

Abstract
Background and Purpose
The persistence of deleterious effects of hyperglycaemia even after glucose normalization is referred to as ‘metabolic memory’. However, similar persistent effects of the metabolic consequences of a high fat diet (HFD) have not been described.
Experimental Approach
Rats were given a normal pellet diet (NPD) or a HFD for 3 months. The animals from the HFD group were then returned to the NPD to observe the long-term effects of insulin resistance. Endothelial dysfunction was assessed by carbachol-mediated vasorelaxation and eNOS phosphorylation.
Key Results
As expected, HFD consumption resulted in insulin resistance and endothelial dysfunction. Phosphorylation of eNOS at S1177 was decreased in HFD rats, compared with that in the NPD group. Rats on 3 months of HFD showed glucose intolerance and impaired insulin sensitivity and were then switched back to NPD (REV group). Levels of cholesterol and triglyceride, and adiposity returned to normal in REV rats. However, endothelium-dependent vascular responses to carbachol which were impaired in HFD rats, continued to be impaired in REV rats. Similarly, decreased eNOS phosphorylation after HFD was not improved after 1 or 6 months of REV.
Conclusions and Implications
Our data indicate that returning to NPD did not improve the insulin sensitivity or the endothelial dysfunction induced by HFD. Although some biochemical parameters responsible for insulin resistance and endothelial dysfunction were normalized, molecular and vascular abnormalities, involving NO, persisted for several months, highlighting the long-lasting effects of metabolic memory.
Tables of Links
| TARGETS |
|---|
| Enzymes |
| eNOS |
| Set7/9, histone methyltransferase |
| LIGANDS |
|---|
| Carbachol |
| Cholesterol |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Endothelial dysfunction precedes many cardiovascular diseases including hypertension, atherosclerosis and coronary artery disease, which are also characterized by insulin resistance (McVeigh and Cohn, 2003; Huang, 2009). Similarly, metabolic disorders, such as type-2 diabetes mellitus (T2DM) and obesity characterized by insulin resistance, also involve endothelial dysfunction (Reaven, 1988; Kim et al., 2006; Del Turco et al., 2013). Mechanisms contributing to insulin resistance and endothelial dysfunction include glucotoxicity, lipotoxicity and inflammation. Many molecular, cellular, physiological and clinical studies indicate the direct relationship between insulin resistance and endothelial dysfunction, which reinforces the link between metabolic and cardiovascular diseases.
High fat diet (HFD) consumption, a major risk factor for the development of obesity is closely linked to insulin resistance and endothelial dysfunction (Storlien et al., 1986; Boden and Shulman, 2002; Bakker et al., 2009; Karpe et al., 2012). Impairment of the insulin-induced phosphoinositol-3 kinase pathway and augmentation of the MAPK decreases the production of vasodilator NO and secretion of vasoconstrictor enodthelin-1 respectively. This imbalance in vasodilator to vasoconstrictor levels indeed causes endothelial dysfunction (Arcaro et al., 2002; Caballero, 2003; DeFronzo, 2009; Eringa et al., 2013).
The long-term influence of early metabolic control in development and progression of diabetic micro and macro vascular complications is supported by several clinical studies (LeRoith et al., 2005; Ihnat et al., 2007; Ceriello et al., 2009). Recent clinical trials have suggested that normalization of glucose levels were not able to suppress risk of cardiovascular complications. The persistence of these deleterious effects of hyperglycaemia in cells, even after glucose normalization is referred as ‘hyperglycaemic memory’ (Paneni et al., 2013). Several reports demonstrate a similar association of metabolic memory with metabolic disorders. High glucose-induced activation of p65 subunit of NF-κB persisted even after glucose normalization in endothelial cells. High glucose-induced ROS resulted in up-regulation of histone methyltransferases (Set7/9) which results in lysine H3 methylation on the p65 gene promoter, leading to vascular inflammation (El-Osta et al., 2008). Glucose normalization did not suppress p53 transcriptional activity and expression of downstream proteins like PTEN, p21, PUMA and TIGAR (Schisano et al., 2011). Further, up-regulated mitochondrial protein p66shc was not reversed by glucose normalization in endothelial cells and diabetic mice (Paneni et al., 2012). Taken together, these reports suggest that, even after glycaemic control, the deleterious effects of hyperglycaemia and vascular complications persist. However, a similar persistence of HFD-induced metabolic impairment, after removal of the HFD remains to be investigated.
The evidence of long-term effects of saturated free fatty acids was provided by Gao et al. showing that insulin-induced glucose uptake was persistently impaired even after removal of fatty media (palmitate) from cultured monocytes (Gao et al., 2009). This could be explained if we assume that, as with transient hyperglycaemia, elevated free fatty acids also generate a metabolic memory. To check this hypothesis, we investigated effects of long-term HFD consumption followed by diet reversal (REV) on insulin resistance and endothelial dysfunction in rats.
Methods
Model development
All experiments were approved by the Institutional Animal Ethics Committee (IAEC approval no. 12/52, NIPER) and performed in accordance with the guidelines of Committee for Control and Supervision of Experimentation on Animals (CPCSEA), India on animal experimentation. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 125 animals were used in the experiments described here.
Six- to 8-week-old male Sprague–Dawley rats (160–180 g) were procured from the Central Animal Facility of the Institute. They were maintained under standard environmental condition (temperature 20 ± 2°C, humidity 50 ± 10% and 12 h light and dark cycle) with food and water ad libitum. All the animals were housed three per cage and maintained on normal pellet diet (NPD) for 1 week before dietary manipulation, for acclimatization. Insulin resistance was then induced in rats by feeding HFD (5.3 kcal·g−1, carbohydrate 17%, protein 25%, fat 58% kcal; see Supporting Information, Table 1 for details), while the control rats were fed with NPD (3.8 kcal·g−1, carbohydrate 67%, protein 21%, fat 12% kcal; Ashirwad Industries, Tripari, India) for 3 months. To study metabolic memory, subsequent to HFD feeding for 3 months, HFD-fed rats were returned to NPD and followed for another 6 months. Age-matched NPD and HFD controls were maintained throughout the study for comparison.
