

Abstract
A small library of truncated/lipid-conjugated neuromedin U (NmU) analogs was synthesized and tested in vitro using an intracellular calcium signaling assay. The selected, most active analogs were then tested in vivo, and showed potent anorexigenic effects in a diet-induced obese (DIO) mouse model. The most promising compound, NM4-C16 was effective in a once-weekly-dose regimen. Collectively, our findings suggest that short, lipidated analogs of NmU are suitable leads for the development of novel anti-obesity therapeutics.
Keywords: Neuromedin U receptor agonists, Antiobesity agents, Lipid-conjugated peptides, Obesity
Graphical abstract
1. Introduction
Obesity is a leading preventable cause of death worldwide, with increasing prevalence in adults and children. This has become one of the most serious public health problems of the 21st century [1]. Generally, obesity increases the likelihood of various diseases, particularly heart disease, type 2 diabetes, obstructive sleep apnea, certain types of cancer, and osteoarthritis [2]. Currently, therapeutics capable of reducing appetite and increasing energy expenditure are of high interest, and gut peptides which regulate energy homeostasis represent attractive leads [3–5]. Neuromedin U (NmU) is an endogenous peptide, highly conserved across species, that is implicated in a number of physiological processes including nociception, stress, inflammation, blood pressure, feeding, energy homeostasis, and glycemic control [6,7]. NmU is widely distributed in the body both peripherally and centrally [8–11], and its function is mediated by two G-protein coupled receptors (GPCRs): NMU1 and NMU2 [6,12–18]. NMU1 is predominantly expressed in the peripheral tissues, particularly the gastrointestinal tract, pancreas, uterus and testes [16,18], whereas NMU2 is mainly expressed in the central nervous system (CNS) with the highest levels in the hypothalamus, hippocampus, spinal cord and paraventricular nucleus [11,14,15]. In humans, NMU gene variants have been linked to excess body weight [19].
The bioactivity of NmU is mediated mainly through its C-terminal conserved region: F1L2F3R4P5R6N7-amide [6,20]. To date, several structure–activity relationship (SAR) studies of this fragment have been carried out, establishing structural requirements for potent agonistic activity [21–28]. Generally, R6 and N7-amide are necessary for binding and activation respectively of the avian peripheral receptor and substitution of F3→Y results in improved bioactivity. N-terminal modifications with pyroglutamic acid, succinic acid and glutaric acid are permissive, showing improved aminopeptidase resistance and increased agonistic activity in contractility assays [26]. Using the same assays it was also determined that substitution of either, F1, F3, R4, P5, R6, or N7-amide with glycine or their D-amino acid counterparts results in decreased bioactivity [29]. At murine NMU receptors, residues R4 and R6 are necessary for the agonistic activity with R4 being critical [30]. Moreover, the C-terminal N7-amide moiety is required for intracellular Ca2+ signaling by both recombinant human NMU1 [13] and murine NMU1 and NMU2 [30]. However recently published data for modified human full-length and truncated NmU analogs [31] showed that the N-terminal N7-amide may be replaced by D-norleucine-amide, giving additional selectivity toward NMU2 (compounds K and Q). Similar selectivity toward NMU2 is also observed for R6→homoArg replacement in combination with F1→W or F1→W and P5→A substitutions (compounds I, J, O and P). Notably, other receptor-selective analogs of NmU were also recently described [28]. The selectivity toward NMU2 was achieved by simultaneous modifications in positions F1, F3 and R4. Similar selectivity toward NMU1 was exerted by modifications of L2 at both its side chain and α–amine-group. The modification of P5 can also have variable outcome in terms of bioactivity. For example, in compound 1b [28] such modifications always lead to analogs with decreased activity regardless of substituent (e.g. Hyp, compound S4a; L-pipecolic acid, compound S4b; and 1-aminocyclopropane-1-carboxylic acid, compound S4c). On the other hand in certain modification schemes they seem beneficial (compounds J and P) [31].
Peripheral administration of NmU reduces food intake and body weight in rodents and birds [15,32–35], as well as increasing locomotor activity and core body temperature in rodents [34] with effects primarily being mediated by NMU2 [36]. Moreover, NmU-overexpression in mice results in a lean phenotype with improved glucose homeostasis [37]. In contrast, NmU-deficient mice develop obesity characterized by hyperphagia, reduced energy expenditure and hyperglycemia [38]. Notably, the chronic administration of NmU does not cause tachyphylaxis and significantly improves glucose tolerance in diet-induced obese (DIO) mice [39]. The unfavorable pharmacokinetic properties of NmU (the half-life of NmU after subcutaneous (s.c.) injection is less than 5 min [39]) were improved by conjugation with polyethylene glycol (PEG) [40] or human serum albumin (HSA) [41] showing in both cases long-lasting, potent anorectic, and blood glucose-normalizing activity. Although mentioned in a patent [31], full length lipid-conjugated (NMU24) and lipid/PEG40-bis-conjugated (NMU25) analogs of neuromedin U showed limited activity. Interestingly, centrally administered NmU-related neuropeptide neuromedin S (NmS), which shares the C-terminal sequence with NmU and activates the same receptors, exerts even greater anorexigenic effect through the corticotropin releasing hormone (CRH) and α-melanocyte-stimulating hormone systems [36,42].
2. Results
Lipidated and additionally stabilized short peptides derived from hormones have been shown to be suitable agents for hormone-replacement therapy [43–46]. To test whether NmU could be modified/derivatized to yield a similar type of compound(s), we synthesized a small library (Table 1) of truncated NmU analogs utilizing various stabilization protocols [47], including: cyclization, lipidation, introduction of α, α-disubstituted amino acids, a retro-inverso-approach, and a combination of these. All peptides were synthesized as C-terminal amides using a standard Fmoc protocol [48] and characterized by analytical RP-HPLC and MALDI-MS (Table 1 & Figure S1). Generally lipidation was achieved by N-terminal conjugation of palmitic acid. However in the case of retro-inverso derivatives and analog NM4A we used iminodiacetic acid mono-N-palmityl amide (IdaNHPal, see Figure 1), which we previously found to be a useful lipidation moiety [44,45]. The double-lipidated analogs, NM4A-C16 and NM4-C16, were synthesized by reductive alkylation using a previously described protocol [49]. As starting materials NM4A and Ahx-Aib-FLFRPRN-amide were employed respectively. Reaction(s) were carried out “in solution” (1,4-dioxane:CH3OH:H2O/5:4:1) with an excess of 1-hexadecanal (50 eq) and sodium cyanoborohydride (NaBH3CN, 100 eq) as the reductive agent. The cyclic analogs NM11-NM16 were synthesized from their linear counterparts (NM8-NM10) using a published S-alkylation protocol [50]. Reaction(s) were carried out in a 50% solution of DMSO in DMF in the presence of cesium carbonate (Cs2CO3) and tetrabutylammonium iodide (TBAI), producing all of the expected analogs but with disappointingly low yields (<5%).
Table 1.
Sequences, analytical and in vitro activity data obtained for NM analogs.
| Peptide | Sequence | Composition | MW Calc / Found | RT (min) | NMU1
|
NMU2
|
||||
|---|---|---|---|---|---|---|---|---|---|---|
| pEC50 | Emax (% hNmU) | Emax at 10 μM as % hNmU Emax | pEC50 | Emax (% hNmU) | Emax at 10 μM as % hNmU Emax | |||||
| hNmU | FRVDEEFQSPFASQSRGY -FL- Phe -Arg- Pro-R-Asn | C141H203N41O38 | 3080.41 / 3080.91 | 32.23 | 8.97±0.12 | 100 | 9.10±0.16 | 100 | ||
| NM1 | Ac -FL- Phe -Arg- Pro-R-Asn | C47H71N16O9 | 990.17 / 990.96 | 30.74 | 9.24±0.12 | 45.5±4.3 | - | 9.47±0.17 | 135.2±0.2 | - |
| NM2 | Aib -FL- Phe -Arg- Pro-R-Asn | C49H76N16O9 | 1033.24 / 1034.14 | 25.77 | 9.25±0.22 | 66.3±0.6 | - | 9.67±0.19 | 69.9±0.1 | - |
| NM3 | Pal-Aib -FL- Phe -Arg- Pro-R-Asn | C65H106N16O10 | 1271.65 / 1272.79 | 64.44 | 6.85±0.08 | 87.8±1.4 | - | 6.97±0.30 | 126.2±0.2 | - |
| NM31 | Pal-Aib -FL- Phe-NMeR- Pro-R-NMeN | C67H110N16O10 | 1299.71 / 1300.86 | 57.24* | - | - | 13.8±2.0 | - | - | 17.9±1.5 |
| NM32 | Pal-Aib -FL- NMeF -Arg- Pro-R-NMeN | C67H110N16O10 | 1299.71 / 1300.98 | 57.16* | - | - | 19.1±1.2 | - | - | 15.2±3.8 |
| NM33 | Pal-Aib -FL- αMeF -Arg- Pro-R-NMeN | C67H110N16O10 | 1299.71 / 1300.89 | 56.22* | - | - | 20.6±1.9 | - | - | 19.2±2.0 |
