

Abstract
Our previous study suggested that early dantrolene treatment reduced amyloid plaque burden and nearly abolished learning and memory loss in a triple transgenic Alzheimer's disease (3xTg-AD) mouse model. In this study, we investigated the long term treatment of dantrolene on amyloid and tau neuropathology, brain volume and cognitive function in aged 3xTg-AD mice. Fifteen month old 3xTg-AD mice and wild type controls were treated with oral dantrolene (5 mg/kg) or vehicle control twice a week for 6 months. Learning and memory were examined using the Morris Water Maze at 21 and again at 22 months of age. After the behavioral testing, hippocampal and cortical brain volumes were calculated with magnetic resonance imaging and motor function was evaluated using the rotorod. The amyloid burden and tau neurofibrillary tangles in the hippocampus were determined using immunohistochemistry. We found that dantrolene significantly decreased the intraneuronal amyloid accumulation by as much as 76% compared to its corresponding vehicle control, together with a trend to reduce phosphorylated tau in the hippocampus. No significant differences could be detected in hippocampal or cortical brain volume, motor function or cognition among all experimental groups, indicating that the mice were still pre-symptomatic for Alzheimer's disease. Thus, pre-symptomatic and long term dantrolene treatment significantly decreased the intraneuronal amyloid burden in aged 3xTg-AD mice prior to significant changes in brain volume, or cognition.
Keywords: Dantrolene, Alzheimer's disease, Calcium, MRI, Cognition, Amyloid
INTRODUCTION
Intracellular calcium (Ca2+) homeostasis plays important roles in neuronal growth,1-4 survival and death,5-11 as well as in synaptic and cognitive function.12-16 Recent studies suggest that disruption of intracellular Ca2+ homeostasis plays important roles in the neuropathology of Alzheimer's disease (AD) and associated cognitive dysfunction.5;14;15;17-21 In particular, abnormal Ca2+ release from the endoplasmic reticulum (ER) via either the Inositol 1,4,5-Trisphosphate receptor (InsP3R)18;19;22;23 or the ryanodine receptor (RYR)14-16;24;25 Ca2+ channels may contribute to neuronal death, synapse and cognitive dysfunction. The number and function of RYRs have been demonstrated to be abnormally increased not only in different brain regions of AD mice,14;24;26 but also in AD patients.27 Therefore, reversal of the overactivation of RYRs may be a new therapeutic target for the treatment of AD.16;24;25;28
Dantrolene is a known antagonist of the RYR and is used clinically to treat malignant hyperthermia, muscle spasms and neuroleptic malignant syndrome.28;29 Dantrolene has been shown to inhibit neurodegeneration induced by multiple insults, including ischemia, hypoxemia, seizures and trauma.28;30-33 Dantrolene has also been shown to be neuroprotective in animal models of various types of neurodegenerative diseases.16;24;25;34-38 In various neuronal cultures with AD features, dantrolene also demonstrated neuroprotective effects.28;39 Therefore, dantrolene is theoretically a potential drug to reverse the calcium dysregulation and neuropathology in AD and restore cognitive dysfunction. In fact, our previous study has demonstrated that early and chronic treatment of dantrolene at the beginning of Ca2+ dysregulation in triple transgenic Alzheimer disease (3xTg-AD) mice (with APP, presenilin-1 and tau mutations)40 nearly abolished learning and memory loss in old age, with an associated significant reduction of amyloid burden in the brain.16 These results were supported by two later studies in a different murine AD model.24;25 The current study further investigated whether dantrolene treatment initiated after the onset of amyloid neuropathology in aged 3xTg-AD mice,40;41 could still have therapeutic effects.
METHODS
Animals
3xTg-AD mice have been demonstrated to be an adequate transgenic animal model for testing efficacy of drug treatments in AD.16;42 Animal protocols were approved as required by the University of Pennsylvania Institutional Animal Care and Use Committees (IACUC) and all mice were treated in strict accordance to APS/NIH guidelines. 3xTg-AD mice, harboring thepresinilin−1 (PS1M146V), amyloid precursor protein (APPswe), and tau (p301L) transgenes, were bred using an initial 3xTg-AD breeding pair courtesy of Dr. Frank LaFerla at the University of California at Irvine, CA. The 3xTg-AD mouse model has been shown to develop both amyloid plaque and neurofibrillary tangle neuropathology and cognitive dysfunction in an age-dependent manner.14;40;41 Calcium dysregulation in this mouse model begins at 2 months of age, long before amyloid pathology and cognitive dysfunction are apparent.40;41 Both male and female mice were used in this study and aged-matched C57BL/6 wild type (WT) mice were used as controls.
Dantrolene Treatment and Experimental Groups
Fifteen month old WT and 3xTg-AD mice were fed 5 mg of dantrolene (Sigma, St Louis, MO) dissolved in 2% corn flour in 2.5ml of phosphate buffered saline control (PBS), twice a week, by oral gavages as described in previous studies21;34 for 6 months, until 21 months of age (Fig. 1). Four groups of mice were included in this study: 3xTg-AD mice treated with dantrolene (3xTg-DAN); 3xTg-AD mice treated with vehicle control (3xTg-PBS); C57BL/6 wild type mice treated with dantrolene (WT-DAN) or vehicle control (WT-PBS). No animals exhibited signs of toxicity and no mortality was detected in any of the groups.
FIGURE. 1.
