fluorescence values were reported as F/Fo according to the calculation: [ F = (340 nm)f/(380 nm)f – (340 nm)0/(380 nm)0]. For studies in 0 mM external calcium, the cells were loaded as above in this section. After being loaded, the cells were washed four times with 0 calcium HBSS (supplemented with 200 μM ethylene glycol tetraacetic acid (EGTA)). The remainder of the study was carried out in calcium-free HBSS.
Simultaneous Fura-2 and Mag Fluo-4 Calcium Assay
Simultaneous measurement of sarcoplasmic reticulum and cytosolic calcium were adapted from previous studies for the FlexStation 3 microplate reader and airway smooth muscle cells.24,25 Immortalized human airway smooth muscle cells were cultured on black-walled 96-well plates to 100 % confluence and washed four times with fluorescence buffer (FB) (concentration in mM: 145 NaCl, 5 KCl, 1 Na2HPO4, 0.5 MgCl2, 1 CaCl2, 10 HEPES, 5 glucose, pH 7.4, 37°C). Cells were incubated with 5 μM mag-fluo-4 AM with 0.1% pluronic in FB for 40 minutes. Cells were washed three times and incubated with 5 μM fura-2 AM with 0.05% pluronic for 30 minutes. They were then washed three times with FB and incubated for 30 minutes. Cells were then pretreated with vehicle (0.1% DMSO), benzbromarone (10 μM or 50 μM) or MONNA (10 μM or 50 μM) for 5 minutes after which they were placed immediately in a FlexStation 3 microplate reader (Molecular Devices). Baseline fluorescence was measured and bradykinin or acetylcholine (10 μM final concentration) was pipetted into the wells of the plate. Fluorescence was read every 5 seconds using excitation wavelengths of 340, 380 and 465 simultaneously with emission wavelength of 510. For studies with acetylcholine, M3-overexpressing human airway smooth muscle cells were used.22
Statistics
Data were analyzed using one-way ANOVA with repeated measures, unless two-way ANOVA is stated in results. The Bonferroni correction was applied for multiple comparisons. Statistical significance was established at P < 0.05, and all values are expressed as means ± SE. For organ bath experiments, “n” refers to the number of guinea pig tracheal rings or human airway smooth muscle strips. Sample sizes were selected based upon prior experience with organ bath experiments which typically requires an n= 4–7 to see an effect of antagonist compared to its vehicle control. For mouse lung slices, “n” refers to the number of airways. For cellular assays, “n” refers to a well of a 96 well plate. All data was analyzed using GraphPad Prism software ver 4.0, GraphPad Software, Inc, (La Jolla, CA).
Results
TMEM16A antagonists relax an acetylcholine contraction in guinea pig tracheal rings
Four different TMEM16A antagonists (benzbromarone, T16Ainh-A01, B25 and MONNA) were analyzed for their ability to relax an acetylcholine (Ach)-induced contraction in guinea pig tracheal rings. Rings were treated with vehicle (0.1% DMSO) or cumulative doses of TMEM16A antagonist (10 μM, 50 μM and 100 μM) in 30 minute intervals. 10 μM benzbromarone significantly relaxed an Ach contraction (70.9 ± 6.7% of initial plateau contractile force remaining at 30 min, respectively, **p<0.001, n=6), while T16Ainh-A01, MONNA and B25 failed to show significant relaxation at 10 μM (fig. 1). 50 μM benzbromarone, T16Ainh-A01 and MONNA relaxed an Ach contraction (−10.7 ± 3.1%, 51.8 ± 4.6%, 73.3 ± 8.0% of initial force remaining at 30 min, respectively, **p<0.01, ***p<0.001, n=6). All four TMEM16A antagonists relaxed an Ach contraction at 100 μM (benzbromarone: −40.5 ± 8.2%, T16Ainh-A01: 35.1 ± 4.7%, MONNA: 21.8 ± 9.3%, B25: 21.8 ± 9.3%, ***p<0.001, n=6, two-way ANOVA). Benzbromarone treatment resulted in significantly increased relaxation when compared to all three other TMEM16A antagonists at the 50 and 100 μM concentrations (p<0.001 comparing benzbromarone to other three TMEM16A antagonists).
Figure 1. TMEM16A antagonists relax an established acetylcholine (Ach)-induced contraction in guinea pig tracheal rings.
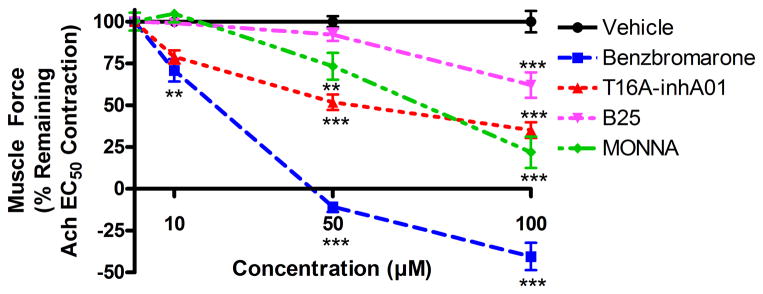
Ach EC50 pre-contracted tracheal rings were treated with cumulatively increasing concentrations of benzbromarone (10 μM, 50 μM and 100 μM) or vehicle (0.1% DMSO) at 30 minute intervals. 10 μM benzbromarone significantly relaxed an Ach contraction (**p<0.01, compared to vehicle control), while T16Ainh-A01, MONNA and B25 failed to show significant relaxation at 10 μM. 50 μM benzbromarone, T16Ainh-A01 and MONNA relaxed an Ach contraction (**p<0.01, ***p<0.001). All four TMEM16A antagonists relaxed an Ach contraction at 100 μM (***p<0.001). (n=6 rings from 5 animals). Relaxation was normalized to DMSO vehicle controls, represented as 100%.