| NM34 | Pal-Aib -FL- Phe-NMeR- Pro-R-NMeN | C67H110N16O10 | 1299.71 / 1301.14 | 56.41* | - | - | 19.6±3.2 | - | - | 18.4±3.2 |
| NM35 | Pal-Aib -FL- NMeF -Arg- Pro-R-NMeN | C67H110N16O10 | 1299.71 / 1300.96 | 55.85* | - | - | 22.9±3.8 | - | - | 67.0±16.5 |
| NM36 | Pal-Aib -FL- αMeF -Arg- Pro-R-NMeN | C67H110N16O10 | 1299.71 / 1301.11 | 55.13* | - | - | 15.9±6.6 | - | - | 28.6±7.9 |
| NM3D1 | CT-IdaNHPal -FL- Phe -Arg- Pro-R-Asn-NT | C65H107N17O10 | 1286.67 / 1287.44 | 50.30* | - | - | - | - | - | - |
| NM3D2 | CT-IdaNHPal-Ahx -FL- Phe -Arg- Pro-R-Asn-NT | C71H118N18O11 | 1399.83 / 1400.21 | 49.76* | - | - | - | - | - | - |
| NM3D3 | CT-IdaNHPal-Ahx-Aib -FL- Phe -Arg- Pro-R-Asn-NT | C75H125N19O12 | 1484.93 / 1485.29 | 50.34* | - | - | - | - | - | - |
| NM4 | Pal-dPEG12-Aib -FL- Phe -Arg- Pro-R-Asn | C92H159N17O23 | 1871.37 / 1872.04 | 58.82 | 8.75±0.15 | 99.3±15.5 | - | 8.72±0.10 | 92.8±2.2 | - |
| NM4A | IdaNHPal-Aib -FL- Phe -Arg- Pro-R-Asn | C69H114N18O11 | 1371.77 / 1372.21 | 49.28* | 7.91±0.45 | 55.7±10.0 | - | 7.99±0.09 | 67.2±0.1 | - |
| NM4A-C16 | C16-IdaNHPal-Aib -FL- Phe -Arg- Pro-R-Asn | C85H146N18O11 | 1596.18 / 1596.36 | 63.19* | ~ 4.9Δ | - | 118.0±29.6 # | ~ 5.0Δ | - | 99.6±17.9 # |
| NM4A-Nic | Nic-IdaNHPal-Aib -FL- Phe -Arg- Pro-R-Asn | C75H117N19O12 | 1476.85 / 1476.89 | 53.64* | 7.32±0.18 | 57.7±14.8 | - | 7.37±0.09 | 52.8±0.2 | - |
| NM4-C16 | (C16)2-Ahx-Aib -FL- Phe -Arg- Pro-R-Asn | C87H151N17O10 | 1595.24 / 1595.28 | 63.70* | ~ 6.3Δ | - | 81.0±3.6 # | ~ 6.1Δ | - | 28.4±4.3 # |
| NM5 | Aib -FL- Phe -Arg- Pro-R-Asn-Aib | C53H83N17O10 | 1118.34 / 1118.62 | 26.22 | 7.23±0.09 | 77.0±4.6 | - | 7.25±0.09 | 104.7±0.2 | - |
| NM6 | Pal-Aib -FL- Phe -Arg- Pro-R-Asn-Aib | C69H113N1O11 | 1356.76 / 1356.82 | 64.48 | - | - | 40.6±2.2 | - | - | 60.7±12.1 |
| NM7 | Pal-dPEG12-Aib -FL- Phe -Arg- Pro-R-Asn-Aib | C96H166N18O24 | 1956.48 / 1956.82 | 58.79 | 6.89±0.12 | 77.8±4.7 | - | 6.97±0.26 | 66.5±0.1 | - |
| NM8 | C-Ahx -FL- Phe -Arg- Pro-R-Asn-Ahx-C | C63H101N19O12S2 | 1380.73 / 1381.99 | 30.65 | 6.79±0.50 | 59.8±0.5 | - | 6.55±0.10 | 53.6±0.1 | - |
| NM9 | C-Ahx -FL- Phe -Arg- Oic-R-Asn-Ahx-C | C67H107N19O12S2 | 1434.82 / 1435.46 | 33.00 | 5.87±0.08 | 50.1±1.3 | - | 6.14±0.13 | 35.5±0.3 | - |
| NM10 | C-Ahx -FL- Phe -Arg- Tic-R-Asn-Ahx-C | C68H103N19O12S2 | 1442.80 / 1443.57 | 33.69 | ~ 5.3Δ | - | 61.0±12.7 | ~ 5.6Δ | - | 97.8±11.8 |
| NM11 | X(C-Ahx -FL- Phe -Arg- Pro-R-Asn-Ahx-C) | C71H107N19O12S2 | 1482.87 / 1484.28 | 33.69 | - | - | 33.7±0.2 | - | - | 60.6±11.6 |
| NM12 | X(C-Ahx -FL- Phe -Arg- Oic-R-Asn-Ahx-C) | C75H113N19O12S2 | 1536.96 / 1538.37 | 35.33 | - | - | 30.00±6.0 | - | - | 70.0±6.0 |
| NM13 | X(C-Ahx -FL- Phe -Arg- Tic-R-Asn-Ahx-C) | C76H109N19O12S2 | 1544.94 / 1545.62 | 35.93 | - | - | 60.4±13.8 | - | - | 39.3±8.8 |
| NM14 | PalS-X(C-Ahx -FL- Phe -Arg- Pro-R-Asn-Ahx-C) | C88H141N19O12S3 | 1753.38 / 1753.97 | 60.53 | 5.87±0.05 | 50.0±3.0 | - | 6.00±0.09 | 53.1±0.3 | - |
| NM15 | PalS-X(C-Ahx -FL- Phe -Arg- Oic-R-Asn-Ahx-C) | C92H147N19O12S3 | 1807.47 / 1807.82 | 60.77 | ~ 5.2Δ | - | 96.4±7.7 | ~ 5.2Δ | - | 118.4±4.8 |
| NM16 | PalS-X(C-Ahx -FL- Phe -Arg- Tic-R-Asn-Ahx-C) | C93H143N19O12S3 | 1815.45 / 1816.37 | 61.51 | ~ 5.1Δ | - | 133.8±22.0 | ~ 5.2Δ | - | 126.6±10.8 |
All peptides were synthesized as C-terminal amides, NM3D1-D3 are all (D)-retro-inverso-analogs where: NT is the N-terminus and CT is the C-terminus of the peptide(s). Abbreviations: Ahx-6-aminohexanoic acid; Aib-aminoisobutyric acid; Ida-iminodiacetic acid, αMeF-α-methyl-L-phenylalanine; NMeR-Nα-methyl-L-arginine; NMeN-N-methyl-L-asparagine; NMeF-N-methyl-L-phenylalanine; Oic-(2S,3aS,7aS)-octahydro-1H-indole-2-carboxylic acid; Tic-(3S)-1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid; Pal-palmitic acid; PalS-X-3,5-bis(methyl-S-cysteinyl)-1-(methyl-S-palmityl)-benzene; X-1,3-bis(methyl-S-cysteinyl)-benzene; NHPal- N-palmitylamide; dPEG12-40-amino-4,7,10,13,16,19,22,25,28,31,34,37-dodecaoxotetradecanoic acid. Analytical RP-HPLC profiles were obtained with an analytical reversed-phase C18 SymmetryShield™ RP18 column, 4.6×250 mm, 5 μm (Waters Corp., Milford, MA) or (*) an analytical reversed-phase XBridge™ BEH300 C4 column, 4.6×150 mm, 3.5 μm (Waters Corp., Milford, MA). pEC50 and Emax values were obtained using a calcium signaling assay where data are mean ± SEM, n≥4.
Emax at 30 μM as % hNmU Emax.
Approximate EC50 values were derived from curves that had insufficient data points at high concentrations of ligand but which did approach the Emax of hNmU. Values are presented to give an indication of likely agonist potency.
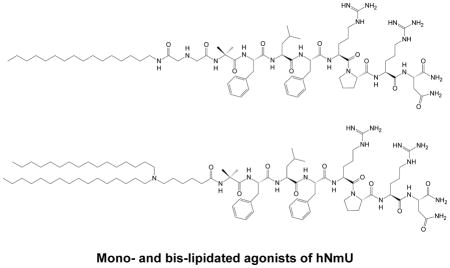
Figure 1.

The most active analogs of NmU synthesized for this study: (A) NM4, (B) NM4A and (C) NM4-C16.
All synthesized NmU-analogs were tested in vitro using a cellular calcium signaling assay [51–53] in HEK293 cells expressing recombinant human NMU1 or NMU2. Examples of concentration-response curves are shown in Figure 2. As a result, we found a lipid-conjugated, truncated analog of NmU, NM4 that possesses high potency at both NMU1 and NMU2, which is similar to that of the native human peptide (hNmU) (Table 1, Figure 1). Notably, NM4 also contains an α, α-disubstituted amino acid (aminoisobutyric acid, Aib) that may confer increased resistance to enzymatic degradation [54].
Figure 2.
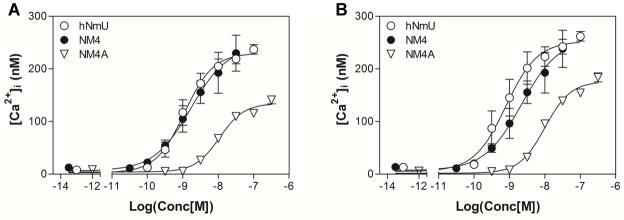
Examples of calcium signaling assay dose-response curves for the maximal changes in intracellular [Ca2+] ([Ca2+]i) performed in fluo-4-loaded HEK293 cells with stable expression of either human NMU1 (A) or human NMU2 (B).
To test whether newly synthesized NmU analogs possess any in vivo anorectic activity, we compared NM4 and NM7 peptides in a diet-induced obese (DIO) mouse model using previously described protocols [39,41]. Initially we focused on single dose experiments comparing NM4 and NM7 at a dose of 5344 nmoles/kg body weight (Figure 3). NM7 was used as an alternative lead compound because it contains two Aib residues that should confer even greater resistance to degradation within the circulation. This could potentially compensate for lower potency and produce responses equivalent to or better than NM4. Since palmitoylated analogues have limited water solubility, we employed a phospholipid-based, commercially available drug delivery system, PUREBRIGHT® SL-220 (NOF America Corp., White Plains, NY) that is suitable for delivery of lipidated peptides [44,45]. Direct comparison revealed that in single dose experiment(s) NM4 showed significant anorectic activity (Figure 3A) that was dose-dependent (Figure 3B). However, NM4 contains a dPEG12 linker (40-amino-4,7,10,13,16,19,22,25,28,31, 34,37-dodecaoxotetradecanoic acid, Peptides International, Inc., Louisville, KY), which is particularly expensive and contributes significantly to the cost of peptide production. Therefore we sought a less expensive alternative(s), which resulted in the discovery of the “PEG-free” NM4A analog (Figure 1) that utilized an N-terminal iminodiacetic acid mono-N-palmityl amide (IdaNHPal). This analog has similar bioactivity to NM4 but is ~10 times less expensive to produce. Subsequent in vivo testing revealed that NM4A is at least 3.5 times more active than NM4 and that its anorectic effect after a single dose (1603 nmoles/kg; 2.2 mg/kg) may last up to ~20 days (Figure 3C). NM4A was characterized further in chronic in vivo experiments. Ad libitum-fed male DIO C57BL/6 mice, which were maintained on a high fat diet (Cat# D12492, Research Diets, Inc., New Brunswick, NJ), were treated subcutaneously (s.c.) with either vehicle or NM4A (2.2 mg/kg; 1603 nmoles/kg) either every 2, 4 or 7 days for a period of 21 days. As shown in Figure 4A, NM4A exerts potent anorectic effects when injected every 2 or every 4 days, with less frequent dosing showing limited activity in this chronic administration regime. Notably s.c. administration of NM4A at the abovementioned doses caused initial gastric emptying within 24 hours of administration but such effects subsided with time. To indicate whether NM4A would be a useful adjunct in obese subjects who are dieting, we tested its efficiency in a once weekly regimen at a dose of 2.2 mg/kg. In this case, ad libitum fed male DIO C57BL/6 mice were kept on either the high fat diet (FD) or regular (normal) diet (ND) during the administration of NM4A. The data suggest that NM4A is beneficial under such circumstances, showing an additional decrease in body weight (Figure 4B).
Figure 3.
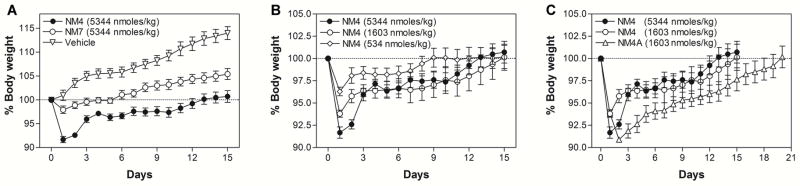
Single dose in vivo experiments performed in the DIO mouse model. (A) Comparison of anorectic effects of NM4 and NM7. (B) NM4 dose-response experiments. (C) Comparison of NM4 and NM4A anorectic activity.
Figure 4.