Estimation of biochemical parameters
Samples of venous blood (400 μL, with heparin; 200 IU mL-1 of blood) were collected for the estimation of glucose (GOD-POD), triglycerides (GPO-POD), and total cholesterol (CHOD-POD) using commercially available spectrophotometric kits (Accurex Biomedical Pvt. Ltd., Mumbai, India). The remaining plasma samples were then stored at −20°C until insulin was measured, by elisa using rat insulin as standard (DRG Diagnostics GmbH, Marburg, Germany). Insulin resistance was quantified by the homeostatic model assessment (HOMA-IR) as described earlier (Matthews et al., 1985; Shinohara et al., 2002).
I.p. glucose tolerance test (IPGTT)
IPGTT was performed as described previously (Karpe et al., 2012). In brief, after 3 months of HFD feeding, six rats of each group were fasted for 8 h before being subjected to IPGTT by giving i.p. glucose load (2 g·kg−1 i.p.). The blood samples were taken at 0 (just before), 15, 30, 60, 120 min after glucose load from all animals for estimation of glucose.
Vascular reactivity studies
Animals were killed under ketamine-xylazine anaesthesia, the thoracic aorta was removed and placed in ice-cold oxygenated Krebs–Henseleit solution (KHS). The aorta was cleaned from adhering adventitious tissue, taking care not to stretch the aorta or damage the endothelium while cleaning. Intact aortic rings (3–4 mm in length) were cut from the cleaned aorta. KHS comprised of (in mmol·L−1): NaCl: 118; KCl: 4.7; MgSO4.7H2O: 1.2; CaCl2.2H2O: 2.6; KH2PO4: 1.2; NaHCO3: 25; glucose: 5.5; pH 7.4. Aortic rings were carefully mounted, using stainless steel hooks, on the isometric force transducer in awater-jacketed organ bath chamber. A resting tension of 1 g was applied to the aortic ring. Each ring was allowed to equilibrate in 25 mL KHS (37°C) bubbled with carbogen (5% CO2 + 95% O2) for 120 min. KHS was changed every 15 minutes during this equilibration period. At the beginning of each experiment, aortic rings were primed with 80 μmol·L−1 KCl (depolarizing solution) to check the viability of tissue. Isometric tension responses were recorded using an automatic organ bath (Panlab, Harvard Apparatus; Panlab s.I.u., Barcelona, Spain) connected to the Power Lab data acquisition system (ADInstruments Pty Ltd., Bella Vista, NSW, Australia). Cumulative concentration–response curves to carbachol (Sigma-Aldrich, MO USA) were obtained in both animal groups. To record carbachol-mediated relaxations, aortic rings were precontracted with 10 nmol·L−1 phenylephrine (Sigma-Aldrich) and responses to increasing concentration of carbachol (10 nmol·L−1 to 10 μmol·L−1) were obtained. Aortic rings that failed to produce more than 70% relaxation were considered endothelium denuded. At the end of the experiment, aortic rings were blotted dry and their length and weights were measured to calculate tension normalized to cross-sectional area (mg·mm−2) using the following formula:
 |
(density of vascular smooth muscle cell: 1.05 mg·mm−3)
Western blotting
Protein isolation and Western blotting was performed as described previously (Karpe et al., 2012). In brief, frozen samples of rat thoracic aorta were thawed, minced into small pieces, and homogenized in lysis buffer. Samples were loaded and analysed by SDS-PAGE in 10% polyacrylamide gels. After electrophoresis, proteins were electrotransferred to nitrocellulose membrane (Pall Corporation, FL, USA). The blot membrane was then incubated in a blocking buffer 5% BSA in Tris buffer saline for 2 h at room temperature and then probed with antibody directed against phospho-eNOS at S1177 (1:2000) (Santa Cruz Biotechnology, California, USA #sc-12972) at room temperature. Proteins were detected with the enhanced chemiluminescence system and ECL Hyperfilm (Amersham Pharmacia Biotech, UK). Actin (Santa Cruz Biotechnology) expression levels were used to normalize the results.
Immunohistochemistry (IHC) for S1177 phosphorylation of eNOS in thoracic aorta
Rats were anaesthetized and aortae were removed and stored in 10% formol saline. Paraffin blocks were prepared after completing the routine processing. Sections were prepared from paraffin blocks and deparafinized with xylene, followed by antigen retrieval by heating in citrate buffer (10 mM). The following goat polyclonal primary antibodies were used: anti-eNOS (S1177) (1:50). Streptavidin peroxidase (STV–HRP) system (Vectastain) was used to amplify the signals followed by detection with diaminobenzidine as a chromogen. Slides were counterstained with haematoxylin, dehydrated with alcohols and xylene and mounted in DPX. Histological images were captured by a charged-coupled device camera attached with the Olympus microscope (Model BX 51, Olympus, Tokyo, Japan). ImageJ software (NIH, Bethesda, MD, USA) was used to quantify the images by first converting them to greyscale and then inverting. The integrated pixel density was then taken and divided by the surface area measured. The integrated pixel density of the negative control sections (secondary antibody only) was also measured and their respective average integrated pixel density per squared micrometer (background) was subtracted for appropriate phospho-protein measurements. Measurements from 10–12 aortic fields were used per quantitation.
Data analysis
Results are expressed as mean ± SEM and n refers to number of animals in a particular group. Statistical analysis was performed using GraphPad Prism, version 5.01 (GraphPad Software, Inc., La Jolla, CA, USA). Comparison between two groups was evaluated using Student's t-test. Multiple group comparisons were performed using two-way ANOVA. Post hoc analysis was performed using Bonferroni's test. Results were considered significant if P < 0.05.
