

Abstract
Background and Purpose
The lymphatic system maintains tissue homeostasis by unidirectional lymph flow, maintained by tonic and phasic contractions within subunits, ‘lymphangions’. Here we have studied the effects of the inflammatory cytokine IL-1β on tonic contraction of rat mesenteric lymphatic muscle cells (RMLMC).
Experimental Approach
We measured IL-1β in colon-conditioned media (CM) from acute (AC-CM, dextran sodium sulfate) and chronic (CC-CM, T-cell transfer) colitis-induced mice and corresponding controls (Con-AC/CC-CM). We examined tonic contractility of RMLMC in response to CM, the cytokines h-IL-1β or h-TNF-α (5, 10, 20 ng·mL−1), with or without COX inhibitors [TFAP (10−5 M), diclofenac (0.2 × 10−5 M)], PGE2 (10−5 M)], IL-1-receptor antagonist, Anakinra (5 μg·mL−1), or a selective prostanoid EP4 receptor antagonist, GW627368X (10−6 and 10−7 M).
Key Results
Tonic contractility of RMLMC was reduced by AC- and CC-CM compared with corresponding control culture media, Con-AC/CC-CM. IL-1β or TNF-α was not found in Con-AC/CC-CM, but detected in AC- and CC-CM. h-IL-1β concentration-dependently decreased RMLMC contractility, whereas h-TNF-α showed no effect. Anakinra blocked h-IL-1β-induced RMLMC relaxation, and with AC-CM, restored contractility to RMLMC. IL-1β increased COX-2 protein and PGE2 production in RMLMC.. PGE2 induced relaxations in RMLMC, comparable to h-IL-1β. Conversely, COX-2 and EP4 receptor inhibition reversed relaxation induced by IL-1β.
Conclusions and Implications
The IL-1β-induced decrease in RMLMC tonic contraction was COX-2 dependent, and mediated by PGE2. In experimental colitis, IL-1β and tonic lymphatic contractility were causally related, as this cytokine was critical for the relaxation induced by AC-CM and pharmacological blockade of IL-1β restored tonic contraction.
Tables of Links
| LIGANDS |
|---|
| Anakinra, IL-1 receptor antagonist |
| Diclofenac |
| GW627368X |
| IL-1β |
| IL-4 |
| IL-13 |
| PGE2 (dinoprostone) |
| TNF-α |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,cAlexander et al., 2013a,b,c,,).
Introduction
The common major characteristic and cardinal clinical symptom of inflammatory bowel disease (IBD), Crohn's disease (CD) and ulcerative colitis (UC) is a severe, and chronic, relapsing intestinal inflammation (Baumgart and Sandborn, 2012; Ordas et al., 2012). Although the precise aetiology of IBD is still unknown, the intestinal microvasculature (arterioles, capillaries, venules) has been identified as a major contributor to the initiation and perpetuation of intestinal inflammatory processes by triggering leukocyte recruitment and extravasation (Chidlow et al., 2007; Danese, 2011). However, the role of the lymphatic components of the vascular system in the pathophysiology of IBD still remains unclear (Van Kruiningen and Colombel, 2008).
The lymphatic system maintains tissue homeostasis by returning interstitial fluid, digested lipids and perivascularly infiltrated immune cells to the venous side of the bloodstream. Lymph uptake in the intestine begins within the villi, where initial lymphatics facilitate the passive transport of cells and fluids across a single layer of lymphatic endothelial cells (LECs), which are covered by a discontinuous basement membrane (Alitalo, 2011). Subsequently, lymph drains through precollector vessels into collecting lymphatic vessels, which are invested with a muscle cell layer, a continuous basement membrane and unidirectional valves, which prevent lymph backflow (Alitalo, 2011). In collecting lymphatics, propulsive lymph flow is generated and regulated by complex interactions of phasic, and sustained, tonic contractions of the lymphatic muscle within a complicated network of connected valve-containing contractile subunits, called ‘lymphangions’ (Smith, 1949; Mislin, 1961; Mislin and Rathenow, 1962; Zawieja et al., 1993). Recent studies have shown that force and frequency of lymphatic contractions can be positively and negatively influenced by a variety of autocrine and paracrine signals, including nervous innervation, NO, prostanoids, chemokines and cytokines (Becker et al., 2014). Among these, inflammatory cytokines have been strongly associated with IBD pathophysiology, as the persistent and uncontrolled imbalance between pro- and anti-inflammatory cytokines is a central event of the overwhelming immune response in CD and UC (Neurath, 2014). Thereby, inflammatory cytokines (including IL-1β and TNF-α) affect multiple functions of the vascular system by initiating diverse downstream inflammatory pathways (Hatoum et al., 2003; Danese, 2011). One important inflammation-induced vascular effect mediated only by IL-1β is the up-regulation of the two major angio- and lymphangiogenic mediators, VEGF-C and -D, following NF-κB activation (Ristimaki et al., 1998; Watari et al., 2008). Both events have consistently been implicated in inflammation-induced angiogenesis and lymphangiogenesis (Huggenberger et al., 2011; Achen and Stacker, 2012; Kim et al., 2012a).
Although these alterations display structural reorganization of the lymphatic system in response to inflammation, important functional changes in lymph transport have also been recognized. Whereas many studies reveal an increase in lymph flow as an acute response to inflammation (which serves to balance an increased cell and fluid load), others have reported a reduction in lymph transport performance under chronic conditions (Huggenberger et al., 2010; von der Weid and Muthuchamy, 2010; Lachance et al., 2013). Some of our recent work has shown that, in a model of intestinal inflammation, lymph flow from the ileum fell significantly within 24 h of induction and remained depressed for days (Cromer et al., 2015). Additionally, increasing evidence has demonstrated an important capacity of inflammatory cytokines to depress lymphatic pumping function, consistent with reports of reduced pumping in different states of inflammation (von der Weid and Muthuchamy, 2010; Aldrich and Sevick-Muraca, 2013). This effect has been shown in animal models of intestinal inflammation, which revealed impairment of the innate active contractile capacity within collecting mesenteric lymphatics (Wu et al., 2006; Mathias and von der Weid, 2013). In these models, diminished phasic and tonic contractile responses in collecting lymphatic vessels were correlated with lymphatic dysfunction, lymphatic vessel dilation and mucosal inflammation (Von Der Weid and Rehal, 2010). Accordingly, cytokines, including ILs and TNF-α, have been proposed to mediate these effects, and IL-1β in particular has been the focus of several recent experimental studies. Using in vitro and in vivo models of lymphatic transport function, it has been shown that IL-1β potently inhibits lymphatic propulsion and thus lymph flow, through a failure of lymphatic contractility (Hanley et al., 1989; Aldrich and Sevick-Muraca, 2013).
Although the earlier studies provide important insights into the roles of inflammatory cytokines on lymphatic transport function, they did not unequivocally differentiate between a direct influence of these mediators on lymphatic muscle cells, or indirect effects on lymphatic endothelial or resident and infiltrated inflammatory cells, on lymphatic pumping. Given the close association of immune/inflammatory cells with the mesenteric-collecting lymphatics, this differentiation is important (Bridenbaugh et al., 2013; Chatterjee and Gashev, 2014). As an important link between vascular and immune responses to inflammation, the understanding of interstitial fluid and especially antigen and immune cell clearance, via lymph transport, underscores the importance to gain further mechanistic insights into these phenomena. Although currently available lymphatic muscle cell in vitro models do not reproduce all of the phasic contractile responses seen in vivo, our studies show that rat lymphatic muscle cells exhibit tonic contraction and show many similar contractile responses to those reported in vivo.
In this study, we have demonstrated that exposure to the inflammatory cytokine IL-1β, which is known to contribute to IBD aetiology, depressed tonic lymphatic muscle contraction in an in vitro model of lymphatic muscle function. We also demonstrated that this effect can be reproduced using conditioned media (CM) from acute and chronic murine models of experimental colitis, which is IL-1β dependent and associated with a COX-2 and PGE2 pathway.
Methods
Ethical approval
All animal care and experimental procedures were reviewed and approved by the Institutional Animal Care and Use Committee of LSUHSC-S. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 16 animals were used in the experiments described here.
Mice
We used 6- to 8-week-old male C57BL/6J and recombinase activating gene-1-deficient (Rag-1 −/−, B6.129S7-Rag1) mice, which lack T cells and B cells. All mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and kept in a controlled environmental room at 25°C with a 12/12 h light/dark cycle at the animal care facility at LSUHSC-S, with free access to standard pellet diet and tap water before induction of colitis.
Induction of experimental colitis
We used two complementary models of experimental colitis: the acute model of erosive injury in the dextran sodium sulfate (DSS)-induced colitis and the T-cell-dependent model of chronic colitis following adoptive transfer of CD4+CD45RBhigh T-cell transfer. Acute colitis was induced in C57BL/6J mice by feeding 3% DSS (MW 36–50 kDa; ICN Biomedicals, Costa Mesa, CA, USA) in drinking water for 7 days. Chronic colitis was induced by reconstituting Rag-1 −/− mice with CD4+CD45RBhigh T cells, as previously described (Ostanin et al., 2009).
Cells
Rat mesenteric lymphatic muscle cells (RMLMC) were obtained as previously described (Chakraborty et al., 2011), and cultured in DMEM (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% FBS (Atlanta Biologicals, Flowery Branch, GA, USA), 1% penicillin-streptomycin-amphotericin B (PSA, Coring Cellgro, Herndon, VA, USA) and 200 μM glutamine (HyClone Laboratories, Logan, UT, USA). These cells retain their contractile apparatus and regulation and were used from passage 12 to 28. Cells were cultured at 37°C in 7.5% CO2. Cell culture medium was changed weekly and cells split every 14 days at a 1:5 ratio.