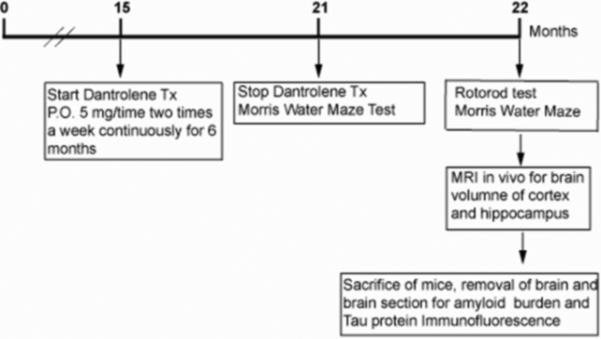
Summary of the timeline for the experimental protocol. P.O., oral; MRI, Magnetic resonance imaging.Tx, treatment
Morris Water Maze (MWM) Behavioral Testing
Cognitive function was assessed in all four experimental groups at 21 and again at 22 months of age using the MWM tests, as we previously described.8;16;43;44 The water in the 1.5-m diameter pool was maintained at a temperature between 25 °C and 29 °C and opacified with titanium (IV) dioxide throughout the entire training and testing period to obscure the submerged 15-cm diameter platform. After each trial, each mouse was dried and warmed before returning it to its cage. The time gap between trials was a minimum of 30 to 45 min. Mice were tracked with a video camera mounted above the pool, which fed images to a computer that was installed with IMAQ PCI-1407 software (National Instruments, Austin, TX). During cued training, the mice were trained to escape from the MWM for 5 days with 4 trails per day. The mice were allotted 60 s to find the flagged submerged platform, and then allowed to remain on the platform for 10 to 15 s. Those animals that did not find the platform in 60 s were gently guided to it, and allowed to remain there for 10 to 15 s before removal. There were no visual cues and the platform location was fixed and starting points were varied. Place tests were used to determine reference learning by working out the spatial relationship between distant cues and the escape platform (submerged, no flag), which remained in the same location for all place trials. The starting points were random and the time to reach the platform was recorded for each trial. The mice received two blocks of trials each day for 5 days. Probe tests, to determine memory retention, were conducted 1 h after the last trial of the reference learning (place) testing. For this test, the platform was removed from the pool and the swim path was recorded for 60 s for each mouse. The starting location was always in the “opposite” quadrant. The ratio of the means of the percent time spent in the target quadrant compared to opposite quadrant was calculated as an indication of memory retention.
Rotorod Tests
Before the MWM, we examined the locomotor ability of all mice using the rotorod test, as described previously.16 (Fig. 1) All mice received two 60 s training trials on the rotorod (IITC Series 8, Life Sciences, Woodland hills, CA) at 9 rpm with a 30 min interval between trials. The animals then underwent three test trials for a maximum of 120 s with variable speed, ranging from 4 to 40 rpm. The time spent on the rotorod was recorded for each mouse as an indicator for motor skill. The interval between trials was 60 min to assure that mice had enough rest between trials.
Magnetic Resonance Imaging
In vivo Magnetic resonance imaging (MRI) exams were performed (Fig. 1) on a 9.4 T horizontal bore scanner equipped with 12 cm ID 40 gauss/cm gradients and interfaced to a Direct Drive console (Agilent, Palo Alto, CA). Animals were prepared for the study by induction of general anesthesia using 2% isoflurane in oxygen delivered via a nose cone. Rectal temperature probe and respiration pillow were then mounted on the animal and the animal was positioned in a 20 mm ID quadrature RF coil (M2M, Cleveland, OH). After positioning the RF coil in the magnet, respiration and core body temperature monitoring was initiated (SAII, Stony Brook, NY). A regulated warm air source was directed over the animal while it was in the magnet in order to maintain core body temperature at 37± 1°C.
Following RF calibration and generation of scout images, a contiguous axial series of images spanning the central region of the brain was generated using the following acquisition parameters: TE= 20 msec, TR = 1500, field of view 16×16 mm, slice thickness 0.25 mm, slices = 40-48, matrix = 192×192, averages = 8. Respiration gating was employed during the study such that data was only acquired during the quiescent phase of the respiration cycle. The brain volume in hippocampus and cortex were quantified using MRI and compared among all experimental groups.
Immunohistochemistry and Image Analysis
Immediately after the MRI experiments, mice were euthanized (Fig.1) under 3-4% isoflurane by simultaneous intracardiac perfusion through the left ventricle with ice cold PBS and right atria exsanguinations. The left side of each brain was post-fixed in 4% paraformaldehyde overnight at 4°C prior to paraffin embedding. As described previously,45;46 coronal brain sections of 10 μm thickness were deparaffinized and hydrated through a series of graded alcohol steps and washed in phosphate-buffered saline. Antigen retrieval was performed by heating in Antigen Unmasking Solution (Vector Labs, Burlingame, CA) in a decloaking chamber (Biocare Medical, Concord, CA) for 2 min. Sections were incubated in 5% hydrogen peroxide to block endogenous peroxidase activity. Using the Vectastain mouse-on-mouse kit (Vector Labs), sections were blocked with mouse IgG blocking reagent for 1 hour. Slides were incubated overnight at 4°C with primary antibody. Antibody 6E10 (1:600; Covance, Dedham, MA) was used to detect intracellular amyloid. 6E10 showed cross-reactivity with full-length APP, soluble APP cleaved at alpha site, C99, and A beta. Antibody AT180 (1:400, Thermo Scientific, Rockford, IL) was used for human hyper-phosphorylated tau protein at the Thr231 and Ser235 residues. Negative control sections were incubated in blocking solution that did not contain primary antibody. Sections were washed in Tris-buffered saline buffer (50mM Tris, 150mM NaCl, pH 7.8), incubated in goat F(ab’)2 anti-mouse IgG Conjugate (1:1000 for 6E10 and 1:200 for AT180) (Vector Laboratories, Burlingame, CA) for 1 h. For AT180, the sections were stained using the DAB (3, 3'-diaminobenzidine) HRP (horseradish peroxidase) substrate and coverslipped with permount (ThermoFisher, Scientific, Waltham, MA). The 6E10 sections were coverslipped with Prolong Gold antifade reagent with DAPI (cell stain) (Life Technologies, Eugene, OR).
Images were acquired and quantified using IP lab 4.0 software (BioVision technologies, Chester Springs, PA,) linked to an Olympus IX70 microscope (Olympus corporation, Tokyo, Japan) equipped with a Cooke SensiCam camera (Cooke Corporation, Romulus, MI). Brain sections corresponding to the Atlas of the Developing Mouse Brain,47 (positioned approximately 2000-2200 mm away from frontal pole) throughout anatomic region of interest (intermediate hippocampal CA1 region) were captured from each animal under 4X, 10X, 20X and 40X magnification. Three sections were chosen from each animal and analyzed for 6E10 positive cells and plaque numbers in the hippocampal CA1 region. Two persons blinded to the treatments counted the total number of 6E10 positive cells in the hippocampal CA1 region and amyloid plague in all regions of the hippocampus on the slide, including the dentate gyrus. Similarly, the AT180 positive cells were determined in the hippocampal CA1 close to the dorsal subiculum. The areas of hippocampal CA1 region was defined according to the Atlas of the Developing Mouse Brain,47 and the area measured using IPLab Suite v3.7 imaging processing and analysis software (Biovision Technologies, Exton, PA). The means were calculated from three consecutive slides per animal.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism v5.04 software and the detailed statistical methods and animal numbers are explained in the figure legends. P < 0.05 was accepted as statistically significant.