Results
HFD feeding results in glucose intolerance and insulin resistance
It has been well established that animals given a HFD develop features of insulin resistance. In initial characterization studies, we developed our model of HFD-induced insulin resistance. Three months of HFD feeding resulted in increased body weight gain (Figure 1A). Fasting plasma glucose levels (Figure 1B), measured at 1 month intervals over the 3 months, was increased in HFD rats. Similarly fasting plasma insulin levels (Figure 1C) of HFD rats measured at the end of 3 months showed a marked increase over the levels in NPD rats. Next we checked the glucose disposal, using a IPGTT performed at the end of 3 months of HFD feeding. As expected, HFD-fed rats displayed greater glucose intolerance than the NPD rats (Figure 1D–E). This suggested impaired insulin sensitivity in HFD-fed rats and explained the mild hyperglycaemia and compensatory hyperinsulinaemia resulting in the higher HOMA index in the HFD rats (Figure 1F). Plasma triglyceride levels (Supporting Information Fig. S1a) and plasma total cholesterol levels (Supporting Information Fig. S1b) of HFD-fed rats were significantly higher than in NPD-fed rats. Further, the adiposity index, derived from the weight of different fat pads (epididymis, retroperitoneal, visceral and mesentery) was significantly increased after HPD, compared with NPD rats (Supporting Information Fig. S1c). Overall, these results confirm that HFD-fed rats displayed the characteristics of insulin resistance.
Figure 1.
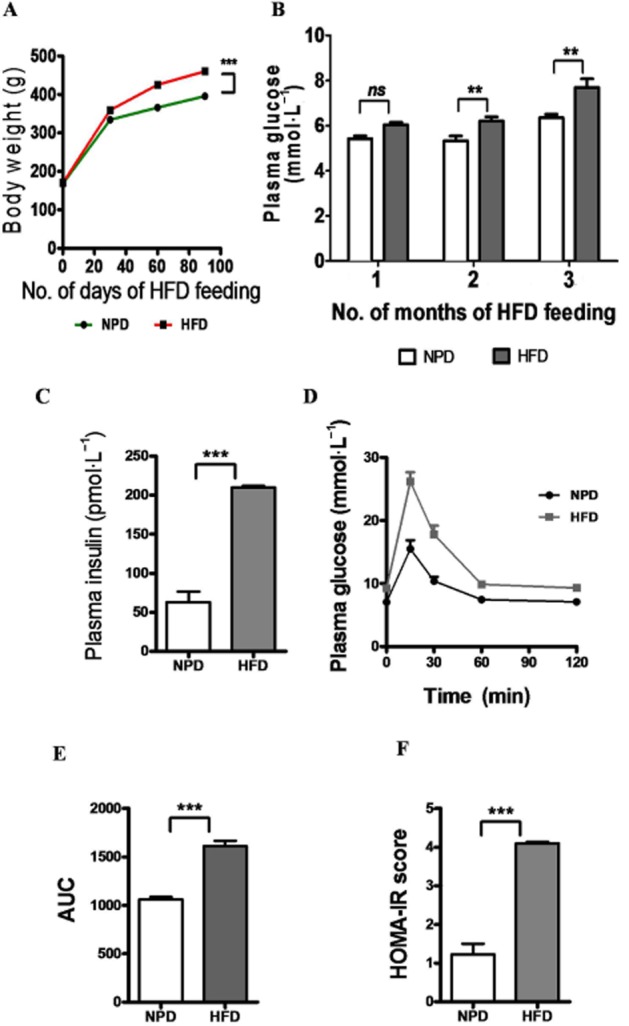
Characterization of HFD-induced insulin resistance. (A) Growth curve demonstrating body weight gain of rats, assessed at 30 day intervals in each group. (B and C) Bar graphs of the increased fasting plasma glucose and insulin levels, respectively, of HFD-fed animals. (D and E) glucose tolerance test (IPGTT), with representative bar graph showing AUC values, confirms decreased insulin sensitivity with HFD intake. (F) HOMA-IR scores indicate compensatory hyperinsulinaemia and insulin-resistant state of HFD-fed animals. Data shown are means ± SEM, n = 6–8 rats per group. **P < 0.01, ***P < 0.001; significantly different as indicated.
Insulin resistance causes endothelial dysfunction in HFD rats
Endothelial functioning in this insulin-resistant state was determined by measuring vascular responses to carbachol. A significant impairment of carbachol-mediated vasorelaxation was observed in aortic rings from HFD rats, comapred with that in rings from NPD rats (P < 0.001, Figure 2A). To gain further insight into endothelial dysfunction, the levels of the active form of eNOS (phosphorylated at S1177) was measured by IHC and Western blot analysis. IHC analysis of thoracic aortic sections revealed diminished levels of the active form of eNOS in the endothelial lining (Figure 2B–C). Further, Western blot analysis confirmed that the eNOS phosphorylation levels were lower in aorta obtained from HFD rats than NPD rats (Figure 2E–F).
Figure 2.

HFD consumption alters endothelium-dependent vasorelaxant responses in thoracic aorta and causes endothelial dysfunction. (A) Cumulative concentration–response curve to carbachol after pre-contraction with phenylephrine suggests impaired endothelium-dependent vasorelaxation in thoracic aortic rings is due to decreased bioavailability of NO. (B) Phosphorylation of eNOS (S1177), shown by IHC, in the EC of the aortas was decreased by HFD consumption. Phospho-eNOS S1177 was stained in the aortas of NPD (a and b) and HFD-fed (c and d) rats (magnification: 100×). b and d indicate inverted images of the respective photomicrographs. Arrows point to positively staining ECs. 10–12 random fields, including those in a–d, were analysed from the aortas of NPD- and HFD-fed rats for eNOS phosphorylation. C, Quantitation of eNOS phosphorylation at S1177. D-E, shows Western blot of S1177 phosphorylation of eNOS in NPD- and HFD-fed animals. Data shown are means ± SEM, n = 6–8 rats per group. *P < 0.05, ***P < 0.001; significantly different as indicated.
Long-term HFD consumption induces persistent glucose intolerance and decreased insulin sensitivity after REV
Long-term effects of HFD consumption on morphological and biochemical parameters and insulin sensitivity were studied after returning the HFD rats to NPD (REV group). The growth rate of the REV rats was slightly suppressed to that of age-matched HFD-fed rats (Figure 3A). Biochemical measurements made under fasting conditions, at 1, 2 4 and 6 months after REV, demonstrated that plasma glucose (Figure 3B) and plasma insulin (Figure 3C) levels were still significantly higher in REV rats, compared with NPD-fed rats. Similarly, IPGTT also indicated that glucose intolerance and impaired insulin sensitivity persisted after cessation of HFD feeding for up to 6 months, indicating the effects of metabolic memory (Figure 3D–E). The HOMA-IR score which quantitates insulin resistance, was still elevated, relative to that of NPD rats, after 6 months of REV and cessation of HFD feeding (Figure 3F).
