

Abstract
Hypoxia is known to promote vasodilation of coronary vessels through several mediators including cardiac-derived adenosine and endothelium-derived prostanoids and nitric oxide. To date, the impact of endogenous glycogen depletion in vascular smooth muscle and the resultant alterations in cellular energy state (e.g., AMP-activated protein kinase, AMPK) on the contractile response to G protein-coupled receptor agonists (e.g., serotonin, 5-HT) has not yet been studied. In the present study, ex vivo exposure of endothelium-denuded human saphenous vein rings to hypoxic and glucose-deprived conditions during KCl-induced contractions for 30 min resulted in a marked depletion of endogenous glycogen by ~80% (from ~1.78 μmol/g under normoxia to ~0.36 μmol/g under hypoxia). Importantly, glycogen-depleted HSV rings, which were maintained under hypoxia/reoxygenation and glucose-deprived conditions, exhibited significant increases in basal AMPK phosphorylation (~6-fold ↑) and 5-HT-induced AMPK phosphorylation (~19-fold ↑) with an accompanying suppression of 5-HT-induced maximal contractile response (~68% ↓), compared with respective controls. Exposure of glycogen-depleted HSV rings to exogenous D-glucose, but not the inactive glucose analogs, prevented the exaggerated increase in 5-HT-induced AMPK phosphorylation and restored 5-HT-induced maximal contractile response. In addition, the ability of exogenous D-glucose to rescue cellular stress and impaired contractile function occurred through GLUT1-mediated but insulin/GLUT4-independent mechanisms. Together, the present findings from clinically-relevant human saphenous vein suggest that the loss of endogenous glycogen in vascular smooth muscle and the resultant accentuation of AMPK phosphorylation by GPCR agonists may constitute a yet another mechanism of metabolic vasodilation of coronary vessels in ischemic heart disease.
Keywords: vascular smooth muscle, hypoxia, glycogen, AMPK, serotonin
1. Introduction
Glycogen, a principal storage form of carbohydrate, serves as a readily mobilizable source of energy to control vital functions in striated muscles (e.g., skeletal muscle and myocardium) and vascular smooth muscle even under anaerobic conditions [1–4]. Dysregulation of glycogen turnover manifests in skeletal muscle and cardiovascular abnormalities, and it results from deficiency of glycogen-metabolizing enzymes [5] or mutation of AMP-activated protein kinase γ2/3 (AMPK γ2/3) subunit [6, 7]. For instance, in patients with inherited deficiency of lysosomal acid α-glucosidase, failure of glycogen breakdown results in lysosomal glycogen storage disease (type II, Pompe disease), which is characterized by muscular hypotonia, cardiomegaly, aortic/basilar artery aneurysm, and aortic stiffness [8–10]. In patients with naturally occurring mutations in AMPKγ2 subunit (encoded by PRKAG2 gene), dysregulated metabolism results in cytoplasmic glycogen accumulation and hypertrophic cardiomyopathy [7]. While the genetic defects in these intracellular enzymes culminate in glycogen storage disease phenotype, metabolic stresses such as hypoxia and contraction promote glycogen depletion in striated and smooth muscles [2, 4, 11, 12]. Studies with striated muscles have revealed the impact of glycogen depletion toward ATP loss, AMPK activation, and altered functional response [11, 12]. Nevertheless, the relationship between glycogen content and AMPK activation and their regulatory roles toward smooth muscle contraction have thus far not been examined in vascular beds from any species, including the conduits used in patients undergoing coronary artery bypass grafting (CABG) surgery, under normoxic or hypoxic conditions.
Endogenous glycogen serves as an ‘energy reserve’ for vascular smooth muscle contraction [2, 3]. Seminal studies by several investigators over the past three decades have provided compelling evidence for active glycogen turnover (glycogen synthesis and its degradation) in vascular smooth muscle using porcine carotid artery as a model system [2, 3, 13, 14]. The metabolic pathways such as glycogenolysis and glycolysis contribute to the formation of ATP, an ‘immediate energy source’ for contracting smooth muscle. Of importance, hypoxic conditions result in a marked depletion of glycogen in arterial smooth muscle [2, 4, 15]. While glycogen depletion is known to decrease ‘cellular energy state’ (↓ATP) in vascular smooth muscle [4], it remains unknown as to how this will influence the activation state of AMPK (‘an energy sensor’) during agonist-induced contractions. Notably, AMPK functions as a sensor of cellular energy reserve (glycogen content) and cellular energy state (AMP/ATP or ADP/ATP ratio) [16, 17]. This is evident from the structural features of heterotrimeric AMPK complexes that comprise a catalytic subunit (α) and two regulatory subunits (β and γ). AMPKβ subunit contains a glycogen-binding domain, the association of which to glycogen results in AMPK inhibition [16, 18]. Stress-induced glycogen depletion and ATP loss would promote AMPK activation. The increase in AMPK activity occurs due to the replacement of ATP on AMPKγ subunit by ADP and AMP, and the consequent increase in AMPKα phosphorylation (at Thr172 residue) and allosteric activation [17]. Thus, it is important to determine the critical relationship between glycogen content and AMPK activation in intact vascular smooth muscle during agonist-induced contractions under normoxic and hypoxic conditions.
Under metabolic stress, exogenous glucose (an energy substrate) has been shown to suppress AMPK activation in striated muscles [19, 20]. The uptake of glucose in VSMCs occurs through plasma membrane-localized glucose transporter-1 (GLUT1) or insulin-dependent GLUT4-mediated mechanism [21–24]. While glucose uptake and glycolysis would promote ATP formation to diminish AMPK activity, the interplay between endogenous glycogen and exogenous glucose toward altered AMPK phosphorylation remains to be examined in vascular smooth muscle during agonist-induced contraction.
Recently, we and several other investigators have shown that metabolic stress [25], metformin [26], and AICAR [26–28] inhibits agonist-induced smooth muscle contraction through AMPK activation in endothelium-denuded arterial ring preparations from rodent or porcine models. In addition, previous studies have shown that serotonin, a platelet-derived vasoactive agonist, dilates coronary resistance vessels but induces constriction of coronary conduit vessels [cited in [29]. In the present study, we hypothesize that in saphenous vein conduits from human subjects, glycogen depletion by acute hypoxia activates AMPK thereby compromising serotonin-induced smooth muscle contraction. The objectives of the present study are to determine: i) the effects of hypoxia on glycogen content, AMPK phosphorylation, and serotonin-induced smooth muscle contraction; and ii) the effects of hypoxia-reoxygenation on basal and serotonin-induced changes in AMPK phosphorylation and contraction in the absence or presence of exogenous glucose during isometric contractions in human saphenous vein.
2. Materials and methods
2.1. Materials
Serotonin (5-hydroxytryptamine hydrochloride), phenylephrine hydrochloride, acetylcholine chloride, D-glucose, L-glucose, and 2-deoxy-D-glucose were purchased from Sigma-Aldrich (St. Louis, MO). The primary antibodies for AMPKα, phospho-AMPKαThr172, ERK1/2, phospho-ERK1/2, Akt, and phospho-AktSer473 were purchased from Cell Signaling Technology (Danvers, MA). The primary antibodies for GLUT1, GLUT4, and β-actin were purchased from Abcam (Cambridge, MA). HRP-conjugated goat anti-rabbit secondary antibody was purchased from Bio-Rad (Hercules, CA). All other chemicals were from Fisher Scientific (Fair Lawn, NJ) or Sigma-Aldrich (St. Louis, MO).
2.2. Human subjects and saphenous vein specimens
The study protocol was approved by the Institutional Review Board at the Georgia Regents University, Charlie Norwood VA Medical Center, Penn State Hershey College of Medicine, and University of Georgia. According to the approved protocol, the leftover saphenous vein specimens from subjects undergoing coronary artery bypass grafting (CABG) surgery were used for the current study. Saphenous vein specimens, which were obtained from a total of 23 subjects, were transported from the operating room to the laboratory in ice-cold Krebs buffer. The specimens from subjects with a history of diabetes were excluded in this study.
2.3. Animals and isolation of thoracic aorta
All animal experiments were performed in accordance with the Charlie Norwood Veterans Affairs Medical Center Institutional Animal Care and Use Committee guidelines, and were approved by the committee. Adult male Wistar rats (Charles River Laboratories, Wilmington, MA) were maintained in a room at a controlled temperature of 23°C with a 12:12-hr dark-light cycle. They had free access to water and standard rodent chow diet. On the day of contractility studies, rats were sacrificed followed by isolation of thoracic aorta as described [26].
2.4. Preparation of human saphenous vein rings and isometric tension measurements
Human saphenous vein (HSV) specimens were immediately placed in a petri-dish containing ice-cold oxygenated Krebs-Henseleit bicarbonate (KHB) buffer (118 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 2.5 mM CaCl2, 25 mM NaHCO3, and 11 mM glucose; pH 7.4). HSV was carefully cleaned free of any adherent fat and connective tissue. Endothelium was removed by gently rubbing the luminal surface using a polyethylene tube. HSV was then cut into 2-mm rings and mounted in the organ bath system for isometric tension measurements, as described previously [26].