Benzbromarone relaxes both an acetylcholine and leukotriene D4-induced contraction in human airway smooth muscle
As benzbromarone was found to be the most effective TMEM16A antagonist in guinea pig tracheal ring organ bath studies, benzbromarone was further tested in human airway smooth muscle. Human airway smooth muscle was contracted with acetylcholine (Ach) EC50 and then treated with vehicle (0.1% DMSO) or cumulative concentrations of benzbromarone in 30 minute intervals. Vehicle treatment resulted in no change of contractile force, while increasing concentrations of benzbromarone caused a relaxation. When muscle was contracted with Ach EC50, treatment with 10 μM and 50 μM benzbromarone resulted in significant relaxation (10 μM: 64.1 ± 10.5%, 50 μM 9.5 ± 20.4% of initial plateau contractile force remaining at 30 min, *p<0.05, **p<0.01, n=8) (fig. 2). Therefore, as benzbromarone was also effective in a human model, it could have clinical efficacy in relaxing acute bronchospasm.
Figure 2. Benzbromarone relaxes an established acetylcholine (Ach)-induced contraction in human airway smooth muscle.

Human airway smooth muscle was contracted with Ach EC50 and then treated with cumulatively increasing concentrations of benzbromarone (Benzb) (10 μM, 50 μM) or with vehicle (0.1% DMSO) at 30 minute intervals. Treatment with 10 μM and 50 μM benzbromarone resulted in significant relaxation (*p<0.05, **p<0.01, n=8 samples from 4 patients).
Human airway smooth muscle was also studied with another clinically relevant Gq-coupled contractile agonist, leukotriene D4 (LTD4). LTD4 exposure resulted in a strong contraction (greater than twice the contractile force induced by an EC50 concentration of Ach (not shown)), which was also relaxed by cumulatively increasing concentrations of benzbromarone. Treatment with 10 μM, 50 μM and 100 μM benzbromarone resulted in significant relaxation (10 μM: 68.5 ± 10.5%, 50 μM 39.9 ± 13.3%, 100 μM: 8.4 ± 12.5% of initial plateau contractile force remaining at 30 min, *p<0.05, **p<0.01, ***p<0.001, n=8) (fig. 3). Therefore, the relaxation by benzbromarone is not specific to an Ach contraction.
Figure 3. Benzbromarone relaxes an established leukotriene D4 (LTD4)-induced contraction in human airway smooth muscle.

Human airway smooth muscle was contracted with LTD4 (20nM) and then treated with cumulatively increasing concentrations of benzbromarone (Benzb) (10 μM, 50 μM, 100 μM) or vehicle (0.1% DMSO) at 30 minute intervals. Treatment with 10 μM, 50 μM and 100 μM benzbromarone resulted in significant relaxation (*p<0.05, **p<0.01, ***p<0.001, n=8 samples from 4 patients).
Benzbromarone relaxes mouse peripheral airways
As we had successfully shown that benzbromarone was effective in guinea pig tracheal muscle and human airway smooth muscle from large central airways, we next sought to investigate whether it was effective in small distal peripheral airways. Mouse peripheral airways were contracted with methacholine and then treated with benzbromarone (fig. 4AB). Benzbromarone 10 μM relaxed 72.4 ± 6.2% of a methacholine contraction in mouse peripheral lung slices (fig 4C). The contraction of a peripheral airway in lung slice by methacholine and its subsequent relaxation by benzbromarone is shown in real time in an accompanying supplemental digital video (supplemental digital content).
Figure 4. Benzbromarone relaxes methacholine (MCh) contraction in mouse peripheral airways.
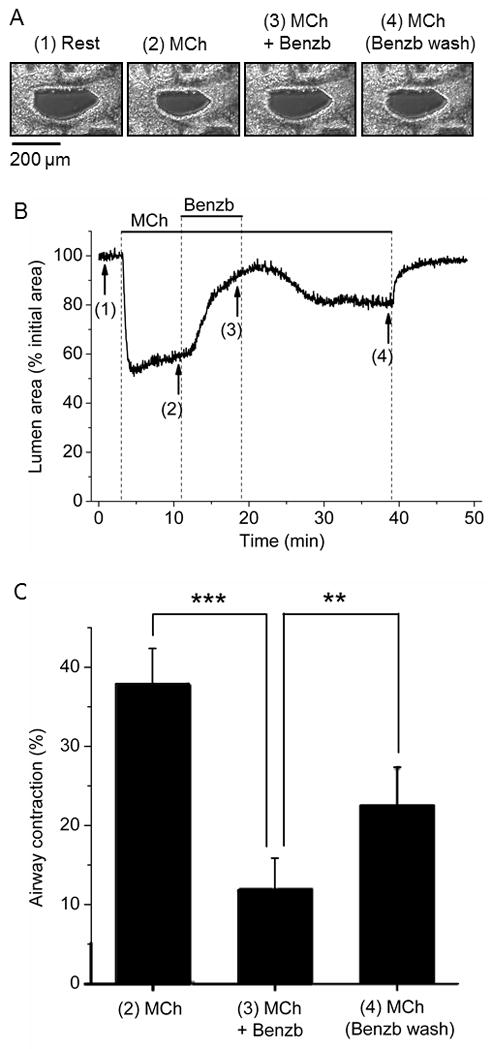
A: Representative images of an airway in a lung slice at rest and after exposure to 0.3 μM MCh, 10 μM benzbromarone (Benzb) + MCh, and subsequent Benzb washout with MCh. The images were taken at the times indicated in numbers under the trace in B.
B: Changes in airway lumen area in response to MCh and Benzb superfused at the times indicated by the lines on top of the graph. The airway contracted in response to MCh, relaxed after addition of Benzb, recontracted after Benzb washout with MCh and relaxed after MCh removal.
C: Summary of the changes in airway contraction in response to MCh, Benzb, and Benzb washout with MCh (n=9 airways from 2 animals, ***p<0.001, **p<0.01).
Benzbromarone potentiates relaxation of β-agonists
Next we sought to determine whether benzbromarone could act synergistically with β-agonists, the first line agents used to treat acute bronchospasm. Benzbromarone 10 μM treatment decreased the isoproterenol EC50 for relaxation from 5.5 to 2.1 nM (n=9, p<0.01, two-way ANOVA), which represents a 2.6 fold potentiation of the relaxation effect of the β-agonist. Benzbromarone 25 μM treatment decreased the isoproterenol EC50 for relaxation from 5.5 to 0.7 nM (n=9, p<0.01, two-way ANOVA), which represents a 7.9 fold potentiation (fig. 5A). These findings indicate that benzbromarone is working synergistically with isoproterenol to relax an Ach-induced contraction of ASM.