Figure 3.

Transient HFD consumption causes persistent insulin resistance. (A) Growth curve demonstrating body weight gain of rats, taken at 60 day intervals in each group, following return to NPD after 3 months of HFD (diet reversal, REV.). (B and C) Plasma glucose and insulin levels indicate sustained hyperglycaemia and hyperinsulinaemia following REV. (D and E) Glucose tolerance test (IPGTT) performed after different durations of REV (1, 2, 4 and 6 months) indicates relatively poor improvement in glucose disposal rate in animals on REV. (F) HOMA-IR scores indicate persistent compensatory hyperinsulinaemia and insulin-resistant state, even after 6 months of REV. Data shown are means ± SEM, n = 6–8 rats per group. **P < 0.01; ***P < 0.001; #P < 0.05, ##P < 0.01, ###P < 0.001; †P < 0.05, ††P < 0.01, †††P < 0.001; significantly different as indicated.
REV normalized lipid levels and adiposity index in HFD rats
Next, we checked the effect of REV on lipid levels in HFD rats. Interestingly, following the change of diet, circulating triglyceride (Figure 4A) and total cholesterol (Figure 4B) levels were normalized in REV rats. Further, the fat pads weights were reduced and eventually normalized after 6 months of NPD in the REV rats (Figure 4C).
Figure 4.
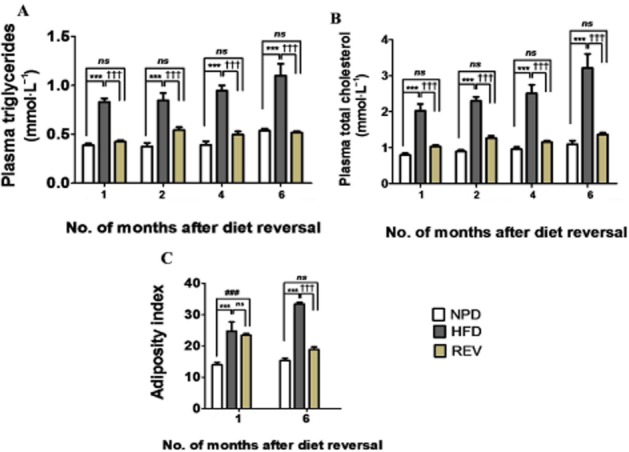
REV normalizes lipid profile and adiposity index. (A and B) Plasma triglyceride and plasma cholesterol levels, after REV for 1, 2, 4 and 6 months indicating normalized lipid profile. (C) Adiposity index (fat pad weights) after REV for 1 and 6 months. Data shown are means ± SEM, n = 6–8 rats per group. ***P < 0.001; ###P < 0.001; †††P < 0.001; significantly different as indicated.
Long-term HFD feeding is associated with persistent endothelial dysfunction and indicates existence of ‘vascular memory’
Endothelial function was studied using vascular reactivity studies performed after 1 month (REV-1) and 6 months (REV-6) of diet reversal. Surprisingly, we found that vasorelaxation responses to carbachol in thoracic aortic rings was significantly impaired in REV-1 animals and was comparable with age-matched HFD-fed rats (Figure 5A). After 6 months of NPD (REV-6 group), the vasorelaxant responses to carbachol had improved but were still impaired, compared with those from the age-matched NPD group (Figure 5B). Interestingly, endothelial dysfunction was further aggravated with increased duration of HFD consumption. Parallel to the functional data obtained, IHC analysis also showed lower level expression of the phosphorylated form of eNOS (Figure 5C–E) in thoracic aorta in REV rats, compared with age-matched NPD-fed rats, suggesting a metabolic memory of HFD consumption. Western blot analysis revealed that eNOS S1177 phosphorylation at S1177 were compromised even after cessation of HFD feeding in REV-1 rats and was the same as that in HFD rats (Figure 6A). However, rats in the REV-6 group did show some improvement in eNOS phosphorylation, in comparison with HFD-fed rats (Figure 6A).
Figure 5.

Transient HFD consumption persistently alters endothelium-dependent vasorelaxant responses in rings of thoracic aorta and induces long-lasting endothelial dysfunction. (A and B) Cumulative concentration–response curve to carbachol after pre-contraction with phenylephrine in thoracic aortic rings suggesting decreased bioavailability of NO even after 1 and 6 months of REV respectively. (C) eNOS phosphorylation at S1177 was determined by IHC in the EC of the aortas and was reduced after 1 month of REV. Phospho-eNOS S1177 was stained in the aortas of NPD (a and b), HFD (c and d) and REV-1 (e and f) rats (magnification: 100×). b and d indicate inverted images of the respective photomicrographs. (D) S1177 eNOS phosphorylation levels were determined by IHC in the EC of the aortas and were reduced even after 6 months of REV. Phospho-eNOS S1177 was stained in the aortas of NPD (a and b), HFD (c and d) and REV-6 (e and f) rats (magnification: 100x). b, d and f indicate inverted images of the respective photomicrographs. Arrows point to positively staining ECs. In both the figures C and D, 10–12 random fields, including those in a–f, were analysed from the aortas of NPD- and HFD-fed rats for eNOS phosphorylation. (E) Quantitation of eNOS phosphorylation at S1177 after different durations of REV. Data shown are means ± SEM, n = 6–8 rats per group. ***P < 0.001; ###P < 0.001; †††P < 0.001; significantly different as indicated.
Figure 6.