CM
CM were prepared using colon tissues from untreated, acute and chronic colitis-induced mice. Mice were anaesthetized by i.p. administration of ketamine (50 mg·mL−1) and xylazine (2.85 mg·mL−1), and killed by cardiac puncture, followed by removal of the entire colon. Colons were longitudinally opened, washed in ice-cold PBS, 1 cm2 of each colon manually chopped and incubated in 10% FBS with 3% PSA supplemented DMEM for 24 h at 37°C. Subsequently, these media were collected, centrifuged at 485 x g for 15 min (min) at 4°C, filtered (0.22 μm, Puradisc 25 AS, Thermo/Fisher) and stored at −80°C.
Collagen gel contraction assay
Preparation of rat tail type 1 collagen
Rat tail type 1 collagen matrices were prepared by a modification of the protocol previously published by Benoit et al. (2008). Briefly, rat tail tendons were manually excised, washed with 100% isopropanol (Thermo/Fisher) and dissolved in sterile 4 mM acetic acid for 24 h at 4°C under constant agitation. Collagen solution was filtered through a 250 μm nylon filter (Spectrum Labs, Rancho Dominguez, CA, USA), centrifuged at 19 x g for 20 min at 4°C and snap frozen. Using a bench-top manifold freeze-dryer (Millrock Technology, Kingston, NY, USA), frozen aliquots were dried and stored at −20°C for future use.
Preparation of RMLMC/collagen gel
Twenty-four hours prior to experiments, freeze-dried collagen was resolubilized in cold 0.012 M hydrochloric acid (HCl) at 2.5 mg·mL−1 final collagen concentration and incubated overnight at 4°C with gentle agitation. On the day of the experiment, 0.8 mL of cold 5× PBS was added to 3.2 mL of dissolved collagen gel and the pH was titrated with 0.5 M sodium hydroxide (NaOH) to 7.4. Cultured RMLMC were washed twice with PBS and then harvested with trypsin-EDTA (Sigma-Aldrich). These cells were centrifuged at 485 x g for 5 min, resuspended in DMEM (supplemented with 10% FBS, PSA and glutamine) and counted. A total of 1.2 × 106 cells (50 000 cells per well) were resuspended in 8 mL of supplemented DMEM. The final RMLMC/collagen mixture (8 mL of cell suspension in 4 mL of collagen gel solution) was seeded in 500 μL aliquots into 24-well plates (Thermo/Fisher) and incubated at 37°C for 1 h, to polymerize. In our tonic contraction, assay gels maintained established levels of tonic contractility and did not exhibit relaxation or ‘fatigue’ once contracted.
Cytokines and CM treatment
RMLMC incorporated into collagen gels were stimulated with 1 mL DMEM (plus 10% FBS, PSA and glutamine) supplemented to a final concentration with cytokines: h-IL-1β (5, 10, 20 ng·mL−1), h-TNF-α (5, 10, 20 ng·mL−1), with or without COX inhibitors, TFAP [10× half maximal inhibitory concentration (IC50) dose = 10−5 M], diclofenac (10× IC50 dose = 0.2 × 10−5 M), PGE2 (10−5 M), IL-1 receptor antagonist, Anakinra (5 μg·mL−1, optimized in a dose–response assay; see Supporting Information Fig. S2), EP4 receptor antagonist [GW627368X (10−6 and 10−7 M)] or Con-AC, Con-CC-, AC-, CC-CM and 10-fold diluted CC-CM (1 mL·well−1). Gels were then gently detached from the edges of the plates to allow unimpeded contraction, and incubated for 4 days at 37°C in 7.5% CO2. In these experiments, RMLMC incorporated into gels attach to collagen fibres in three dimensions and promote progressive gel contraction as a function of time. To monitor time and treatment-dependent changes in contraction, digital photographs of gels were recorded daily over 4 days using a camera (Nikon D40, Tokyo, Japan). Experiments with CM were performed using three mice per group (Con-AC-, AC-, CC-, Con-CC-CM) in duplicate, and CM from each mouse was used in quadruplicate. Gel contractions in four replicate wells (quadruplicates) were averaged for each single n value, with n = 3 per group (i.e. 12 gels analysed for n = 3). Single stimulation/inhibition experiments were performed using five different RMLMC set-ups. Each culture of RMLMC was used to set up a minimum of four replicates per treatment concentration and the average of gel contraction in four wells averaged for each single n value for an n = 5 per treatment concentration (20 gels analysed for n = 5).
Gel contraction image analysis
Gel contraction was defined as the change in gel surface area on day 4 as a fraction of its area measured on day 0, and normalized to internal controls. All measurements were made using the NIH ImageJ analysis program (Schneider et al., 2012). At day 0, gel surface areas were initially equal to the well surface area as immediately after gel polymerization, contraction had not yet begun. Over 4 days, RMLMC contraction reduced the respective gel area, which was found to show the greatest differences at this time point. On day 4, gel surface areas were measured and area changes were determined as the gel area divided by the initial gel surface area on day 0. This value was subtracted from 1 to express it as a fractional change in area and then normalized to the contraction in control gels within each experiment, which was set as 100% contraction (see Supporting Information Fig. S4).
COX-2 and PGE2 induction
RMLMC were split at a ratio of 1:3 into 6-well plates and incubated for 3 days until fully confluent. Media were then replaced with media supplemented with recombinant h-IL-1β (20 ng·mL−1), recombinant h-TNF-α (20 ng·mL−1), Con-, AC- or CC-CM, respectively, for 24 h. Afterwards, supernatant media were collected and stored at −80°C. RMLMC were placed on ice, washed with PBS and detached by scraping in 100 μL reducing sample buffer [45% Milli-Q water, 12% 0.5 M Tris-HCl, pH 6.8, 2% SDS, 15% glycerol, 2% phenol red (Sigma-Aldrich), 10% β-mercaptoethanol and protease inhibitor cocktail (Sigma-Aldrich)]. The cell lysate was homogenized, boiled for 1 min at 100°C and stored at −80°C.
Western blotting for COX-2
Total protein concentration was measured in all samples using a 660 nm protein assay (Thermo/Fisher). Cell lysate samples (40 μg per sample) were separated on 7.5% SDS polyacrylamide gels and transferred to nitrocellulose membranes using a transblot apparatus (Idea Scientific, Minneapolis, MN, USA). Protein transfer was verified with Ponceau-S (Sigma-Aldrich) staining. Membranes were blocked with 5% non-fat milk and were incubated with anti-COX-2 antibody (Cayman Chemical, 1:1000 in 0.1% milk powder) overnight at 4°C. An anti-actin N-terminal antibody (Sigma-Aldrich, 1:2000 in 0.1% milk powder) was used as loading control. Anti-rabbit IgG (whole molecule)-peroxidase antibody (Sigma-Aldrich, 1:1500 in 0.1% milk) was added as secondary antibody for 1 h at 25°C. Blots were visualized using enhanced chemiluminescence (Pierce ECL Western Blotting Substrate, Rockford, IL, USA). Densitometry of the resulting bands was performed using NIH ImageJ analysis program.
PGE2 elisa
PGE2 concentrations in control and cytokine-treated RMLMC supernatants were determined using a commercially available PGE2 elisa kit (ENZO Life Science®, Farmingdale, NY, USA) performed according to the manufacturer's protocol.
BD™ cytometric bead array (CBA)
IL-1β and TNF-α concentrations in CM were measured using the CBA Flex Set System, Cell Signaling Master Buffer Kit, the CBA Mouse IL-1β and CBA TNF-α Flex Sets (all BD Bioscience, San Diego, CA, USA). The assay was performed according to the manufacturer's instructions, and all samples were run in duplicate.
Data analysis
Data are presented as mean ± SEM. When comparing three or more experimental groups, one-way anova with Bonferroni's post hoc testing was used. Differences between two sets of data were determined using the unpaired two-tailed Student's t-test (GraphPad Instat 3 software, San Diego, CA, USA). Comparisons were considered statistically significant at P < 0.05.
Materials
Recombinant human (h) TNF-α and IL-1β were purchased from Thermo/Fisher (Waltham, MA, USA). PGE2 was obtained from Cayman Chemical (Ann Arbor, MI, USA). Diclofenac (as the sodium salt) was purchased from Sigma-Aldrich and the selective COX-1 inhibitor IV [N-(5-amino-2-pyridinyl)-4-trifluoromethylbenzamide] TFAP was purchased from EMD Millipore (Billerica, MA). Anakinra (Kineret®; Sobi, Inc., Waltham, MA, USA), a recombinant IL-1 receptor antagonist (IL-1Ra), was obtained from Sobi, Inc. (Ardmore, PA, USA). The selective competitive antagonist of the prostanoid EP4 receptor, GW627368X, was purchased from Cayman Chemical. All reagents were added to supplemented DMEM prior to the experiments.
Results
Tonic contraction of RMLMC was decreased following stimulation with CM from acute and chronic colitis-induced mice
First, we evaluated whether tissue-derived mediators, originating from acute and chronic colitis-induced mice, could influence the tonic contraction of RMLMC. RMLMC were stimulated with control-conditioned media from either control C57BL/6J (Con-AC-CM) or Rag-1 −/− (Con-CC-CM) mice, and with AC- and CC-CM from acute and chronic colitis-induced mice respectively. We found that after 4 days the tonic contractility (expressed in % of contraction vs. control) of RMLMC was significantly reduced by both AC-CM and CC-CM (Figure 1A and B).
Figure 1.