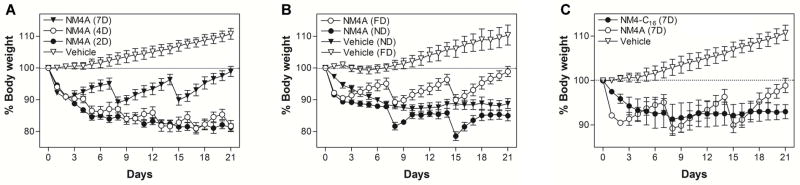
Chronic in vivo experiments performed in the DIO mouse model.. (A) Ad libitum fed male DIO C57BL/6 mice were treated with either vehicle or NM4A (2.2 mg/kg; 1603 nmoles/kg) every 2, 4 or 7 days for 21 days. (B) Ad libitum fed male DIO C57BL/6 mice were treated with either vehicle or NM4A (2.2 mg/kg; 1603 nmoles/kg) once a week for 21 days. After the initial treatment animals were kept either on high-fat diet (FD) or normal diet (ND). (C) Ad libitum fed male DIO C57BL/6 mice were treated with either vehicle or NM4A (2.2 mg/kg; 1603 nmoles/kg) or NM4-C16 (2.6 mg/kg; 1603 nmoles/kg) every 7 days for 21 days.
Considering current trends in obesity/diabetes treatment [55], the development of long-acting analogs of NmU is highly desirable. Such analogs could be used in combination therapies with, for example, incretin-based therapeutic(s) such as semaglutide [56–58], a long-acting glucagon-like peptide 1 (GLP-1) analog that possesses a plasma half-life of 160 h. Since NM4A showed limited activity in a once-weekly regimen we synthesized double-lipidated analogs that should theoretically have improved pharmacokinetic properties. For example, plasma half-life should be increased as a result of the increased possibility/strength of hydrophobic interactions with abundant plasma proteins (albumin, HDL, etc.) [59]. In addition, lipid-conjugation may improve bio-activity by increasing the local concentration of the analog(s) in the hydrophobic environment of lipid rafts within cell plasma membranes [60–62] that are enriched in various GPCRs [63–66]. Therefore, using a reductive-alkylation approach, we synthesized two analogs: NM4A-C16 and NM4-C16, with varying lipidation points/geometry (Figure S2). In vitro testing revealed that only NM4-C16 with a symmetrical lipidation motif that is removed from main body of the NM-peptide, possesses potent bioactivity (albeit reduced compared to NM4A) (Table 1). To indicate whether this approach had yielded an analog that could be used in a once-weekly injection regime, we performed pharmacokinetic studies of NM4A and NM4-C16. The double-lipidated analog, NM4-C16, did indeed have an extended plasma half-life (t1/2=28.2±1.0 h) compared to the single-lipidated NM4A (t1/2=24.0±1.0 h) although the effects of double-lipidation were limited (Figure 5). These results, as well as the relatively short plasma half-life of both, PEG- and HSA-NmU conjugates reported elsewhere [40,41] (t1/2≈11–54 h, depending on species) suggested that a key factor for the in vivo activity of NmU analogs/derivatives may be their resistance to enzymatic proteolysis. Alternatively, loss of their activity may involve partial hydrolysis of the N-terminal N7-amide to α- or β-monocarboxylic acid residues (X-Asn-OH or X-Asp-NH2) via an aminosuccinimide intermediate [22]. Therefore we performed plasma stability studies for both NM4A and NM4-C16 analogs using full length hNmU as a comparator [67–69]. Results (Figure 5C) indicated that both analogs have significant resistance to mouse plasma driven enzymatic degradation, with NM4-C16 being more stable than NM4A. Notably, under the same experimental conditions parental hNmU is quickly degraded falling to less than 1% of initial content within 4 h. Since plasma stability studies do not fully recapitulate physiological conditions (e.g. lack of membrane(s) bound enzymes, different ratios and distribution, etc.) the predictive value of such experiments is rather limited. Nonetheless, the plasma stability of NM4A at t1/2 (24 h) is 79.4±1.8% and plasma stability of NM4-C16 at its t1/2 (28 h) is 98.7±1.9% suggesting that enzymatic resistance may play limited role in performed animal experiments, as at respective t1/2 time points both peptides remain largely intact. Even with a prolonged time of exposure (144 h = 6 days) amounts of both peptides remain significant reaching 39.9±0.4% for NM4A and 60.8±0.4% for NM4-C16.
Figure 5.
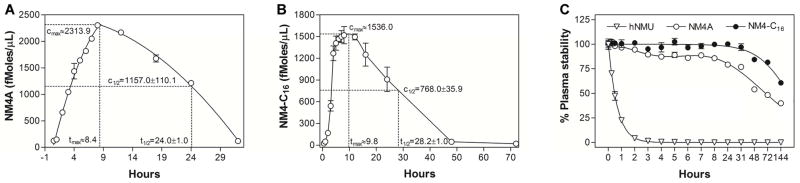
Pharmacokinetics and plasma stability of NM4A and NM4-C16 analogs. For pharmacokinetic studies animals were individually dosed with 10 mg/kg of (A) NM4A or (B) NM4-C16. Plasma stability studies (C) were performed in freshly prepared mouse plasma at 25 μM final concentration(s) of tested analogs.
Further modifications of NM4-C16 are limited due to the high specificity of NMU1 and NMU2 [6,28]. Indeed, our attempts to increase proteolytic resistance of truncated NmU analogs proved largely unsuccessful, with various retro-inverso-, α-carbon-methylated, N-methylated and cyclic analogs being in most cases virtually inactive (Table 1). Nonetheless, data obtained for the analogs NM8-16 suggest that substitution of Pro for more rigid and bulky residues, e.g. Oic or Tic, has an undesirable effect (potency: NM8>NM9>NM10). Moreover, comparison between analogs NM8-10 (linear peptides), NM11-13 (cyclic peptides) and NM14-16 (cyclic/lipidated peptides) also suggests that cyclization is not a viable option as it renders analogs NM11-13 inactive. Interestingly, introduction of a lipid moiety to the cyclic analogs (NM14-16) seems, at least partially, to restore bioactivity. These results are generally in line with published observations that underline the importance of P5 and N7-amide residues for agonistic activity at NMU receptors [21–31]. Considering the inactivity of our retro-inverso-analogs (NM3D1-3) and α-carbon-/N-methylated-variants (NM31-NM35), as well as the limited activity of cyclic derivatives (NM14) and derivatives containing D-amino acids [29,31], further derivatization and/or modifications of NmU for pharmaceutical purposes appears to be extremely difficult since even small changes in the structure may lead to significant loss of bioactivity. Nonetheless, a viable modification seems to be the introduction of a small α, α-disubstituted amino acid(s) (Aib) at the flanking positions of the native active sequence (e.g. FLFRPRN). Notably bis-substituted analogs (e.g. X-Aib-FLFRPRN-Aib) show decreased potency in in vitro assays (NM5<NM2 & NM6<NM3) and very limited activity in vivo (NM7, Figure 3A). Lipidation itself does not appear to have detrimental effects, provided its position is significantly removed from the active portion of the analog(s) (NM4 versus NM2). Although the impact of lipidation on full-length human NmU analogs seem to be more diverse depending on their individual structure [31].
Despite limited in vitro activity of the double-lipidated NM4-C16, this analog was tested in vivo using a once weekly s.c. injection regime at 2.6 mg/kg, which is equimolar to the dose of NM4 used previously (i.e. 1603 nmoles/kg). As shown in Figure 5C, once-weekly administration of NM4-C16 resulted in sustained weight loss in the DIO mouse model. Moreover, in this case we observed very limited gastric emptying in experimental animals and their hematological parameters/blood cell counts were within “normal” range (Table 2). In this particular case, we believe that observed discrepancies between in vitro an in vivo activity may be attributed to limited water solubility of NM4-C16 which limits effective concentrations of the compound in in vitro settings.
Table 2.
Hematological parameters of animals treated with NM4-C16. Ad libitum fed male DIO C57BL/6 mice (high-fat diet) were treated with NM4-C16 (2.6 mg/kg; 1603 nmoles/kg) every 7 days for 21 days. Blood was collected by cardiac puncture and cell counts performed using a HemaVet 950FS Hematology Analyzer (Drew Scientific Inc., Dallas, TX). Control animals received vehicle alone.
| Cells | Count/mL
|
|
|---|---|---|
| Treated | Control | |
| White Blood Cells | 5.73 ± 0.49×106 | 7.35 ± 1.21×106 |
| Neutrophils | 1.02 ± 0.06×106 | 1.30 ± 0.21×106 |
| Lymphocytes | 4.56 ± 0.43×106 | 4.82 ± 0.68×106 |
| Monocytes | 1.46 ± 0.14×105 | 1.07 ± 0.31×105 |
| Red Blood Cells | 9.49 ± 0.31×109 | 9.17 ± 0.17×109 |
| Platelets | 6.95 ± 0.23×108 | 5.97 ± 0.32×108 |
For three groups of animals treated every 2, 4 or 7 days we also evaluated abdominal fat content (Figure 6A). These data indicate that chronic treatment with either, NM4A or NM4-C16 decreases abdominal fat content from 8.1±0.4% (control) to 5.3±0.2%, 4.9±0.3% and 4.5±0.4% for animals treated every 2, 4 and 7 days respectively.
Figure 6.
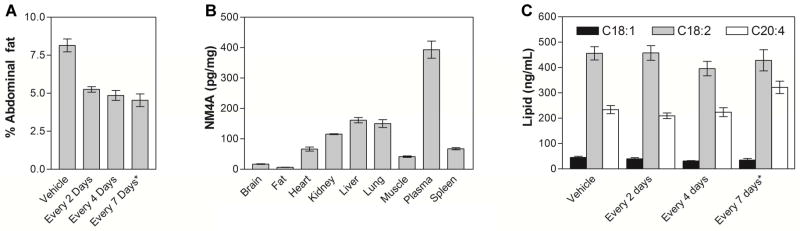
(A) Abdominal fat of chronically treated animals. Ad libitum fed male DIO C57BL/6 mice were treated with either vehicle or NM4A (2.2 mg/kg; 1603 nmoles/kg) every 2 days or NM4A (2.2 mg/kg; 1603 nmoles/kg) every 4 days or (*) NM4-C16 (2.6 mg/kg; 1603 nmoles/kg) every 7 days for 21 days. Subsequently, their abdominal fat was harvested and weighed. Results are expressed as abdominal fat as a % of total body weight. (B) Distribution of NM4A in NM4A chronically treated animals (2.2 mg/kg; every 2 days for 28 days). Subsequently their organs were harvested and analyzed using MDS Sciex QSTAR XL Hybrid Quadrupole Time-of-Flight LC/MS (Applied Biosystems) with methylated NM4A (NM4A-Me) as an internal standard. (C) LPA levels in the plasma of chronically treated animals with either vehicle or NM4A (2.2 mg/kg; 1603 nmoles/kg) every 2 days or NM4A (2.2 mg/kg; 1603 nmoles/kg) every 4 days or (*)NM4-C16 (2.6 mg/kg; 1603 nmoles/kg) every 7 days for 21 days. (18:1)-1-oleoyl-2-hydroxy-sn-glycero-3-phosphatidic acid, (18:2)-1-linoleoyl-2-hydroxy-sn-glycero-3-phosphatidic acid, (20:4)-1-arachidonoyl-2-hydroxy-sn-glycero-3-phosphatidic acid.