RESULTS
Dantrolene Treatment Decreases Amyloid in Hippocampus
Intracellular amyloid, shown by 6E10 positive cells, in the hippocampal CA1 region in the 3xTg mice treated with PBS was remarkably and significantly increased compared to wild type control mice (Fig. 2). However, long term treatment with dantrolene in these aged 3xTg-AD mice significantly reduced the intracellular amyloid by as much as 76 % in the hippocampal CA1 region compared to its vehicle control (Fig. 2). We did not detect intraneuronal amyloid in any of wild type mice and there were no identifiable extracellular amyloid plaques observed in the hippocampus of the 3xTg-AD or WT mice in any experimental group.
FIGURE 2.
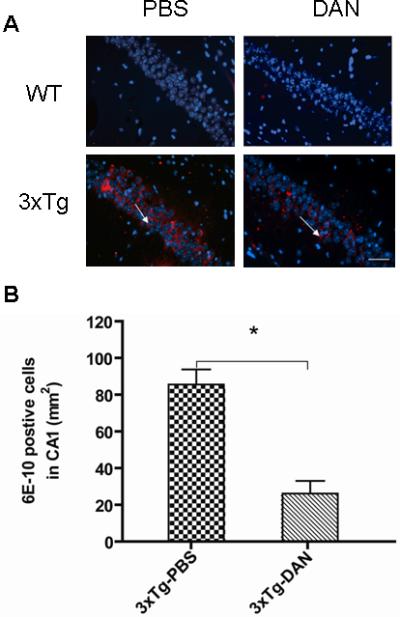
Dantrolene significantly decreases intracellular amyloid in the hippocampus of triple transgenic Alzheimer (3xTG-AD) mice. (A) Representative 6E10 immunoreactivity (red immunofluorescence) in the hippocampal pyramidal cells (blue immunofluorescence). Arrows indicate 6E10 positive cells in the hippocampual CA1 region. Scale bar represents 80 μm. (B) The 3xTg-AD control mice (3xTg-PBS) had statistically significant more intraneuronal amyloid accumulation than 3xTg-AD mice treated with dantrolene (3xTg-DAN) (*P<0.05). All data represent mean ± SEM from 5 mice (N=5) in each group and analyzed by one-way ANOVA followed by Bonferroni post hoc tests.
Effects of Dantrolene Treatment on Phosphorylated Tau Protein in Hippocampus
We detected a trend for a reduction in AT180 positive cells in the hippocampal CA1 region in the dantrolene treated 3xTG-AD compared to 3xTg-AD controls (by as much as 30%), which did not reach statistical significance (Fig. 3 B). As expected, there were no detectable phosphorylated tau protein (AT180) positive cells in the wild type control mice treated with either dantrolene or its vehicle control (Fig. 3 A, B).
FIGURE 3.
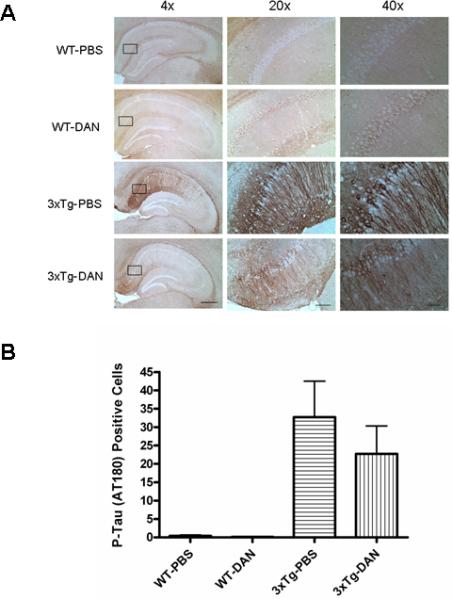
The effect of long term dantrolene treatment on phosphorylated tau protein levels in wild type and triple transgenic Alzheimers (3xTg-AD) mice. (A) Brightfield micrographs of immunohistochemical staining showing the distribution of phosphorylated tau proteins (AT180 antibody) in the CA1 region of the hippocampus. Scale bars for 4x, 20X and 40X magnification represent 400, 80 and 40μm respectively. Rectangles cover the areas used for higher magnification (20X and 40X) examination. (B) No statistical difference was found in the number of phosphorylated tau (AT180) positive neurons in hippocampal CA1 region in the 3xTg-AD mice controls (3xTg-PBS) compared to those treated with dantrolene (3xTg-DAN). No tau positive neurons were found in wild type mice treated with either dantrolene (WT-DAN) or vehicle control, PBS (WT-PBS). All data represent mean ± SEM from 5 mice in each group and analyzed by one-way ANOVA followed by Bonferroni post hoc tests.
Effects of Late Dantrolene Treatment on Brain Volume
There were no significant differences in cortical or hippocampal brain volume, determined by in vivo MRI, among all experimental groups (Fig. 4)
FIGURE 4.
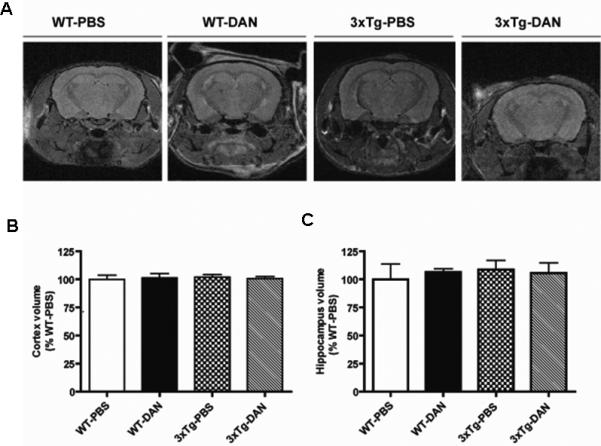
Dantrolene treatment did not change cortical or hippocampal volume in Alzheimer transgenic mice. (A) Representative images of brain magnetic resonance imaging (MRI) depict slices of a T2 weighted morphological MRI images used for cortical and hippocampual measurements from 22 month old wild type mice treated with either dantrolene (WT-DAN) or vehicle control PBS (WT-PBS) and 3xTg-AD mice treated with dantrolene (3xTg-DAN) or PBS (3xTg-PBS). (B, C) Statistical analyses and comparison of the brain volume of the cortex (B) and hippocampus (C) measured by MRI from all groups. Data represent mean ± SEM and were analyzed by one-way ANOVA followed by Bonferroni post hoc tests.