After passing the stainless steel wires through the lumen, HSV ring preparations were suspended from the isometric force displacement transducers (Model FT03; Grass Technologies, West Warwick, RI) and kept immersed in 10 ml of KHB buffer in the organ bath system (8 chambers, Radnoti Glass Technology, Monrovia, CA). The KHB buffer was bubbled continuously with a gas mixture of 95% O2 and 5% CO2 and maintained at 37°C. Isometric tension was measured as changes in millinewtons (mN) of force using Octal Bridge Amplifier and PowerLab 8/35 data-acquisition system and recorded using LabChart Pro V7 software (AD Instruments, Colorado Springs, CO).
Each ring was gradually stretched to a basal tension of 9.8 mN (1 g) over a period of ~1.5 hour and then equilibrated for an additional 2 hours. During this equilibration period, KHB buffer was changed every 30 min. After equilibration, the contractile response to 80 mM KCl was determined followed by washes with KHB buffer until the passive basal tension is restored. HSV ring preparations from 16 subjects (out of a total of 23 subjects), which showed a contractile response of > 1 g tension with 80 mM KCl, were included in the study. The remaining HSV ring preparations from 7 subjects were discarded. To verify endothelial denudation, HSV rings were pre-contracted with phenylephrine (1 μM) until the attainment of plateau phase, followed by addition of acetylcholine (1 μM). The absence of a relaxant response to acetylcholine confirmed the denudation of HSV rings. Subsequently, HSV rings were washed with KHB buffer until the passive basal tension is restored.
Rat aortic ring preparations were subjected to endothelial denudation and equilibration in a similar manner.
2.5. Experimental protocols for isometric tension measurements in HSV rings under normoxia or hypoxia/reoxygenation conditions (Fig. 1)
Fig. 1.

Schematic of the protocols that utilize endothelium-denuded ring preparations ex vivo to determine: A) the effects of hypoxia on glycogen content under glucose-deprived conditions (0 mM D-glucose); and B) effects of H/R on basal and 5-HT-induced changes in smooth muscle contractility and AMPK/ERK/Akt phosphorylation in the presence of: i) 0 mM D-glucose, ii) 5.5 mM D-glucose or glucose analogs, or iii) 5.5 mM D-glucose with insulin and/or GLUT4 inhibitor, as described under ‘Materials and methods’. KCl, potassium chloride; H/R, hypoxia/reoxygenation; 5-HT, serotonin; IB, immunoblotting.
Endothelium-denuded HSV ring preparations were maintained in KHB buffer containing 5.5 mM D-glucose under normoxic conditions (95% O2 and 5% CO2). In parallel, HSV rings were subjected to hypoxia (95% N2/5% CO2) for 30 min. During this 30 min hypoxia, HSV rings were stimulated with 80 mM KCl initially in the presence of 5.5 mM D-glucose (20 min) and then under glucose-deprived conditions (10 min), as described previously [2, 15]. To determine the changes in endogenous glycogen content after acute hypoxia (versus normoxia), glycogen measurements were performed in HSV tissues as described in section 2.6.
In addition, after acute hypoxia, HSV rings were subjected to reoxygenation (95% O2 and 5% CO2) for 30 min under the following conditions: i) 0 mM D-glucose, ii) 5.5 mM D-glucose or glucose analogs, iii) 5.5 mM D-glucose with insulin, or iv) 5.5 mM D-glucose with insulin and GLUT4 inhibitor, as described in the respective figure legends. HSV rings were then challenged with cumulative concentrations of serotonin (5-HT, 10−9 to 10−4 M). 5-HT was used in the present study because it has previously been shown to induce an increase in vessel tension higher than norepinephrine [30]. After contractility studies, the respective rings were subjected to extraction procedures for immunoblot analysis as described in sections 2.7. and 2.8.
Select studies included the use of rat aortic ring preparations for isometric tension measurements and immunoblot analysis, as described in Fig. 2.
Fig. 2.

Effects of hypoxia/reoxygenation (H/R) on smooth muscle contractility and AMPK phosphorylation in rat aortic smooth muscle ex vivo. Endothelium-denuded rat aortic rings were maintained under normoxic or H/R conditions as described under ‘Materials and methods’. A–B) The changes in Emax and pEC50 values for 5-HT-induced contractions were determined. The data shown are the mean ± SEM values obtained with aortic rings from 3 rats. ##p < 0.01, ###p < 0.001 compared with control (+ 5-HT, Norm); **p < 0.01 compared with 0 mM D-glucose (+ 5-HT, H/R). The changes in AMPKαThr172 phosphorylation were then determined. The representative immunoblots for phospho-AMPKαThr172, AMPKα, and β-actin are shown in panel C.
2.6. Measurement of glycogen content in HSV tissues after acute hypoxia
After acute hypoxia, HSV rings were immediately rinsed in ice-cold distilled water, blotted to dryness, weighed, snap-frozen in liquid nitrogen, and stored at −80°C until analysis. The total glycogen content in the respective HSV tissues was determined using the glycogen assay kit (Sigma) according to the manufacturer’s protocol. In brief, HSV tissues were homogenized in 200 μl of ice-cold distilled water, using TissueLyser LT (Qiagen, Valencia, CA) at a setting of 50 Hz for 10 min with samples being placed on ice intermittently. The homogenates were boiled for 5 min to inactivate the enzymes and then centrifuged at 13,000 x g for 5 min. The sample supernatants (2 μl) and glycogen standards (0.04 – 0.4 μg) were made up to 50 μl using hydrolysis assay buffer in a 96-well fluorescence assay plate. Hydrolysis enzyme mix (1 μl) was added to each of the samples and glycogen standards, and mixed well. In parallel, hydrolysis enzyme mix was not added to another replicate of each of the samples to eliminate the background signal due to glucose. After 30 min incubation at room temperature, 50 μl of development enzyme mix (48.8 μl development buffer + 1 μl development enzyme mix + 0.2 μl fluorescent peroxidase substrate) was added to each of the hydrolyzed or non-hydrolyzed reaction samples and standards, and mixed well. The reaction plate was incubated for 30 min at room temperature and the fluorescence intensity (λex = 535/λem = 587) was measured using a SpectraMax M3 multi-mode microplate reader (Molecular Devices, Sunnyvale, CA). The total tissue glycogen content was expressed as μmol glucosyl units/g tissue weight.
2.7. Extraction and quantification of proteins in HSV tissues
After contractility studies, HSV rings were immediately rinsed in ice-cold fresh phosphate-buffered saline, blotted to dryness, snap-frozen in liquid nitrogen, and stored at −80°C until analysis. HSV ring tissues were then thawed and homogenized in 100 μl RIPA lysis buffer containing protease and phosphatase inhibitors (Thermo Scientific, Rockford, IL) using TissueLyser LT (Qiagen, Valencia, CA) at a setting of 50 Hz for 10 min with samples being placed on ice intermittently. The homogenates were incubated at 4°C for 1 hour on a rotator and centrifuged at 1000 x g for 10 min at 4°C to remove tissue debris. The supernatants were mixed with 2x laemmli sample buffer at a ratio of 1:1 followed by heating at 67.5°C for 10 min. Proteins were quantified using Bio-Rad DC assay kit (Bio-Rad, Hercules, CA).
2.8. Immunoblot analysis
HSV tissue samples (20 μg protein each) were electrophoresed using pre-cast 4–12% NuPage mini-gels (Life Technologies, Carlsbad, CA), and the resolved proteins were transferred to nitrocellulose membranes (Hybond C, GE Healthcare Life Sciences, Piscataway, NJ). The membranes were blocked in 5% bovine serum albumin, and probed with the primary antibodies specific for phospho-AMPKαThr172, AMPKα, phospho-ERK1/2, ERK1/2, phospho-AktSer473, Akt, GLUT1, or GLUT4. β-actin was used as an internal control. After extensive washes, the immunoreactivity was detected using specific HRP-conjugated secondary antibodies followed by enhanced chemiluminescence (GE Healthcare Life Sciences). The protein bands were quantified by densitometric analysis using Image J.
2.9. Total RNA extraction/quantification and assessment of RNA quality/integrity
HSV specimens were submerged in 10 volumes of RNAlater RNA stabilization reagent solution and incubated overnight at 4°C. Subsequently, tissues were removed from the reagent and transferred to −80°C for storage. HSV tissues were then homogenized in QIAzol Lysis Reagent containing phenol and guanidine thiocyanate using TissueLyser LT (Qiagen, Valencia, CA) at a setting of 50 Hz for 10 min with samples being placed on ice intermittently. Total RNA was extracted using RNeasy universal mini kit (Qiagen, Valencia, CA). The isolated RNAs were treated with RNase-free DNase I (Qiagen, Valencia, CA) to remove contamination due to genomic DNA. 1 μl each of the RNA samples was applied directly onto the system optics of NanoDrop 2000 spectrophotometer (Thermo Scientific, Wilmington, DE) for RNA quantification at 260 nm followed by assessment of its purity. The purity of RNA samples was verified by measurement of absorbance ratios at 260/280 nm and 260/230 nm. The absorbance ratios of the RNA samples were > 2.0 at 260/280 nm and at 260/230 nm, thereby precluding contaminants due to proteins and thiocyanates/organic compounds, respectively. The integrity of the RNA was verified by conventional agarose gel electrophoresis and also by Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA), as described in our previous studies [23].