To examine the body distribution of NM-analogs we performed limited ADME studies in animals that underwent chronic treatment with NM4A every 2 days for 28 days (2.2 mg/kg; 1603 nmoles/kg). As shown in Figure 6B, the highest content of NM4A was detected in the plasma of experimental animals, with significant amounts also accumulating in the liver, lungs and kidneys. Notably NM4A does not appear to enter the brain, suggesting that its biological effects are transduced via NMU receptors present in the periphery.
The therapeutic use of NmU analogs in obesity and diabetes will require prolonged treatment regimes. Since NmU has cardiovascular effects [70,71] as well as being indicated in cancer [72–74], it is of great interest whether prolonged exposure to our analogs may have related side effects. To assess such possible effects we measured levels of lysophosphatidic acid (LPA, (20:4)-1-arachidonoyl-2-hydroxy-sn-glycero-3-phosphatidic acid; (18:2)-1-linoleoyl-2-hydroxy-sn-glycero-3-phosphatidic acid; and (18:1)-1-oleoyl-2-hydroxy-sn-glycero-3-phosphatidic acid) in the plasma of animals treated frequently (1603 nmoles/kg; every 2 or 4 or 7 days for 21 days) with NM4A or NM4-C16 using a previously described LC-ESI-MS/MS-based method [75–78]. LPA is a ubiquitously present bioactive phospholipid which plays important roles in diverse cellular processes such as cell migration, proliferation and differentiation by acting on its cognate GPCRs [79]. The LPA signaling network is prominent in atherosclerosis development [80–84], cancer initiation, progression and metastasis [85–96] and has also been linked to insulin resistance [97–99] making it well suited for assessment of possible side effects of our therapeutics. As shown in Figure 6C, prolonged treatment of DIO mice with NM4A does not increase plasma levels of LPA suggesting that therapy should have no negative effects on obese patients that are prone to atherosclerosis and diabetes development. Since LPA is also indicated in cancer, these results also suggest that use in obese cancer patients or people who are not aware they have cancer, should be relatively safe, which may be of concern due to the connection of NmU with breast and pancreatic cancers [72–74]. In the case of NM4-C16 slightly elevated levels of (20:4) were observed which may be of concern since this particular lipid was shown to enhance progression of atherosclerosis in animal model [82].
To examine whether lipidation and the position of the lipid addition has an impact on the secondary structure of the NM-analogs, we performed FTIR [100] studies of selected compounds that are listed in Supplemental Material (Figure S2 & S3). Results are summarized in Table 3. An examination of the secondary structure of the full length hNmU by infrared spectrometry indicated that the peptide has a broad absorbance spectrum in the amide I conformation band centered at 1657 cm−1 (Figure S4). Deconvolution of this broad absorbance revealed strong contributions from α-helix and loop-turn components (Table 3). The turn components were adjacent to the α-helical absorbance suggesting that they represented strong 310-helix (type III turn propensity) conformations typical of peptides with Aib residues [101]. The well-defined β-sheet peak centered at 1629 cm−1 was also present. Shorter versions of the parent sequence either had a significant disordered structure as seen with NM1 and its palmitoylated derivative NM3 or significant β-sheet components as seen with the spectrum of NM2 and NM4C13 (Figure S4; Table 3). With the exception of NM4C13, all NM4 N-terminal derivatives had a strong helix–turn conformation suggesting that the addition of N-terminal lipid, PEG or related components helped stabilize the helix and turn propensity of the truncated hNMU sequence. These results further suggest that the N-terminal lipid derivatization not only stabilizes the short peptide structure but may provide an additional hydrophobic anchoring of the active conformation to the active site.
Table 3.
Analysis of peptides secondary structure by FTIR spectroscopy.
| Peptide* | % Conformation
|
|||
|---|---|---|---|---|
| α-helix | β-sheet | loop-turn | disordered | |
| hNMU | 30.36 | 26.20 | 24.57 | 18.87 |
| NM1 | 30.35 | 15.95 | 21.79 | 31.91 |
| NM2 | 26.36 | 31.56 | 27.57 | 11.51 |
| NM3 | 35.24 | 15.95 | 21.79 | 31.91 |
| NM4 | 30.40 | 16.95 | 31.69 | 20.96 |
| Ac-NM2 | 30.97 | 25.95 | 34.66 | 20.96 |
| NM4C13 | 26.46 | 19.36 | 34.62 | 19.56 |
| NM4A | 29.84 | 20.75 | 21.69 | 27.72 |
| NM4A-COOH | 27.77 | 16.72 | 25.92 | 29.59 |
| NM4-Me | 34.05 | 15.27 | 33.08 | 17.60 |
| NM4A-Nic | 29.47 | 18.34 | 29.27 | 22.92 |
| NM4A-C16 | 45.93 | 20.49 | 20.17 | 13.41 |
| NM4-C16 | 30.31 | 16.97 | 27.72 | 25.00 |
self-films of peptides (1.2 mM) hydrated with HFIP:D2O 10 mM buffer pD=7.4 (v:v, 4:6) were analyzed for secondary conformation based on secondary structural analysis using spectral deconvolution and curve fitting.
3. Discussion
In recent years obesity became a global health problem, and the development of anti-obesity therapeutics attracted much attention [55]. Our recent study aimed at the development of short, lipidated peptide analogs, derived from NmU, that could be used as such therapeutics. Using various peptides’-stabilization-protocols, including: cyclization, lipidation, introduction of α, α-disubstituted amino acids, a retro-inverso-approach, and a combination of these, we synthesized a group of truncated NmU analogs, that underwent an extensive in vitro and in vivo testing. As a result, three potent truncated/lipidated agonists on NmU were found: NM4, NM4A and NM4-C16. Unlike previously described PEG-, and HSA-conjugates that require an NmU-carrier conjugation step, these analogs can be efficiently synthesized in large quantities and lipid conjugated using SPPS technology giving the final product with a high yield. In addition, they possess short, 8 amino acids long core sequence, which is approximately 1/3 of native hNmU. Taking into account the molecular weight of reported conjugates [40,41] our analogs are approximately 40 times smaller allowing for delivery of larger quantities of active ingredient (per mole count) in relatively smaller doses. Moreover, use of smaller, palmitoylated peptide(s) may be advantageous over conjugates with macromolecular carriers due to possible side effects that may be associated with the latter. Namely, conjugates with HSA may lead to generation of an antibody-based immune response. Hypersensitivity to macromolecular-PEGs has also been reported [102] triggering, for example, withdrawal of the PEGylated erythropoietic agent Omontys® from the market. Importantly, our double-lipidated analog NM4-C16, was active in vivo in a once weekly s.c. injection regimen, which suggests it could be used in combination therapies with a long-acting glucagon-like peptide 1 (GLP-1) analog(s) during treatment of diabetic patients. Since administration of GLP-1 derivatives also leads to weight loss, it is reasonable to assume possible synergistic effects. However, as NmU is implicated in both insulin secretion [103,104], and glucose homeostasis [39], successful application of such combination therapy will certainly require an extensive further experimentation. Generally, our results demonstrated utility of lipidation approach in modification of short NmU derivatives producing physiologically active derivatives. As the core sequence of these analogs was only minimally altered with proteolytic-resistance-inducing substituents (Aib residue at N-terminus), lipid-conjugation appears to be particularly well suited for the synthesis of therapeutically relevant NmU agonists. Nonetheless, further derivatization and/or modification of NmU for pharmaceutical applications appears to be particularly difficult since even small changes in the structure generally lead to significant loss of bioactivity. Moreover, lingering problem of receptor selectivity (NMU1 versus NMU2) remains largely unsolved, at least in the context of pharmaceutical applications. However, synthesis of NMU1- and NMU2-receptor-specific agonists was recently reported [28], which certainly stimulate further studies in this promising therapeutic area.
4. Conclusions
New family of truncated/lipidated agonists of neuromedin U was synthesized, characterized, and tested both, in vitro and in vivo using a DIO mouse model. Administration of selected peptides in various regimens resulted in significant weight loss in experimental animals. We believe that these analogs may serve as leads for the development of anti-obesity therapeutics.
5. Experimental section
5.1. Peptide synthesis
All peptides were synthesized as C-terminal cysteamine-amides by the solid phase method using a CEM Liberty automatic microwave peptide synthesizer (CEM Corporation Inc., Matthews, NC), applying 9-fluorenylmethyloxycarbonyl (Fmoc) chemistry and commercially available amino acid derivatives and reagents (EMD Biosciences, San Diego, CA and Chem-Impex International, Inc., Wood Dale, IL). Cysteamine 2-Chlorotrityl Resin (EMD Biosciences, San Diego, CA) was used as a solid support. Peptides were cleaved from resin using modified reagent K (TFA 94% (v/v); phenol, 2% (w/v); water, 2% (v/v); TIS, 1% (v/v); EDT, 1% (v/v); 2 h) and precipitated by addition of ice-cold diethyl ether. Subsequently, peptides were purified by preparative reverse-phase high performance liquid chromatography (RP-HPLC) and their purity evaluated by matrix-assisted laser desorption ionization spectrometry (MALDI-MS) as well as analytical RP-HPLC.
5.2. Synthesis of NM4A-C16, NM4-C16 and NM4A-Me by reductive alkylation
The analogs NM4A-C16, NM4-C16 and NM4A-Me (for FTIR and PK studies only) were synthesized from NM4A, Ahx-Aib-FLFRPRN-amide and NM4A respectively. Peptides were dissolved in a mixture of 1,4-dioxane, methanol and water (5:4:1) and mixed with proper carbonyl compound (50 eq, 30 min). For analogs NM4A-C16 and NM4-C16 commercially available 1-hexadecanal was used (Cayman Chemical Company, Ann Arbor, MI), and in the case of NM4A-Me formalin was employed. Subsequently, acetic acid was added (100 eq) and the reaction mixture was placed in an ice-bath and mixed for an additional 20 min (magnetic stirrer). Subsequently freshly prepared water solution of NaBH3CN (100 eq) was added dropwise with vigorous mixing and afterwards the reaction mixture was agitated for an additional 3 hours. The reaction mixture was then further acidified with acetic acid (500 eq), diluted with water (1:1) and freeze-dried. Obtained solid residue(s) were purified by preparative RP-HPLC and their purity evaluated by MALDI-MS as well as analytical RP-HPLC.
5.3. Synthesis of cyclic analogs NM11-16
Cyclization of the linear analogs NM8-NM10 was performed in a 50% solution of DMSO in DMF using 1,3-bis(bromomethyl)benzene (NM11-NM13) or 1-(palmityl-S-methyl)-3,5-bis(bromomethyl)-benzene (NM14-NM16). Briefly, peptides (2 eq.) were dissolved at final concentrations of 5 mg/ml. Subsequently anhydrous cesium carbonate (Cs2CO3, 20 eq.), tetrabutylammonium iodide, (TBAI, 4 eq.) and proper bis(bromomethyl)-benzene-derivative (1 eq) were added. The solution was vigorously mixed on a magnetic stirrer and the progress of the reaction monitored by analytical RP-HPLC. Subsequently peptides were purified and characterized as described in the peptide synthesis section.