Figure 5. Benzbromarone treatment decreases the EC50 dose of isoproterenol or albuterol required for relaxation.

A: When guinea pig tracheal rings were contracted with acetylcholine (Ach) EC50 and then treated with cumulatively increasing concentrations of isoproterenol, a dose dependent relaxation was observed. At the 0.5 nM concentration of isoproterenol, vehicle (0.1% DMSO), 10 μM benzbromarone (Benzb) or 25 μM Benzb was added. The EC50 for relaxation with vehicle treated guinea pig tracheal rings was 5.5 nM isoproterenol. Benzb 10 μM treatment decreased the isoproterenol EC50 for relaxation from 5.5 to 2.1 nM (n=9, p<0.01). Benzb 25 μM treatment decreased the isoproterenol EC50 for relaxation from 5.5 to 0.7 nM (n=9 rings from 4 animals, p<0.01 two-way ANOVA).
B. Treatment with a combination of isoproterenol 1 nM and Benzb 10 μM (n=12) shows a significant decrease in airway smooth muscle force compared to treatments with either compound alone (Relaxation not normalized to DMSO vehicle control, isoproterenol study done in presence of DMSO vehicle of benzbromarone), (*p<0.05 compared to vehicle, $p<0.001 compared to isoproterenol 1 nM alone (n=10), #p<0.05 compared to Benzb 10 μM alone (n=9)).
C. When human airway smooth muscle was contracted with Ach EC50 and then treated with cumulatively increasing concentrations of albuterol, a dose dependent relaxation was observed. At the 0.5 nM concentration of albuterol, vehicle (0.1% DMSO) or 25 μM benzbromarone was added. The EC50 for relaxation with vehicle was 62.9 nM albuterol. Benzb 25 μM treatment decreased the albuterol EC50 for relaxation from 62.9 to 23.2 nM (p<0.05 two-way ANOVA, n=5 samples from 3 patients).
To further demonstrate that benzbromarone is working synergistically with isoproterenol, the percent of remaining contractile force was calculated after 1 nM of isoproterenol alone (a subtherapeutic dose in the organ bath model), after 10 μM benzbromarone alone, or after the combination of 1 nM isoproterenol/10 μM benzbromarone. The percent of the remaining contractile force with 1 nM isoproterenol alone was 88.9 ± 3.3% and with 10 μM benzbromarone alone was 79.6 ± 6.7%, while the percent remaining contractile force with the combination was 49.8 ± 8.0% (fig. 5B). These results suggest a synergistic relationship, as the percent relaxation of the combination of the two drugs (50.2%) is greater than the sum of each treated separately (11.1 + 20.4 = 31.5%).
For a further clinical correlation of this conclusion from guinea pig studies, studies with human airway smooth muscle and albuterol were carried out. Benzbromarone 25 μM decreased the EC50 of albuterol from 62.9 nM to 23.2 nM (n=5, p<0.05, two-way ANOVA), which represents a 2.7 fold shift in relaxation (fig. 5C). Therefore, this relationship is also observed in human tissue with a selective β2 agonist commonly used for intraoperative bronchospasm.
Benzbromarone hyperpolarizes human airway smooth muscle cells
In order to elucidate the contributing cellular mechanisms involved in the ASM relaxation induced by benzbromarone, human airway smooth muscle cells in culture were used. FLIPR membrane potentiometric dye was used to assess whether benzbromarone changed membrane potential. Potassium gluconate 40 mM was used as a positive control as a depolarizing agent, and NS1619 10 μM (K+ channel opener) was used as a positive control as a hyperpolarizing agent. Benzbromarone 50 μM caused a hyperpolarization reflected by a decrease in RFU −54.8 ± 5.9, while Eact (TMEM16A agonist) 50 μM caused a depolarization reflected by an increase in RFU 115.3 ± 4.4 (***p<0.001, n=8–12) (fig. 6).
Figure 6. Benzbromarone hyperpolarizes while Eact depolarizes human airway smooth muscle cells.
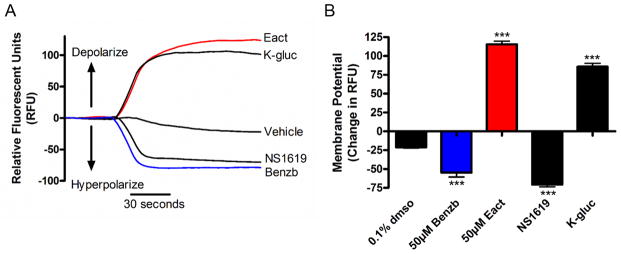
A: Representative tracings of continuous fluorescence recordings of membrane potential using FLIPR potentiometric indicator. Eact and potassium gluconate (K-gluc) depolarize, while NS1619 (K+-channel opener) and benzbromarone hyperpolarize human airway smooth muscle cells. K-gluc and NS1619 were used as controls as they are known to depolarize and hyperpolarize, respectively.
B: Quantitative change in relative fluorescent units (RFU) showing a decrease in RFU with NS1619 (n=12) and benzbromarone (n=9), and an increase with K-gluc (n=10) and Eact (n=9) (***p<0.001).