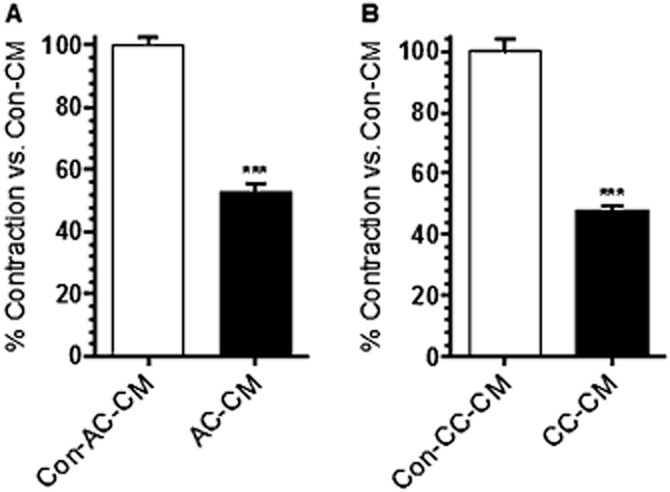
AC- and CC-CM decreased RMLMC contractility. (A) RMLMC treated with AC-CM for 4 days showed a significantly lower contractility (expressed in % of contraction vs. control) compared with control (Con-AC-CM, C57BL/6J mice) treated RMLMC. (B) CC-CM decreased RMLMC contractility at day 4 significantly compared with control (Con-CC-CM from Rag-1 −/− mice) treated RMLMC; n = 3, ***P < 0.001 versus control; Student's t-test; data are mean ± SEM.
IL-1β and TNF-α were increased in CM from murine models of acute and chronic colitis
Because IL-1β and TNF-α have been suggested to contribute to experimental and human colitis and IL-1β has also been proposed to depress lymphatic transport capacity, we tested whether IL-1β and TNF-α were present in CM from acute and chronic colitis-induced mice. Although IL-1β and TNF-α were not detectable in Con-AC/CC-CM, IL-1β and TNF-α were clearly present in AC-CM (Figure 2A and C) and in CC-CM (Figure 2B and D).
Figure 2.
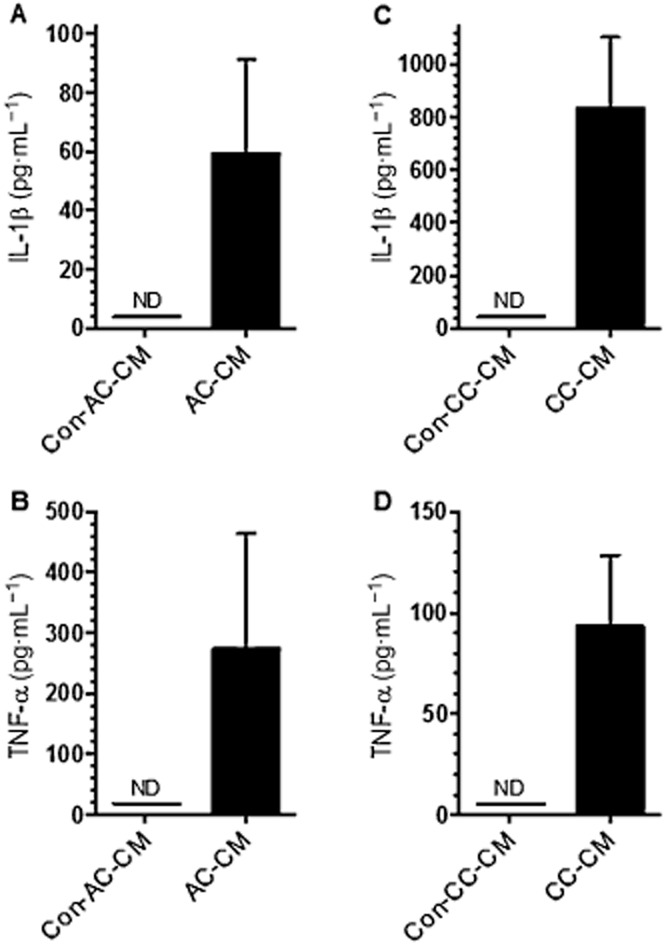
Concentration of IL-1β and TNF-α in CM measured by cytometric bead array. The concentration (pg·mL−1) of IL-1β (A and C) and TNF-α (B and D) was measured in CM from AC-CM and CM from untreated controls (Con-AC-CM) as well as in CC-CM and respective untreated controls (Con-CC-CM) n = 4, ND, not detectable; data are mean ± SEM.
IL-1β, but not TNF-α, decreased tonic RMLMC contractility
Although IL-1β and TNF-α have been associated with changes in the lymphatic vasculature under inflammatory conditions, the direct effect of these cytokines on tonic mesenteric lymphatic muscle cell contraction has not been previously evaluated. Therefore, we examined the effect of recombinant h-TNF-α (20 ng·mL−1) and h-IL-1β (20 ng·mL−1) on isolated RMLMC contractility, using the in vitro collagen gel contraction assay; Figure 3A shows a representative photograph of the collagen gel contraction assay, in which RMLMC were incubated with IL-1β (20 ng·mL−1) or TNF-α (20 ng·mL−1) over the period of 4 days. As shown in Figure 3B, h-TNF-α had no significant inhibitory effect, whereas h-IL-1β significantly impaired RMLMC contractility at day 4. Competitive inhibition of IL-1β from binding to IL-1 type 1 receptor (IL-1R1) using Anakinra (5 μg·mL−1) maintained RMLMC contractility in gels co-treated with h-IL-1β (Figure 3B). To test whether the observed effects were dose dependent, we next examined different concentrations of h-IL-1β and found that 5 ng·mL−1, 10 ng·mL−1 and 20 ng·mL−1, all significantly impaired RMLMC/collagen gel contraction, showing a concentration-dependent effect of IL-1β on RMLMC contractility (Figure 3C). We then stimulated RMLMC with various doses of h-TNF-α (5, 10 and 20 ng·mL−1), but none of these treatments had a significant influence on gel contractility (Figure 3D). We further tested if IL-1β + TNF-α could exhibit synergistic effects and found an enhanced relaxing effect, when cells we co-treated with IL-1β and TNF-α (see Supporting Information Fig. S3).
Figure 3.
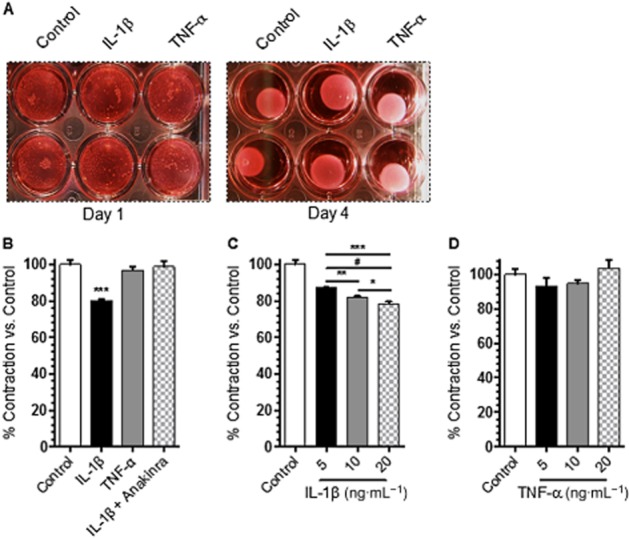
IL-1β decreased RMLMC contractility. (A) A representative photograph of RMLMC/collagen gels incubated with IL-1β (20 ng·mL−1) and TNF-α (20 ng·mL−1) over the period of 4 days. (B) IL-1β (20 ng·mL−1), but not TNF-α (20 ng·mL−1), decreased RMLMC contractility (expressed in % of contraction vs. control) while IL-1 receptor antagonist (Anakinra 5 μg·mL−1) preserved RMLMC contractility in the presence of IL-1β. (C) IL-1β concentration-dependently decreased RMLMC contraction. (D) Different concentrations of TNF-α had no effect on RMLMC contraction. Contractility of RMLMC was measured after 4 days of incubation with cytokines; n = 5, ***P < 0.001 versus control, **P < 0.01 IL-1β 5 versus 10 ng·mL−1, #P < 0.001 IL-1β 5 versus 20 ng·mL−1, *P < 0.05 IL-1β 10 versus 20 ng·mL−1; one-way anova, with Bonferroni's post hoc test; data are mean ± SEM.
IL-1 receptor antagonist restored RMLMC contractility depressed by CM from acute colitis-induced mice
To further examine the influence of IL-1β on the relaxation effect seen in RMLMC after stimulation with AC- and CC-CM, we added Anakinra to test whether this treatment could restore lymphatic muscle contraction. We treated RMLMC with Con-AC-, AC-, Con-CC- and CC-CM supplemented with Anakinra at a dose of 5 μg·mL−1. As shown in Figure 4, when Anakinra blocked IL-1 receptors, AC-CM-stimulated RMLMC showed a nearly complete restoration of normal contractility, compared with AC-CM alone (Figure 4A). However, blockade of the IL-1β receptor in RMLMC stimulated with CC-CM did not reverse the decrease in contractility produced by CC-CM (Figure 4B). This failure of Anakinra to restore contractility to CC-CM stimulated RMLMC could have been due to the greatly increased levels of IL-1β in CC-CM (14-fold levels in AC-CM). We therefore used the same concentration of Anakinra (5 μg·mL−1), which reflects a frequently used, appropriate and therapeutic concentration, with 10-fold diluted CC-CM, where IL-1β levels were expected to be similar to levels found in AC-CM. Under these conditions, Anakinra (5 μg·mL−1) was able to restore RMLMC contractility suppressed by the diluted CC-CM, compared with the corresponding control treatment (Figure 4C). To exclude potential relaxing effects caused by Anakinra itself, we also tested its effects on RMLMC contractility and found no significant differences between Anakinra and control treatments ( Figures 4A and B).
Figure 4.