5.4. Analytical RP-HPLC
Analytical RP-HPLC was performed on a Varian ProStar 210 HPLC system equipped with ProStar 325 Dual Wavelength UV-Vis detector with the wavelengths set at 220 nm and 280 nm (Varian Inc., Palo Alto, CA). Mobile phases consisted of solvent A, 0.1% TFA in water, and solvent B, 0.1% TFA in acetonitrile. Analyses of peptides were performed with either an analytical reversed-phase C4 XBridge™ BEH300 column, 4.6×150 mm, 3.5 μm (Waters, Milford, MA), or an analytical reversed-phase C18 SymmetryShield™ column, 4.6×250 mm, 5 μm (Waters, Milford, MA) applying linear gradient of solvent B from 0 to 100% over 100 min (flow rate: 1 ml/min).
5.5. Cellular calcium signaling assay
HEK293 cells expressing either the neuromedin U 1 receptor or neuromedin U 2 receptor (HEK-NMU1 and HEK-NMU2 respectively)56 were grown to approximately 90% confluence in ELISA strip plates (96-well format) pre-coated with poly-D-lysine (0.1% w/v) and loaded for 45 min at 37 °C with 2 μM fluo-4-AM in Krebs’-HEPES buffer with bovine serum albumin (KHB-BSA; composition: 10 mM HEPES; 4.2 mM NaHCO3; 11.7 mM D-glucose; 1.18 mM MgSO4·7H2O; 1.18 mM KH2PO4; 4.69 mM KCl; 118 mM NaCl; 1.3 mM CaCl2·2H2O; 0.1% (w/v) BSA; pH 7.4). Monolayers were then washed and equilibrated for 5 min at 37 °C in 100 μl KHB-BSA for subsequent recording of fluorescence as an index of intracellular [Ca2+] ([Ca2+]i) using a microplate reader (NOVOstar; BMG LABTECH, Aylesbury, U.K.). Briefly, 20 μl of KHB-BSA, with or without ligand, was added into the well (200 μL/s) and fluorescence determined at 0.5 s intervals by excitation at 485 nm and collection of emitted light at 520 nm. Changes in fluorescence above basal levels (before ligand addition) were determined. When required, [Ca2+]i was calculated using the formula: [Ca2+]i = Kd (F−Fmin)/(Fmax−F), with the Kd of fluo-4 taken as 350 nM [53]. Fmax was obtained by removal of buffer and addition of KHB-BSA buffer containing 4 mM [Ca2+] and ionomycin (2 μM) to representative wells and the fluorescence measured for 10 min. Fmin was then derived by replacing buffer with Ca2+-free KHB-BSA buffer containing 2 mM EGTA and fluorescence measured for 10 min [51]. For the determination of concentration-response relationships, the maximal change in [Ca2+]i was calculated following ligand addition. Concentration-response curves were then fitted using a four-parameter logistic equation to determine EC50 values (GraphPad Software Inc., CA).
5.6. Animal experiments
All animal experiments were approved by the UCLA Animal Care and Use Committee (ARC#1999-173-23) and conformed to local and national guidelines. Ad libitum fed male DIO C57BL/6 mice (n=8 per group) kept on D12492, a high-fat diet composed of 60% Kcal from fat (Research Diets, Inc., New Brunswick, NJ) were weighed and individually dosed (s.c., 30 min prior to the onset of the dark phase of the light cycle) with either vehicle or NM4A (2.2 mg/kg; 1603 nmoles/kg) every 2 days or NM4A (2.2 mg/kg; 1603 nmoles/kg) every 4 days or NM4A (2.2 mg/kg; 1603 nmoles/kg) every 7 days or NM4-C16 (2.6 mg/kg; 1603 nmoles/kg) every 7 days for 21 days and their body weight were measured daily. Due to limited water solubility, the lipidated analogs NM4A and NM4-C16 were formulated in a phospholipid-based, commercially available drug delivery system, PURE-BRIGHT® SL-220 (NOF America Corp., White Plains, NY). Briefly, the calculated amount of the desired peptide (1 eq) and 10 equivalents of PUREBRIGHT® SL-220 (1 mg : 10 mg) were solubilized in a minimal volume of 90% absolute ethyl alcohol in water. Subsequently the solution was aliquoted into Eppendorf tubes and freeze-dried. The solid residue was re-solubilized in an appropriate amount of PBS directly before use by vortexing.
5.6.1. Body distribution analysis
Ad libitum fed male DIO C57BL/6 mice that underwent chronic treatment with NM4A (2.2 mg/kg; 1603 nmoles/kg; dosing every 2 days for 28 days) were sacrificed and their organs/tissues harvested and weighted. Subsequently 4 volumes of a DMSO/ACN mixture (1:1) containing 0.1% of TFA, were added and samples homogenized using a rod homogenizer (max. speed for 30 s). The homogenate(s) were transferred to 1.5 mL Eppendorf tubes and centrifuged for 10 min at 13000 rpm. Obtained supernatant(s) were analyzed the Agilent 6460 Triple Quadrupole LC/MS System (Agilent Technologies, Santa Clara, CA) with methylated NM4A (NM4A-Me) as an internal standard.
5.6.2. Pharmacokinetic (PK) studies
C57BL/6 mice were weighted and individually dosed with either NM4A or NM4-C16 at 10 mg/kg dose. Subsequently small samples of blood were collected at the indicated time-points and centrifuged (3000 rpm/10 min). Obtained plasma samples were transferred into the 0.5 mL centrifuge tubes and immediately diluted with 4 volumes of a DMSO/ACN mixture (1:1) containing 0.1% of TFA. Subsequently samples were centrifuged at 13000 rpm for 10 min and obtained supernatants analyzed using the Agilent 6460 Triple Quadrupole LC/MS System (Agilent Technologies, Santa Clara, CA) with methylated NM4A (NM4A-Me) as an internal standard.
5.7. Plasma stability studies
Tested analog(s) (10 mM stock solutions in DMSO) were added to freshly prepared mouse plasma (1 μL per 400 μL of plasma, c=25 μM) and incubated at 37 °C. Subsequently small samples of plasma (10 μL) were collected at the indicated time-points and immediately diluted with 200 μL of a DMSO/ACN mixture (1:1) containing 0.1% of TFA. Samples were then centrifuged at 13000 rpm for 10 min and obtained supernatants analyzed using the Agilent 6460 Triple Quadrupole LC/MS System (Agilent Technologies, Santa Clara, CA) with methylated NM4A (NM4A-Me) as an internal standard.
5.8. LPA measurements
LPA was determined by LC-MS/MS as previously described [75–77]. Briefly, plasma samples (100 μL) were extracted using 3cc, 60 mg, Waters Oasis HLB solid phase extraction cartridges. The extracted samples were evaporated under inert gas to dryness using low heat (30–37 °C), and reconstituted with 75 μL of methanol with vortexing. The extracts were transferred to Eppendorf tubes and centrifuged to remove debris. The supernatants were analyzed using the Agilent 6460 Triple Quadrupole LC/MS System (Agilent Technologies, Santa Clara, CA) and quantified using Agilent Mass Hunter QQQ Quantitative Analysis software.
5.9. Determination of peptide secondary structure by FTIR
Since the NM peptides used in this study have a lipo-peptide like character, samples were measured as self-films dried onto a germanium ATR (Attenuated Total Reflectance) sampling crystal. Infrared spectra were recorded at 25 °C using a Bruker Vector 22 FTIR spectrometer equipped with a deuterated triglycine sulfate (DTGS) detector, averaged over 256 scans at a gain of 4 and a resolution of 2 cm−1. FTIR spectra of the peptide samples were obtained from peptide self-films prepared by dissolving peptides in hexafluoroisopropanol and then air drying the solution onto 50×20×2 mm 45° ATR crystals fitted for the Bruker spectrometer (Pike Technologies, Madison, WI). The dried peptide self-film was then hydrated by exposing the sample to nitrogen gas saturated with vapor from deuterated 10 mM sodium phosphate buffer (pH=7.4) for one hour prior to spectral acquisition. The relative proportions of α-helix, turn-loop, β-sheet, and disordered conformations of solution and multilayer IR spectra were determined by Fourier self-deconvolution for band narrowing and area calculations of component peaks of the FTIR spectra using curve-fitting software supplied by Galactic Software (GRAMS/AI, version 8.0; Thermo Electron Corp., Waltham, MA). The frequency limits for the different structures were: α-helix (1662–1645 cm−1), β-sheet (1637–1613 cm−1 and 1710–1682 cm−1), turns (1682–1662 cm−1), and disordered or random (1650–1637 cm−1) [100].
Supplementary Material
Figure S1. An example of analytical RP-HPLC profile and corresponding MS-spectra obtained for analog NM4-C16.
Figure S2. Structures of neuromedin analogs tested in FTIR experiments-Series 1.
Figure S3. Structures of neuromedin analogs tested in FTIR experiments-Series 2.
Figure S4. FTIR spectra of self-films of Series 1 & 2 peptides hydrated with HFIP : D2O 10 mM buffer pD=7.4 (v:v, 4:6).
Highlights.
Truncated analogues of neuromedin U are active in vivo.
Lipidated derivatives of NmU are viable drug candidates for obesity treatment.
Position of lipidation influences affinity of NmU derivatives to NMU1 and NMU2 receptors.
The pharmacokinetic properties of selected lipidated NmU derivatives were studied in murine model.
Acknowledgments
This project was partially supported by funds from the Adams and Burnham endowments provided by the Dean’s Office of the David Geffen School of Medicine at UCLA (PR), the UCSD/UCLA NIDDK Diabetes Research Center grant 5P30DK063491 (JPW), the NIH/NIAID award 5U19AI067769 (EDM and WHM) and the NIH/NHLBI grant 5P01HL030568 (MN).
Appendix A. Supplementary data
Supplementary data (representative analytical RP-HPLC profile and corresponding MALDI-MS spectra, structures of NM analogs tested in FTIR experiments and corresponding FTIR spectra) associated with this article can be found, in the online version, at…..