300 ng of total RNA isolated from HSV tissues was then reverse transcribed to cDNA by Superscript First Strand RT-PCR system (Life Technologies, Grand island, NY) using oligo(dT) primers. Quantitative real-time PCR (qPCR) analysis was performed using Applied Biosystems 7900HT Fast Real-Time PCR system and QuantiTect SYBR Green PCR kit (Qiagen, Valencia, CA) with the primer sets, as described [23]. A SYBR Green master mix of the following reaction components was prepared to the indicated end-concentration: 0.3 μl forward primer (0.3 μM), 0.3 μl reverse primer (0.3 μM), 5.0 μl 2X QuantiTect SYBR Green PCR master mix (Qiagen), 3.4 μl RNase-DNase free water, and 1 μl cDNA as PCR template to obtain a final reaction volume of 10 μl. qPCR thermal conditions were: 95°C for 15 min, 40 cycles of 94°C for 15 sec, 53°C for 30 sec, and 72°C for 30sec, followed by a default melting curve program. The specificity of qPCR products was documented with Applied Biosystems disassociation curve analysis, which resulted in single product-specific melting temperatures. In addition, electrophoresis on a 2% agarose gel resulted in a single product with the desired length for a specific primer pair. As a positive control, human reference cDNA (Clontech, Mountain View, CA) was used to verify the amplified PCR products for the chosen genes that included five class I glucose transporter (GLUT) isoforms, and β2-microglobulin (B2M), as described [23].
2.10. Statistical analysis
Results are expressed as means ± SEM values. The n value represents the number of subjects. To determine Emax and EC50 values (−log EC50 or pEC50), nonlinear regression analysis was performed using GraphPad Prism software (version 6.01, GraphPad Software, Inc., La Jolla, CA). Statistical analyses of the data among groups were performed by paired t-test (parametric) or one-way repeated measures ANOVA followed by Bonferroni t test. Values of p < 0.05, 0.01, 0.001 were considered statistically significant.
3. Results
3.1. Exposure of rat aortic rings to hypoxia/reoxygenation (H/R) and glucose-deprived conditions results in the suppression of 5-HT-induced smooth muscle contractility with an accompanying exaggerated AMPK phosphorylation: reversal by exogenous D-glucose
Previous studies with porcine carotid artery have shown that hypoxic conditions lead to a significant depletion of endogenous glycogen by ~50–60% [2, 15]. Nevertheless, the consequence of glycogen depletion on the contractile response induced by G-protein coupled receptor agonists (e.g., serotonin, 5-HT) has not been examined in vascular beds from any species.
In our initial studies, endothelium-denuded rat aortic rings were subjected to hypoxia (as described in section 2.5, Fig. 1) to deplete endogenous glycogen [2, 15]. Aortic rings were then maintained under reoxygenated and glucose-deprived conditions for 30 min. As shown in Fig. 2A–B, subsequent stimulation with 5-HT resulted in a significant decrease in maximal contractility by 41.4 ± 1.6 % (versus 100% control under normoxic conditions, n = 3, p < 0.001) with significant changes in pEC50 values (5.4 ± 0.1 versus control value of 6.0 ± 0.1, p < 0.01). Inclusion of exogenous D-glucose (5.5 mM) during reoxygenation conditions led to a marked restoration of 5-HT-induced maximal contractile response (p < 0.05).
After contractility studies, aortic rings were subjected to immunoblot analysis. As shown in Fig. 2C, the rings that were exposed to H/R with glucose-deprived conditions showed an exaggerated AMPK phosphorylation by 15.4-fold (lane 3), compared with normoxic control conditions (lane 1; normalized to 1.0). Inclusion of exogenous D-glucose (5.5 mM) during H/R resulted in 3.9-fold increase in AMPK phosphorylation (lane 2) compared with normoxia (lane 1). Upon stimulation with 5-HT, there was a further enhancement in AMPK phosphorylation to 29-fold under H/R conditions with glucose deprivation (lane 6). Similar results were obtained with 5.5 mM 2-deoxyglucose, an inactive analog of D-glucose (lane 7). However, inclusion of exogenous D-glucose (5.5 mM) during H/R followed by 5-HT stimulation led to 6.9-fold increase in AMPK phosphorylation (lane 5). Thus, exposure of normal rat aortic smooth muscle to H/R and glucose-deprived conditions resulted in exaggerated AMPK phosphorylation before or after 5-HT stimulation; and this enhancement in AMPK phosphorylation was prevented by inclusion of exogenous D-glucose.
As a follow-up of these initial findings with normal rat aorta, we performed all subsequent studies with human saphenous vein (HSV) specimens obtained after CABG surgery.
3.2. Saphenous vein specimens and the demographics of subjects
Of the saphenous vein specimens obtained from 21 subjects, the specimens from 16 subjects were used for contractility studies as described in section 2.4. The demographics of these 16 subjects are shown in Table 1. While all subjects were nondiabetic, they had the following risk factors of cardiovascular disease: hypertension (~69%); dyslipidemia (~81%); and smoking (~50%).
Table 1.
Demographics of subjects with no history of diabetes
| Number of subjects, n | 16 |
| Age (years) | 64.5 ± 1.9 |
| Gender (male/female) | 13 / 3 |
| Race (C/AA/H) | 13 / 2 / 1 |
| BMI (Kg/m2) | 27.3 ± 1.4 |
| Height (cm) | 174.6 ± 2.3 |
| Weight (Kg) | 83.7 ± 5.3 |
| Hypertension, n (%) | 11 (68.75%) |
| Dyslipidemia, n (%) | 13 (81.25%) |
| Smoking status, n (%) | 8 (50.0%) |
Note: C, Caucasian; AA, African American; H, Hispanic
3.3. Exogenous D-glucose deprivation does not affect 5-HT-induced smooth muscle contractility in HSV rings under normoxic conditions
As shown in Fig. 3A–B, stimulation of endothelium-denuded HSV rings with 5-HT in the absence of glucose led to an increase in contractility with an Emax value of 92.9 ± 4.4% (versus 100% control; n.s.) and pEC50 value of 6.7 ± 0.1 M (similar to control). Thus, acute exclusion of exogenous D-glucose did not affect smooth muscle contractile response in HSV rings under normoxic conditions.
Fig. 3.
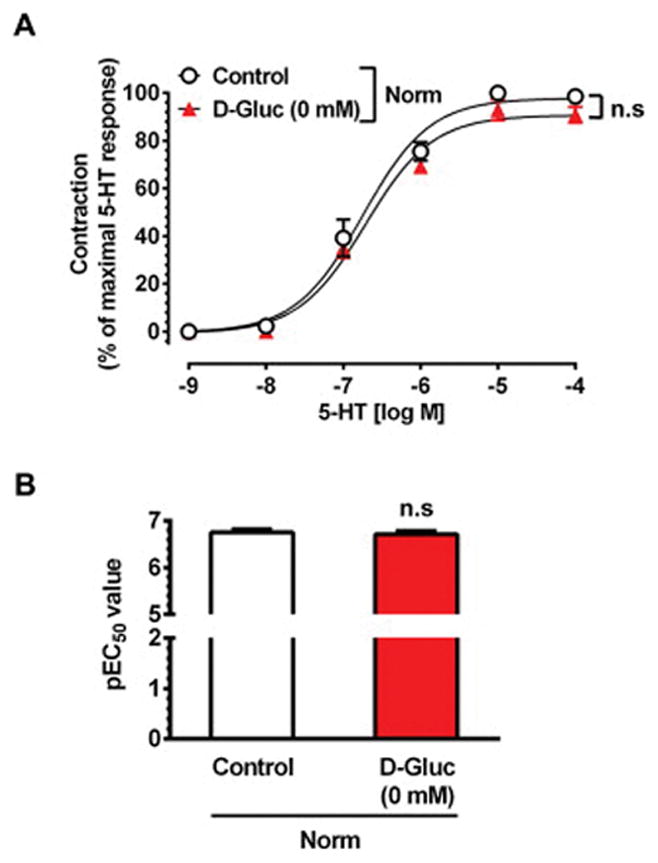
Effect of exogenous D-glucose deprivation on 5-HT-induced smooth muscle contractility in HSV rings under normoxic conditions. Endothelium-denuded HSV rings were equilibrated in KHB buffer (95% O2/5% CO2) containing 5.5 mM D-glucose for 2 hours, and then maintained in the presence (○, control) or absence (▲) of 5.5 mM D-glucose for 30 min. Subsequently, the contractile response to cumulative concentrations of 5-HT were determined. The data shown are the mean ± SEM values obtained with HSV rings from 3 subjects. n.s., not significant.
3.4. Endogenous glycogen depletion by hypoxia suppresses 5-HT-induced smooth muscle contractility in HSV rings
Next, endothelium-denuded HSV rings were exposed to hypoxic conditions as described in 2.5 (Fig. 1). As shown in Fig. 4A, this led to a marked depletion of endogenous glycogen content by ~79.8% (from 1.78 ± 0.04 μmol glucosyl units/g tissue weight under normoxia to 0.36 ± 0.08 μmol glucosyl units/g tissue weight under hypoxia, n = 5, p < 0.001). After glycogen depletion, HSV rings were maintained under reoxygenated and glucose-deprived conditions for 30 min. Fig. 4B–C shows that subsequent exposure to 5-HT led to a significant decrease in maximal contractility by 68.5 ± 6.0 % (versus 100% control, n = 8, p < 0.001) without significant changes in pEC50 values (6.3 ± 0.2 versus control value of 6.8 ± 0.1, n.s.). Thus, glycogen depletion in HSV rings resulted in the suppression of agonist-induced smooth muscle contractility.