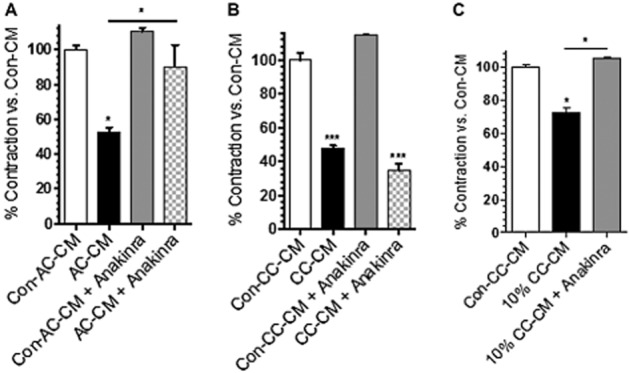
The IL-1 receptor antagonist Anakinra restored contractility of RMLMC treated with AC-CM. (A) Treatment with an IL-1 receptor antagonist inhibited the relaxation effect of AC-CM on RMLMC and restored their physiological contractility. When treated with AC-CM + IL-1 receptor antagonist RMLMC contractility showed no significant difference compared with Con-AC-CM (C57BL/6J mice). (B) Treatment with the IL-1 receptor antagonist did not restore RMLMC contractility suppressed by CC-CM. Contractility in RMLMC treated with CC-CM + IL-1 receptor antagonist showed a significant decrease compared with Con-CC-CM (Rag-1 −/− mice). (C) Anakinra inhibited the relaxation effect of 10-fold diluted (d) CC-CM on RMLMC and restored their physiological contractility. No significant difference in RMLMC contractility treated with 10% (d) CC-CM + IL-1 receptor antagonist; n = 3, *P < 0.05, ***P < 0.001 versus control; one-way anova, with Bonferroni's post hoc test; data are mean ± SEM.
IL-1β, but not TNF-α, increased COX-2 expression in RMLMC
IL-1β and TNF-α can each elicit inflammation-associated vasodilatation by activating endothelial COX-2 followed by the biosynthesis of vasodilator PGs from arachidonic acid (AA), resulting in smooth muscle relaxation. Additionally, IL-1β can produce similar effects on endothelium-denuded blood vessels, consistent with COX-2 production in smooth muscle cells (Beasley, 1999). However, these effects have so far only been addressed in blood vascular smooth muscle cells and not in lymphatic muscle cells. To test whether differences described here between the relaxing properties of IL-1β and TNF-α and between Con- and AC- or CC-CM may reflect differences in COX-2 protein levels in lymphatic cells, we evaluated the ability of these inflammatory cytokines, and CM, to induce COX-2 protein levels in RMLMC using Western blotting. Stimulation with h-TNF-α (20 ng·mL−1) did not induce COX-2 protein levels in RMLMC (Figure 5A). However, h-IL-1β (20 ng·mL−1) significantly increased COX-2 protein levels (measured as scan density values) in RMLMC, by approximately five-fold (Figure 5A). Also, AC- and CC-CM induced COX-2 protein levels in RMLMC after 24 h of treatment, compared with relevant Con-AC/CC-CM (Figure 5B and C).
Figure 5.
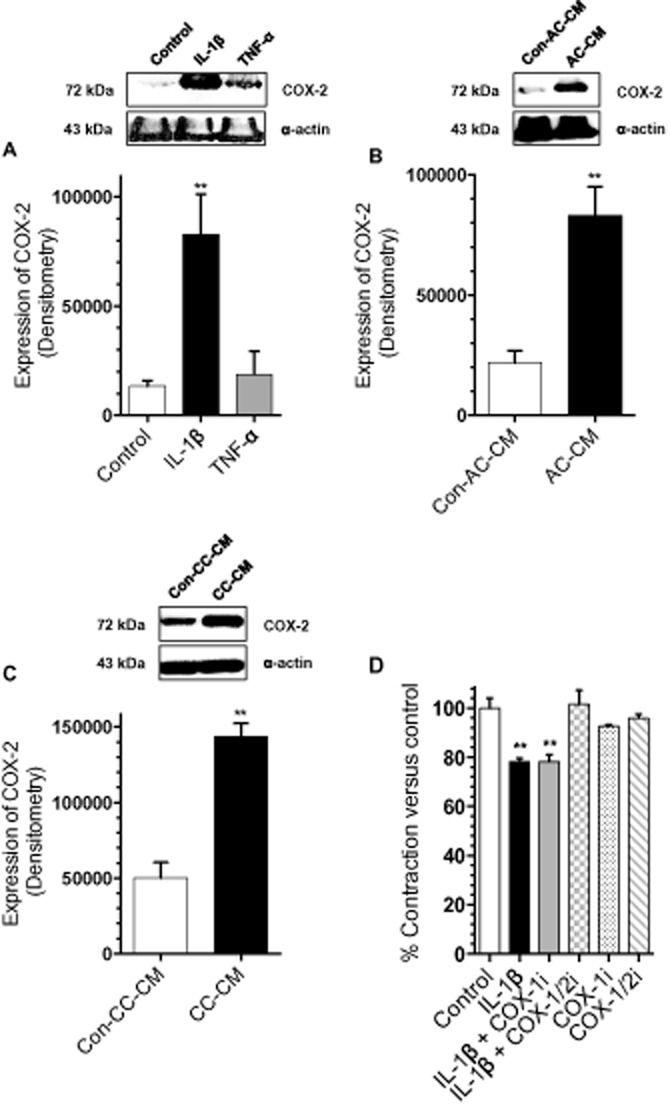
IL-1β increased COX-2 expression in RMLMC while COX-2 inhibition restored RMLMC contractility in the presence of IL-1β. (A) IL-1β (20 ng·mL−1), but not TNF-α (20 ng·mL−1), significantly induced COX-2 expression in RMLMC after 24 h of treatment. (B and C) AC-CM and CC-CM induced COX-2 expression in RMLMC after 24 h of treatment, compared with corresponding Con-AC/CC-CM. (D) COX-2 inhibition restored RMLMC contraction in combination with IL-1β as the COX-1/2 inhibitor (diclofenac), but not the COX-1 inhibitor (TFAP), restored the relaxation effect of IL-1β on RMLMC at day 4: n = 3 for Figure 5A–C, n = 5 for Figure 5D, **P < 0.01 versus control; one-way anova, Bonferroni's post hoc test was used for experiments, shown in Figure 5A and D. Student's t-test was used for experiments in Figure 5B and C; data are mean ± SEM.
Inhibition of COX-2, but not COX-1, restored RMLMCs contractility depressed by IL-1β
Because we found that IL-1β induced COX-2 protein levels in RMLMC, we investigated whether IL-1β-mediated relaxation was dependent on COX-2 activity. To differentiate between mediators derived from COX-1 and COX-2, we used the non-selective COX-1/-2 inhibitor diclofenac, and the selective COX-1 inhibitor TFAP, to attempt to restore RMLMC gel contractility decreased by IL-1β. RMLMC/collagen gels were treated with DMEM (plus 10% FBS, PSA and glutamine) as control or DMEM (plus with 10% FBS, PSA and glutamine) with h-IL-1β (20 ng·mL−1), h-IL-1β (20 ng·mL−1) + TFAP (10−5 M) or IL-1β (20 ng·mL−1) + diclofenac (0.2 × 10−5 M). The non-selective COX-1/-2 inhibitor (diclofenac) blocked the relaxation effects of h-IL-1β, whereas TFAP (the selective COX-1 inhibitor) had no significant effect on IL-1β-stimulated RMLMC contractility (Figure 5D). To rule out possible relaxing effects caused by diclofenac or TFAP itself, we tested the influence of these inhibitors alone on RMLMC contractility. Figure 5D shows no significant differences between diclofenac, TFAP or control treatment.
PGE2 was induced in RMLMC by IL-1β
AA metabolites are important regulators of lymphatic vessel contractility, and lymphatic vessels are capable of producing AA metabolites, which may influence their contractility in an autocrine fashion. COX-2 catalyses the conversion of AA to several PGs, of which PGE2 is the most potent vasodilator. As we had found that COX-2 was highly expressed in RCLSMCs in response to IL-1β, which can increase PG levels. We measured PGE2 in supernatants from RMLMC after stimulation with h-IL-1β (20 ng·mL−1) and h-TNF-α (20 ng·mL−1). Only IL-1β, not TNF-α, significantly increased PGE2 levels (Figure 6A). Although we attempted to quantify PGE2 in CM, we were not able to generate reliable data because of several technical difficulties (e.g. presence of mouse IgG in CM). Thus, we conducted additional experiments to test a possible relaxing effect of the pre-existing PGE2 in CM (see Supporting Information Fig. S1). Our results suggest that the amount of PGE2 in CM had negligible affects.
Figure 6.
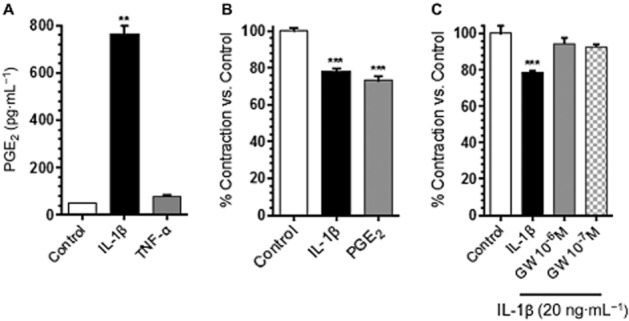
RMLMC produced PGE2 in response to IL-1β stimulation, while treatment with an EP4 receptor antagonist maintained their physiological contractility. (A) PGE2 levels in RMLMC supernatant were increased in response to IL-1β (20 ng·mL−1), but not TNF-α (20 ng·mL−1). (B) RMLMC contraction was significantly inhibited by PGE2 (10−5 M) stimulation (similar to the relaxation effect to IL-1β), compared with control. (C) The EP4 receptor antagonist (GW627368X) restored RMLMC contraction that had been suppressed by IL-1β. GW627368X at 10−7 and 10−6 M reversed the relaxation effect of IL-1β on RMLMC at day 4; n = 3 for Figure 6A, n = 5 for Figure 6B and C, ***P < 0.001 versus control; one-way anova, Bonferroni's post hoc test; data are mean ± SEM.