Benzbromarone attenuates an increase in intracellular calcium induced by histamine or bradykinin
To further elucidate the cellular mechanisms involved in relaxation induced by benzbromarone, the effects on Gq-coupled increases in intracellular calcium were investigated. Human airway smooth muscle cells were loaded with the calcium indicator fura-2 AM, and were pretreated with either vehicle or benzbromarone prior to the addition of histamine or bradykinin, agonists for receptors coupled to Gq. Figure 7A is a representative tracing demonstrating a dose dependent attenuation by benzbromarone of the increase in intracellular calcium by histamine in the presence of 2mM extracellular calcium. Treatment with 10 μM benzbromarone attenuated the peak in fluorescence from 0.83 ± 0.04 in control to 0.56 ± 0.07 RFU ratio (F/Fo) or 68.2 ± 8.4% of control (fig. 7B). The peak in fluorescence was attenuated dose dependently, such that the peak calcium increase was 0.20 ± 0.05 (24.7 ± 5.5% control) with 50 μM benzbromarone, and 0.02 ± 0.003 (or 2.3 ± 0.4% control) with 100 μM benzbromarone (n=6–12, ***p<0.001 compared to vehicle control, ##p<0.001 compared to 10μM, $p<0.01 compared to 50 μM). Similar results were observed when cells were challenged with bradykinin, another agonist of a receptor coupled to Gq (figure 7C). Treatment with 10 μM benzbromarone attenuated the peak in intracellular calcium fluorescence from 0.90 ± 0.06 in control to 0.52 ± 0.04 (57.9 ± 4.4% of control), while treatment of 50 μM (0.12 ± 0.03 or 13.8 ± 2.9% control) and 100 μM (0.03 ± 0.007 or 2.9 ± 0.7% control) showed further significant attenuation. Therefore, benzbromarone attenuates the increase in intracellular calcium, which in the presence of 2mM extracellular calcium is entering the cell cytosol from calcium channels on both the plasma membrane and sarcoplasmic reticulum (SR).
Figure 7. Benzbromarone attenuates an increase in intracellular calcium due to histamine and bradykinin in cultured human airway smooth muscle cells.

A: Representative fura-2 calcium tracings in the presence of 2mM extracellular calcium. Human airway smooth muscle cells were pretreated with either vehicle (0.1% DMSO) or benzbromarone (Benzb) (10 μM, 50 μM, 100 μM) and then 10 μM histamine was pipetted into the wells of the plate. Increases in intracellular calcium was attenuated in a concentration-dependent manner by pretreatment with benzbromarone.
BC: Average peak fluorescence in 2mM extracellular calcium. Cells were pretreated with either vehicle (0.1% DMSO) (n=12) or benzbromarone (10 μM, 50 μM, 100 μM) and then 10 μM histamine (B) (n=9) or bradykinin (C) (n=6). Pretreatment with benzbromarone (10 μM, 50 μM, 100 μM) showed an attenuation of increases in intracellular calcium when compared to vehicle control (***p<0.001). Attenuation was concentration dependent (##p<0.001 compared to 10μM, $p<0.01 compared to 50μM).
D. Representative tracing (as in figure 7A) of intracellular calcium concentrations in the absence of extracellular calcium. Increases in intracellular calcium were attenuated by pretreatment with increasing concentrations of benzbromarone.
EF: Average peak fluorescence (as in figure 7BC) of intracellular calcium induced by histamine or bradykinin in the absence of extracellular calcium. Pretreatment with benzbromarone (50 μM, 100 μM) showed an attenuation of increases in intracellular calcium when compared to vehicle control (***p<0.001) (vehicle and 50 μM Benzb n=10, 10 μM and 100 μM Benzb n=7).
Calcium assays were then repeated in the presence of 0mM external calcium to investigate whether benzbromarone was affecting intracellular calcium flux involving the SR. As demonstrated in figure 7D (compared to fig. 7A), calcium flux was attenuated by removal of external calcium as seen by smaller peak fluorescence as expected. Calcium flux in response to both histamine and bradykinin was not significantly attenuated by 10 μM benzbromarone, but was attenuated by 50 and 100 μM benzbromarone pretreatment (fig. 7E and 7F). With pretreatment of 50 μM benzbromarone, the increase in intracellular calcium with histamine was attenuated from 0.37 ± 0.06 in control to 0.05 ± 0.02 or 14.6 ± 6.3% of control (n=7–10, ***p<0.001). With bradykinin challenge 50 μM benzbromarone attenuated peak fluorescence from 0.51 ± 0.05 in control to 0.03 ± 0.005 or 5.7 ± 1.1% of control (n=7–10, ***p<0.001). No further attenuation was seen by treatment with 100 μM. These results suggest that a component of TMEM16A antagonists’ attenuation of intracellular calcium increases are due to blockade of SR calcium release.
Benzbromarone attenuates SR calcium release
To further demonstrate that benzbromarone is affecting SR calcium release, we measured SR calcium flux with the calcium indicator mag-fluo-4 AM simultaneously with fura-2 AM. Mag-fluo-4 has been used as an indicator for SR calcium release as it has a low affinity for calcium and preferentially detects calcium in the SR where the calcium levels are higher.26 We first verified that mag-fluo-4 reflects SR calcium levels in airway smooth muscle cells. For verification studies we used thapsigargin, an irreversible SR calcium-ATPase inhibitor. SR calcium-ATPase continuously refills SR calcium, therefore inhibition with thapsigargin causes a slow depletion of SR calcium stores. Cells were loaded with both mag-fluo-4 AM and fura-2 AM in calcium free buffer, and thapsigargin was injected. As seen in Figure 8A, thapsigargin caused a decrease of mag-fluo-4 fluorescence representing a loss of calcium from the SR, while simultaneously observing an increase in fura-2 representing an increase in cytosolic calcium as the calcium exits the SR. Cells were then treated with bradykinin, a Gq-coupled agonist known to induce SR calcium release. In vehicle treated cells, bradykinin caused the expected decrease in mag-fluo-4 and increase in fura-2, representing calcium leaving the SR and entering the cytoplasm. In cells treated with thapsigargin, as expected no bradykinin effect was seen as cells had been pre-depleted of SR calcium.
Figure 8. Benzbromarone and MONNA attenuate SR calcium release in human airway smooth muscle cells.

A. Representative tracing of calcium imaging using fura-2 and mag-fluo-4. Cells were pretreated with vehicle (0.1% DMSO) or 1 μM thapsigargin, and then 10 μM bradykinin. Thapsigargin treatment caused a leak of calcium from the SR, represented by a decrease in fluorescence of mag-fluo-4 and increase in fura-2 fluorescence.
B. Representative tracing demonstrating attenuation of SR calcium release with 50 μM benzbromarone treatment. With control vehicle (0.1% DMSO) pretreatment, bradykinin causes a decrease in mag-fluo-4 fluorescence and increase in fura-2, while pretreatment with 50 μM benzbromarone attenuates these changes.