Fig. 4.

Effects of glycogen depletion by hypoxia on basal and 5-HT-induced changes in smooth muscle contractility and AMPK phosphorylation in HSV rings. A) Endothelium-denuded HSV rings were equilibrated in KHB buffer (95% O2/5% CO2) containing 5.5 mM D-glucose for 2 hours. A parallel set of HSV rings were maintained in KHB buffer (95% N2/5% CO2) containing 5.5 mM D-glucose and stimulated with 80 mM KCl for 20 min. This was followed by maintenance in glucose-free KHB buffer (95% N2/5% CO2) and stimulation with 80 mM KCl for 10 min, as described in Fig. 1. Control HSV rings included the maintenance of HSV rings in KHB buffer (95% O2/5% CO2) containing 5.5 mM D-glucose without KCl stimulation for similar time intervals. Glycogen contents in HSV rings were then determined, as described in ‘Materials and methods’. ###p < 0.001 compared with glycogen-enriched HSV (n = 5). B–C) Glycogen-depleted HSV rings were maintained in glucose-free KHB buffer 30 min. The contractile responses to 5-HT were determined in parallel with control rings. The changes in Emax and pEC50 values were then calculated. ###p < 0.001 compared with HSV rings maintained under control conditions (+ 5-HT) (n = 8). D–F) After contractility studies, the same aortic rings were subjected to immunoblot analysis using specific primary antibodies, as described under ‘Materials and methods’. The representative immunoblots are shown in panel D. The data shown in the bar graphs (panels E and F) are the mean ± SEM values (n = 4). *p < 0.05, **p < 0.01, ***p < 0.001 compared with normoxia (− 5-HT); ###p < 0.001 compared with normoxia (+ 5-HT); £££p < 0.001 compared with H/R (− 5-HT).
3.5. Endogenous glycogen depletion by hypoxia augments basal AMPK phosphorylation, which is accentuated upon 5-HT challenge in HSV rings
In vascular smooth muscle, glycogen depletion by hypoxia leads to decreased ATP level [4], which may enhance AMPK activity. To determine the likely reciprocal relationship between glycogen content and AMPK activation, HSV rings that were used for contractility studies (described in section 3.4.) were subjected to immunoblot analysis. Fig. 4D and E show that in glycogen-depleted HSV rings (H/R), there was an increase in basal AMPK phosphorylation by 6.3 ± 1.2-fold (p < 0.01, − 5-HT, lane 3) compared with control (− 5-HT, lane 1). 5-HT stimulation led to a robust increase in AMPK phosphorylation by 19.3 ± 1.4-fold (p < 0.001, + 5-HT, lane 4) compared with control (− 5-HT, lane 1). In parallel studies with normoxic conditions, 5-HT challenge led to an increase in AMPK phosphorylation by 2.5 ± 0.2-fold (p < 0.05, + 5-HT, lane 2) compared with control (− 5-HT, lane 1).
In conjunction with altered AMPK phosphorylation, we also examined the phosphorylation state of ERK since previous studies have shown that 5-HT-induced contractile response is in part mediated by ERK [31]. As shown in Fig. 4D and F, the basal or 5-HT-induced ERK phosphorylation did not differ significantly between normoxic and H/R conditions.
Thus, in glycogen depleted HSV, suppression of agonist-induced smooth muscle contractility was associated with a robust increase in AMPK phosphorylation.
3.6. Exogenous D-glucose, but not 2-deoxy-D-glucose or L-glucose, prevents the suppression of 5-HT-induced smooth muscle contractility in glycogen-depleted HSV rings
Activation of AMPK has been shown to promote ATP-generating catabolic pathways, including glucose uptake and glycolysis [32, 33]. Since glycogen depletion results in increased AMPK phosphorylation in HSV smooth muscle, we determined the effects of exogenous D-glucose on 5-HT-induced contractility. As shown in Fig. 5A–B, in glycogen-depleted HSV rings (H/R) maintained in glucose-deprived conditions, 5-HT stimulation led to a significant decrease in maximal contractility by 69.6 ± 5.7% (versus 100% control, p < 0.001) without significant changes in pEC50 values (6.3 ± 0.2 versus control value of 6.8 ± 0.1, n.s.). Importantly, inclusion of exogenous D-glucose under glycogen-depleted conditions (H/R) prevented the suppression of 5-HT-induced contractile response in HSV smooth muscle.
Fig. 5.
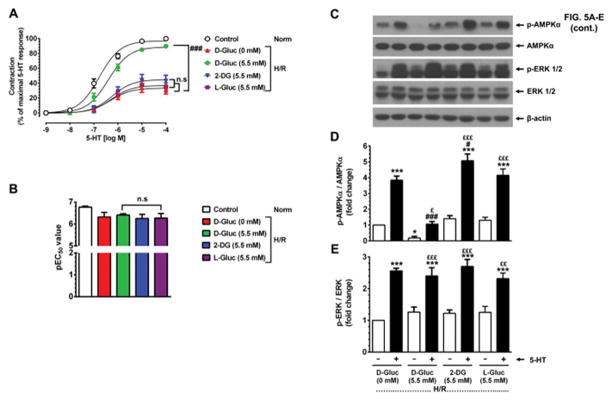
Effects of exogenous D-glucose versus 2-deoxy-D-glucose or L-glucose on basal and 5-HT-induced changes in smooth muscle contractility and AMPK phosphorylation in glycogen-depleted HSV rings. A–B) Glycogen-depleted HSV rings were exposed to no glucose (0 mM, ▲), 5.5 mM D-glucose (●), 5.5 mM 2-deoxy-glucose (▼), or 5.5 mM L-glucose (■) for 30 min in KHB buffer (95% O2/5% CO2). In parallel, control HSV rings were maintained in KHB buffer containing 5.5 mM D-glucose (○). The contractile responses to 5-HT were determined. The changes in Emax and pEC50 values were then calculated. n.s., not significant; ###p < 0.001 compared with 0 mM D-glucose (n = 9). C–E) After contractility studies, aortic rings were subjected to immunoblot analysis using specific primary antibodies. The representative immunoblots are shown in panel C. The data shown in the bar graphs (panels D and E) are the mean ± SEM values (n = 4). *p < 0.05, ***p < 0.001 compared with 0 mM D-glucose (− 5-HT); #p < 0.05, ###p < 0.001 compared with 0 mM D-glucose (+ 5-HT); £p < 0.05, ££p < 0.01, £££p < 0.001 compared with respective glucose analog (− 5-HT).
In a parallel set of HSV rings, 2-deoxy-D-glucose or L-glucose was included instead of D-glucose. While the uptake of 2-deoxyglucose results in its rapid phosphorylation to 2-deoxyglucose-6-phosphate (which inhibits glycolysis), L-glucose does not undergo phosphorylation by hexokinase. Hence, inclusion of 2-deoxy-glucose or L-glucose showed the diminution in 5-HT-induced contractile response similar to that observed under glucose-deprived conditions in glycogen-depleted HSV rings.
3.7. Exogenous D-glucose, but not 2-deoxy-D-glucose or L-glucose, prevents the increase in basal and 5-HT-stimulated AMPK phosphorylation in glycogen-depleted HSV rings
Previous studies have shown that in rats subjected to starvation for 48 hr, refeeding diminishes AMPK activity in liver and adipose tissue. In addition, exogenous glucose decreases AMPK activity ex vivo in skeletal muscle isolated from rats on 16–20 hr fast [19]. To determine whether exogenous glucose supplementation alters cellular energy state in vascular smooth muscle under glycogen-depleted conditions, immunoblot analysis was performed with HSV rings that were used for contractility studies (described in section 3.6.). As shown in Fig. 5C and D, exposure to exogenous D-glucose diminished basal AMPK phosphorylation by 0.8 ± 0.1-fold (p < 0.05, − 5-HT, lane 3) compared with glucose-deprived conditions (− 5-HT, lane 1). In addition, after exposure to exogenous D-glucose, stimulation with 5-HT resulted in an increase in AMPK phosphorylation by 1.1 ± 0.2-fold (n.s., + 5-HT, lane 4 versus lane 1) much lower than that observed under glucose-deprived conditions, which was 3.8 ± 0.3-fold (p < 0.001, + 5-HT, lane 2 versus lane 1). Unlike D-glucose, supplementation with either 2-deoxyglucose or L-glucose showed the changes in basal and 5-HT-induced AMPK phosphorylation similar to that observed under glucose-deprived conditions. Furthermore, inclusion of D-glucose, 2-deoxyglucose, or L-glucose did not result in significant changes in basal or 5-HT-induced ERK phosphorylation, compared with glucose-deprived condition (Fig. 5C and E). Thus, exposure to exogenously added D-glucose, but not its inactive analogs, during isometric contractions resulted in a marked suppression of exaggerated AMPK phosphorylation in HSV smooth muscle under glycogen-depleted conditions.