Exogenous PGE2, dinoprostone, decreased RMLMC contractility
We tested if direct stimulation of RMLMC with exogenous PGE2 could produce similar effects on tonic contractility as direct stimulation by IL-1β. Earlier studies on isolated lymphatic vessel contractile activity have shown that PGE2-induced maximum inhibition around 30 μM, with a calculated EC50 of 10−5.9 M (Rehal et al., 2009). Ren et al. (1995) reported that rat vascular smooth muscle cells were stimulated by PGE2 with EC50 of 4.9 × 10−6 M. Hence, RMLMC collagen gels were treated with exogenous PGE2 (dinoprostone; 10−5 M). As shown in Figure 6B, dinoprostone induced relaxation effects on RMLMC, similar to those of h-IL-1β.
EP4 receptor blockade restored RMLMC contractility depressed by IL-1β
We further determined the contribution of EP4 receptors in mediating IL-1β-induced decreases in RMLMC contraction. RMLMC were treated with h-IL-1β (20 ng·mL−1) alone or in co-treatment with the EP4 receptor antagonist (GW627368X, 10−7 and 10−6 M, IC50 = 10−6 M) (Wilson et al., 2006; Rehal et al., 2009). After 4 days, co-incubation with the EP4 receptor antagonist blocked the relaxation effects of h-IL-1β stimulation and RMLMC contractility was restored to control values (Figure 6C).
Discussion
We used an in vitro collagen gel contraction assay, to demonstrate crucial pathways for the reduction in tonic RMLMC contraction, mediated by IL-1β. We demonstrated that this cytokine was essential for producing the relaxation effects caused by CM from acute experimental colitis, as IL-1β receptor blockade restored tonic contraction. Direct measurement of IL-1β, direct effects of recombinant IL-1β and the pharmacological blockade of IL-1β receptors with Anakinra further supported these findings. Despite potential influences of testing the effect of CM (mouse) and recombinant cytokines (human) on cells from a different species (rat), our results remain consistent with IL-1β as a potent contraction-limiting cytokine. The results presented here indicate that IL-1β stimulation up-regulated COX-2 expression in RMLMC, to increase local levels of the vasodilator PGE2. We further showed that this effect is cytokine specific as TNF-α neither altered COX-2 expression nor had any effects on the tonic contraction of RMLMC.
Lymph transport in vivo is a complicated system involving lymphatic pumping, interstitial and venous pressures, lymph formation and lymphatic vascular resistance. So, the mechanisms that alter lymph flow during inflammation can alter any of these transport mechanisms and are variably dependent on the type of inflammation. Additionally because lymphatic muscle contains both smooth and striated muscle contractile and regulatory components, they exhibit both tonic and phasic contractile characteristics (Muthuchamy et al., 2003). These different functional and molecular components work together and interact in a complex process that is not fully understood. Additionally many (although not all) inflammatory agents inhibit phasic contractile frequency and pumping, leaving lymph outflow resistance as one of the principal elements regulating lymph transport. This is one of the first descriptions of an in vitro assay of tonic lymphatic muscle contractile function. To date no one has developed a cultured lymphatic muscle cell model that reproduces phasic contractile function. This issue is also true for other striated muscle cell types. Thus, although these cultures of lymphatic muscle cells are not a model of phasic lymphatic contractility, they do represent a model of lymphatic muscle tonic activity. Our model is currently the only such lymphatic model available.
Inflammatory cytokines, including IL-1β and TNF-α, are expressed within the inflamed gut in animal models of colitis, which are characterized by a reduced lymphatic function (Egger et al., 2000; Neurath, 2014). Accordingly, our data on AC- and CC-CM showed that IL-1β and TNF-α were generated in substantial levels. However, little is known about the direct mechanisms and effects of inflammatory cytokines on lymphatic muscle cell contractile function in collecting lymphatic vessels. Thus, to assess the role of these cytokines in regulating tonic lymphatic muscle cell contraction, we determined the effects of AC- and CC-CM on RMLMC contractility. Previously, Hanley et al. (1989) reported an inhibition of fluid pumping and tone in isolated mesenteric lymphatic vessels in response to IL-1β, an effect, which they attributed to the production of COX metabolites. Cytokine-induced dysregulation of lymphatic contractile function could in part account for the tonic and phasic contractile inhibition observed in mesenteric lymphatic vessels from an experimental model of ileitis. Wu et al. (2006) reported that the lymphatic vessel diastolic diameter increased with the severity of inflammation (reflecting a loss of tonic contraction), and that this effect could be seen in vivo and in vitro, thus indicating a substantial influence of intestinal inflammation on tonic contraction in mesenteric lymphatic vessels.
To further ascertain the effects seen in response to CM, we tested the direct effect of exogenous inflammatory cytokines on RMLMC tonic contractility. Because recombinant h-IL-1β and TNF-α have revealed sufficient cross-reactivity and can stimulate cell lines of rat origin, we used h-IL-1β and TNF-α in our model to further achieve a more clinically relevant and translational application (Kehrer et al., 1988). IL-1β, but not TNF-α, showed a dose-dependent inhibition of RMLMC contraction. Although IL-1β stimulation alone did not produce the same level of RMLMC contractility compared with that following AC- or CC-CM, we believe this is due to other mediators present in AC- and CC-CM, which contribute to RMLMC relaxation (Becker et al., 2014). However, our data clearly demonstrate that IL-1β is both necessary and sufficient to induce lymphatic muscle relaxation. Further studies are warranted to elucidate other mediators present in AC- and CC-CM that either contribute to lymphatic relaxation alone or enhance IL-1β-induced relaxation. Cytokines modulate vascular smooth muscle function through binding to specific receptors expressed on their surface (Sprague and Khalil, 2009). In our model, IL-1β receptor blockade with Anakinra restored RMLMC contractility suppressed by AC- and d-CC-CM. The lack of effect of Anakinra on undiluted CC-CM could be due to insufficient IL-1 receptor blockade by the used dose of 5 μg·mL−1 as CC-CM contained 14-fold more IL-1β than AC-CM. However, the same dose of Anakinra did restore RMLMC contractility suppressed by a 10-fold dilution of CC-CM, supporting the possibility that IL-1β is one of the critical lymphatic ‘relaxant’ cytokines released during experimental colitis. Treatment with an IL-1 receptor antagonist has been shown to reduce the severity of experimental immune complex colitis in rats (Cominelli et al., 1990). Our findings indicate that IL-1 receptor blockade may also partly reduce the inflammatory responses by restoring lymphatic transport capacity.
We demonstrated that IL-1β markedly induced COX-2 protein levels in RMLMC, as has been observed in several other cell types, including vascular and bronchial smooth muscle cells (Vigano et al., 1997; Beasley, 1999). Furthermore, IL-1β-induced inhibition of the tonic contractility in RMLMC was restored by the non-selective COX inhibitor diclofenac, whereas the selective COX-1 inhibitor TFAP alone had no effect on RMLMC contractility, reduced by IL-1β. This suggests that inhibition of COX-2-, but not of COX-1, was responsible for restoring tonic RMLMC contractility. Together, these results demonstrated that IL-1β inhibition of RMLMC contraction is COX-2 dependent. It is also likely that this effect was related to generation of COX-2 products, in particular, PGE2. Vascular and bronchial smooth muscle cells produce PGE2 in response to treatment with inflammatory cytokines (Feng et al., 1993; Belvisi et al., 1997) and lymphatic vessels themselves are also capable of producing PGs (Johnston and Gordon, 1981). Karnezis et al. (2012) have shown that LECs have the capacity to produce PGE2, although it was unclear whether lymphatic muscle cells could be a possible source of PG production. In the present study, we detected high levels of PGE2 in RMLMC culture media treated with IL-1β, showing that PGE2 was produced directly by lymphatic muscle cells. Although these data do not rule out other PGs, and other PG sources, being involved in these responses, our results are the first demonstration of PGE2 production by mesenteric lymphatic muscle cells in response to IL-1β, in the absence of endothelial cells.
Studies on isolated mesenteric lymphatic vessels have shown that direct application of PGE2 can induce lymphatic vessel relaxation, which could be mediated by the EP receptors, EP1-4 (Hata and Breyer, 2004; Rehal et al., 2009). We found that EP4 receptor mRNA was present in RMLMC (data not shown). The decrease in lymphatic muscle contraction could thus be produced by direct PGE2 receptor binding in an autocrine fashion. Studies on isolated mesenteric lymphatic vessels from TNBS-treated animals also showed that the EP4 receptor was the main receptor involved in the PGE2-induced decrease in lymphatic pumping, and the downstream signalling involved the cAMP/PKA pathway (von der Weid and Muthuchamy, 2010). Our experiments showed that EP4 receptors played a major role in IL-1β-induced decrease in RMLMC tonic contraction as a selective EP4 receptor antagonist blocked the relaxation effect of Il-1β and restored RMLMC contractility. This suggests that PGE2 mediates IL-1β-induced decrease in RMLMC contraction via binding to EP4 receptors. Collectively, these data demonstrate that RMLMC are capable of generating and responding to PGE2 in the absence of endothelial cells and confirmed PGE2 as one of major regulators of lymphatic vessel contractility during inflammation.