CE. Pretreatment with benzbromarone (10 μM, 50 μM) or MONNA (10 μM, 50 μM) showed an attenuation of increases in intracellular calcium induced by 10 μM bradykinin (C) (**p<0.01, ***p<0.001, n=10 DMSO, n=15 Benzb and MONNA) or 10 μM acetylcholine (Ach) (E) (***p<0.001, n=6 DMSO, n=8 Benzb and MONNA) when compared to vehicle control.
DF. Pretreatment with benzbromarone (10 μM, 50 μM) or MONNA (10 μM, 50 μM) showed an attenuation of decreases in SR calcium induced by 10 μM bradykinin (D) (*p<0.05, **p<0.01, ***p<0.001, n=10 DMSO, n=15 Benzb and MONNA) or 10 μM Ach (F) (*p<0.05, ***p<0.001, n=6 DMSO, n=8 Benzb and MONNA) when compared to vehicle control.
After verifying the technique, the effect of pretreatment with benzbromarone and MONNA were investigated. As seen in the tracing in figure 8B, benzbromarone attenuated the increase in fura-2 and decrease in mag-fluo-4. Under control conditions with vehicle pretreatment, bradykinin treatment resulted in an increase in fura-2 fluorescence (fig. 8C) and a simultaneous 21.1 ± 2.2% decrease in mag-fluo-4 fluorescence (fig. 8D). Pretreatment with either benzbromarone (10 μM: 12.0 ± 0.8%, 50 μM: 6.2 ± 1.2%) or MONNA (10 μM: 14.5 ± 1.8%, 50 μM: 9.5 ± 2.0%) significantly attenuated this change (fig. 8D), confirming that TMEM16A antagonists were blocking SR calcium release.
Studies were then repeated in human airway smooth muscle cells overexpressing the M3 muscarinic receptor in order to investigate whether this mechanism would also apply to relaxation of an acetylcholine contraction as observed in our organ bath studies. Acetylcholine treatment resulted in an increase in fura-2 fluorescence (fig. 8E) and a simultaneous 12.0 ± 1.8% decrease in mag-fluo-4 fluorescence (fig. 8F), and pretreatment with either benzbromarone (10 μM: 5.7 ± 2.6%, 50 μM: 2.0 ± 0.7%) or MONNA (10 μM: 2.3 ± 0.6%, 50 μM: 1.5 ± 0.4%) significantly attenuated this change (fig. 8F). All studies were done with simultaneous recording of fura-2 fluorescence (fig. 8C and 8E).
Discussion
There are several reasons why peri-operative bronchospasm is a major concern for anesthesiologists. While most cases of acute bronchospasm are treated effectively by deepening the anesthetic and using inhaled bronchodilators, there is the potential for catastrophic patient outcomes when it does occur. This is corroborated by evidence derived from the ASA closed-claims database which shows that while these cases represent only a small number of total cases in the database (2%), when they do occur 90% of these cases are associated with death or severe brain injury.27 Given the fact that 10–30% of people carry the diagnosis of asthma,1 that up to 34% of these patients are poorly controlled,28 and that in this latter group nearly 1 in 5 patients experience intra-operative bronchospasm2,3 the likelihood of encountering this problem in the peri-operative period is extremely high. This problem is further compounded by the fact that many patients with chronic bronchospastic disease are tolerant or refractory to β-adrenoceptor agonists.29 All of these factors underscore the importance of expanding the anesthesiologist’s pharmacological armamentarium to include novel compounds that relax acute bronchospasm in the peri-operative period and can be used in concert with existing drugs to provide synergistic relaxation.
We believe one such drug class are antagonists of the TMEM16A receptor of the calcium-activated chloride channel family. In the present study we demonstrate four different TMEM16A antagonists relax acetylcholine (Ach)-induced contractions in guinea pig tracheal rings and that benzbromarone relaxes both Ach- and LTD4-induced contractions in human airway smooth muscle (ASM). While previous studies have shown that TMEM16A antagonists can prevent a methacholine contraction with pretreatment,13,14,30 this is the first study to demonstrate that they can relieve a pre-existing contraction to a variety of contractile mediators in both small and large airways and work synergistically with β-agonists. Additionally, we show mechanistic evidence that benzbromarone hyperpolarizes the plasma membrane of human ASM cells and attenuates intracellular calcium flux.
While Huang et al demonstrated that pretreatment with benzbromarone attenuated a methacholine-induced contraction in human ASM, and Zhang et al used an ovalbumin-sensitized model to demonstrate that benzbromarone attenuates methacholine-induced contractions, the experimental approach in both of these studies does not mirror the clinical reality that anesthesiologists encounter in the peri-operative period. In clinical scenarios, the need to relieve a pre-existing bronchospasm is a more common occurrence, yet both of these studies utilized an experimental paradigm that gave a pretreatment prior to a contraction to attenuate the initiation of a contraction. Our study is unique in that we treated with benzbromarone after the ASM was pre-contracted and in a plateau phase of contraction. We specifically designed the experiments this way to mimic clinical bronchospasm. As such, this study is the first to show benzbromarone holds promise as both a prophylactic treatment to prevent bronchospasm and perhaps more importantly for peri-operative physicians, TMEM16A antagonism is an effective tool to acutely relieve ongoing bronchospasm.
The four different TMEM16A antagonists varied in their dose-dependent ability to relax ASM. This could be due to simply different potencies of each drug, but benzbromarone actually has the highest IC50 of all of the antagonists (9.97 μM) and demonstrated the greatest degree of relaxation. However, it is difficult to directly compare the IC50s of the four TMEM16A antagonists since the original measurements were done in different recombinant models using different high throughput screening methods.
An alternative explanation for the differences in relaxation efficacies seen between these four TMEM16A antagonists is differences in their specificities for TMEM16A versus other chloride modulating proteins which may also contribute to ASM contractile tone. After the initial identification of these four novel TMEM16A antagonists, each group determined the selectivity for TMEM16A versus other chloride channel proteins. For example, 10 μM benzbromarone effectively blocks TMEM16A and TMEM16B, but not CFTR.14 The effect of benzbromarone on other chloride channels such as bestrophin or the CLC family of chloride transporters was not investigated. MONNA inhibited TMEM16A but not CFTR, CLC or bestrophins. TMEM16B was not tested in the study.31 B25 was tested for lack of effect at the CFTR channel,17 while no specificity information was done with T16Ainh-A01.15 It is possible that a component of benzbromarone’s effect is due to blockade of other chloride channels in addition to TMEM16A, or another off target effect that promotes relaxation.