3.8. Exogenous D-glucose prevents the suppression of 5-HT-induced smooth muscle contractility through insulin/GLUT4-independent mechanism in glycogen-depleted HSV rings
Previously, we and several other investigators have shown that glucose uptake is mediated by GLUT1 in aortic smooth muscle cells [21–23]. In addition, insulin stimulation promotes GLUT4-mediated glucose uptake in different vascular beds [34]. We therefore examined whether insulin regulates D-glucose-induced changes in smooth muscle contractility in glycogen-depleted HSV rings in the absence of presence of GLUT4 inhibitor, indinavir. Previous studies have shown that indinavir at 10 to 25 μM concentration inhibits GLUT4- but not GLUT1-mediated glucose uptake [24, 35].
Fig. 6A–B shows that in glycogen-depleted HSV rings (H/R) maintained in glucose-deprived conditions, 5-HT stimulation led to a significant diminution in maximal contractility by 68.8 ± 4.5 (versus 100% control, p < 0.001) without changes in pEC50 values (6.5 ± 0.2 versus control value of 6.9 ± 0.1, n.s.). Exposure to exogenous D-glucose under glycogen-depleted conditions (H/R) prevented the suppression of 5-HT-induced contractile response. In a parallel set of glycogen-depleted HSV rings, inclusion of either insulin or GLUT4 inhibitor (indinavir)-plus-insulin along with exogenous D-glucose showed the changes in 5-HT-induced contractile response similar to that observed with exogenous D-glucose alone.
Fig. 6.

Effects of exogenous D-glucose, in the absence or presence of insulin and GLUT4 inhibitor, on basal and 5-HT-induced changes in smooth muscle contractility and AMPK phosphorylation in glycogen-depleted HSV rings. A–B) Glycogen-depleted HSV rings were exposed to no glucose (0 mM, ▲), 5.5 mM D-glucose (●), 100 nM insulin plus 5.5 mM D-glucose (◆), or 25 μM indinavir (GLUT4 inhibitor) plus 100 nM insulin plus 5.5 mM D-glucose (■) for 30 min in KHB buffer (95% O2/5% CO2). In parallel, control HSV rings were maintained in KHB buffer containing 5.5 mM D-glucose (○) for a similar time interval. The contractile responses to 5-HT were determined. The changes in Emax and pEC50 values were then calculated. ###p < 0.001 compared with 0 mM D-glucose (n = 3). C–F) After contractility studies, aortic rings were subjected to immunoblot analysis using specific primary antibodies. The representative immunoblots are shown in panel C. The data shown in the bar graphs (panels D, E, and F) are the mean ± SEM values (n = 3). *p < 0.05, ***p < 0.001 compared with 0 mM D-glucose (− 5-HT); ###p < 0.001 compared with 0 mM D-glucose (+ 5-HT); £p < 0.05, £££p < 0.001 compared with respective treatment conditions (− 5-HT); $$$p < 0.001 compared with insulin plus D-glucose (− 5-HT); ¢¢¢p < 0.001 compared with indinavir plus insulin plus D-glucose (− 5-HT).
3.9. Exogenous D-glucose prevents the increase in basal and 5-HT-stimulated AMPK phosphorylation independent of insulin and GLUT4 in glycogen-depleted HSV rings
Fig. 6C–D shows that exogenous D-glucose diminished basal AMPK phosphorylation by 0.8 ± 0.1-fold (p < 0.05, − 5-HT, lane 3) compared with glucose-deprived conditions (− 5-HT, lane 1). In addition, after exposure to exogenous D-glucose, stimulation with 5-HT led to an increase in AMPK phosphorylation by 1.1 ± 0.3-fold (n.s., + 5-HT, lane 4 versus lane 1) much lower than that observed under glucose-deprived conditions, which was 4.0 ± 0.5-fold (p < 0.001, + 5-HT, lane 2 versus lane 1). Supplementation of D-glucose with either insulin (lane 5 and 6) or indinavir-plus-insulin (lanes 7 and 8) showed the changes in basal and 5-HT-induced AMPK phosphorylation similar to that observed with exogenous D-glucose alone (lanes 3 and 4). Furthermore, exposure to insulin or indinavir-plus-insulin in the presence of D-glucose did not result in significant changes in basal or 5-HT-induced ERK phosphorylation, compared with glucose-deprived condition (Fig. 6C and E). Although insulin did not influence D-glucose regulation of AMPKαThr172 phosphorylation, it increased Akt phosphorylation that was further augmented by 5-HT challenge during isometric contractions (Fig. 6C and F). Thus, exposure to exogenously added D-glucose during isometric contractions led to a marked suppression of exaggerated AMPK phosphorylation independent of insulin/GLUT4 in HSV smooth muscle under glycogen-depleted conditions.
3.10. Expression of Class I GLUT mRNAs and proteins in HSV tissues
Using total RNAs isolated from HSV smooth muscle, the present study has examined the relative expression levels of Class I GLUT mRNAs in HSV by qPCR analysis using GLUT isoform specific primer pairs as described [23]. As shown in Fig. 7A, the expression of GLUT1, -3, and -4 mRNA was observed in HSV. The expression levels of other two Class I GLUT mRNAs (GLUT2 and GLUT14) were in undetectable amounts. Fig. 7B shows the expression of GLUT1 and GLUT4 proteins in HSV. Although GLUT3 mRNA was observed, GLUT3 protein was not detectable in HSV.
Fig. 7.
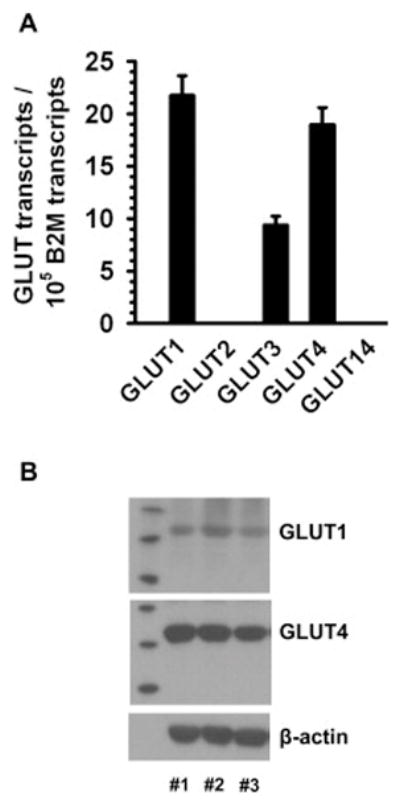
Expression of Class I GLUT mRNAs and proteins in HSV rings. A) Total RNAs isolated from HSV were subjected to qPCR analysis for Class I GLUT isoforms, as described. The bar graph compares the relative expression levels of Class I GLUT isoform mRNAs. The GLUT isoform mRNAs were expressed as mean ± SEM values of transcripts/105 B2M transcripts. The data shown are representative of triplicate determinations from 2 different subjects. B) HSV lysates were subjected to immunoblot analysis using primary antibodies specific for GLUT1 and GLUT4. β-actin was used as an internal control. The blots shown are representative of HSV specimens from 3 different subjects.
4. Discussion
The present study demonstrates for the first time that the conduit vessel (e.g., saphenous vein), commonly used in CABG surgery, is susceptible to changes in cellular energy state and thereby alterations in contractile function upon exposure to acute hypoxia-reoxygenation (H/R) conditions. Using ex vivo preparations of human saphenous vein smooth muscle, the critical role of the key components of carbohydrate metabolism (e.g., endogenous glycogen and exogenous glucose) is envisaged in the context of altered energy state (e.g., AMPK phosphorylation) and serotonin (5-HT)-induced contractile response. The principal findings from endothelium-denuded human saphenous vein include: i) a marked depletion of endogenous glycogen content by ~80% upon exposure to acute hypoxia during isometric contractions; ii) the consequent increases in basal and 5-HT-induced AMPK phosphorylation with an accompanying suppression of 5-HT-induced contraction during H/R under glucose-deprived conditions; and iii) the ability of exogenous D-glucose to rescue cellular stress and impaired contractile function (independent of insulin), as revealed by GLUT1-mediated effects to prevent exaggerated AMPK phosphorylation and restore the diminution in contractile response by 5-HT during H/R. Together, these findings strongly suggest that endogenous glycogen is a critical energy source to subserve agonist-induced smooth muscle contraction in the vessel wall under normoxic conditions.
Vascular smooth muscle tone is under the control of cellular signaling events that mediate an increase in force generation (vasoconstriction) or a reduction in force generation (vasorelaxation). The increase in contractile force is mediated by a rise in cytosolic free Ca2+, Ca2+-calmodulin complex formation, myosin light chain kinase (MLCK) activation, MLC phosphorylation, and subsequent myosin-actin interaction for cross-bridge cycling [36]. In addition to MLCK-mediated MLC phosphorylation, activation of RhoA/Rho-associated kinase and the consequent phosphorylation of myosin phosphatase targeting subunit 1 (MYPT1) are known to maintain MLC phosphorylation state. Notably, previous studies have shown that AMPK activation in vascular smooth muscle inhibits MLCK or RhoA/Rho-associated kinase activity, thereby suppressing agonist-induced MLC phosphorylation and contractile force [37, 38]. The present findings from endothelium-denuded human saphenous vein suggest that the loss of endogenous glycogen during acute hypoxic insult and the resultant exaggerated increase in AMPK phosphorylation by GPCR agonists may therefore compromise contractile function and promote an apparent vasorelaxant response (Fig. 8).