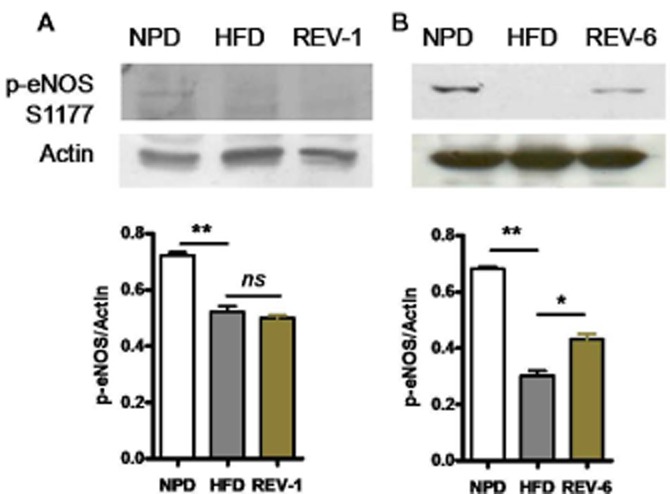
REV does not improve eNOS phosphorylation. (A and B) Western blot of S1177 phosphorylation of eNOS in aorta in REV-1 and REV-6 groups. The return to NPD did not increase eNOS phosphorylation indicating persistent endothelial dysfunction. Data shown are means ± SEM, n = at least three sets of independent experiments. *P < 0.05, **P < 0.01; significantly different as indicated.
Discussion
This study was designed to answer some simple questions about the persistence of insulin resistance and endothelial dysfunction, after cessation of HFD. We demonstrated that HFD feeding just for 3 months induced insulin resistance and endothelial dysfunction, which persisted up to 6 months after returning to the NPD. Our results provide the first evidence for the existence of a metabolic memory with increase in insulin and glucose levels in the HFD-induced model of insulin resistance.
Insulin resistance is a characteristic finding in obesity, type 2 diabetes and components of the cardio-metabolic syndrome, including hypertension and dyslipidaemia, which collectively contribute to a substantial risk for cardiovascular disease (Del Turco et al., 2012; Muniyappa and Sowers, 2013). Association of insulin resistance with endothelial dysfunction, even in the absence of overt diabetes is well recognised (Hsueh et al., 2003; Rask-Madsen et al., 2007). The most significant clinical observations regarding metabolic memory came from the inaugural Diabetes Control and Complications Trial, which was followed by the Epidemiology of Diabetes Interventions and Complications Study, which suggested that the deleterious end-organ effects, that occurred in both conventional and intensified glycaemic control groups, persisted for more than 5 years after the patients had returned to normal glycaemic control (The Writing Team for the Diabetes et al., 2002; Nathan DM et al., 2005). Metabolic memory is known to persist despite intensive treatment (Ihnat et al., 2007; Ceriello et al., 2009) and the duration of poor glycaemic control before initiation of good glycaemic control appears to play a key role in the outcome of reinstitution of good glycaemic control (Kowluru, 2003). Transient hyperglycaemia has been shown to promote gene-activating epigenetic changes and signalling events critical in the development and progression of vascular complications (Roy et al., 1990; Cooper and El-Osta, 2010; Siebel et al., 2010). Mono-methylation, by the histone methyltransferases Set7/9, of Lys4 of histoneH3 (H3K4me) of the p65 subunit of the NF-κB complex results in persistent expression of inflammatory genes and extacellular matrix proteins (El-Osta et al., 2008). Further, decreased promoter occupancy of the chromatin histone H3 Lys9 methyltransferase Suv39h1, lysine-specific demethylase1 and their associated epigenetic markers, increased histone H3 lysine-9 trimethylation (H3K9me3), decreased H3K4 methylation (H3K4me), respectively, have been identified as playing key roles in sustained inflammatory gene expression in diabetic vascular cells (Reddy et al., 2008; Villeneuve et al., 2008; 2010,). Elevation of saturated fatty acids like palmitic acid, apart from promoting insulin resistance, could also mediate metabolic memory associated with insulin resistance (Gao et al., 2009).
NO is the most important vasoactive mediator of endothelium, produced by conversion of L-arginine to L-citrulline and NO by eNOS (Mount et al., 2007; Kolluru et al., 2010). Phosphorylation at S1177 of eNOS is a well-characterized event that increases eNOS activity and NO production by twofold (Fulton et al., 2001). eNOS activity has opposing outcomes, depending on the vascular tissue levels of tetrahydrobiopterin (BH4), a cofactor for eNOS. Deficiency of BH4 leads to the production by eNOS of superoxide rather than NO (Bauersachs and Schafer, 2005). Consistent with the reports mentioned earlier, our data show that even after cessation of HFD, the eNOS phosphorylation at S1177 was not restored to normal levels. This suggests a relative inactivation of eNOS and consequently reduced NO bioavailability. It has been well established that ACh-induced relaxation in the thoracic aorta and other conduit vessels is almost entirely mediated by NO (Karpe and Tikoo, 2014). In particular relevance to these reports, our findings of diminished vasorelaxation to carbachol in aortic rings, lasting for up to 6 months after cessation of HFD, would be compatible with the decreased phosphorylation and function of eNOS that we have observed. A recent report suggests that dysfunction in the SIRT1/AMPK/eNOS pathway contributed to cellular metabolic memory of high glucose and led to diabetic retinopathy. Restoration of this pathway by metformin suppressed this memory (Zheng et al., 2012). Previously, we reported that HFD-induced insulin resistance markedly reduced aortic expression of the SIRT1/AMPK/eNOS pathway, contributing to endothelial dysfunction (Karpe and Tikoo, 2014). The persistent decrease in the activation of eNOS and endothelial-dependent vasorelaxation in REV animals might also be due to compromised SIRT1 and AMPK activity, a possibility that merits further investigation.