Effects of Late Dantrolene Treatment on Behavior
Cognitive function was assessed using the MWM. There were no differences in motor or visual acuity in the aged 3xTg-AD or WT mice among all groups as demonstrated in the cued training (data not shown). In addition, no motor impairment was found in any of the experimental groups using the rotorod assay (Fig. 5 A, B). We could detect no significant differences in reference learning, determined by the escape latency during the place trials (Fig. 5 C, D), or in retention memory, as determined by the probe tests (Fig. 5 E, F), among all experimental groups at either 21 or 22 months of age. These results suggested that 3xTgAD mice used in this study were pre-symptomatic, as learning was not impaired (Fig 5C, D). Synaptic dysfunction is an early event in this model and is associated with intracellular amyloid-beta accumulation in pre-symptomatic mice. We have found that the 3xTgAD mice are living longer and that the AD neuropathology and cognitive decline have become delayed in progressive generations of this model.
FIGURE 5.
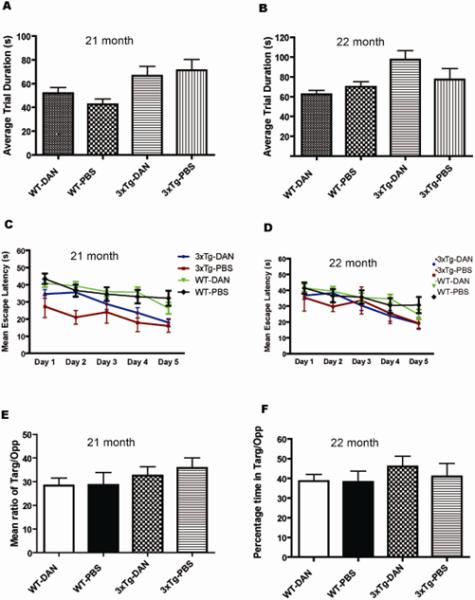
Effects of dantrolene treatment on behavior. (A, B) No significant differences were found in the average trial duration on the rotorod, which was used to assess motor function for each of the experimental groups. (C - F) Learning and memory testing using the Morris Water Maze (MWM). Fifteen month old mice were treated with dantrolene for 6 months and their reference memory was tested at both 21 months (C) and 22 months of age (D). There were no differences detected in the mean escape latency at either time point. (E, F). No differences were found at either age in the probe test, which measured the mean of the ratio of the percent time spent in the target quadrant to the opposite quadrant when the hidden platform was removed. Experimental groups included wild type mice treated with dantrolene (WT-DAN) (n=14) or PBS (WT-PBS) (n=12) and triple transgenic Alzheimer disease mice treated with dantrolene (3xTg-DAN) (n=14) or PBS (3xTg-PBS) (n=16) All data represent mean ± SEM and were analyzed by two-way ANOVA with repeated measures.
DISCUSSION
This study demonstrates that long-term oral treatment with dantrolene in aged 3xTg-AD mice significantly decreased intraneuronal amyloid accumulation in the hippocampus. However, it is not clear whether these effects are due to an inhibition in the production of amyloid or an increased clearance of amyloid. The striking finding of this study is that dantrolene treatment starting well after the initiation of amyloid neuropathology still significantly reduced the intraneuronal amyloid load, supporting the finding from previous studies showing that dantrolene inhibited the production of pathological amyloid 25 and subsequent aggregation.16;25 Studies from this 3xTgAD mouse model and humans have found that the amyloid-beta peptide accumulates intracellularly 48. Western blot assays in this model using 6E1040 have shown that the intracellular accumulation of 6E10 is associated with amyloid-beta. The human tau protein is also expressed intracellularly in this model. These animals are born with both transgenes but their intracellular detection does not start until 6-12 months, reflective of the disease process. It is controversial as to whether extracellular amyloid plaques are the root cause of cognitive dysfunction or a side product in AD. While some studies demonstrated the relationship between amyloid pathology and cognitive dysfunction, others have not.49-51;51 The number of plaques in AD patients does not seem to correlate with cognitive performance.52;53 In contrast, the total amyloid load may be a better measure.54 Interestingly, intraneuronal amyloid production and aggregation may play an important role in AD neuropathology and may be a potential biomarker for the onset of AD.55 Early intraneuronal amyloid accumulation in the hippocampus may contribute to cognitive dysfunction at a later time point in the 3xTg-AD mice.55 In addition, amyloid accumulation may function upstream of tau, promoting tau pathology and subsequent neuronal death and/or synaptic dysfunction.56 Amyloid oligomers may cause tau-dependent microtubule disassembly,57 impairment of normal mitochondrial transport along microtubes,58 dendritic microtubule severing,59 dysregulation of the cell cycle,60 and impairment of long-term potentiation.61 All of these may eventually result in impaired synaptic function and subsequent cognitive dysfunction.56 If intraneuronal amyloid accumulation plays an important role in AD neuropathology and contributes to cognitive dysfunction,51;55;62 then treatment with dantrolene may decrease the intraneuronal amyloid load in an AD brain as demonstrated in the current study, and contribute to the amelioration of synaptic and cognition dysfunction shown in previous studies.16;24;25 The striking finding in this study is that dantrolene decreased intraneuronal accumulation in presymptomatic AD mice, suggesting it may have the potential to prevent symptomatic AD. Further studies are needed to determine if the reduction of intraneuronal amyloid accumulation by dantrolene in an AD vulnerable brain contributes to memory and learning improvement in other AD transgenic mice or in patients. Additionally, it is known that ER stress plays an important role in the neuropathology of AD.63-65 Abnormal Ca2+ release from the ER, via overactivation of either InsP3 or ryanodine receptors, contributes to ER stress,28;66;67 especially in AD.5;18;65;67-70 Therefore, inhibiting the over-activation of these receptors16;24;25;28,19;71;72 and thereby ameliorating the detrimental ER stress, may be a potential therapy for AD. Recent results from translational studies16;24;25 are encouraging regarding the use of dantrolene, and other drugs that stabilize the intracellular calcium homeostasis, for the treatment of AD. Further studies are important and urgent to test this class of drug in AD patients.73;74
While long term treatment of dantrolene in aged 3XTg-AD mice significantly decreased presympotomatic intraneuronal amyloid load, this study is limited by the difficulty in evaluating the possible therapeutic effects of dantrolene on cognitive dysfunction due to the lack of obvious memory or learning impairment in these aged AD mice. It will be important to investigate the possible therapeutic role of dantrolene treatment in ameliorating cognitive dysfunction in a more aggressive AD animal model in the future. Particularly, future studies need to investigate whether dantrolene can be a drug to stop the progression of cognitive dysfunction in AD, not just relieve symptoms.73 The treatment of frank AD has not been effective thus far and the field is moving towards treatment during the pre-mild cognitive impairment (pre-MCI) stage. Pre-MCI is primarily characterized by reduced hippocampal volume and amyloid beta accumulation, well before the onset of behavioral symptoms 75;76. While it is difficult to predict pre-MCI in humans, 3xTg-AD mice eventually acquire the AD phenotype and is thus as a good model of an AD vulnerable brain. The prolonged progression and delay in the AD phenotype, which we see here, is a good model of pre-MCI. While the delay in the disease progression in our 3xTg-AD mice may be related to genetic drift toward the background strains,77 we find that it just takes longer than expected with each generation to develop the AD phenotype. However, the significant appearance of positive 6E10 and AT180 cells in the hippocampus CA1 (Fig. 2 and 3) argues against significant gene drift in our 3xTg-AD mice.