Fig. 8.
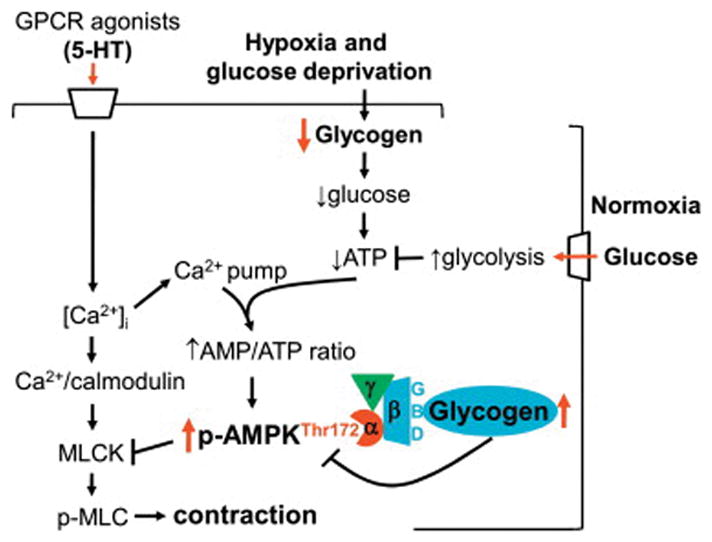
Endogenous glycogen depletion by hypoxia and glucose deprivation inhibits serotonin (5-HT)-induced smooth muscle contraction through exaggerated AMPK phosphorylation, which would occur via: i) diminished association of glycogen with the glycogen-binding domain (GBD) of AMPKβ subunit; and ii) ↑AMP/ATP ratio upon coordinated inhibition of ATP synthesis (by glycogen depletion) and increase in ATP consumption (by [Ca2+]i elevating agents that activate ATP-driven Ca2+ pumps).
Previous studies by several investigators have underscored the obligatory role of endogenous glycogen toward oxidative metabolism during vascular smooth muscle contraction [2, 3]. The glycogen content in vascular smooth muscle has been shown to be in the range of ~14 μmol/g in resistance vessels (e.g., cow mesenteric artery) and ~2.6 to ~3.9 μmol/g in conduit vessels (e.g., porcine carotid artery and human peripheral artery) [cited in [3]. In the present study using clinically-relevant conduit vessel (e.g., human saphenous vein), glycogen content in vascular smooth muscle is found to be ~1.8 μmol/g. In conjunction with previous reports that demonstrate the ability of arterial smooth muscle to synthesize and store glycogen [3], venous smooth muscle glycogen storage may also result from de novo synthesis of glycogen through activation of key glycogenic enzymes (e.g., glycogen synthase). Of importance, earlier studies with porcine carotid artery have estimated that the endogenous glycogen would be completely depleted in ~38 minutes during contractions in the absence of exogenous substrates [3]. In the present study, acute exposure of contracting human saphenous vein to hypoxia (30 minutes) under glucose-deprived conditions results in glycogen depletion by ~80%, suggesting glycogen utilization toward oxidative metabolism in venous smooth muscle. These findings implicate hypoxia as a key determinant of endogenous glycogen reserve, the loss of which would affect cellular energy state in vascular smooth muscle.
Previously, Rubin et al. have reported that metabolic stress [due to hypoxia and 2-deoxyglucose (which inhibits glycolysis)] enhances AMPK activity in porcine carotid artery smooth muscle [25]. Although the likely changes in glycogen content have not been examined in these earlier studies, the present findings reveal that metabolic stress (due to hypoxia and glucose-deprived conditions) is associated with glycogen depletion and thereby enhanced AMPK phosphorylation in vascular smooth muscle. Several lines of evidence suggest that glycogen depletion results in: i) diminished association of glycogen with the glycogen-binding domain in AMPKβ subunit [16, 18, 39]; and ii) decreased ATP level (thereby enhancing AMP/ATP or ADP/ATP ratio), both of which would facilitate AMPK activation. It is noteworthy that in classical insulin-responsive tissues (e.g., skeletal muscle), high glycogen content is associated with a decrease in AMPK activation, whereas metabolic stress-induced glycogen depletion results in enhanced AMPK activity [11, 40]. Together, these findings strongly suggest that vascular smooth muscle recapitulates some of the characteristics of skeletal muscle at least in regard to the essential link between glycogen depletion and AMPK activation during contraction with superimposed metabolic stress.
Next, Fogarty et al. have previously pointed out that inhibition of ATP synthesis (by phenformin) and increased ATP consumption (by A23187, a Ca2+ ionophore) in HeLa cells would act in an additive manner to increase ADP/ATP ratio (a surrogate for AMP/ATP ratio), thereby enhancing AMPK activity [41]. In line with these seminal findings, our recent studies have shown that in metformin-treated rat aortic smooth muscle, stimulation with phenylephrine (a [Ca2+]i-elevating agent) augments AMP/ATP ratio thereby resulting in a robust increase in AMPK phosphorylation [26]. In HSV smooth muscle subjected to glycogen depletion (under hypoxic- and glucose-deprived conditions that would inhibit ATP synthesis), stimulation with serotonin (5-HT, another [Ca2+]i-elevating agent) results in an exaggerated increase in AMPK phosphorylation (present study). Together, these findings suggest that under the conditions of diminished mitochondrial ATP synthesis (e.g., biguanide treatment, hypoxia, and glycogen depletion/glucose deprivation), challenge with [Ca2+]i-elevating agents has the potential to enhance ATP consumption by activating ATP-driven Ca2+ pumps, thereby accounting for exaggerated AMPK phosphorylation. Although the ATP cost for Ca2+ pump activity has not been reported in vascular smooth muscle, the energy utilized for MLC phosphorylation and cross-bridge cycling (actomyosin ATPase) is ~175-fold less than that observed in skeletal muscle [42]. Notably, studies with skeletal muscle have shown that Ca2+ pump activity plays a major role in ATP consumption compared with cross-bridge cycling, which accounts for only ~20% of total ATP consumption during contraction [43].
Since hypoxia-induced loss of glycogen in vascular smooth muscle results in enhanced activation of AMPK by serotonin, experimental approaches that allow reoxygenation and exogenous glucose availability should alleviate metabolic stress by suppressing exaggerated AMPK activation. Notably, glucose uptake and metabolism in vascular smooth muscle cells is dependent on glucose transporters (e.g., GLUT1 or GLUT4) and insulin receptor signaling, as evidenced in different vascular beds [21–24, 34, 44]. For instance, studies by several investigators including our previous findings demonstrate that in aortic smooth muscle cells glucose uptake is mediated by plasma membrane-localized GLUT1 independent of insulin [21–23]. In addition, Marcus et al. have shown that in renal microvascular smooth muscle cells, glucose uptake occurs through GLUT4 recruitment, which is dependent on insulin [34]. In the present study, HSV smooth muscle exhibits the expression of both GLUT1 and GLUT4 at the level of mRNA and protein. However, the extent to which exogenous D-glucose per se prevents exaggerated AMPK phosphorylation in HSV smooth muscle remains essentially the same in the presence or absence of insulin or GLUT4 inhibitor, indinavir. Together, these findings suggest that GLUT1-mediated glucose uptake and metabolism would allow increased ATP formation to prevent AMPK activation and metabolic stress. Although insulin receptor stimulation does not affect AMPKαThr172 phosphorylation in vascular smooth muscle (present study), skeletal muscle and hepatocytes [45], it has been shown to phosphorylate AMPKαSer485/491 thereby inhibiting AMPK activity in skeletal muscle and hepatocytes [45]. In this regard, insulin-induced Akt activation may mediate AMPKαSer485/491 phosphorylation to inhibit AMPK activity [45]. In conjunction with these findings, insulin-induced Akt phosphorylation (present study) [44, 46] is likely to inhibit AMPK activity [45] and promote glycogen synthesis [44] in vascular smooth muscle. Future studies should determine how insulin regulates signaling events (e.g., Akt and AMPK activity) in a temporal manner to stimulate glycogen metabolism in the vessel wall.
Previous studies by several investigators including our recent findings demonstrate that metabolic stress [25], metformin [26], or AICAR [26–28] inhibits agonist-induced smooth muscle contraction through AMPK activation. While it is known that increased intracellular AMP/ATP ratio [26] or ZMP [28] contributes to AMPK activation in vascular smooth muscle, the present findings reveal that depletion of endogenous glycogen followed by serotonin challenge results in exaggerated AMPK activation, thereby leading to diminished contractile response. As a corollary, provision of exogenous energy substrate (D-glucose) prevents exaggerated activation of AMPK and restores serotonin-induced contractile response. Thus, altered cellular energy state (e.g., ↓ glycogen → ↑ AMPK) in vascular smooth muscle per se may account for coronary vasodilation independent of endothelium. It is noteworthy that metabolic vasodilation is commonly attributed to a number of vasodilators of endothelial and myocardial origin (e.g., nitric oxide, prostanoids, adenosine, and potassium) [47]. In addition, withdrawal of vasoconstrictor action of endothelin by NO and prostanoids contributes to coronary vasodilation during exercise [48]. Furthermore, serotonin, which induces vasoconstriction in coronary conduit vessels, evokes metabolic vasodilation in coronary resistance vessels [cited in [29]. From the present observations, it is apparent that the extent of endogenous glycogen depletion in vascular smooth muscle during metabolic stress may constitute a yet another key determinant of coronary vasodilation as a function of different vascular beds. Further studies are clearly warranted that should determine the impact of glycogen depletion on vasodilator reserve [49] in conduit versus resistance vessels in ischemic heart disease.