Since benzbromarone was the most effective relaxant in guinea pig studies, it was chosen for translational studies utilizing human airway smooth muscle. Our present studies also uncovered an important mechanistic distinction regarding TMEM16A mediated relaxation. While Huang et al showed that benzbromarone pretreatment was effective against a methacholine contraction, they failed to show a positive relaxant effect against a KCl contraction, and concluded that benzbromarone’s effect is specific to the G protein-coupled muscarinic receptor.14 However, we previously showed that pretreatment with either tannic acid or benzbromarone attenuated a substance P-induced contraction in guinea pig tracheal rings,13 and the current studies additionally show effectiveness against an LTD4 contraction in human ASM. Therefore, this manuscript clearly establishes the relaxant capacity of TMEM16A blockade against a variety of contractile mediators.
The capacity of TMEM16A antagonists to relieve LTD4 mediated contractile pathways cannot be understated. Leukotrienes, specifically LTD4, are potent bronchoconstrictors and anti-leukotriene drugs have been used to treat asthma since the 1990s. LTD4 binds to the CysLT receptor 1 (CysLT1) to mediate mucus secretion and edema in addition to bronchoconstriction.32 Anti-leukotriene drugs such as montelukast work through antagonist of the CysLT1 receptor, and have been shown to be clinically effective. If TMEM16A antagonists are also effective against LTD4 contractions (as in the current study) they may work synergistically with antileukotriene drugs at downstream effector molecules associated with the G protein coupled receptor pathway. Indeed, additional studies elaborating benzbromarone’s potential for modulating mucus secretion and edema involved in leukotriene signaling are underway.
In this study we also demonstrated that benzbromarone is effective in peripheral as well as central airways. Given the observation that contractile and relaxant pathways may differ at different anatomical locations throughout the trachea-bronchial tree,33 we included an experimental approach to illustrate TMEM16A antagonism mediates pro-relaxant effects on ASM throughout the lung. Our organ bath studies utilize guinea pig or human samples from large central airways, but we also tested benzbromarone’s effect on mouse peripheral airways. These latter studies are important because peripheral airway constriction is an important (but under-recognized) component of clinical bronchospasm. In the periphery, ASM completely encircles the airway lumen and constriction can cause significant obstruction to airflow and even distal alveolar collapse.34–36
This study also examined the important role of potential synergistic drug effects between airway relaxants. Our study shows that TMEM16A antagonists behave synergistically with the gold-standard agent to treat bronchospasm, β-agonists. Benzbromarone significantly potentiated the relaxation induced by isoproterenol and albuterol. As increasing doses of β-agonists can lead to tachyphylaxis, drugs that work synergistically to both decrease chronic doses and rescue bronchospasm refractory to β-agonists are invaluable. β-agonists work through the G protein coupled β2-adrenoreceptor, which activates adenylyl cyclase, increases cAMP and activates protein kinase A (PKA).37,38 PKA has many downstream targets including myosin light chain kinase and the SR ryanodine receptor, as well as BKCa channels.22 Therefore, β-agonists also work through both modulation of intracellular calcium flux (through blockade of the SR ryanodine receptor) and modulation of plasma membrane potential (hyperpolarization through activation of BKCa channels, important potassium channels in ASM). TMEM16A antagonists may be working synergistically by amplifying the blockade of SR calcium release and augmenting the hyperpolarization through blockade of chloride flux at the plasma membrane.
ASM cells have a resting membrane potential of approximately −60mV and the opening of a TMEM16A channel should cause depolarization while blockade would cause hyperpolarization. Cellular studies confirmed that benzbromarone hyperpolarized the human ASM cell. The importance of membrane potential in relaxation of ASM has been questioned. When calcium channel blockers failed to showed effectiveness for asthma despite efficacy in vascular smooth muscle, drugs targeting changes in membrane potential were abandoned.39 Therefore, although hyperpolarization favors relaxation it may be only one contributing factor in relaxation through TMEM16A blockade. A more important role for TMEM16A may be found intracellularly. It has been proposed that blockade of chloride flux may not only prevent depolarization of the ASM cell at the plasma membrane, but may also prevent calcium release from the SR due to the role of chloride in balancing the charge debt created by the movement of calcium across the SR.7 In our study we demonstrate that benzbromarone attenuates calcium flux from both the plasma membrane and SR. This unique dual action suggests multiple downstream targets of TMEM16A antagonism or suggests that TMEM16A may be present on the SR.
In conclusion, we have identified four different TMEM16A antagonists that demonstrate bronchorelaxant properties. In particular the TMEM16A antagonist benzbromarone, has been shown to relax both central and peripheral airways, work synergistically with β-agonists, and is effective at relaxing a contraction induced by Ach and LTD4. Benzbromarone works in part through membrane hyperpolarization and attenuation of calcium flux both at the plasma membrane and SR, suggesting a novel role of chloride flux in ASM relaxation.
Supplementary Material
Real time image of the contraction of a mouse peripheral airway in lung slice. Methacholine (MCh) and benzbromarone (Benzb) are present when the label appears in the video otherwise the lung slice is superfused with HBSS alone. Slice is contracted with MCh, and subsequently relaxed by addition of benzbromarone in the presence of MCh. Benzbromarone was then washed out with subsequent re-contraction with MCh, and finally MCh washed to return to resting state.
Acknowledgments
The authors thank Changjoon Justin Lee, Ph.D., Brain Science Institute Korea Institute of Science and Technology (KIST), Seoul, Korea, for the gift of MONNA.
This work was supported by the Foundation for Anesthesia and Education Research Mentored Research Training Grant - Basic Science, Stony Wold-Herbert Fund Research Grant, NIH GM008464 (CWE), NIH GM065281 (CWE).