Previously, we have demonstrated that inflammatory cytokines suppressed several LEC functions (Chaitanya et al., 2010). In response to IL-1β and TNF-α, LECs showed alterations in barrier function, up-regulation of cell adhesion molecules and changes in proliferation rate, all of which are believed to play central roles in the interstitial fluid and immune cell uptake, within the initial lymphatics (Johnson et al., 2006; Chaitanya et al., 2010). The effects of inflammatory cytokines on both lymphatic endothelial and muscle cell types will also alter antigen and cytokine delivery to the node and thus inflammatory/immune reactions. Because IL-1β increased permeability and decreased lymph transport in mesenteric lymphatic vessels, one possible consequence could be the interstitial retention of fluids (oedema), lipids as well as immune cells and even the retrograde drainage from lymph from the vessel back into the interstitial space (Cromer et al., 2014). This is compatible with one of our recent reports, in which we demonstrated that transgenic FoxC2 +/− mice, which are characterized by diminished intestinal lymph flow including valvular insufficiency, are highly susceptible to acute DSS-induced colitis and show signs of lymphatic transport failure as displayed by structural (dilated torturous lymphatic vessels) and functional (greater submucosal oedema, higher immune cell burden) changes in the intestinal lymphatic vasculature (Becker et al., 2015). Additionally, Prox1+/− mice, which are characterized by lymphatic leakage, displayed an abnormal accumulation of lipids within the intestinal wall, partly reproducing the unique feature of fat wrapping in patients with CD (Harvey et al., 2005; von der Weid et al., 2011).
The shared molecular target of functional alterations in lymphatic muscle cells is their contractile apparatus, specifically their myosin light and heavy chains (MLC, MHC) and the corresponding phosphorylation status responsible for purposive muscle contraction. The influence of cytokines in general and specifically IL-1β on myosin light chain phosphorylation has previously been addressed. Although the critical influence of IL-1β on gastrointestinal motility disorders is long recognized, it has only recently been shown that IL-1β reduces the activity of important regulatory proteins such as CPI-17 (a phosphorylation-dependent inhibitor of myosin phosphatase), resulting in decreased phosphorylation of MLC (Cao et al., 2005; Hu et al., 2007; Ohama et al., 2007). Although this concept has been successfully transferred to vascular smooth muscle cells, so far little is known about the molecular pathways, in which cytokines alter lymphatic muscle contractility at a molecular level (Kim et al., 2012b). It is worth mentioning that even lymphatic muscle cells from different origins of the lymphatic vasculature (thoracic duct vs. mesentery) show significant differences in their contractile behaviour and MHC isoforms, which may further complicate the translation of results obtained from other than lymphatic muscle cells, to the lymphatic contractile apparatus and its molecular response to inflammatory stimuli (Muthuchamy et al., 2003; Gashev et al., 2004).
One possible limitation of our study is the focus on pro-inflammatory cytokines (IL-1β and TNF-α), which does not take the important role of Th2 and Th17 cytokines into account. Various studies have recently shown differential results, when reporting an increase in intestinal smooth muscle contractility, triggered by CD3+ T-cell-derived Th2 cytokines (IL-4, IL-13), a decrease in intestinal contraction induced by the Th17 cytokine IL-17 and a dual (activation vs. inhibition) dose-dependent modulation of airway smooth muscle cells by IL-4 (Akiho et al., 2005b; Ohta et al., 2008; Mori et al., 2014). These findings are especially interesting as both hypo- as well as hyper-contractility of intestinal smooth muscle cells are displayed in clinical symptoms of IBD patients (e.g. strictures and constipation), while it is long recognized that human and experimental IBD are mainly accompanied by an impaired mesenteric lymphatic activity and diminished lymphatic transport function (Von Der Weid and Rehal, 2010). In line with the Il-1β / COX2 / PGE2 pathway, described in this study, Akiho et al. (2005a) showed an increase in PGE2 levels after exposure to the Th2 cytokines IL-4 and IL-13, which resulted in an enhanced intestinal muscle hyper-contractility. These results may indicate that the PGE2-triggered increase in contractility can be induced by both Th1 and Th2 cytokines and that its relaxing effect is similar in different muscle cells. However, based on structural and functional differences, the results obtained from smooth muscle cells of origins other than the lymphatic vasculature have yet to be translated to the unique lymphatic muscle function and thus may lead towards further investigations in this field.
In summary, we have demonstrated a pathway in which IL-1β-induced PGE2 production via up-regulation of COX-2 expression inhibits RMLMC tonic contraction. This study established one potential causal relationship between inflammatory cytokines and lymphatic muscle cells in the regulation of tonic lymphatic contractility in experimental colitis. Thus, IL-1β may play an important role in the pathophysiology of experimental colitis and human IBD, by depressing lymphatic transport capacity, leading to retention of inflammatory cells and mediators in the intestine, exacerbating the inflammatory state in a cascade reaction. Our findings indicate that lymphatic muscle contractility can be impaired independently of endothelial cells, suggesting that impaired lymphatic transport function represents an important component of the complex pathophysiology of experimental and human IBD. Overall, these data expand our understanding of the molecular mechanisms mediating lymphatic pump failure in experimental colitis and provide valuable mechanistic insights, which can exploit the lymphatic system as a target for the management of IBD.
Acknowledgments
We thank Merilyn Jennings, Josh Bryan and Courtney Parker of LSUHSC-S for their technical assistance. This work supported by a fellowship from the Malcom Feist Cardiovascular Research Endowment, LSU Health Science Center – Shreveport (M. A.), a grant from the Department of Defense (W81XWH-11-1-0577, ‘Lymphatic Vascular Based Therapy in IBD’, J. S. A.), the National Institute of General Medical Sciences COBRE Grant (P30-GM110703, I. T.) and a German Research Foundation (DFG) grant (BE 5619/1-1, F. B.).
Glossary
- AA
arachidonic acid
- AC-CM
acute colitis-conditioned media
- CBA
cytometric beads array
- CC-CM
chronic colitis-conditioned media
- CD
Crohn's disease
- CM
conditioned media
- Con-AC-CM
control acute colitis-conditioned media
- Con-CC-CM
control chronic colitis-conditioned media
- DSS
dextran sodium sulfate
- IBD
inflammatory bowel disease
- LECs
lymphatic endothelial cells
- RMLMC
rat mesenteric lymphatic muscle cells
- TFAP
N-(5-amino-2-pyridinyl)-4-trifluoromethylbenzamide
- UC
ulcerative colitis
Author contributions
All experiments were undertaken at Louisiana State University Health Sciences Center – Shreveport, Louisiana. J. S. A. conceived and designed the research. M. A., F. B., M. W. and D. O. performed the experiments. M. A., M. W. and I. T. analysed the data. M. A., F. B., M. W., I. T., Y. W. and J. S. A. interpreted the results of the experiments. D. C. Z. and M. M. contributed essential reagents or tools. M. A., F. B., F. N. E. G., I. T., P. Y. W. and J. S. A. drafted the manuscript. All authors contributed to the critical review of drafts of the manuscript and gave final approval of the version to be published. M. A. and F. B. contributed equally to this work.
Conflict of interest
The authors declare no conflicts of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 PGE2 decreased rat mesenteric lymphatic muscle cell (RMLMC) contractility resistant to Anakinra treatment. RMLMC treated with PGE2 (10−5 M) for 4 days showed a significantly lower contractility (expressed in % of contraction vs. control) compared with control-treated RMLMC, while Anakinra (5 μg·mL−1) treatment showed no significant effect on the PGE2-induced relaxation. n = 4, **P < 0.01 versus control, one-way anova, with Bonferroni's post hoc test; data are mean ± SEM.
Figure S2 Different doses of Anakinra restored IL-1β-induced decreased rat mesenteric lymphatic muscle cell (RMLMC) contractility. RMLMC treated with IL-1β (20 ng·mL−1) for 4 days showed a significantly lower contractility (expressed in % of contraction vs. control) compared with control-treated RMLMC while co-treatment with Anakinra (0.5, 5, 50 μg·mL−1) restored RMLMC relaxation. n = 5, ***P < 0.001 versus control, one-way anova, with Bonferroni's post hoc test; data are mean ± SEM.
Figure S3 IL-1β + TNF-α exhibit synergistic effects in decreasing RMLMC contractility. IL-1β (20 ng·mL−1), but not TNF-α (20 ng·mL−1), decreased RMLMC contractility (expressed in % of contraction vs. control) while IL-1β (20 ng·mL−1) + TNF-α (20 ng·mL−1) decrease RMLMC contractility to a greater extent than IL-1β (20 ng·mL−1) alone. IL-1 receptor antagonist (Anakinra 5 μg·mL−1) maintained RMLMC contractility in the presence of IL-1β or IL-1β + TNF-α at day 4. n = 4, ***P < 0.001 versus control, ***P < 0.01 IL-1β (20 ng·mL−1) versus IL-1β (20 ng·mL−1) + TNF-α (20 ng·mL−1). One-way anova, with Bonferroni's post hoc test; data are mean ± SEM.
Figure S4 Equation for gel contraction. exp. represents the values of the treated gels, whereas con. represents the values for the untreated internal control gels. t0 = day 0, t4 = day 4. As we observed slightly different contractile response within our control groups, an own control for each single experimental treatment was set. These individual controls were set as 100% and the results of parallel conducted treatment studies were normalized to this internal control. When, for example, the control group produced a 70% contraction and a treatment a 35% contraction, then the data are presented as 100% contraction for control and 50% contraction for the treatment group.