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Barness LA, Opitz JM, Gilbert-Barness E. Obesity: genetic, molecular, and environmental aspects. Am J Med Genet A. 2007;143A:3016–3034. doi: 10.1002/ajmg.a.32035. [DOI] [PubMed] [Google Scholar]
- 2.Haslam DW, James WP. Obesity. Lancet. 2005;366:1197–1209. doi: 10.1016/S0140-6736(05)67483-1. [DOI] [PubMed] [Google Scholar]
- 3.Karra E, Batterham RL. The role of gut hormones in the regulation of body weight and energy homeostasis. Mol Cell Endocrinol. 2010;316:120–128. doi: 10.1016/j.mce.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 4.Moran TH, Dailey MJ. Minireview: Gut peptides: targets for antiobesity drug development? Endocrinology. 2009;150:2526–2530. doi: 10.1210/en.2009-0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wolnerhanssen B, Beglinger C. Therapeutic potential of gut peptides. Forum Nutr. 2010;63:54–63. doi: 10.1159/000264393. [DOI] [PubMed] [Google Scholar]
- 6.Brighton PJ, Szekeres PG, Willars GB. Neuromedin U and its receptors: structure, function, and physiological roles. Pharmacol Rev. 2004;56:231–248. doi: 10.1124/pr.56.2.3. [DOI] [PubMed] [Google Scholar]
- 7.Mitchell JD, Maguire JJ, Davenport AP. Emerging pharmacology and physiology of neuromedin U and the structurally related peptide neuromedin S. Br J Pharmacol. 2009;158:87–103. doi: 10.1111/j.1476-5381.2009.00252.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Augood SJ, Keast JR, Emson PC. Distribution and characterisation of neuromedin U-like immunoreactivity in rat brain and intestine and in guinea pig intestine. Regul Pept. 1988;20:281–292. doi: 10.1016/0167-0115(88)90063-8. [DOI] [PubMed] [Google Scholar]
- 9.Ballesta J, Carlei F, Bishop AE, Steel JH, Gibson SJ, Fahey M, Hennessey R, Domin J, Bloom SR, Polak JM. Occurrence and developmental pattern of neuromedin U-immunoreactive nerves in the gastrointestinal tract and brain of the rat. Neuroscience. 1988;25:797–816. doi: 10.1016/0306-4522(88)90037-1. [DOI] [PubMed] [Google Scholar]
- 10.Domin J, Ghatei MA, Chohan P, Bloom SR. Characterization of neuromedin U like immunoreactivity in rat, porcine, guinea-pig and human tissue extracts using a specific radioimmunoassay. Biochem Biophys Res Commun. 1986;140:1127–1134. doi: 10.1016/0006-291x(86)90752-7. [DOI] [PubMed] [Google Scholar]
- 11.Graham ES, Turnbull Y, Fotheringham P, Nilaweera K, Mercer JG, Morgan PJ, Barrett P. Neuromedin U and Neuromedin U receptor-2 expression in the mouse and rat hypothalamus: effects of nutritional status. J Neurochem. 2003;87:1165–1173. doi: 10.1046/j.1471-4159.2003.02079.x. [DOI] [PubMed] [Google Scholar]
- 12.Fujii R, Hosoya M, Fukusumi S, Kawamata Y, Habata Y, Hinuma S, Onda H, Nishimura O, Fujino M. Identification of neuromedin U as the cognate ligand of the orphan G protein-coupled receptor FM-3. J Biol Chem. 2000;275:21068–21074. doi: 10.1074/jbc.M001546200. [DOI] [PubMed] [Google Scholar]
- 13.Hedrick JA, Morse K, Shan L, Qiao X, Pang L, Wang S, Laz T, Gustafson EL, Bayne M, Monsma FJ., Jr Identification of a human gastrointestinal tract and immune system receptor for the peptide neuromedin U. Mol Pharmacol. 2000;58:870–875. doi: 10.1124/mol.58.4.870. [DOI] [PubMed] [Google Scholar]
- 14.Hosoya M, Moriya T, Kawamata Y, Ohkubo S, Fujii R, Matsui H, Shintani Y, Fukusumi S, Habata Y, Hinuma S, Onda H, Nishimura O, Fujino M. Identification and functional characterization of a novel subtype of neuromedin U receptor. J Biol Chem. 2000;275:29528–29532. doi: 10.1074/jbc.M004261200. [DOI] [PubMed] [Google Scholar]
- 15.Howard AD, Wang R, Pong SS, Mellin TN, Strack A, Guan XM, Zeng Z, Williams DL, Jr, Feighner SD, Nunes CN, Murphy B, Stair JN, Yu H, Jiang Q, Clements MK, Tan CP, McKee KK, Hreniuk DL, McDonald TP, Lynch KR, Evans JF, Austin CP, Caskey CT, Van der Ploeg LH, Liu Q. Identification of receptors for neuromedin U and its role in feeding. Nature. 2000;406:70–74. doi: 10.1038/35017610. [DOI] [PubMed] [Google Scholar]
- 16.Raddatz R, Wilson AE, Artymyshyn R, Bonini JA, Borowsky B, Boteju LW, Zhou S, Kouranova EV, Nagorny R, Guevarra MS, Dai M, Lerman GS, Vaysse PJ, Branchek TA, Gerald C, Forray C, Adham N. Identification and characterization of two neuromedin U receptors differentially expressed in peripheral tissues and the central nervous system. J Biol Chem. 2000;275:32452–32459. doi: 10.1074/jbc.M004613200. [DOI] [PubMed] [Google Scholar]
- 17.Shan L, Qiao X, Crona JH, Behan J, Wang S, Laz T, Bayne M, Gustafson EL, Monsma FJ, Jr, Hedrick JA. Identification of a novel neuromedin U receptor subtype expressed in the central nervous system. J Biol Chem. 2000;275:39482–39486. doi: 10.1074/jbc.C000522200. [DOI] [PubMed] [Google Scholar]
- 18.Szekeres PG, Muir AI, Spinage LD, Miller JE, Butler SI, Smith A, Rennie GI, Murdock PR, Fitzgerald LR, Wu H, McMillan LJ, Guerrera S, Vawter L, Elshourbagy NA, Mooney JL, Bergsma DJ, Wilson S, Chambers JK. Neuromedin U is a potent agonist at the orphan G protein-coupled receptor FM3. J Biol Chem. 2000;275:20247–20250. doi: 10.1074/jbc.C000244200. [DOI] [PubMed] [Google Scholar]
- 19.Hainerova I, Torekov SS, Ek J, Finkova M, Borch-Johnsen K, Jorgensen T, Madsen OD, Lebl J, Hansen T, Pedersen O. Association between neuromedin U gene variants and overweight and obesity. J Clin Endocrinol Metab. 2006;91:5057–5063. doi: 10.1210/jc.2006-1442. [DOI] [PubMed] [Google Scholar]
- 20.Minamino N, Kangawa K, Matsuo H. Neuromedin U-8 and U-25: novel uterus stimulating and hypertensive peptides identified in porcine spinal cord. Biochem Biophys Res Commun. 1985;130:1078–1085. doi: 10.1016/0006-291x(85)91726-7. [DOI] [PubMed] [Google Scholar]
- 21.Hashimoto T, Kurosawa K, Sakura N. Structure-activity relationships of neuromedin U. II. Highly potent analogs substituted or modified at the N-terminus of neuromedin U-8. Chem Pharm Bull (Tokyo) 1995;43:1154–1157. doi: 10.1248/cpb.43.1154. [DOI] [PubMed] [Google Scholar]
- 22.Kawai T, Shibata A, Kurosawa K, Sato Y, Kato S, Ohki K, Hashimoto T, Sakura N. Structure-activity relationships of neuromedin U. V. study on the stability of porcine neuromedin U-8 at the C-terminal asparagine amide under mild alkaline and acidic conditions. Chem Pharm Bull (Tokyo) 2006;54:659–664. doi: 10.1248/cpb.54.659. [DOI] [PubMed] [Google Scholar]
- 23.Kurosawa K, Sakura N, Hashimoto T. Structure-activity relationships of neuromedin U. III. Contribution of two phenylalanine residues in dog neuromedin U-8 to the contractile activity. Chem Pharm Bull (Tokyo) 1996;44:1880–1884. doi: 10.1248/cpb.44.1880. [DOI] [PubMed] [Google Scholar]
- 24.Okimura K, Sakura N, Ohta S, Kurosawa K, Hashimoto T. Contractile activity of porcine neuromedin U-25 and various neuromedin U-related peptide fragments on isolated chicken crop smooth muscle. Chem Pharm Bull (Tokyo) 1992;40:1500–1503. doi: 10.1248/cpb.40.1500. [DOI] [PubMed] [Google Scholar]
- 25.Sakura N, Ohta S, Uchida Y, Kurosawa K, Okimura K, Hashimoto T. Structure-activity relationships of rat neuromedin U for smooth muscle contraction. Chem Pharm Bull (Tokyo) 1991;39:2016–2020. doi: 10.1248/cpb.39.2016. [DOI] [PubMed] [Google Scholar]
- 26.Sakura N, Kurosawa K, Hashimoto T. Structure-activity relationships of neuromedin U. I. Contractile activity of dog neuromedin U-related peptides on isolated chicken crop smooth muscle. Chem Pharm Bull (Tokyo) 1995;43:1148–1153. doi: 10.1248/cpb.43.1148. [DOI] [PubMed] [Google Scholar]
- 27.Sakura N, Kurosawa K, Hashimoto T. Structure-activity relationships of neuromedin U. IV. Absolute requirement of the arginine residue at position 7 of dog neuromedin U-8 for contractile activity. Chem Pharm Bull (Tokyo) 2000;48:1166–1170. doi: 10.1248/cpb.48.1166. [DOI] [PubMed] [Google Scholar]
- 28.Takayama K, Mori K, Taketa K, Taguchi A, Yakushiji F, Minamino N, Miyazato M, Kangawa K, Hayashi Y. Discovery of selective hexapeptide agonists to human neuromedin u receptors types 1 and 2. J Med Chem. 2014;57:6583–6593. doi: 10.1021/jm500599s. [DOI] [PubMed] [Google Scholar]
- 29.Hashimoto T, Masui H, Uchida Y, Sakura N, Okimura K. Agonistic and antagonistic activities of neuromedin U-8 analogs substituted with glycine or D-amino acid on contractile activity of chicken crop smooth muscle preparations. Chem Pharm Bull (Tokyo) 1991;39:2319–2322. doi: 10.1248/cpb.39.2319. [DOI] [PubMed] [Google Scholar]
- 30.Funes S, Hedrick JA, Yang S, Shan L, Bayne M, Monsma FJ, Jr, Gustafson EL. Cloning and characterization of murine neuromedin U receptors. Peptides. 2002;23:1607–1615. doi: 10.1016/s0196-9781(02)00097-9. [DOI] [PubMed] [Google Scholar]
- 31.Marsh DJ, Pessi A, Bednarek MA, Bianchi E, Ingallinella P, Peier AM. EP 1 999 143 B1. Neuromedin U receptor agonists and uses thereof. 2011 Jul 13;:1–70. Ref Type: Patent.