Although a previous study demonstrated the aggravating effects of dantrolene on amyloid pathology, neurodegeneration and synapse damage in AD mice,21 several recent studies16;24;25 consistently showed the beneficial effects of dantrolene to ameliorate memory loss and associated reduction of amyloid burden in various transgenic AD mice. These recent reports16;24;25 are significant and are encouraging for further studies on dantrolene therapy for AD, especially for our mild cognitively impaired and AD patients. Considerable experience has been accumulated on the clinical use of dantrolene to treat other disorders, such as malignant hyperthermia, muscle spasm and neuroleptic malignant syndrome.28;29 The few side effects from the chronic use of dantrolene including drowsy and sleepy, weakness etc., and are relatively tolerable in patients.29;78 Likewise, our aged mice showed no motor or other side effects from 6 months treatment with dantrolene. Should the efficacy of dantrolene treatment for AD be proven, it could be implemented in AD patients quickly.
The neuroprotective effects of dantrolene are clearly dose-dependent in in vitro and in vivo models 28;36;79;80. There has been controversy as to whether or not dantrolene can easily pass through the blood brain barrier. Our studies,16;36 together with others,34 suggested that dantrolene passed the BBB to a limited degree. Therefore, an important strategy to improve dantrolene treatment in AD in the future will be to increase its concentration in the brain while decreasing its concentration in the periphery to maximize its effects on the brain and minimize any side effects. Another strategy to improve drug therapy for AD will be to develop new drugs that selectively inhibit ryanodine receptors and easily get into the brain, without significant peripheral side effects.
MRI has been increasingly used in AD mice as a new research tool considering its advantage of noninvasiveness and repeated in vivo measures to monitor the progress of neuropathology. 81. MRI has been used to measure in vivo changes of brain volume,82, amyloid load83, white matter84. Cognitive dysfunction in AD mice seems to be closely associated to the volume of the hippocampus.82 Hippocampal and cortical volumes can be clearly viewed and accurately measured by the MRI technique in this study (Fig. 4). We did not detect amyloid plaques in the different brain regions, consistent with the absence of extracellular plaques in the brain by immunohistochemistry. The brain volume, especially in the hippocampal formation, has been demonstrated to be either decreased85;86 or increased87 in AD patients. The decrease in hippocampal volume may be caused by neurodegeneration, while an increase in the brain volume is presumably due to progressive gliosis and amyloidosis87. The lack of amyloid plaque and changes in hippocampus volume determined by MRI in the aged 3xTg-AD mice in the current study correlated to the absence of cognitive dysfunction, suggesting that these aged 3xTg-AD mice are still presymptomatic, despite the significant increase in intraneuronal amyloid accumulation and slight elevation of phosphorylated tau protein. Our results suggest that MRI is a useful tool to monitor the dynamic changes of neuropathology in the brain of AD mice
In summary, long-term treatment with dantrolene for pre-symptomatic Alzheimer's disease is effective for the reduction in the accumulation of intraneuronal amyloid and may be a potential therapeutic treatment for pre-clinical AD.
ACKNOWLEDGMENTS
The research work was performed in the lab of Dr. Huafeng Wei and should be attributed to the Department of Anesthesiology, University of Pennsylvania, Philadelphia, PA. The authors appreciate the valuable comments and support from Drs. Roderic Eckenhoff and Lee A. Fleisher from the Department of Anesthesiology and Critical Care, University of Pennsylvania, Philadelphia, PA. This project has been funded by National Institute of General Medical Science, National Institute of Health, Baltimore, Maryland, R01 grant (1-R01GM084979-01, 3R01GM084979-02S1 to H.W.). March of Dimes Birth Defects (12-FY08-167 to H.W.), White Plains, New York; the Research Fund at the Department of Anesthesiology at the University of Pennsylvania, and the MRI studies were performed in the Small Animal Imaging Facility in the Department of Radiology at the University of Pennsylvania, Philadephia, PA.
REFERENCES
- 1.Carey MB, Matsumoto SG. Spontaneous calcium transients are required for neuronal differentiation of murine neural crest. Dev Biol. 1999;215:298–313. doi: 10.1006/dbio.1999.9433. [DOI] [PubMed] [Google Scholar]
- 2.Fiedler MJ, Nathanson MH. The type I inositol 1,4,5-trisphosphate receptor interacts with protein 4.1N to mediate neurite formation through intracellular Ca waves. Neurosignals. 2011;19:75–85. doi: 10.1159/000324507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weissman TA, Riquelme PA, Ivic L, Flint AC, Kriegstein AR. Calcium waves propagate through radial glial cells and modulate proliferation in the developing neocortex. Neuron. 2004;43:647–661. doi: 10.1016/j.neuron.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 4.Zhao X, Yang Z, Liang G, et al. Dual Effects of Isoflurane on Proliferation, Differentiation, and Survival in Human Neuroprogenitor Cells. Anesthesiology. 2013;118:537–549. doi: 10.1097/ALN.0b013e3182833fae. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci. 2008;31:454–463. doi: 10.1016/j.tins.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blackshaw S, Sawa A, Sharp AH, Ross CA, Snyder SH, Khan AA. Type 3 inositol 1,4,5-trisphosphate receptor modulates cell death. FASEB J. 2000;14:1375–1379. doi: 10.1096/fj.14.10.1375. [DOI] [PubMed] [Google Scholar]
- 7.Ferreiro E, Oliveira CR, Pereira C. Involvement of endoplasmic reticulum Ca2+ release through ryanodine and inositol 1,4,5-triphosphate receptors in the neurotoxic effects induced by the amyloid-beta peptide. J Neurosci Res. 2004;76:872–880. doi: 10.1002/jnr.20135. [DOI] [PubMed] [Google Scholar]