References
- Achen MG, Stacker SA. Vascular endothelial growth factor-D: signaling mechanisms, biology, and clinical relevance. Growth Factors. 2012;30:283–296. doi: 10.3109/08977194.2012.704917. [DOI] [PubMed] [Google Scholar]
- Akiho H, Deng Y, Blennerhassett P, Kanbayashi H, Collins SM. Mechanisms underlying the maintenance of muscle hypercontractility in a model of postinfective gut dysfunction. Gastroenterology. 2005a;129:131–141. doi: 10.1053/j.gastro.2005.03.049. [DOI] [PubMed] [Google Scholar]
- Akiho H, Lovato P, Deng Y, Ceponis PJ, Blennerhassett P, Collins SM. Interleukin-4- and -13-induced hypercontractility of human intestinal muscle cells-implication for motility changes in Crohn's disease. Am J Physiol Gastrointest Liver Physiol. 2005b;288:G609–G615. doi: 10.1152/ajpgi.00273.2004. [DOI] [PubMed] [Google Scholar]
- Aldrich MB, Sevick-Muraca EM. Cytokines are systemic effectors of lymphatic function in acute inflammation. Cytokine. 2013;64:362–369. doi: 10.1016/j.cyto.2013.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G Protein-Coupled Receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Catalytic Receptors. Br J Pharmacol. 2013b;170:1676–1705. doi: 10.1111/bph.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013c;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alitalo K. The lymphatic vasculature in disease. Nat Med. 2011;17:1371–1380. doi: 10.1038/nm.2545. [DOI] [PubMed] [Google Scholar]
- Baumgart DC, Sandborn WJ. Crohn's disease. Lancet. 2012;380:1590–1605. doi: 10.1016/S0140-6736(12)60026-9. [DOI] [PubMed] [Google Scholar]
- Beasley D. COX-2 and cytosolic PLA2 mediate IL-1beta-induced cAMP production in human vascular smooth muscle cells. Am J Physiol. 1999;276:H1369–H1378. doi: 10.1152/ajpheart.1999.276.4.H1369. [DOI] [PubMed] [Google Scholar]
- Becker F, Yi P, Al-Kofahi M, Ganta VC, Morris J, Alexander JS. Lymphatic dysregulation in intestinal inflammation: new insights into inflammatory bowel disease pathomechanisms. Lymphology. 2014;47:3–27. [PubMed] [Google Scholar]
- Becker F, Potepalov S, Shehzahdi R, Bernas M, Witte M, Abreo F, et al. Downregulation of FoxC2 increased susceptibility to experimental colitis: influence of lymphatic drainage function? Inflamm Bowel Dis. 2015;21:1282–1296. doi: 10.1097/MIB.0000000000000371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belvisi MG, Saunders MA, Haddad E, Hirst SJ, Yacoub MH, Barnes PJ, et al. Induction of cyclo-oxygenase-2 by cytokines in human cultured airway smooth muscle cells: novel inflammatory role of this cell type. Br J Pharmacol. 1997;120:910–916. doi: 10.1038/sj.bjp.0700963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit C, Gu Y, Zhang Y, Alexander JS, Wang Y. Contractility of placental vascular smooth muscle cells in response to stimuli produced by the placenta: roles of ACE vs. non-ACE and AT1 vs. AT2 in placental vessel cells. Placenta. 2008;29:503–509. doi: 10.1016/j.placenta.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridenbaugh EA, Wang W, Srimushnam M, Cromer WE, Zawieja SD, Schmidt SE, et al. An immunological fingerprint differentiates muscular lymphatics from arteries and veins. Lymphat Res Biol. 2013;11:155–171. doi: 10.1089/lrb.2013.0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao W, Fiocchi C, Pricolo VE. Production of IL-1beta, hydrogen peroxide, and nitric oxide by colonic mucosa decreases sigmoid smooth muscle contractility in ulcerative colitis. Am J Physiol Cell Physiol. 2005;289:C1408–C1416. doi: 10.1152/ajpcell.00073.2005. [DOI] [PubMed] [Google Scholar]
- Chaitanya GV, Franks SE, Cromer W, Wells SR, Bienkowska M, Jennings MH, et al. Differential cytokine responses in human and mouse lymphatic endothelial cells to cytokines in vitro. Lymphat Res Biol. 2010;8(3):155–164. doi: 10.1089/lrb.2010.0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S, Nepiyushchikh Z, Davis MJ, Zawieja DC, Muthuchamy M. Substance P activates both contractile and inflammatory pathways in lymphatics through the neurokinin receptors NK1R and NK3R. Microcirculation. 2011;18:24–35. doi: 10.1111/j.1549-8719.2010.00064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee V, Gashev AA. Mast cell-directed recruitment of MHC class II positive cells and eosinophils towards mesenteric lymphatic vessels in adulthood and elderly. Lymphat Res Biol. 2014;12:37–47. doi: 10.1089/lrb.2013.0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chidlow JH, Jr, Shukla D, Grisham MB, Kevil CG. Pathogenic angiogenesis in IBD and experimental colitis: new ideas and therapeutic avenues. Am J Physiol Gastrointest Liver Physiol. 2007;293:G5–G18. doi: 10.1152/ajpgi.00107.2007. [DOI] [PubMed] [Google Scholar]
- Cominelli F, Nast CC, Clark BD, Schindler R, Lierena R, Eysselein VE, et al. Interleukin 1 (IL-1) gene expression, synthesis, and effect of specific IL-1 receptor blockade in rabbit immune complex colitis. J Clin Invest. 1990;86:972–980. doi: 10.1172/JCI114799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cromer W, Wang W, Zawieja SD, von der Weid PY, Newell-Rogers MK, Zawieja DC. Colonic insult impairs lymph flow, increases cellular content of the lymph, alters local lymphatic microenvironment, and leads to sustained inflammation in the rat ileum. Inflamm Bowel Dis. 2015;21(7):1553–1563. doi: 10.1097/MIB.0000000000000402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cromer WE, Zawieja SD, Tharakan B, Childs EW, Newell MK, Zawieja DC. The effects of inflammatory cytokines on lymphatic endothelial barrier function. Angiogenesis. 2014;17:395–406. doi: 10.1007/s10456-013-9393-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danese S. Role of the vascular and lymphatic endothelium in the pathogenesis of inflammatory bowel disease: ‘brothers in arms’. Gut. 2011;60:998–1008. doi: 10.1136/gut.2010.207480. [DOI] [PubMed] [Google Scholar]
- Egger B, Bajaj-Elliott M, MacDonald TT, Inglin R, Eysselein VE, Buchler MW. Characterisation of acute murine dextran sodium sulphate colitis: cytokine profile and dose dependency. Digestion. 2000;62:240–248. doi: 10.1159/000007822. [DOI] [PubMed] [Google Scholar]
- Feng L, Sun W, Xia Y, Tang WW, Chanmugam P, Soyoola E, et al. Cloning two isoforms of rat cyclooxygenase: differential regulation of their expression. Arch Biochem Biophys. 1993;307:361–368. doi: 10.1006/abbi.1993.1601. [DOI] [PubMed] [Google Scholar]
- Gashev AA, Davis MJ, Delp MD, Zawieja DC. Regional variations of contractile activity in isolated rat lymphatics. Microcirculation. 2004;11:477–492. doi: 10.1080/10739680490476033. [DOI] [PubMed] [Google Scholar]
- Hanley CA, Elias RM, Movat HZ, Johnston MG. Suppression of fluid pumping in isolated bovine mesenteric lymphatics by interleukin-1: interaction with prostaglandin E2. Microvasc Res. 1989;37:218–229. doi: 10.1016/0026-2862(89)90039-3. [DOI] [PubMed] [Google Scholar]
- Harvey NL, Srinivasan RS, Dillard ME, Johnson NC, Witte MH, Boyd K, et al. Lymphatic vascular defects promoted by Prox1 haploinsufficiency cause adult-onset obesity. Nat Genet. 2005;37:1072–1081. doi: 10.1038/ng1642. [DOI] [PubMed] [Google Scholar]
- Hata AN, Breyer RM. Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol Ther. 2004;103:147–166. doi: 10.1016/j.pharmthera.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Hatoum OA, Miura H, Binion DG. The vascular contribution in the pathogenesis of inflammatory bowel disease. Am J Physiol Heart Circ Physiol. 2003;285:H1791–H1796. doi: 10.1152/ajpheart.00552.2003. [DOI] [PubMed] [Google Scholar]
- Hu W, Mahavadi S, Li F, Murthy KS. Upregulation of RGS4 and downregulation of CPI-17 mediate inhibition of colonic muscle contraction by interleukin-1beta. Am J Physiol Cell Physiol. 2007;293:C1991–C2000. doi: 10.1152/ajpcell.00300.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huggenberger R, Ullmann S, Proulx ST, Pytowski B, Alitalo K, Detmar M. Stimulation of lymphangiogenesis via VEGFR-3 inhibits chronic skin inflammation. J Exp Med. 2010;207:2255–2269. doi: 10.1084/jem.20100559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huggenberger R, Siddiqui SS, Brander D, Ullmann S, Zimmermann K, Antsiferova M, et al. An important role of lymphatic vessel activation in limiting acute inflammation. Blood. 2011;117:4667–4678. doi: 10.1182/blood-2010-10-316356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LA, Clasper S, Holt AP, Lalor PF, Baban D, Jackson DG. An inflammation-induced mechanism for leukocyte transmigration across lymphatic vessel endothelium. J Exp Med. 2006;203(12):2763–2777. doi: 10.1084/jem.20051759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston MG, Gordon JL. Regulation of lymphatic contractility by arachidonate metabolites. Nature. 1981;293:294–297. doi: 10.1038/293294a0. [DOI] [PubMed] [Google Scholar]