- 32.Kamisoyama H, Honda K, Saneyasu T, Sugahara K, Hasegawa S. Central administration of neuromedin U suppresses food intake in chicks. Neurosci Lett. 2007;420:1–5. doi: 10.1016/j.neulet.2007.03.062. [DOI] [PubMed] [Google Scholar]
- 33.Kojima M, Haruno R, Nakazato M, Date Y, Murakami N, Hanada R, Matsuo H, Kangawa K. Purification and identification of neuromedin U as an endogenous ligand for an orphan receptor GPR66 (FM3) Biochem Biophys Res Commun. 2000;276:435–438. doi: 10.1006/bbrc.2000.3502. [DOI] [PubMed] [Google Scholar]
- 34.Nakazato M, Hanada R, Murakami N, Date Y, Mondal MS, Kojima M, Yoshimatsu H, Kangawa K, Matsukura S. Central effects of neuromedin U in the regulation of energy homeostasis. Biochem Biophys Res Commun. 2000;277:191–194. doi: 10.1006/bbrc.2000.3669. [DOI] [PubMed] [Google Scholar]
- 35.Shousha S, Nakahara K, Miyazato M, Kangawa K, Murakami N. Endogenous neuromedin U has anorectic effects in the Japanese quail. Gen Comp Endocrinol. 2005;140:156–163. doi: 10.1016/j.ygcen.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 36.Peier A, Kosinski J, Cox-York K, Qian Y, Desai K, Feng Y, Trivedi P, Hastings N, Marsh DJ. The antiobesity effects of centrally administered neuromedin U and neuromedin S are mediated predominantly by the neuromedin U receptor 2 (NMUR2) Endocrinology. 2009;150:3101–3109. doi: 10.1210/en.2008-1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kowalski TJ, Spar BD, Markowitz L, Maguire M, Golovko A, Yang S, Farley C, Cook JA, Tetzloff G, Hoos L, Del Vecchio RA, Kazdoba TM, McCool MF, Hwa JJ, Hyde LA, Davis H, Vassileva G, Hedrick JA, Gustafson EL. Transgenic overexpression of neuromedin U promotes leanness and hypophagia in mice. J Endocrinol. 2005;185:151–164. doi: 10.1677/joe.1.05948. [DOI] [PubMed] [Google Scholar]
- 38.Hanada R, Teranishi H, Pearson JT, Kurokawa M, Hosoda H, Fukushima N, Fukue Y, Serino R, Fujihara H, Ueta Y, Ikawa M, Okabe M, Murakami N, Shirai M, Yoshimatsu H, Kangawa K, Kojima M. Neuromedin U has a novel anorexigenic effect independent of the leptin signaling pathway. Nat Med. 2004;10:1067–1073. doi: 10.1038/nm1106. [DOI] [PubMed] [Google Scholar]
- 39.Peier AM, Desai K, Hubert J, Du X, Yang L, Qian Y, Kosinski JR, Metzger JM, Pocai A, Nawrocki AR, Langdon RB, Marsh DJ. Effects of peripherally administered neuromedin U on energy and glucose homeostasis. Endocrinology. 2011;152:2644–2654. doi: 10.1210/en.2010-1463. [DOI] [PubMed] [Google Scholar]
- 40.Ingallinella P, Peier AM, Pocai A, Marco AD, Desai K, Zytko K, Qian Y, Du X, Cellucci A, Monteagudo E, Laufer R, Bianchi E, Marsh DJ, Pessi A. PEGylation of Neuromedin U yields a promising candidate for the treatment of obesity and diabetes. Bioorg Med Chem. 2012;20:4751–4759. doi: 10.1016/j.bmc.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 41.Neuner P, Peier AM, Talamo F, Ingallinella P, Lahm A, Barbato G, Di MA, Desai K, Zytko K, Qian Y, Du X, Ricci D, Monteagudo E, Laufer R, Pocai A, Bianchi E, Marsh DJ, Pessi A. Development of a neuromedin U-human serum albumin conjugate as a long-acting candidate for the treatment of obesity and diabetes. Comparison with the PEGylated peptide. J Pept Sci. 2013 doi: 10.1002/psc.2582. [DOI] [PubMed] [Google Scholar]
- 42.Ida T, Mori K, Miyazato M, Egi Y, Abe S, Nakahara K, Nishihara M, Kangawa K, Murakami N. Neuromedin s is a novel anorexigenic hormone. Endocrinology. 2005;146:4217–4223. doi: 10.1210/en.2005-0107. [DOI] [PubMed] [Google Scholar]
- 43.Flinn N, Coppard S, Toth I. Oral absorption studies of lipidic conjugates of thyrotropin releasing hormone (TRH)1 and luteinizing hormone-releasing hormone (LHRH)1. Int J Pharm. 1996;137:33–39. [Google Scholar]
- 44.Preza GC, Ruchala P, Pinon R, Ramos E, Qiao B, Peralta MA, Sharma S, Waring A, Ganz T, Nemeth E. Minihepcidins are rationally designed small peptides that mimic hepcidin activity in mice and may be useful for the treatment of iron overload. J Clin Invest. 2011;121:4880–4888. doi: 10.1172/JCI57693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ramos E, Ruchala P, Goodnough JB, Kautz L, Preza GC, Nemeth E, Ganz T. Minihepcidins prevent iron overload in a hepcidin-deficient mouse model of severe hemochromatosis. Blood. 2012;120:3829–3836. doi: 10.1182/blood-2012-07-440743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang J, Chow D, Heiati H, Shen WC. Reversible lipidization for the oral delivery of salmon calcitonin. J Control Release. 2003;88:369–380. doi: 10.1016/s0168-3659(03)00008-7. [DOI] [PubMed] [Google Scholar]
- 47.Biron E, Chatterjee J, Ovadia O, Langenegger D, Brueggen J, Hoyer D, Schmid HA, Jelinek R, Gilon C, Hoffman A, Kessler H. Improving oral bioavailability of peptides by multiple N-methylation: somatostatin analogues. Angew Chem Int Ed Engl. 2008;47:2595–2599. doi: 10.1002/anie.200705797. [DOI] [PubMed] [Google Scholar]
- 48.Fields GB, Noble RL. Solid phase peptide synthesis utilizing 9-fluorenylmethoxycarbonyl amino acids. Int J Pept Protein Res. 1990;35:161–214. doi: 10.1111/j.1399-3011.1990.tb00939.x. [DOI] [PubMed] [Google Scholar]
- 49.Ruchala P, Picur B, Lisowski M, Cierpicki T, Wieczorek Z, Siemion IZ. Synthesis, conformation, and immunosuppressive activity of CLX and its analogues. Biopolymers. 2003;70:497–511. doi: 10.1002/bip.10422. [DOI] [PubMed] [Google Scholar]
- 50.Micewicz ED, Luong HT, Jung CL, Waring AJ, McBride WH, Ruchala P. Novel dimeric Smac analogs as prospective anticancer agents. Bioorg Med Chem Lett. 2014;24:1452–1457. doi: 10.1016/j.bmcl.2014.02.024. [DOI] [PubMed] [Google Scholar]
- 51.Bootman MD, Roderick HL. Using calcium imaging as a readout of GPCR activation. Methods Mol Biol. 2011;746:277–296. doi: 10.1007/978-1-61779-126-0_15. [DOI] [PubMed] [Google Scholar]
- 52.Brighton PJ, Szekeres PG, Wise A, Willars GB. Signaling and ligand binding by recombinant neuromedin U receptors: evidence for dual coupling to Galphaq/11 and Galphai and an irreversible ligand-receptor interaction. Mol Pharmacol. 2004;66:1544–1556. doi: 10.1124/mol.104.002337. [DOI] [PubMed] [Google Scholar]
- 53.Yamasaki-Mann M, Demuro A, Parker I. cADPR stimulates SERCA activity in Xenopus oocytes. Cell Calcium. 2009;45:293–299. doi: 10.1016/j.ceca.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yamaguchi H, Kodama H, Osada S, Kato F, Jelokhani-Niaraki M, Kondo M. Effect of alpha,alpha-dialkyl amino acids on the protease resistance of peptides. Biosci Biotechnol Biochem. 2003;67:2269–2272. doi: 10.1271/bbb.67.2269. [DOI] [PubMed] [Google Scholar]
- 55.Greenwood HC, Bloom SR, Murphy KG. Peptides and their potential role in the treatment of diabetes and obesity. Rev Diabet Stud. 2011;8:355–368. doi: 10.1900/RDS.2011.8.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Devaraj S, Maitra A. Pancreatic safety of newer incretin-based therapies: are the “-tides” finally turning? Diabetes. 2014;63:2219–2221. doi: 10.2337/db14-0545. [DOI] [PubMed] [Google Scholar]
- 57.Gotfredsen CF, Molck AM, Thorup I, Nyborg NC, Salanti Z, Knudsen LB, Larsen MO. The human GLP-1 analogs liraglutide and semaglutide: absence of histopathological effects on the pancreas in nonhuman primates. Diabetes. 2014;63:2486–2497. doi: 10.2337/db13-1087. [DOI] [PubMed] [Google Scholar]
- 58.Monami M, Dicembrini I, Marchionni N, Rotella CM, Mannucci E. Effects of glucagon-like peptide-1 receptor agonists on body weight: a meta-analysis. Exp Diabetes Res. 2012;2012:672658. doi: 10.1155/2012/672658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Madsen K, Knudsen LB, Agersoe H, Nielsen PF, Thogersen H, Wilken M, Johansen NL. Structure-activity and protraction relationship of long-acting glucagon-like peptide-1 derivatives: importance of fatty acid length, polarity, and bulkiness. J Med Chem. 2007;50:6126–6132. doi: 10.1021/jm070861j. [DOI] [PubMed] [Google Scholar]
- 60.Ingallinella P, Bianchi E, Ladwa NA, Wang YJ, Hrin R, Veneziano M, Bonelli F, Ketas TJ, Moore JP, Miller MD, Pessi A. Addition of a cholesterol group to an HIV-1 peptide fusion inhibitor dramatically increases its antiviral potency. Proc Natl Acad Sci U S A. 2009;106:5801–5806. doi: 10.1073/pnas.0901007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Doyle JR, Harwood BN, Krishnaji ST, Krishnamurthy VM, Lin WE, Fortin JP, Kumar K, Kopin AS. A two-step strategy to enhance activity of low potency peptides. PLoS One. 2014;9:e110502. doi: 10.1371/journal.pone.0110502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Avadisian M, Gunning PT. Extolling the benefits of molecular therapeutic lipidation. Mol Biosyst. 2013;9:2179–2188. doi: 10.1039/c3mb70147f. [DOI] [PubMed] [Google Scholar]
- 63.Pike LJ. Lipid rafts: bringing order to chaos. J Lipid Res. 2003;44:655–667. doi: 10.1194/jlr.R200021-JLR200. [DOI] [PubMed] [Google Scholar]
- 64.Insel PA, Head BP, Ostrom RS, Patel HH, Swaney JS, Tang CM, Roth DM. Caveolae and lipid rafts: G protein-coupled receptor signaling microdomains in cardiac myocytes. Ann N Y Acad Sci. 2005;1047:166–172. doi: 10.1196/annals.1341.015. [DOI] [PubMed] [Google Scholar]
- 65.Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1:31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 66.Chini B, Parenti M. G-protein coupled receptors in lipid rafts and caveolae: how, when and why do they go there? J Mol Endocrinol. 2004;32:325–338. doi: 10.1677/jme.0.0320325. [DOI] [PubMed] [Google Scholar]
- 67.Eldridge JA, Milewski M, Stinchcomb AL, Crooks PA. Synthesis and in vitro stability of amino acid prodrugs of 6-beta-naltrexol for microneedle-enhanced transdermal delivery. Bioorg Med Chem Lett. 2014;24:5212–5215. doi: 10.1016/j.bmcl.2014.09.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kang S, Watanabe M, Jacobs JC, Yamaguchi M, Dahesh S, Nizet V, Leyh TS, Silverman RB. Synthesis of mevalonate- and fluorinated mevalonate prodrugs and their in vitro human plasma stability. Eur J Med Chem. 2014;90C:448–461. doi: 10.1016/j.ejmech.2014.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Machado A, Fazio MA, Miranda A, Daffre S, Machini MT. Synthesis and properties of cyclic gomesin and analogues. J Pept Sci. 2012;18:588–598. doi: 10.1002/psc.2439. [DOI] [PubMed] [Google Scholar]