Footnotes
Disclosure: The authors declare no competing interests
Meetings: Presented in part orally October 12, 2014 at the ASA Annual Meeting 2014 in New Orleans, Louisiana, First Place Residents’ Research Essay Contest
References
- 1.Somasundaram K, Ball J. Medical emergencies: pulmonary embolism and acute severe asthma. Anaesthesia. 2013;68 (Suppl 1):102–16. doi: 10.1111/anae.12051. [DOI] [PubMed] [Google Scholar]
- 2.Pizov R, Brown RH, Weiss YS, Baranov D, Hennes H, Baker S, Hirshman CA. Wheezing during induction of general anesthesia in patients with and without asthma. A randomized, blinded trial. Anesthesiology. 1995;82:1111–6. doi: 10.1097/00000542-199505000-00004. [DOI] [PubMed] [Google Scholar]
- 3.El-Metainy S, Ghoneim T, Aridae E, Abdel Wahab M. Incidence of perioperative adverse events in obese children undergoing elective general surgery. Br J Anaesth. 2011;106:359–63. doi: 10.1093/bja/aeq368. [DOI] [PubMed] [Google Scholar]
- 4.Liccardi G, Salzillo A, Sofia M, D’Amato M, D’Amato G. Bronchial asthma. Curr Opin Anaesthesiol. 2012;25:30–7. doi: 10.1097/ACO.0b013e32834e7b2e. [DOI] [PubMed] [Google Scholar]
- 5.Martinez FD. Serious adverse events and death associated with treatment using long-acting beta-agonists. Clin Rev Allergy Immunol. 2006;31:269–78. doi: 10.1385/CRIAI:31:2:269. [DOI] [PubMed] [Google Scholar]
- 6.Suissa S, Ariel A. US Food and Drug Administration-mandated trials of long-acting beta-agonists safety in asthma: will we know the answer? Chest. 2013;143:1208–13. doi: 10.1378/chest.12-2881. [DOI] [PubMed] [Google Scholar]
- 7.Hirota S, Trimble N, Pertens E, Janssen LJ. Intracellular Cl- fluxes play a novel role in Ca2+ handling in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2006;290:L1146–53. doi: 10.1152/ajplung.00393.2005. [DOI] [PubMed] [Google Scholar]
- 8.Janssen LJ, Sims SM. Ca(2+)-dependent Cl- current in canine tracheal smooth muscle cells. Am J Physiol. 1995;269:C163–9. doi: 10.1152/ajpcell.1995.269.1.C163. [DOI] [PubMed] [Google Scholar]
- 9.Bao R, Lifshitz LM, Tuft RA, Bellve K, Fogarty KE, ZhuGe R. A close association of RyRs with highly dense clusters of Ca2+-activated Cl- channels underlies the activation of STICs by Ca2+ sparks in mouse airway smooth muscle. J Gen Physiol. 2008;132:145–60. doi: 10.1085/jgp.200709933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caputo A, Caci E, Ferrera L, Pedemonte N, Barsanti C, Sondo E, Pfeffer U, Ravazzolo R, Zegarra-Moran O, Galietta LJ. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science. 2008;322:590–4. doi: 10.1126/science.1163518. [DOI] [PubMed] [Google Scholar]
- 11.Schroeder BC, Cheng T, Jan YN, Jan LY. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell. 2008;134:1019–29. doi: 10.1016/j.cell.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galietta LJ. The TMEM16 protein family: a new class of chloride channels? Biophys J. 2009;97:3047–53. doi: 10.1016/j.bpj.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gallos G, Remy KE, Danielsson J, Funayama H, Fu XW, Chang HY, Yim P, Xu D, Emala CW., Sr Functional expression of the TMEM16 family of calcium-activated chloride channels in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2013;305:L625–34. doi: 10.1152/ajplung.00068.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang F, Zhang H, Wu M, Yang H, Kudo M, Peters CJ, Woodruff PG, Solberg OD, Donne ML, Huang X, Sheppard D, Fahy JV, Wolters PJ, Hogan BL, Finkbeiner WE, Li M, Jan YN, Jan LY, Rock JR. Calcium-activated chloride channel TMEM16A modulates mucin secretion and airway smooth muscle contraction. Proc Natl Acad Sci U S A. 2012;109:16354–9. doi: 10.1073/pnas.1214596109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Namkung W, Phuan PW, Verkman AS. TMEM16A inhibitors reveal TMEM16A as a minor component of calcium-activated chloride channel conductance in airway and intestinal epithelial cells. J Biol Chem. 2011;286:2365–74. doi: 10.1074/jbc.M110.175109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oh SJ, Hwang SJ, Jung J, Yu K, Kim J, Choi JY, Hartzell HC, Roh EJ, Lee CJ. MONNA, a Potent and Selective Blocker for TMEM16A/Anoctamin-1. Mol Pharmacol. 2013;84:726–735. doi: 10.1124/mol.113.087502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar S, Namkung W, Verkman AS, Sharma PK. Novel 5-substituted benzyloxy-2-arylbenzofuran-3-carboxylic acids as calcium activated chloride channel inhibitors. Bioorg Med Chem. 2012;20:4237–44. doi: 10.1016/j.bmc.2012.05.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Namkung W, Yao Z, Finkbeiner WE, Verkman AS. Small-molecule activators of TMEM16A, a calcium-activated chloride channel, stimulate epithelial chloride secretion and intestinal contraction. FASEB J. 2011;25:4048–62. doi: 10.1096/fj.11-191627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ellis JL, Undem BJ. Role of cysteinyl-leukotrienes and histamine in mediating intrinsic tone in isolated human bronchi. Am J Respir Crit Care Med. 1994;149:118–22. doi: 10.1164/ajrccm.149.1.8111568. [DOI] [PubMed] [Google Scholar]
- 20.Mukherjee S, Trice J, Shinde P, Willis RE, Pressley TA, Perez-Zoghbi JF. Ca2+ oscillations, Ca2+ sensitization, and contraction activated by protein kinase C in small airway smooth muscle. J Gen Physiol. 2013;141:165–78. doi: 10.1085/jgp.201210876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gosens R, Stelmack GL, Dueck G, McNeill KD, Yamasaki A, Gerthoffer WT, Unruh H, Gounni AS, Zaagsma J, Halayko AJ. Role of caveolin-1 in p42/p44 MAP kinase activation and proliferation of human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2006;291:L523–34. doi: 10.1152/ajplung.00013.2006. [DOI] [PubMed] [Google Scholar]