From the translational perspective, the significance of endogenous glycogen content and/or AMPK activation state toward cardiovascular protective effects has thus far been studied in atrial muscle [20] and saphenous vein smooth muscle (present study) and endothelial cells [50], using specimens obtained from patients undergoing CABG surgery. In atrial muscle, ischemic conditions activate AMPK without affecting endogenous glycogen content (~0.33 μmol/g), whereas exposure to insulin and glucose suppresses ischemia-induced AMPK activation [20]. As noted earlier, in the present study with HSV smooth muscle, hypoxic- and glucose-deprived conditions result in a marked depletion of endogenous glycogen (from ~1.78 μmol/g under normoxia to ~0.36 μmol/g under hypoxia) with an accompanying AMPK activation. In addition, glucose exposure prevents exaggerated AMPK activation independent of insulin in HSV smooth muscle (present study), suggesting that atrial muscle and vascular smooth muscle may exhibit distinct differences in the extent of endogenous glycogen metabolism and dependency on exogenously-challenged insulin toward alterations in cellular energy state. With regard to isolated HSV endothelial cells from patients with type 2 diabetes, recent studies demonstrate that increased formation of mitochondrial reactive oxygen species results in enhanced activation of AMPK, which may serve as a defense mechanism against oxidative stress [50]. It is quite apparent that, in conjunction with previous studies on glycogen metabolism in ischemic myocardium and skeletal muscle in different animal models [12, 51], preservation of endogenous glycogen stores may be beneficial for improved ventricular function and maintenance of contractile function in skeletal muscle and vascular smooth muscle.
The limitations of the present study include the use of conduit vessels from subjects with co-morbities including hypertension and dyslipidemia. These disease conditions are likely to alter the vascular responsiveness in the saphenous vein. To address these confounding variables, saphenous vein ring preparations were subjected to several washing steps and equilibration conditions ex vivo prior to the initiation of contractility studies, as described. More importantly, we included select studies using normal rat aorta to verify the relationship between altered contractility and AMPK phosphorylation under hypoxia/reoxygenation conditions.
In conclusion, the present findings from clinically-relevant human saphenous vein suggest that the loss of endogenous glycogen in vascular smooth muscle and the resultant accentuation of AMPK phosphorylation by GPCR agonists may constitute a novel mechanism of coronary vasodilation as an adaptive response in ischemic heart disease.
Acknowledgments
This work was supported by the National Heart, Lung, and Blood Institute/National Institutes of Health Grant (R01-HL-097090), Georgia Regents University Translational Cardiovascular Research Pilot Funding Program, Pennsylvania Department of Health Tobacco Settlement Fund, and University of Georgia Research Foundation Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Roach PJ, Depaoli-Roach AA, Hurley TD, Tagliabracci VS. Glycogen and its metabolism: some new developments and old themes. Biochem J. 2012;441:763–87. doi: 10.1042/BJ20111416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lynch RM, Paul RJ. Compartmentation of glycolytic and glycogenolytic metabolism in vascular smooth muscle. Science. 1983;222:1344–6. doi: 10.1126/science.6658455. [DOI] [PubMed] [Google Scholar]
- 3.Allen TJ, Hardin CD. Influence of glycogen storage on vascular smooth muscle metabolism. Am J Physiol Heart Circ Physiol. 2000;278:H1993–2002. doi: 10.1152/ajpheart.2000.278.6.H1993. [DOI] [PubMed] [Google Scholar]
- 4.Levin M, Leppanen O, Evaldsson M, Wiklund O, Bondjers G, Bjornheden T. Mapping of ATP, glucose, glycogen, and lactate concentrations within the arterial wall. Arterioscler Thromb Vasc Biol. 2003;23:1801–7. doi: 10.1161/01.ATV.0000092872.54026.8D. [DOI] [PubMed] [Google Scholar]
- 5.Huijing F. Glycogen metabolism and glycogen-storage diseases. Physiol Rev. 1975;55:609–58. doi: 10.1152/physrev.1975.55.4.609. [DOI] [PubMed] [Google Scholar]
- 6.Milan D, Jeon JT, Looft C, Amarger V, Robic A, Thelander M, et al. A mutation in PRKAG3 associated with excess glycogen content in pig skeletal muscle. Science. 2000;288:1248–51. doi: 10.1126/science.288.5469.1248. [DOI] [PubMed] [Google Scholar]
- 7.Arad M, Seidman CE, Seidman JG. AMP-activated protein kinase in the heart: role during health and disease. Circ Res. 2007;100:474–88. doi: 10.1161/01.RES.0000258446.23525.37. [DOI] [PubMed] [Google Scholar]
- 8.Geel TM, McLaughlin PM, de Leij LF, Ruiters MH, Niezen-Koning KE. Pompe disease: current state of treatment modalities and animal models. Mol Genet Metab. 2007;92:299–307. doi: 10.1016/j.ymgme.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 9.Laforet P, Petiot P, Nicolino M, Orlikowski D, Caillaud C, Pellegrini N, et al. Dilative arteriopathy and basilar artery dolichoectasia complicating late-onset Pompe disease. Neurology. 2008;70:2063–6. doi: 10.1212/01.wnl.0000313367.09469.13. [DOI] [PubMed] [Google Scholar]
- 10.Nemes A, Soliman OI, Geleijnse ML, Anwar AM, van der Beek NA, van Doorn PA, et al. Increased aortic stiffness in glycogenosis type 2 (Pompe’s disease) Int J Cardiol. 2007;120:138–41. doi: 10.1016/j.ijcard.2006.07.215. [DOI] [PubMed] [Google Scholar]
- 11.Wojtaszewski JF, Jorgensen SB, Hellsten Y, Hardie DG, Richter EA. Glycogen-dependent effects of 5-aminoimidazole-4-carboxamide (AICA)-riboside on AMP-activated protein kinase and glycogen synthase activities in rat skeletal muscle. Diabetes. 2002;51:284–92. doi: 10.2337/diabetes.51.2.284. [DOI] [PubMed] [Google Scholar]
- 12.Lagerstrom CF, Walker WE, Taegtmeyer H. Failure of glycogen depletion to improve left ventricular function of the rabbit heart after hypothermic ischemic arrest. Circ Res. 1988;63:81–6. doi: 10.1161/01.res.63.1.81. [DOI] [PubMed] [Google Scholar]
- 13.Hardin CD, Roberts TM. Differential regulation of glucose and glycogen metabolism in vascular smooth muscle by exogenous substrates. J Mol Cell Cardiol. 1997;29:1207–16. doi: 10.1006/jmcc.1996.0356. [DOI] [PubMed] [Google Scholar]
- 14.Barron JT, Kopp SJ. Phosphorylase b kinase and phosphorylase a phosphatase activities in contracting vascular smooth muscle: stimulation by fatty acid. Biochim Biophys Acta. 1991;1073:550–4. doi: 10.1016/0304-4165(91)90229-a. [DOI] [PubMed] [Google Scholar]
- 15.Zhang C, Paul RJ. Excitation-contraction coupling and relaxation in porcine carotid arteries are specifically dependent on glucose. Am J Physiol. 1994;267:H1996–2004. doi: 10.1152/ajpheart.1994.267.5.H1996. [DOI] [PubMed] [Google Scholar]
- 16.McBride A, Ghilagaber S, Nikolaev A, Hardie DG. The glycogen-binding domain on the AMPK beta subunit allows the kinase to act as a glycogen sensor. Cell Metab. 2009;9:23–34. doi: 10.1016/j.cmet.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–62. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moffat C, Ellen Harper M. Metabolic functions of AMPK: aspects of structure and of natural mutations in the regulatory gamma subunits. IUBMB Life. 2010;62:739–45. doi: 10.1002/iub.387. [DOI] [PubMed] [Google Scholar]
- 19.Saha AK, Xu XJ, Lawson E, Deoliveira R, Brandon AE, Kraegen EW, et al. Downregulation of AMPK accompanies leucine- and glucose-induced increases in protein synthesis and insulin resistance in rat skeletal muscle. Diabetes. 2010;59:2426–34. doi: 10.2337/db09-1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carvalho G, Pelletier P, Albacker T, Lachapelle K, Joanisse DR, Hatzakorzian R, et al. Cardioprotective effects of glucose and insulin administration while maintaining normoglycemia (GIN therapy) in patients undergoing coronary artery bypass grafting. J Clin Endocrinol Metab. 2011;96:1469–77. doi: 10.1210/jc.2010-1934. [DOI] [PubMed] [Google Scholar]