Abdominal obesity is linked to endothelial dysfunction indirectly through insulin resistance and directly by the production of adipokines and pro-inflammatory cytokines, which in turn produce oxidative stress leading to a reduced NO availability (Jonk et al., 2007; Virdis et al., 2012). Further, it has also been reported that unfolded protein response activation occurs in the endothelial cells of non-diabetic obese adults with vascular endothelial dysfunction (Kaplon et al., 2013). These reports suggest a role for adiposity in mediating both insulin resistance and endothelial dysfunction. However in the present study, animals initially fed on HFD (3 months), when returned to NPD for 6 months (REV-6), exhibited characteristics of insulin resistance and endothelial dysfunction even when body fat mass was lost (as indicated by decreased adiposity index). One explanation of this finding would be that lipid metabolites and hyperglycaemia contribute to the persistence of the detrimental effects of HFD. HFD induces increase in lipid levels and mild hyperglycaemia. Both lipids and hyperglycaemia have been shown to activate TLR4 signalling and inflammation (Dasu and Jialal, 2011). DAGs and ceramides are considered as lipotoxic molecules involved in pathogenesis of insulin resistance (Amati et al., 2011). DAG causes activation of PKCε leading to serine phosphorylation of IRS-1 and insulin resistance (Jornayvaz and Shulman, 2012). Free fatty acids induce ceramide synthesis by providing substrates for its synthesis and also through de novo synthesis via TLR4 (Holland et al., 2011). In turn, ceramides (i) activate phosphatase, PP2A to inhibit Akt; (ii) activate JNK and IκKβ kinsases to induce transcription factor Jun and NFκB; and (iii) inhibit mitochondrial respiration, eventually leading to insulin resistance (Summers, 2006). On the other hand, ceramide prevents the insulin-induced phosphorylationof eNOS at S1177, eNOS dimer to monomer formation and NO levels in bovine aortic endothelial cells (Zhang et al., 2012). Zhang et al. have reported either genetic or pharmacological inhibition of ceramide improves endothelial function and eNOS phosphorylation in the arteries of fat-fed mice (Zhang et al., 2012). Taken together, both hyperglycaemia and lipid metabolites could thus play predominant roles in mediating the deleterious effects of high fat feeding.
Apart from elevated lipid levels and hyperglycaemia, a family history of pre-diabetes or diabetes determines trans-generational effects of diabetes and its complications. Several studies suggest that insulin resistance persisted in young subjects with a family history of diabetes, when compared with age-matched subjects without a history of diabetes. In addition, endothelium-dependent vasodilation was impaired in first-degree relatives of T2DM patients, relative to age-matched control subjects, (Ciccone et al. 2014). Our results demonstrated the long-term effects of HFD feeding on insulin resistance and endothelial dysfunction. Interestingly, in the HFD-induced insulin-resistant rats, the metabolic memory persisted after animals were returned to NPD. The most plausible explanation of the metabolic memory would be epigenetic alterations, which could be responsible for the metabolic memory and trans-generational pathogenesis of the disease. Although several reports indicate involvement of epigenetic changes (Pirola et al., 2010; Reddy and Natarajan, 2011), we failed to observe any significant change in global histone acetylation and phosphorylation (data not shown). However, our data does not rule out changes in histone acetylation/phosphorylation and other epigenetic alterations at promoters of specific genes responsible for the vascular consequences of abnormal glucose metabolism.
It will be pertinent to mention here that HFD feeding caused robust increases in insulin, lipid levels and mild hyperglycaemia, supporting our belief that our animal model mimics a pre-diabetic condition, rather than a diabetic animal model (pre-diabetic glucose levels: 1.2–1.3 g·L−1 vs. diabetic: >2.5g·L−1). We cannot rule out a role for mild hyperglycaemia in metabolic memory in this animal model, as previous studies have shown that the role of hyperglycaemia in metabolic memory are performed in the presence of mild to very high glucose conditions (15–25 mM in vitro and >2.5 g·L−1 in vivo) and in the absence of insulin resistance (El-Osta et al., 2008; Zheng et al., 2012). Moreover, our study shows the combined effect of high glucose and insulin on vascular memory. Further studies are required to explore the mechanism(s) by which hyperlipedaemia, hyperinsulinaemia and hyperglycaemia could mediate metabolic memory.
Acknowledgments
The authors thank the National Institute of Pharmaceutical Education and Research, S.A.S. Nagar, India for providing necessary facilities and funding to pursue this work.
Glossary
- HFD
high fat diet
- NPD
normal pellet diet
- REV
diet reversal
- REV-1
1 month of diet reversal
- REV-6
6 months of diet reversal
- T2DM
type-2 diabetes mellitus
Author contributions
D. S. P. T. developed and validated the model, performed biochemical and functional studies and wrote the paper. P. A. K. designed the experiments, performed Western blotting studies and wrote the paper. K. T. designed and supervised the experiments and approved the final version of paper.
Conflicts of interests
None.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 Effect of HFD consumption on lipid levels and adiposity.
{kind=link}
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amati F, Dube JJ, Alvarez-Carnero E, Edreira MM, Chomentowski P, Coen PM, et al. Skeletal muscle triglycerides, diacylglycerols, and ceramides in insulin resistance: another paradox in endurance-trained athletes? Diabetes. 2011;60:2588–2597. doi: 10.2337/db10-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcaro G, Cretti A, Balzano S, Lechi A, Muggeo M, Bonora E, et al. Insulin causes endothelial dysfunction in humans sites and mechanisms. Circulation. 2002;105:576–582. doi: 10.1161/hc0502.103333. [DOI] [PubMed] [Google Scholar]