- 8.Zhao Y, Liang G, Chen Q, et al. Anesthetic-induced neurodegeneration mediated via inositol 1,4,5-trisphosphate receptors. J Pharmacol Exp Ther. 2010;333:14–22. doi: 10.1124/jpet.109.161562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao YL, Xiang Q, Shi QY, et al. GABAergic excitotoxicity injury of the immature hippocampal pyramidal neurons' exposure to isoflurane. Anesth Analg. 2011;113:1152–1160. doi: 10.1213/ANE.0b013e318230b3fd. [DOI] [PubMed] [Google Scholar]
- 10.Smaili SS, Pereira GJ, Costa MM, et al. The role of calcium stores in apoptosis and autophagy. Curr Mol Med. 2013;13:252–265. doi: 10.2174/156652413804810772. [DOI] [PubMed] [Google Scholar]
- 11.Sattler R, Tymianski M. Molecular mechanisms of calcium-dependent excitotoxicity. J Mol Med. 2000;78:3–13. doi: 10.1007/s001090000077. [DOI] [PubMed] [Google Scholar]
- 12.Adasme T, Haeger P, Paula-Lima AC, et al. Involvement of ryanodine receptors in neurotrophin-induced hippocampal synaptic plasticity and spatial memory formation. Proc Natl Acad Sci U S A. 2011;108:3029–3034. doi: 10.1073/pnas.1013580108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bordji K, Becerril-Ortega J, Buisson A. Synapses, NMDA receptor activity and neuronal Abeta production in Alzheimer's disease. Rev Neurosci. 2011;22:285–294. doi: 10.1515/RNS.2011.029. [DOI] [PubMed] [Google Scholar]
- 14.Chakroborty S, Goussakov I, Miller MB, Stutzmann GE. Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. J Neurosci. 2009;29:9458–9470. doi: 10.1523/JNEUROSCI.2047-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berridge MJ. Calcium signalling and Alzheimer's disease. Neurochem Res. 2011;36:1149–1156. doi: 10.1007/s11064-010-0371-4. [DOI] [PubMed] [Google Scholar]
- 16.Peng J, Liang G, Inan S, et al. Dantrolene ameliorates cognitive decline and neuropathology in Alzheimer triple transgenic mice. Neurosci Lett. 2012;516:274–279. doi: 10.1016/j.neulet.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bezprozvanny I. Calcium signaling and neurodegenerative diseases. Trends in Molecular Medicine. 2009;15:89–100. doi: 10.1016/j.molmed.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheung KH, Shineman D, Muller M, et al. Mechanism of Ca2+ disruption in Alzheimer's disease by presenilin regulation of InsP(3) receptor channel gating. Neuron. 2008;58:871–883. doi: 10.1016/j.neuron.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheung KH, Mei L, Mak DO, et al. Gain-of-function enhancement of IP3 receptor modal gating by familial Alzheimer's disease-linked presenilin mutants in human cells and mouse neurons. Sci Signal. 2010;3:ra22. doi: 10.1126/scisignal.2000818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Demuro A, Parker I, Stutzmann GE. Calcium signaling and amyloid toxicity in Alzheimer disease. J Biol Chem. 2010;285:12463–12468. doi: 10.1074/jbc.R109.080895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang H, Sun S, Herreman A, De SB, Bezprozvanny I. Role of presenilins in neuronal calcium homeostasis. J Neurosci. 2010;30:8566–8580. doi: 10.1523/JNEUROSCI.1554-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muller M, Cheung KH, Foskett JK. Enhanced ROS generation mediated by Alzheimer's disease presenilin regulation of InsP3R Ca2+ signaling. Antioxid Redox Signal. 2011;14:1225–1235. doi: 10.1089/ars.2010.3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liang G, Wang QJ, Li Y, et al. A presenilin-1 mutation renders neurons vulnerable to isoflurane toxicity. Anesth Analg. 2008;106:492–500. doi: 10.1213/ane.0b013e3181605b71. [DOI] [PubMed] [Google Scholar]
- 24.Chakroborty S, Briggs C, Miller MB, et al. Stabilizing ER Ca(2+) Channel Function as an Early Preventative Strategy for Alzheimer's Disease. PLoS One. 2012;7:e52056. doi: 10.1371/journal.pone.0052056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oules B, Del PD, Greco B, et al. Ryanodine receptor blockade reduces amyloid-beta load and memory impairments in Tg2576 mouse model of Alzheimer disease. J Neurosci. 2012;32:11820–11834. doi: 10.1523/JNEUROSCI.0875-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stutzmann GE, Smith I, Caccamo A, Oddo S, LaFerla FM, Parker I. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer's disease mice. J Neurosci. 2006;26:5180–5189. doi: 10.1523/JNEUROSCI.0739-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bruno AM, Huang JY, Bennett DA, Marr RA, Hastings ML, Stutzmann GE. Altered ryanodine receptor expression in mild cognitive impairment and Alzheimer's disease. Neurobiol Aging. 2012;33:1001.e1–1001.e6. doi: 10.1016/j.neurobiolaging.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Inan S, Wei H. The cytoprotective effects of dantrolene: a ryanodine receptor antagonist. Anesth Analg. 2010;111:1400–1410. doi: 10.1213/ANE.0b013e3181f7181c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krause T, Gerbershagen MU, Fiege M, Weisshorn R, Wappler F. Dantrolene--a review of its pharmacology, therapeutic use and new developments. Anaesthesia. 2004;59:364–373. doi: 10.1111/j.1365-2044.2004.03658.x. [DOI] [PubMed] [Google Scholar]
- 30.Berg M, Bruhn T, Frandsen A, Schousboe A, Diemer NH. Kainic acid-induced seizures and brain damage in the rat: role of calcium homeostasis. Journal of Neuroscience Research. 1995;40:641–646. doi: 10.1002/jnr.490400509. [DOI] [PubMed] [Google Scholar]
- 31.Makarewicz D, Zieminska E, Lazarewicz JW. Dantrolene inhibits NMDA-induced 45Ca uptake in cultured cerebellar granule neurons. Neurochem Int. 2003;43:273–278. doi: 10.1016/s0197-0186(03)00012-3. [DOI] [PubMed] [Google Scholar]
- 32.Muehlschlegel S, Sims JR. Dantrolene: mechanisms of neuroprotection and possible clinical applications in the neurointensive care unit. Neurocrit Care. 2009;10:103–115. doi: 10.1007/s12028-008-9133-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Niebauer M, Gruenthal M. Neuroprotective effects of early vs. late administration of dantrolene in experimental status epilepticus. Neuropharmacology. 1999;38:1343–1348. doi: 10.1016/s0028-3908(99)00059-3. [DOI] [PubMed] [Google Scholar]