- 22.Townsend EA, Zhang Y, Xu C, Wakita R, Emala CW. Active components of ginger potentiate beta-agonist-induced relaxation of airway smooth muscle by modulating cytoskeletal regulatory proteins. Am J Respir Cell Mol Biol. 2014;50:115–24. doi: 10.1165/rcmb.2013-0133OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Danielsson J, Yim P, Rinderspacher A, Fu XW, Zhang Y, Landry DW, Emala CW. Chloride channel blockade relaxes airway smooth muscle and potentiates relaxation by beta-agonists. Am J Physiol Lung Cell Mol Physiol. 2014;307:L273–82. doi: 10.1152/ajplung.00351.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shmigol AV, Eisner DA, Wray S. Simultaneous measurements of changes in sarcoplasmic reticulum and cytosolic. J Physiol. 2001;531:707–13. doi: 10.1111/j.1469-7793.2001.0707h.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murtazina DA, Chung D, Ulloa A, Bryan E, Galan HL, Sanborn BM. TRPC1, STIM1, and ORAI influence signal-regulated intracellular and endoplasmic reticulum calcium dynamics in human myometrial cells. Biol Reprod. 2011;85:315–26. doi: 10.1095/biolreprod.111.091082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kojima A, Kitagawa H, Omatsu-Kanbe M, Matsuura H, Nosaka S. Sevoflurane protects ventricular myocytes against oxidative stress-induced cellular Ca2+ overload and hypercontracture. Anesthesiology. 2013;119:606–20. doi: 10.1097/ALN.0b013e318292ee52. [DOI] [PubMed] [Google Scholar]
- 27.Woods BD, Sladen RN. Perioperative considerations for the patient with asthma and bronchospasm. Br J Anaesth. 2009;103 (Suppl 1):i57–65. doi: 10.1093/bja/aep271. [DOI] [PubMed] [Google Scholar]
- 28.Miedinger D, Neukomm E, Chhajed PN, Schnyder A, Naef M, Ackermann M, Leuppi JD. The use of the Asthma Control Test in general practice and its correlation with asthma control according to the GINA guidelines. Curr Med Res Opin. 2011;27:2301–8. doi: 10.1185/03007995.2011.630722. [DOI] [PubMed] [Google Scholar]
- 29.Abramson MJ, Walters J, Walters EH. Adverse effects of beta-agonists: are they clinically relevant? Am J Respir Med. 2003;2:287–97. doi: 10.1007/BF03256657. [DOI] [PubMed] [Google Scholar]
- 30.Zhang CH, Li Y, Zhao W, Lifshitz LM, Li H, Harfe BD, Zhu MS, ZhuGe R. The transmembrane protein 16A Ca(2+)-activated Cl- channel in airway smooth muscle contributes to airway hyperresponsiveness. Am J Respir Crit Care Med. 2013;187:374–81. doi: 10.1164/rccm.201207-1303OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oh SJ, Hwang SJ, Jung J, Yu K, Kim J, Choi JY, Hartzell HC, Roh EJ, Lee CJ. MONNA, a potent and selective blocker for transmembrane protein with unknown function 16/anoctamin-1. Mol Pharmacol. 2013;84:726–35. doi: 10.1124/mol.113.087502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scott JP, Peters-Golden M. Antileukotriene agents for the treatment of lung disease. Am J Respir Crit Care Med. 2013;188:538–44. doi: 10.1164/rccm.201301-0023PP. [DOI] [PubMed] [Google Scholar]
- 33.Ebina M, Yaegashi H, Chiba R, Takahashi T, Motomiya M, Tanemura M. Hyperreactive site in the airway tree of asthmatic patients revealed by thickening of bronchial muscles. A morphometric study. Am Rev Respir Dis. 1990;141:1327–32. doi: 10.1164/ajrccm/141.5_Pt_1.1327. [DOI] [PubMed] [Google Scholar]
- 34.Ebina M, Yaegashi H, Takahashi T, Motomiya M, Tanemura M. Distribution of smooth muscles along the bronchial tree. A morphometric study of ordinary autopsy lungs. Am Rev Respir Dis. 1990;141:1322–6. doi: 10.1164/ajrccm/141.5_Pt_1.1322. [DOI] [PubMed] [Google Scholar]
- 35.Terzano C, Laurendi G, Capoccetta G, Rapisarda M, Pacilio R, Petroianni A. The smooth muscle and airway hyperresponsiveness. Eur Rev Med Pharmacol Sci. 2003;7:9–26. [PubMed] [Google Scholar]
- 36.Burgel PR. The role of small airways in obstructive airway diseases. Eur Respir Rev. 2011;20:23–33. doi: 10.1183/09059180.00010410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morgan SJ, Deshpande DA, Tiegs BC, Misior AM, Yan H, Hershfeld AV, Rich TC, Panettieri RA, An SS, Penn RB. beta-Agonist-mediated relaxation of airway smooth muscle is protein kinase A-dependent. J Biol Chem. 2014;289:23065–74. doi: 10.1074/jbc.M114.557652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Billington CK, Ojo OO, Penn RB, Ito S. cAMP regulation of airway smooth muscle function. Pulm Pharmacol Ther. 2013;26:112–20. doi: 10.1016/j.pupt.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Janssen LJ, Killian K. Airway smooth muscle as a target of asthma therapy: history and new directions. Respir Res. 2006;7:123. doi: 10.1186/1465-9921-7-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Real time image of the contraction of a mouse peripheral airway in lung slice. Methacholine (MCh) and benzbromarone (Benzb) are present when the label appears in the video otherwise the lung slice is superfused with HBSS alone. Slice is contracted with MCh, and subsequently relaxed by addition of benzbromarone in the presence of MCh. Benzbromarone was then washed out with subsequent re-contraction with MCh, and finally MCh washed to return to resting state.