- Bakker W, Eringa EC, Sipkema P, Van Hinsbergh VWM. Endothelial dysfunction and diabetes: roles of hyperglycemia, impaired insulin signaling and obesity. Cell Tissue Res. 2009;335:165–189. doi: 10.1007/s00441-008-0685-6. [DOI] [PubMed] [Google Scholar]
- Bauersachs J, Schafer A. Tetrahydrobiopterin and eNOS dimer/monomer ratio – a clue to eNOS uncoupling in diabetes? Cardiovasc Res. 2005;65:768–769. doi: 10.1016/j.cardiores.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Bendale DS, Karpe PA, Chhabra R, Shete SP, Shah H, Tikoo K. 17beta Oestradiol prevents cardiovascular dysfunction in postmenopausal metabolic syndrome by affecting SIRT1/AMPK/H3 acetylation. Br J Pharmacol. 2013;170:779–795. doi: 10.1111/bph.12290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boden G, Shulman GI. Free fatty acids in obesity and type 2 diabetes: defining their role in the development of insulin resistance and -cell dysfunction. Eur J Clin Invest. 2002;32:14–23. doi: 10.1046/j.1365-2362.32.s3.3.x. [DOI] [PubMed] [Google Scholar]
- Caballero AE. Endothelial dysfunction in obesity and insulin resistance: a road to diabetes and heart disease. Obes Res. 2003;11:1278–1289. doi: 10.1038/oby.2003.174. [DOI] [PubMed] [Google Scholar]
- Ceriello A, Ihnat MA, Thorpe JE. The metabolic memory: is more than just tight glucose control necessary to prevent diabetic complications? J Clin Endocrinol Metab. 2009;94:410–415. doi: 10.1210/jc.2008-1824. [DOI] [PubMed] [Google Scholar]
- Ciccone M, Scicchitano P, Cameli M, Cecere A, Cortese F. Endothelial function in pre-diabetes, diabetes and diabetic cardiomyopathy: a review. J Diabetes Metab. 2014;5:364. [Google Scholar]
- Cooper ME, El-Osta A. Epigenetics mechanisms and implications for diabetic complications. Circ Res. 2010;107:1403–1413. doi: 10.1161/CIRCRESAHA.110.223552. [DOI] [PubMed] [Google Scholar]
- Dasu MR, Jialal I. Free fatty acids in the presence of high glucose amplify monocyte inflammation via toll-like receptors. Am J Physiol Endocrinol Metab. 2011;300:E145–E154. doi: 10.1152/ajpendo.00490.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFronzo RA. Insulin resistance, lipotoxicity, type 2 diabetes and atherosclerosis: the missing links. The Claude Bernard Lecture 2009. Diabetologia. 2009;53:1270–1287. doi: 10.1007/s00125-010-1684-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Turco S, Gaggini M, Daniele G, Basta G, Folli F, Sicari R, et al. Insulin resistance and endothelial dysfunction: a mutual relationship in cardiometabolic risk. Curr Pharm Des. 2012;19:2420–2431. doi: 10.2174/1381612811319130010. [DOI] [PubMed] [Google Scholar]
- Del Turco S, Gaggini M, Daniele G, Basta G, Folli F, Sicari R, et al. Insulin resistance and endothelial dysfunction: a mutual relationship in cardiometabolic risk. Curr Pharm Des. 2013;19:2420–2431. doi: 10.2174/1381612811319130010. [DOI] [PubMed] [Google Scholar]
- El-Osta A, Brasacchio D, Yao D, Pocai A, Jones PL, Roeder RG, et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med. 2008;205:2409–2417. doi: 10.1084/jem.20081188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eringa EC, Serne EH, Meijer RI, Schalkwijk CG, Houben AJHM, Stehouwer CDA, et al. Endothelial dysfunction in (pre) diabetes: characteristics, causative mechanisms and pathogenic role in type 2 diabetes. Rev Endocr Metab Disord. 2013;14:1–10. doi: 10.1007/s11154-013-9239-7. [DOI] [PubMed] [Google Scholar]
- Fulton D, Gratton JP, Sessa WC. Post-translational control of endothelial nitric oxide synthase: why isn't calcium/calmodulin enough? J Pharmacol Exp Ther. 2001;299:818–824. [PubMed] [Google Scholar]
- Gao D, Bailey CJ, Griffiths HR. Metabolic memory effect of the saturated fatty acid, palmitate, in monocytes. Biochem Biophys Res Commun. 2009;388:278–282. doi: 10.1016/j.bbrc.2009.07.160. [DOI] [PubMed] [Google Scholar]
- Holland WL, Bikman BT, Wang LP, Yuguang G, Sargent KM, Bulchand S, et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J Clin Invest. 2011;121:1858–1870. doi: 10.1172/JCI43378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsueh WA, Quinones MJ. Role of endothelial dysfunction in insulin resistance. Am J Cardiol. 2003;92:10–17. doi: 10.1016/s0002-9149(03)00611-8. [DOI] [PubMed] [Google Scholar]
- Huang PL. eNOS, metabolic syndrome and cardiovascular disease. Trends Endocrinol Metab. 2009;20:295–302. doi: 10.1016/j.tem.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihnat MA, Thorpe JE, Ceriello A. Hypothesis: the metabolic memory, the new challenge of diabetes. Diabet Med. 2007;24:582–586. doi: 10.1111/j.1464-5491.2007.02138.x. [DOI] [PubMed] [Google Scholar]
- Jonk AM, Houben AJHM, de Jongh RT, Serné EH, Schaper NC, Stehouwer CDA. Microvascular dysfunction in obesity: a potential mechanism in the pathogenesis of obesity-associated insulin resistance and hypertension. Physiology. 2007;22:252–260. doi: 10.1152/physiol.00012.2007. [DOI] [PubMed] [Google Scholar]
- Jornayvaz FR, Shulman GI. Diacylglycerol activation of protein kinase Cepsilon and hepatic insulin resistance. Cell Metab. 2012;15:574–584. doi: 10.1016/j.cmet.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplon RE, Chung E, Reese L, Cox-York K, Seals DR, Gentile CL. Activation of the unfolded protein response in vascular endothelial cells of nondiabetic obese adults. J Clin Endocrinol Metab. 2013;98:E1505–E1509. doi: 10.1210/jc.2013-1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpe PA, Tikoo K. Heat shock prevents insulin resistance-induced vascular complications by augmenting angiotensin-(1–7) signaling. Diabetes. 2014;63:1124–1139. doi: 10.2337/db13-1267. [DOI] [PubMed] [Google Scholar]
- Karpe PA, Gupta J, Marthong RF, Ramarao P, Tikoo K. Insulin resistance induces a segmental difference in thoracic and abdominal aorta: differential expression of AT1 and AT2 receptors. J Hypertens. 2012;30:132–146. doi: 10.1097/HJH.0b013e32834d9efe. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J-A, Montagnani M, Koh KK, Quon MJ. Reciprocal relationships between insulin resistance and endothelial dysfunction molecular and pathophysiological mechanisms. Circulation. 2006;113:1888–1904. doi: 10.1161/CIRCULATIONAHA.105.563213. [DOI] [PubMed] [Google Scholar]