- 34.Chen X, Tang TS, Tu H, et al. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 3. J Neurosci. 2008;28:12713–12724. doi: 10.1523/JNEUROSCI.3909-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu J, Tang TS, Tu H, et al. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 2. J Neurosci. 2009;29:9148–9162. doi: 10.1523/JNEUROSCI.0660-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wei H, Perry DC. Dantrolene is cytoprotective in two models of neuronal cell death. J Neurochem. 1996;67:2390–2398. doi: 10.1046/j.1471-4159.1996.67062390.x. [DOI] [PubMed] [Google Scholar]
- 37.Yano T, Nakayama R, Imaizumi T, Terasaki H, Ushijima K. Dantrolene ameliorates delayed cell death and concomitant DNA fragmentation in the rat hippocampal CA1 neurons subjected to mild ischemia. Resuscitation. 2001;50:117–125. doi: 10.1016/s0300-9572(00)00369-5. [DOI] [PubMed] [Google Scholar]
- 38.Chen X, Wu J, Lvovskaya S, Herndon E, Supnet C, Bezprozvanny I. Dantrolene is neuroprotective in Huntington's disease transgenic mouse model. Mol Neurodegener. 2011;6:81. doi: 10.1186/1750-1326-6-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guo Q, Sopher BL, Furukawa K, et al. Alzheimer's presenilin mutation sensitizes neural cells to apoptosis induced by trophic factor withdrawal and amyloid beta-peptide: involvement of calcium and oxyradicals. J Neurosci. 1997;17:4212–4222. doi: 10.1523/JNEUROSCI.17-11-04212.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oddo S, Caccamo A, Shepherd JD, et al. Triple-transgenic model of Alzheimer's disease with plaques and tangles: Intracellular A beta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 41.Billings LM, Green KN, McGaugh JL, LaFerla FM. Learning decreases A beta*56 and tau pathology and ameliorates behavioral decline in 3xTg-AD mice. J Neurosci. 2007;27:751–761. doi: 10.1523/JNEUROSCI.4800-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kitazawa M, Cheng D, LaFerla FM. Chronic copper exposure exacerbates both amyloid and tau pathology and selectively dysregulates cdk5 in a mouse model of AD. J Neurochem. 2009;108:1550–1560. doi: 10.1111/j.1471-4159.2009.05901.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liang G, Ward C, Peng J, Zhao Y, Huang B, Wei H. Isoflurane causes greater neurodegeneration than an equivalent exposure of sevoflurane in the developing brain of neonatal mice. Anesthesiology. 2010;112:1325–1334. doi: 10.1097/ALN.0b013e3181d94da5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Y, Liang G, Wang S, Meng Q, Wang Q, Wei H. Effect of fetal exposure to isoflurane on postnatal memory and learning in rats. Neuropharmacology. 2007;53:942–950. doi: 10.1016/j.neuropharm.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang JX, Mardini F, Caltagarone BM, et al. Anesthesia in presymptomatic Alzheimer's disease: a study using the triple-transgenic mouse model. Alzheimers Dement. 2011;7:521–531. doi: 10.1016/j.jalz.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang JX, Mardini F, Janik LS, et al. Modulation of murine Alzheimer pathogenesis and behavior by surgery. Ann Surg. 2013;257:439–448. doi: 10.1097/SLA.0b013e318269d623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paxinos G, Halliday G, Watson C, Koutcherov Y, Wang H. Atlas of the Developing Mouse Brain. First ed. Academic Press; 2007. [Google Scholar]
- 48.LaFerla FM, Green KN, Oddo S. Intracellular amyloid-beta in Alzheimer's disease. Nat Rev Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- 49.Castellani RJ, Lee HG, Siedlak SL, et al. Reexamining Alzheimer's disease: evidence for a protective role for amyloid-beta protein precursor and amyloid-beta. J Alzheimers Dis. 2009;18:447–452. doi: 10.3233/JAD-2009-1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kimura R, Devi L, Ohno M. Partial reduction of BACE1 improves synaptic plasticity, recent and remote memories in Alzheimer's disease transgenic mice. J Neurochem. 2010;113:248–261. doi: 10.1111/j.1471-4159.2010.06608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ondrejcak T, Klyubin I, Hu NW, Barry AE, Cullen WK, Rowan MJ. Alzheimer's disease amyloid beta-protein and synaptic function. Neuromolecular Med. 2010;12:13–26. doi: 10.1007/s12017-009-8091-0. [DOI] [PubMed] [Google Scholar]
- 52.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 53.Samuel W, Terry RD, DeTeresa R, Butters N, Masliah E. Clinical correlates of cortical and nucleus basalis pathology in Alzheimer dementia. Arch Neurol. 1994;51:772–778. doi: 10.1001/archneur.1994.00540200048015. [DOI] [PubMed] [Google Scholar]
- 54.Cummings BJ, Cotman CW. Image analysis of beta-amyloid load in Alzheimer's disease and relation to dementia severity. Lancet. 1995;346:1524–1528. doi: 10.1016/s0140-6736(95)92053-6. [DOI] [PubMed] [Google Scholar]
- 55.Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal A beta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- 56.Bloom GS. Amyloid-beta and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014;71:505–508. doi: 10.1001/jamaneurol.2013.5847. [DOI] [PubMed] [Google Scholar]
- 57.King ME, Kan HM, Baas PW, Erisir A, Glabe CG, Bloom GS. Tau-dependent microtubule disassembly initiated by prefibrillar beta-amyloid. J Cell Biol. 2006;175:541–546. doi: 10.1083/jcb.200605187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vossel KA, Zhang K, Brodbeck J, et al. Tau reduction prevents Abeta-induced defects in axonal transport. Science. 2010;330:198. doi: 10.1126/science.1194653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zempel H, Luedtke J, Kumar Y, et al. Amyloid-beta oligomers induce synaptic damage via Tau-dependent microtubule severing by TTLL6 and spastin. EMBO J. 2013;32:2920–2937. doi: 10.1038/emboj.2013.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Seward ME, Swanson E, Norambuena A, et al. Amyloid-beta signals through tau to drive ectopic neuronal cell cycle re-entry in Alzheimer's disease. J Cell Sci. 2013;126:1278–1286. doi: 10.1242/jcs.1125880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shipton OA, Leitz JR, Dworzak J, et al. Tau protein is required for amyloid {beta}-induced impairment of hippocampal long-term potentiation. J Neurosci. 2011;31:1688–1692. doi: 10.1523/JNEUROSCI.2610-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Deane R, Sagare A, Hamm K, et al. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J Clin Invest. 2008;118:4002–4013. doi: 10.1172/JCI36663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Siman R, Flood DG, Thinakaran G, Neumar RW. Endoplasmic reticulum stress-induced cysteine protease activation in cortical neurons: effect of an Alzheimer's disease-linked presenilin-1 knock-in mutation. J Biol Chem. 2001;276:44736–43. doi: 10.1074/jbc.M104092200. [DOI] [PubMed] [Google Scholar]