- 21.Kaiser N, Sasson S, Feener EP, Boukobza-Vardi N, Higashi S, Moller DE, et al. Differential regulation of glucose transport and transporters by glucose in vascular endothelial and smooth muscle cells. Diabetes. 1993;42:80–9. doi: 10.2337/diab.42.1.80. [DOI] [PubMed] [Google Scholar]
- 22.Adhikari N, Basi DL, Carlson M, Mariash A, Hong Z, Lehman U, et al. Increase in GLUT1 in smooth muscle alters vascular contractility and increases inflammation in response to vascular injury. Arterioscler Thromb Vasc Biol. 2011;31:86–94. doi: 10.1161/ATVBAHA.110.215004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pyla R, Poulose N, Jun JY, Segar L. Expression of conventional and novel glucose transporters, GLUT1, -9, -10, and -12, in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2013;304:C574–89. doi: 10.1152/ajpcell.00275.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park JL, Loberg RD, Duquaine D, Zhang H, Deo BK, Ardanaz N, et al. GLUT4 facilitative glucose transporter specifically and differentially contributes to agonist-induced vascular reactivity in mouse aorta. Arterioscler Thromb Vasc Biol. 2005;25:1596–602. doi: 10.1161/01.ATV.0000170137.41079.ab. [DOI] [PubMed] [Google Scholar]
- 25.Rubin LJ, Magliola L, Feng X, Jones AW, Hale CC. Metabolic activation of AMP kinase in vascular smooth muscle. J Appl Physiol. 2005;98:296–306. doi: 10.1152/japplphysiol.00075.2004. [DOI] [PubMed] [Google Scholar]
- 26.Pyla R, Osman I, Pichavaram P, Hansen P, Segar L. Metformin exaggerates phenylephrine-induced AMPK phosphorylation independent of CaMKKβ and attenuates contractile response in endothelium-denuded rat aorta. Biochem Pharmacol. 2014;92:266–279. doi: 10.1016/j.bcp.2014.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goirand F, Solar M, Athea Y, Viollet B, Mateo P, Fortin D, et al. Activation of AMP kinase alpha1 subunit induces aortic vasorelaxation in mice. J Physiol. 2007;581:1163–71. doi: 10.1113/jphysiol.2007.132589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ford RJ, Rush JW. Endothelium-dependent vasorelaxation to the AMPK activator AICAR is enhanced in aorta from hypertensive rats and is NO and EDCF dependent. Am J Physiol Heart Circ Physiol. 2011;300:H64–75. doi: 10.1152/ajpheart.00597.2010. [DOI] [PubMed] [Google Scholar]
- 29.Bache RJ, Stark RP, Duncker DJ. Serotonin selectively aggravates subendocardial ischemia distal to a coronary artery stenosis during exercise. Circulation. 1992;86:1559–65. doi: 10.1161/01.cir.86.5.1559. [DOI] [PubMed] [Google Scholar]
- 30.Lorusso R, Pentiricci S, Raddino R, Scarabelli TM, Zambelli C, Villanacci V, et al. Influence of type 2 diabetes on functional and structural properties of coronary artery bypass conduits. Diabetes. 2003;52:2814–20. doi: 10.2337/diabetes.52.11.2814. [DOI] [PubMed] [Google Scholar]
- 31.Banes A, Florian JA, Watts SW. Mechanisms of 5-hydroxytryptamine(2A) receptor activation of the mitogen-activated protein kinase pathway in vascular smooth muscle. J Pharmacol Exp Ther. 1999;291:1179–87. [PubMed] [Google Scholar]
- 32.Carling D. AMP-activated protein kinase: balancing the scales. Biochimie. 2005;87:87–91. doi: 10.1016/j.biochi.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 33.Towler MC, Hardie DG. AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res. 2007;100:328–41. doi: 10.1161/01.RES.0000256090.42690.05. [DOI] [PubMed] [Google Scholar]
- 34.Marcus RG, England R, Nguyen K, Charron MJ, Briggs JP, Brosius FC., 3rd Altered renal expression of the insulin-responsive glucose transporter GLUT4 in experimental diabetes mellitus. Am J Physiol. 1994;267:F816–24. doi: 10.1152/ajprenal.1994.267.5.F816. [DOI] [PubMed] [Google Scholar]
- 35.Rudich A, Konrad D, Torok D, Ben-Romano R, Huang C, Niu W, et al. Indinavir uncovers different contributions of GLUT4 and GLUT1 towards glucose uptake in muscle and fat cells and tissues. Diabetologia. 2003;46:649–58. doi: 10.1007/s00125-003-1080-1. [DOI] [PubMed] [Google Scholar]
- 36.Webb RC. Smooth muscle contraction and relaxation. Adv Physiol Educ. 2003;27:201–6. doi: 10.1152/advan.00025.2003. [DOI] [PubMed] [Google Scholar]
- 37.Horman S, Morel N, Vertommen D, Hussain N, Neumann D, Beauloye C, et al. AMP-activated protein kinase phosphorylates and desensitizes smooth muscle myosin light chain kinase. J Biol Chem. 2008;283:18505–12. doi: 10.1074/jbc.M802053200. [DOI] [PubMed] [Google Scholar]
- 38.Wang S, Liang B, Viollet B, Zou MH. Inhibition of the AMP-activated protein kinase-alpha2 accentuates agonist-induced vascular smooth muscle contraction and high blood pressure in mice. Hypertension. 2011;57:1010–7. doi: 10.1161/HYPERTENSIONAHA.110.168906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McBride A, Hardie DG. AMP-activated protein kinase--a sensor of glycogen as well as AMP and ATP? Acta Physiol (Oxf) 2009;196:99–113. doi: 10.1111/j.1748-1716.2009.01975.x. [DOI] [PubMed] [Google Scholar]
- 40.Hayashi T, Hirshman MF, Fujii N, Habinowski SA, Witters LA, Goodyear LJ. Metabolic stress and altered glucose transport: activation of AMP-activated protein kinase as a unifying coupling mechanism. Diabetes. 2000;49:527–31. doi: 10.2337/diabetes.49.4.527. [DOI] [PubMed] [Google Scholar]
- 41.Fogarty S, Hawley SA, Green KA, Saner N, Mustard KJ, Hardie DG. Calmodulin-dependent protein kinase kinase-beta activates AMPK without forming a stable complex: synergistic effects of Ca2+ and AMP. Biochem J. 2010;426:109–18. doi: 10.1042/BJ20091372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walker JS, Wingard CJ, Murphy RA. Energetics of crossbridge phosphorylation and contraction in vascular smooth muscle. Hypertension. 1994;23:1106–12. doi: 10.1161/01.hyp.23.6.1106. [DOI] [PubMed] [Google Scholar]
- 43.Zhang SJ, Andersson DC, Sandstrom ME, Westerblad H, Katz A. Cross bridges account for only 20% of total ATP consumption during submaximal isometric contraction in mouse fast-twitch skeletal muscle. Am J Physiol Cell Physiol. 2006;291:C147–54. doi: 10.1152/ajpcell.00578.2005. [DOI] [PubMed] [Google Scholar]
- 44.Artwohl M, Brunmair B, Furnsinn C, Holzenbein T, Rainer G, Freudenthaler A, et al. Insulin does not regulate glucose transport and metabolism in human endothelium. Eur J Clin Invest. 2007;37:643–50. doi: 10.1111/j.1365-2362.2007.01838.x. [DOI] [PubMed] [Google Scholar]
- 45.Valentine RJ, Coughlan KA, Ruderman NB, Saha AK. Insulin inhibits AMPK activity and phosphorylates AMPK Ser(4)(8)(5)/(4)(9)(1) through Akt in hepatocytes, myotubes and incubated rat skeletal muscle. Arch Biochem Biophys. 2014;562:62–9. doi: 10.1016/j.abb.2014.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao Y, Biswas SK, McNulty PH, Kozak M, Jun JY, Segar L. PDGF-induced vascular smooth muscle cell proliferation is associated with dysregulation of insulin receptor substrates. Am J Physiol Cell Physiol. 2011;300:C1375–85. doi: 10.1152/ajpcell.00670.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Farouque HM, Worthley SG, Meredith IT. Effect of ATP-sensitive potassium channel inhibition on coronary metabolic vasodilation in humans. Arterioscler Thromb Vasc Biol. 2004;24:905–10. doi: 10.1161/01.ATV.0000125701.18648.48. [DOI] [PubMed] [Google Scholar]
- 48.Tune JD. Withdrawal of vasoconstrictor influences in local metabolic coronary vasodilation. Am J Physiol Heart Circ Physiol. 2006;291:H2044–6. doi: 10.1152/ajpheart.00653.2006. [DOI] [PubMed] [Google Scholar]
- 49.Bache RJ. Vasodilator reserve: a functional assessment of coronary health. Circulation. 1998;98:1257–60. doi: 10.1161/01.cir.98.13.1257. [DOI] [PubMed] [Google Scholar]
- 50.Mackenzie RM, Salt IP, Miller WH, Logan A, Ibrahim HA, Degasperi A, et al. Mitochondrial reactive oxygen species enhance AMP-activated protein kinase activation in the endothelium of patients with coronary artery disease and diabetes. Clin Sci (Lond) 2013;124:403–11. doi: 10.1042/CS20120239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reynolds THt, Brozinick JT, Jr, Rogers MA, Cushman SW. Mechanism of hypoxia-stimulated glucose transport in rat skeletal muscle: potential role of glycogen. Am J Physiol. 1998;274:E773–8. doi: 10.1152/ajpendo.1998.274.5.E773. [DOI] [PubMed] [Google Scholar]