- Kolluru GK, Siamwala JH, Chatterjee S. eNOS phosphorylation in health and disease. Biochimie. 2010;92:1186–1198. doi: 10.1016/j.biochi.2010.03.020. [DOI] [PubMed] [Google Scholar]
- Kowluru RA. Effect of reinstitution of good glycemic control on retinal oxidative stress and nitrative stress in diabetic rats. Diabetes. 2003;52:818–823. doi: 10.2337/diabetes.52.3.818. [DOI] [PubMed] [Google Scholar]
- LeRoith D, Fonseca V, Vinik A. Metabolic memory in diabetes‘focus on insulin. Diabetes Metab Res Rev. 2005;21:85–90. doi: 10.1002/dmrr.530. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVeigh GE, Cohn JN. Endothelial dysfunction and the metabolic syndrome. Curr Diab Rep. 2003;3:87–92. doi: 10.1007/s11892-003-0059-0. [DOI] [PubMed] [Google Scholar]
- Mount PF, Kemp BE, Power DA. Regulation of endothelial and myocardial NO synthesis by multi-site eNOS phosphorylation. J Mol Cell Cardiol. 2007;42:271–279. doi: 10.1016/j.yjmcc.2006.05.023. [DOI] [PubMed] [Google Scholar]
- Muniyappa R, Sowers JR. Role of insulin resistance in endothelial dysfunction. Rev Endocr Metab Disord. 2013;14:5–12. doi: 10.1007/s11154-012-9229-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan DM, Cleary PA, Backlund JY, Genuth SM, Lachin JM, Orchard TJ, et al. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. NEJM. 2005;353:2643–2653. doi: 10.1056/NEJMoa052187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paneni F, Mocharla P, Akhmedov A, Costantino S, Osto E, Volpe M, et al. Gene silencing of the mitochondrial adaptor p66(Shc) suppresses vascular hyperglycemic memory in diabetes. Circ Res. 2012;111:278–289. doi: 10.1161/CIRCRESAHA.112.266593. [DOI] [PubMed] [Google Scholar]
- Paneni F, Volpe M, Luscher TF, Cosentino F. SIRT1, p66(Shc), and Set7/9 in vascular hyperglycemic memory: bringing all the strands together. Diabetes. 2013;62:1800–1807. doi: 10.2337/db12-1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirola L, Balcerczyk A, Okabe J, El-Osta A. Epigenetic phenomena linked to diabetic complications. Nat Rev Endocrinol. 2010;6:665–675. doi: 10.1038/nrendo.2010.188. [DOI] [PubMed] [Google Scholar]
- Rask-Madsen C, King GL. Mechanisms of disease: endothelial dysfunction in insulin resistance and diabetes. Nat Clin Pract Endocrinol Metab. 2007;3:46–56. doi: 10.1038/ncpendmet0366. [DOI] [PubMed] [Google Scholar]
- Reaven GM. Role of insulin resistance in human disease. Diabetes. 1988;37:1595–1607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- Reddy MA, Natarajan R. Epigenetic mechanisms in diabetic vascular complications. Cardiovasc Res. 2011;90:421–429. doi: 10.1093/cvr/cvr024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy MA, Villeneuve LM, Wang M, Lanting L, Natarajan R. Role of the lysine-specific demethylase 1 in the proinflammatory phenotype of vascular smooth muscle cells of diabetic mice. Circ Res. 2008;103:615–623. doi: 10.1161/CIRCRESAHA.108.175190. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Roy S, Sala R, Cagliero E, Lorenzi M. Overexpression of fibronectin induced by diabetes or high glucose: phenomenon with a memory. PNAS. 1990;87:404–408. doi: 10.1073/pnas.87.1.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schisano B, Tripathi G, McGee K, McTernan PG, Ceriello A. Glucose oscillations, more than constant high glucose, induce p53 activation and a metabolic memory in human endothelial cells. Diabetologia. 2011;54:1219–1226. doi: 10.1007/s00125-011-2049-0. [DOI] [PubMed] [Google Scholar]
- Siebel AL, Fernandez AZ, El-Osta A. Glycemic memory associated epigenetic changes. Biochem Pharmacol. 2010;80:1853–1859. doi: 10.1016/j.bcp.2010.06.005. [DOI] [PubMed] [Google Scholar]
- Storlien LH, James DE, Burleigh KM, Chisholm DJ, Kraegen EW. Fat feeding causes widespread in vivo insulin resistance, decreased energy expenditure, and obesity in rats. Am J Physiol Endocrinol Metab. 1986;251:E576–E583. doi: 10.1152/ajpendo.1986.251.5.E576. [DOI] [PubMed] [Google Scholar]
- Summers SA. Ceramides in insulin resistance and lipotoxicity. Prog Lipid Res. 2006;45:42–72. doi: 10.1016/j.plipres.2005.11.002. [DOI] [PubMed] [Google Scholar]
- The Writing Team for the Diabetes C, Complications Trial/Epidemiology of Diabetes I, Complications Research G. Effect of intensive therapy on the microvascular complications of type 1 diabetes mellitus. JAMA. 2002;287:2563–2569. doi: 10.1001/jama.287.19.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeneuve LM, Reddy MA, Lanting LL, Wang M, Meng L, Natarajan R. Epigenetic histone H3 lysine 9 methylation in metabolic memory and inflammatory phenotype of vascular smooth muscle cells in diabetes. PNAS. 2008;105:9047–9052. doi: 10.1073/pnas.0803623105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeneuve LM, Kato M, Reddy MA, Wang M, Lanting L, Natarajan R. Enhanced levels of microRNA-125b in vascular smooth muscle cells of diabetic db/db mice lead to increased inflammatory gene expression by targeting the histone methyltransferase Suv39h1. Diabetes. 2010;59:2904–2915. doi: 10.2337/db10-0208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virdis A, Fritsch Neves M, Duranti E, Bernini G, Taddei S. Microvascular endothelial dysfunction in obesity and hypertension. Curr Pharm Des. 2012;19:2382–2389. doi: 10.2174/1381612811319130006. [DOI] [PubMed] [Google Scholar]
- Zhang QJ, Holland WL, Wilson L, Tanner JM, Kearns D, Cahoon JM, et al. Ceramide mediates vascular dysfunction in diet-induced obesity by PP2A-mediated dephosphorylation of the eNOS-Akt complex. Diabetes. 2012;61:1848–1859. doi: 10.2337/db11-1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z, Chen H, Li J, Li T, Zheng B, Zheng Y, et al. Sirtuin 1-mediated cellular metabolic memory of high glucose via the LKB1/AMPK/ROS pathway and therapeutic effects of metformin. Diabetes. 2012;61:217–228. doi: 10.2337/db11-0416. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Effect of HFD consumption on lipid levels and adiposity.