- 70.Rahman AA, Shahid IZ, Pilowsky PM. Neuromedin U causes biphasic cardiovascular effects and impairs baroreflex function in rostral ventrolateral medulla of spontaneously hypertensive rat. Peptides. 2013;44:15–24. doi: 10.1016/j.peptides.2013.03.017. [DOI] [PubMed] [Google Scholar]
- 71.Tanida M, Satomi J, Shen J, Nagai K. Autonomic and cardiovascular effects of central neuromedin U in rats. Physiol Behav. 2009;96:282–288. doi: 10.1016/j.physbeh.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 72.Wu Y, McRoberts K, Berr SS, Frierson HF, Jr, Conaway M, Theodorescu D. Neuromedin U is regulated by the metastasis suppressor RhoGDI2 and is a novel promoter of tumor formation, lung metastasis and cancer cachexia. Oncogene. 2007;26:765–773. doi: 10.1038/sj.onc.1209835. [DOI] [PubMed] [Google Scholar]
- 73.Ketterer K, Kong B, Frank D, Giese NA, Bauer A, Hoheisel J, Korc M, Kleeff J, Michalski CW, Friess H. Neuromedin U is overexpressed in pancreatic cancer and increases invasiveness via the hepatocyte growth factor c-Met pathway. Cancer Lett. 2009;277:72–81. doi: 10.1016/j.canlet.2008.11.028. [DOI] [PubMed] [Google Scholar]
- 74.Rani S, Corcoran C, Shiels L, Germano S, Breslin S, Madden S, McDermott MS, Browne BC, O’Donovan N, Crown J, Gogarty M, Byrne AT, O’Driscoll L. Neuromedin U: a candidate biomarker and therapeutic target to predict and overcome resistance to HER-tyrosine kinase inhibitors. Cancer Res. 2014;74:3821–3833. doi: 10.1158/0008-5472.CAN-13-2053. [DOI] [PubMed] [Google Scholar]
- 75.Buga GM, Navab M, Imaizumi S, Reddy ST, Yekta B, Hough G, Chanslor S, Anantharamaiah GM, Fogelman AM. L-4F alters hyperlipidemic (but not healthy) mouse plasma to reduce platelet aggregation. Arterioscler Thromb Vasc Biol. 2010;30:283–289. doi: 10.1161/ATVBAHA.109.200162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chattopadhyay A, Navab M, Hough G, Gao F, Meriwether D, Grijalva V, Springstead JR, Palgnachari MN, Namiri-Kalantari R, Su F, Van Lenten BJ, Wagner AC, Anantharamaiah GM, Farias-Eisner R, Reddy ST, Fogelman AM. A novel approach to oral apoA-I mimetic therapy. J Lipid Res. 2013;54:995–1010. doi: 10.1194/jlr.M033555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Imaizumi S, Grijalva V, Navab M, Van Lenten BJ, Wagner AC, Anantharamiah GM, Fogelman AM, Reddy ST. L-4F differentially alters plasma levels of oxidized fatty acids resulting in more anti-inflammatory HDL in mice. Drug Metab Lett. 2010;4:139–148. doi: 10.2174/187231210791698438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jesionowska A, Cecerska E, Dolegowska B. Methods for quantifying lysophosphatidic acid in body fluids: a review. Anal Biochem. 2014;453:38–43. doi: 10.1016/j.ab.2014.02.021. [DOI] [PubMed] [Google Scholar]
- 79.Aoki J, Inoue A, Okudaira S. Two pathways for lysophosphatidic acid production. Biochim Biophys Acta. 2008;1781:513–518. doi: 10.1016/j.bbalip.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 80.Schober A, Siess W. Lysophosphatidic acid in atherosclerotic diseases. Br J Pharmacol. 2012;167:465–482. doi: 10.1111/j.1476-5381.2012.02021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dohi T, Miyauchi K, Ohkawa R, Nakamura K, Kurano M, Kishimoto T, Yanagisawa N, Ogita M, Miyazaki T, Nishino A, Yaginuma K, Tamura H, Kojima T, Yokoyama K, Kurata T, Shimada K, Daida H, Yatomi Y. Increased lysophosphatidic acid levels in culprit coronary arteries of patients with acute coronary syndrome. Atherosclerosis. 2013;229:192–197. doi: 10.1016/j.atherosclerosis.2013.03.038. [DOI] [PubMed] [Google Scholar]
- 82.Zhou Z, Subramanian P, Sevilmis G, Globke B, Soehnlein O, Karshovska E, Megens R, Heyll K, Chun J, Saulnier-Blache JS, Reinholz M, van ZM, Weber C, Schober A. Lipo-protein-derived lysophosphatidic acid promotes atherosclerosis by releasing CXCL1 from the endothelium. Cell Metab. 2011;13:592–600. doi: 10.1016/j.cmet.2011.02.016. [DOI] [PubMed] [Google Scholar]
- 83.Chen C, Ochoa LN, Kagan A, Chai H, Liang Z, Lin PH, Yao Q. Lysophosphatidic acid causes endothelial dysfunction in porcine coronary arteries and human coronary artery endothelial cells. Atherosclerosis. 2012;222:74–83. doi: 10.1016/j.atherosclerosis.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bot M, Bot I, Lopez-Vales R, van de Lest CH, Saulnier-Blache JS, Helms JB, David S, van Berkel TJ, Biessen EA. Atherosclerotic lesion progression changes lysophosphatidic acid homeostasis to favor its accumulation. Am J Pathol. 2010;176:3073–3084. doi: 10.2353/ajpath.2010.090009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mills GB, Moolenaar WH. The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer. 2003;3:582–591. doi: 10.1038/nrc1143. [DOI] [PubMed] [Google Scholar]
- 86.Leblanc R, Peyruchaud O. New insights in the autotaxin/LPA axis in cancer development and metastasis. Exp Cell Res. 2014 doi: 10.1016/j.yexcr.2014.11.010. [DOI] [PubMed] [Google Scholar]
- 87.Leblanc R, Lee SC, David M, Bordet JC, Norman DD, Patil R, Miller D, Sahay D, Ribeiro J, Clezardin P, Tigyi GJ, Peyruchaud O. Interaction of platelet-derived autotaxin with tumor integrin alphaVbeta3 controls metastasis of breast cancer cells to bone. Blood. 2014;124:3141–3150. doi: 10.1182/blood-2014-04-568683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Peyruchaud O, Leblanc R, David M. Pleiotropic activity of lysophosphatidic acid in bone metastasis. Biochim Biophys Acta. 2013;1831:99–104. doi: 10.1016/j.bbalip.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 89.Willier S, Butt E, Grunewald TG. Lysophosphatidic acid (LPA) signalling in cell migration and cancer invasion: a focussed review and analysis of LPA receptor gene expression on the basis of more than 1700 cancer microarrays. Biol Cell. 2013;105:317–333. doi: 10.1111/boc.201300011. [DOI] [PubMed] [Google Scholar]
- 90.Tsujiuchi T, Hirane M, Dong Y, Fukushima N. Diverse effects of LPA receptors on cell motile activities of cancer cells. J Recept Signal Transduct Res. 2014;34:149–153. doi: 10.3109/10799893.2013.876042. [DOI] [PubMed] [Google Scholar]
- 91.Tsujiuchi T, Araki M, Hirane M, Dong Y, Fukushima N. Lysophosphatidic acid receptors in cancer pathobiology. Histol Histopathol. 2014;29:313–321. doi: 10.14670/HH-29.313. [DOI] [PubMed] [Google Scholar]
- 92.Du J, Sun C, Hu Z, Yang Y, Zhu Y, Zheng D, Gu L, Lu X. Lysophosphatidic acid induces MDA-MB-231 breast cancer cells migration through activation of PI3K/PAK1/ERK signaling. PLoS One. 2010;5:e15940. doi: 10.1371/journal.pone.0015940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gotoh M, Fujiwara Y, Yue J, Liu J, Lee S, Fells J, Uchiyama A, Murakami-Murofushi K, Kennel S, Wall J, Patil R, Gupte R, Balazs L, Miller DD, Tigyi GJ. Controlling cancer through the autotaxin-lysophosphatidic acid receptor axis. Biochem Soc Trans. 2012;40:31–36. doi: 10.1042/BST20110608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jeong KJ, Cho KH, Panupinthu N, Kim H, Kang J, Park CG, Mills GB, Lee HY. EGFR mediates LPA-induced proteolytic enzyme expression and ovarian cancer invasion: inhibition by resveratrol. Mol Oncol. 2013;7:121–129. doi: 10.1016/j.molonc.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Park SY, Jeong KJ, Panupinthu N, Yu S, Lee J, Han JW, Kim JM, Lee JS, Kang J, Park CG, Mills GB, Lee HY. Lysophosphatidic acid augments human hepatocellular carcinoma cell invasion through LPA1 receptor and MMP-9 expression. Oncogene. 2011;30:1351–1359. doi: 10.1038/onc.2010.517. [DOI] [PubMed] [Google Scholar]
- 96.Panupinthu N, Lee HY, Mills GB. Lysophosphatidic acid production and action: critical new players in breast cancer initiation and progression. Br J Cancer. 2010;102:941–946. doi: 10.1038/sj.bjc.6605588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang C, Klett EL, Coleman RA. Lipid signals and insulin resistance. Clin Lipidol. 2013;8:659–667. doi: 10.2217/clp.13.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Savage DB, Petersen KF, Shulman GI. Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol Rev. 2007;87:507–520. doi: 10.1152/physrev.00024.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Coleman RA, Mashek DG. Mammalian triacylglycerol metabolism: synthesis, lipolysis, and signaling. Chem Rev. 2011;111:6359–6386. doi: 10.1021/cr100404w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Byler DM, Susi H. Examination of the secondary structure of proteins by deconvolved FTIR spectra. Biopolymers. 1986;25:469–487. doi: 10.1002/bip.360250307. [DOI] [PubMed] [Google Scholar]
- 101.Silva RA, Yasui SC, Kubelka J, Formaggio F, Crisma M, Toniolo C, Keiderling TA. Discriminating 3(10)- from alpha-helices: vibrational and electronic CD and IR absorption study of related Aib-containing oligopeptides. Biopolymers. 2002;65:229–243. doi: 10.1002/bip.10241. [DOI] [PubMed] [Google Scholar]
- 102.Shah S, Prematta T, Adkinson NF, Ishmael FT. Hypersensitivity to polyethylene glycols. J Clin Pharmacol. 2013;53:352–355. doi: 10.1177/0091270012447122. [DOI] [PubMed] [Google Scholar]
- 103.Kaczmarek P, Malendowicz LK, Pruszynska-Oszmalek E, Wojciechowicz T, Szczepankiewicz D, Szkudelski T, Nowak KW. Neuromedin U receptor 1 expression in the rat endocrine pancreas and evidence suggesting neuromedin U suppressive effect on insulin secretion from isolated rat pancreatic islets. Int J Mol Med. 2006;18:951–955. [PubMed] [Google Scholar]
- 104.Kaczmarek P, Malendowicz LK, Fabis M, Ziolkowska A, Pruszynska-Oszmalek E, Sassek M, Wojciechowicz T, Szczepankiewicz D, Andralojc K, Szkudelski T, Strowski MZ, Nowak KW. Does somatostatin confer insulinostatic effects of neuromedin u in the rat pancreas? Pancreas. 2009;38:208–212. doi: 10.1097/MPA.0b013e31818d9095. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. An example of analytical RP-HPLC profile and corresponding MS-spectra obtained for analog NM4-C16.
Figure S2. Structures of neuromedin analogs tested in FTIR experiments-Series 1.
Figure S3. Structures of neuromedin analogs tested in FTIR experiments-Series 2.
Figure S4. FTIR spectra of self-films of Series 1 & 2 peptides hydrated with HFIP : D2O 10 mM buffer pD=7.4 (v:v, 4:6).