- 64.Lindholm D, Wootz H, Korhonen L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006;13:385–392. doi: 10.1038/sj.cdd.4401778. [DOI] [PubMed] [Google Scholar]
- 65.Katayama T, Imaizumi K, Manabe T, Hitomi J, Kudo T, Tohyama M. Induction of neuronal death by ER stress in Alzheimer's disease. J Chem Neuroanat. 2004;28:67–78. doi: 10.1016/j.jchemneu.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 66.Kiviluoto S, Vervliet T, Ivanova H, et al. Regulation of inositol 1,4,5-trisphosphate receptors during endoplasmic reticulum stress. Biochim Biophys Acta. 2013;1833:1612–1624. doi: 10.1016/j.bbamcr.2013.01.026. [DOI] [PubMed] [Google Scholar]
- 67.Wei H, Xie Z. Anesthesia, calcium homeostasis and Alzheimer's disease. Curr Alzheimer Res. 2009;6:30–35. doi: 10.2174/156720509787313934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mattson MP. Pathways towards and away from Alzheimer's disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Guo Q, Fu WM, Sopher BL, et al. Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nat Med. 1999;5:101–106. doi: 10.1038/4789. [DOI] [PubMed] [Google Scholar]
- 70.Cardenas C, Miller RA, Smith I, et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell. 2010;142:270–283. doi: 10.1016/j.cell.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stutzmann GE. Calcium dysregulation, IP3 signaling, and Alzheimer's disease. Neuroscientist. 2005;11:110–115. doi: 10.1177/1073858404270899. [DOI] [PubMed] [Google Scholar]
- 72.Stutzmann GE, Caccamo A, LaFerla FM, Parker I. Dysregulated IP3 signaling in cortical neurons of knock-in mice expressing an Alzheimer's-linked mutation in presenilin1 results in exaggerated Ca2+ signals and altered membrane excitability. J Neurosci. 2004;24:508–513. doi: 10.1523/JNEUROSCI.4386-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Osborn GG, Saunders AV. Current treatments for patients with Alzheimer disease. J Am Osteopath Assoc. 2010;110:S16–S26. [PubMed] [Google Scholar]
- 74.McKeage K. Memantine: a review of its use in moderate to severe Alzheimer's disease. CNS Drugs. 2009;23:881–897. doi: 10.2165/11201020-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 75.Duara R, Loewenstein DA, Greig MT, et al. Pre-MCI and MCI: neuropsychological, clinical, and imaging features and progression rates. Am J Geriatr Psychiatry. 2011;19:951–960. doi: 10.1097/JGP.0b013e3182107c69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jack CR, Jr., Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 2010;9:119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hirata-Fukae C, Li HF, Hoe HS, et al. Females exhibit more extensive amyloid, but not tau, pathology in an Alzheimer transgenic model. Brain Res. 2008;1216:92–103. doi: 10.1016/j.brainres.2008.03.079. [DOI] [PubMed] [Google Scholar]
- 78.Ward A, Chaffman MO, Sorkin EM. Dantrolene. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic use in malignant hyperthermia, the neuroleptic malignant syndrome and an update of its use in muscle spasticity. Drugs. 1986;32:130–168. doi: 10.2165/00003495-198632020-00003. [DOI] [PubMed] [Google Scholar]
- 79.Wei H, Leeds P, Chen RW, et al. Neuronal apoptosis induced by pharmacological concentrations of 3-hydroxykynurenine: characterization and protection by dantrolene and Bcl-2 overexpression. J Neurochem. 2000;75:81–90. doi: 10.1046/j.1471-4159.2000.0750081.x. [DOI] [PubMed] [Google Scholar]
- 80.Zhang L, Andou Y, Masuda S, Mitani A, Kataoka K. Dantrolene protects against ischemic, delayed neuronal death in gerbil brain. Neurosci Lett. 1992;158:105–108. doi: 10.1016/0304-3940(93)90623-s. [DOI] [PubMed] [Google Scholar]
- 81.Jack CR, Jr., Marjanska M, Wengenack TM, et al. Magnetic resonance imaging of Alzheimer's pathology in the brains of living transgenic mice: a new tool in Alzheimer's disease research. Neuroscientist. 2007;13:38–48. doi: 10.1177/1073858406295610. [DOI] [PubMed] [Google Scholar]
- 82.Yin J, Turner GH, Coons SW, Maalouf M, Reiman EM, Shi J. Association of amyloid burden, brain atrophy and memory deficits in aged apolipoprotein epsilon4 mice. Curr Alzheimer Res. 2014;11:283–290. doi: 10.2174/156720501103140329220007. [DOI] [PubMed] [Google Scholar]
- 83.Wengenack TM, Jack CR, Jr., Garwood M, Poduslo JF. MR microimaging of amyloid plaques in Alzheimer's disease transgenic mice. Eur J Nucl Med Mol Imaging. 2008;35(Suppl 1):S82–S88. doi: 10.1007/s00259-007-0706-9. [DOI] [PubMed] [Google Scholar]
- 84.Kastyak-Ibrahim MZ, Di Curzio DL, Buist R, et al. Neurofibrillary tangles and plaques are not accompanied by white matter pathology in aged triple transgenic-Alzheimer disease mice. Magn Reson Imaging. 2013;31:1515–1521. doi: 10.1016/j.mri.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 85.Redwine JM, Kosofsky B, Jacobs RE, et al. Dentate gyrus volume is reduced before onset of plaque formation in PDAPP mice: a magnetic resonance microscopy and stereologic analysis. Proc Natl Acad Sci U S A. 2003;100:1381–1386. doi: 10.1073/pnas.242746599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Oberg J, Spenger C, Wang FH, et al. Age related changes in brain metabolites observed by 1H MRS in APP/PS1 mice. Neurobiol Aging. 2008;29:1423–1433. doi: 10.1016/j.neurobiolaging.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 87.Maheswaran S, Barjat H, Rueckert D, et al. Longitudinal regional brain volume changes quantified in normal aging and Alzheimer's APP × PS1 mice using MRI. Brain Res. 2009;1270:19–32. doi: 10.1016/j.brainres.2009.02.045. [DOI] [PubMed] [Google Scholar]