- Karnezis T, Shayan R, Caesar C, Roufail S, Harris NC, Ardipradja K, et al. VEGF-D promotes tumor metastasis by regulating prostaglandins produced by the collecting lymphatic endothelium. Cancer Cell. 2012;21:181–195. doi: 10.1016/j.ccr.2011.12.026. [DOI] [PubMed] [Google Scholar]
- Kehrer P, Turnill D, Dayer JM, Muller AF, Gaillard RC. Human recombinant interleukin-1 beta and -alpha, but not recombinant tumor necrosis factor alpha stimulate ACTH release from rat anterior pituitary cells in vitro in a prostaglandin E2 and cAMP independent manner. Neuroendocrinology. 1988;48:160–166. doi: 10.1159/000125004. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Kataru RP, Koh GY. Regulation and implications of inflammatory lymphangiogenesis. Trends Immunol. 2012a;33:350–356. doi: 10.1016/j.it.2012.03.006. [DOI] [PubMed] [Google Scholar]
- Kim JI, Urban M, Young GD, Eto M. Reciprocal regulation controlling the expression of CPI-17, a specific inhibitor protein for the myosin light chain phosphatase in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2012b;303:C58–C68. doi: 10.1152/ajpcell.00118.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachance PA, Hazen A, Sevick-Muraca EM. Lymphatic vascular response to acute inflammation. PLoS ONE. 2013;8:e76078. doi: 10.1371/journal.pone.0076078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathias R, von der Weid PY. Involvement of the NO-cGMP-K(ATP) channel pathway in the mesenteric lymphatic pump dysfunction observed in the guinea pig model of TNBS-induced ileitis. Am J Physiol Gastrointest Liver Physiol. 2013;304:G623–G634. doi: 10.1152/ajpgi.00392.2012. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mislin H. Zur funktionsanalyse der Lymphgefässmotorik (Cavia porcellus L.) Rev Suisse Zool. 1961;68:228–238. , (German) [Google Scholar]
- Mislin H, Rathenow D. Experimentelle untersuchungen über die bewegungskoordination der Lymphangione (Cavia porcellus L.) Rev Suisse Zool. 1962;69:334–344. , (German) [Google Scholar]
- Mori D, Watanabe N, Kaminuma O, Murata T, Hiroi T, Ozaki H, et al. IL-17A induces hypo-contraction of intestinal smooth muscle via induction of iNOS in muscularis macrophages. J Pharmacol Sci. 2014;125:394–405. doi: 10.1254/jphs.14060fp. [DOI] [PubMed] [Google Scholar]
- Muthuchamy M, Gashev A, Boswell N, Dawson N, Zawieja D. Molecular and functional analyses of the contractile apparatus in lymphatic muscle. FASEB J. 2003;17:920–922. doi: 10.1096/fj.02-0626fje. [DOI] [PubMed] [Google Scholar]
- Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol. 2014;14:329–342. doi: 10.1038/nri3661. [DOI] [PubMed] [Google Scholar]
- Ohama T, Hori M, Momotani E, Iwakura Y, Guo F, Kishi H, et al. Intestinal inflammation downregulates smooth muscle CPI-17 through induction of TNF-alpha and causes motility disorders. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1429–G1438. doi: 10.1152/ajpgi.00315.2006. [DOI] [PubMed] [Google Scholar]
- Ohta Y, Hayashi M, Kanemaru T, Abe K, Ito Y, Oike M. Dual modulation of airway smooth muscle contraction by Th2 cytokines via matrix metalloproteinase-1 production. J Immunol. 2008;180:4191–4199. doi: 10.4049/jimmunol.180.6.4191. [DOI] [PubMed] [Google Scholar]
- Ordas I, Eckmann L, Talamini M, Baumgart DC, Sandborn WJ. Ulcerative colitis. Lancet. 2012;380:1606–1619. doi: 10.1016/S0140-6736(12)60150-0. [DOI] [PubMed] [Google Scholar]
- Ostanin DV, Bao J, Koboziev I, Gray L, Robinson-Jackson SA, Kosloski-Davidson M, et al. T cell transfer model of chronic colitis: concepts, considerations, and tricks of the trade. Am J Physiol Gastrointest Liver Physiol. 2009;296(2):G135–G146. doi: 10.1152/ajpgi.90462.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehal S, Blanckaert P, Roizes S, von der Weid PY. Characterization of biosynthesis and modes of action of prostaglandin E2 and prostacyclin in guinea pig mesenteric lymphatic vessels. Br J Pharmacol. 2009;158:1961–1970. doi: 10.1111/j.1476-5381.2009.00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Karpinski E, Benishin CG. Prostaglandin E2 contracts vascular smooth muscle and inhibits potassium currents in vascular smooth muscle cells of rat tail artery. J Pharmacol Exp Ther. 1995;275:710–719. [PubMed] [Google Scholar]
- Ristimaki A, Narko K, Enholm B, Joukov V, Alitalo K. Proinflammatory cytokines regulate expression of the lymphatic endothelial mitogen vascular endothelial growth factor-C. J Biol Chem. 1998;273:8413–8418. doi: 10.1074/jbc.273.14.8413. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RO. Lymphatic contractility: a possible mechanism of lymphatic vessels for the transport of lymph. J Exp Med. 1949;90:497–509. doi: 10.1084/jem.90.5.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprague AH, Khalil RA. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem Pharmacol. 2009;78:539–552. doi: 10.1016/j.bcp.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Kruiningen HJ, Colombel JF. The forgotten role of lymphangitis in Crohn's disease. Gut. 2008;57:1–4. doi: 10.1136/gut.2007.123166. [DOI] [PubMed] [Google Scholar]
- Vigano T, Habib A, Hernandez A, Bonazzi A, Boraschi D, Lebret M, et al. Cyclooxygenase-2 and synthesis of PGE2 in human bronchial smooth-muscle cells. Am J Respir Crit Care Med. 1997;155:864–868. doi: 10.1164/ajrccm.155.3.9117018. [DOI] [PubMed] [Google Scholar]
- von der Weid PY, Muthuchamy M. Regulatory mechanisms in lymphatic vessel contraction under normal and inflammatory conditions. Pathophysiology. 2010;17:263–276. doi: 10.1016/j.pathophys.2009.10.005. [DOI] [PubMed] [Google Scholar]
- von der Weid PY, Rehal S. Lymphatic pump function in the inflamed gut. Ann N Y Acad Sci. 2010;1207(Suppl. 1):E69–E74. doi: 10.1111/j.1749-6632.2010.05715.x. [DOI] [PubMed] [Google Scholar]
- von der Weid PY, Rehal S, Ferraz JG. Role of the lymphatic system in the pathogenesis of Crohn's disease. Curr Opin Gastroenterol. 2011;27:335–341. doi: 10.1097/MOG.0b013e3283476e8f. [DOI] [PubMed] [Google Scholar]
- Watari K, Nakao S, Fotovati A, Basaki Y, Hosoi F, Bereczky B, et al. Role of macrophages in inflammatory lymphangiogenesis: enhanced production of vascular endothelial growth factor C and D through NF-kappaB activation. Biochem Biophys Res Commun. 2008;377:826–831. doi: 10.1016/j.bbrc.2008.10.077. [DOI] [PubMed] [Google Scholar]
- Wilson RJ, Giblin GM, Roomans S, Rhodes SA, Cartwright KA, Shield VJ, et al. GW627368X ((N-{2-[4-(4,9-diethoxy-1-oxo-1,3-dihydro-2H-benzo[f]isoindol-2-yl)phenyl]acetyl} benzene sulphonamide): a novel, potent and selective prostanoid EP4 receptor antagonist. Br J Pharmacol. 2006;148:326–339. doi: 10.1038/sj.bjp.0706726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TF, Carati CJ, Macnaughton WK, von der Weid PY. Contractile activity of lymphatic vessels is altered in the TNBS model of guinea pig ileitis. Am J Physiol Gastrointest Liver Physiol. 2006;291:G566–G574. doi: 10.1152/ajpgi.00058.2006. [DOI] [PubMed] [Google Scholar]
- Zawieja DC, Davis KL, Schuster R, Hinds WM, Granger HJ. Distribution, propagation, and coordination of contractile activity in lymphatics. Am J Physiol Heart Circ. 1993;264(4 Pt 2):H1283–H1291. doi: 10.1152/ajpheart.1993.264.4.H1283. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 PGE2 decreased rat mesenteric lymphatic muscle cell (RMLMC) contractility resistant to Anakinra treatment. RMLMC treated with PGE2 (10−5 M) for 4 days showed a significantly lower contractility (expressed in % of contraction vs. control) compared with control-treated RMLMC, while Anakinra (5 μg·mL−1) treatment showed no significant effect on the PGE2-induced relaxation. n = 4, **P < 0.01 versus control, one-way anova, with Bonferroni's post hoc test; data are mean ± SEM.
Figure S2 Different doses of Anakinra restored IL-1β-induced decreased rat mesenteric lymphatic muscle cell (RMLMC) contractility. RMLMC treated with IL-1β (20 ng·mL−1) for 4 days showed a significantly lower contractility (expressed in % of contraction vs. control) compared with control-treated RMLMC while co-treatment with Anakinra (0.5, 5, 50 μg·mL−1) restored RMLMC relaxation. n = 5, ***P < 0.001 versus control, one-way anova, with Bonferroni's post hoc test; data are mean ± SEM.
Figure S3 IL-1β + TNF-α exhibit synergistic effects in decreasing RMLMC contractility. IL-1β (20 ng·mL−1), but not TNF-α (20 ng·mL−1), decreased RMLMC contractility (expressed in % of contraction vs. control) while IL-1β (20 ng·mL−1) + TNF-α (20 ng·mL−1) decrease RMLMC contractility to a greater extent than IL-1β (20 ng·mL−1) alone. IL-1 receptor antagonist (Anakinra 5 μg·mL−1) maintained RMLMC contractility in the presence of IL-1β or IL-1β + TNF-α at day 4. n = 4, ***P < 0.001 versus control, ***P < 0.01 IL-1β (20 ng·mL−1) versus IL-1β (20 ng·mL−1) + TNF-α (20 ng·mL−1). One-way anova, with Bonferroni's post hoc test; data are mean ± SEM.
Figure S4 Equation for gel contraction. exp. represents the values of the treated gels, whereas con. represents the values for the untreated internal control gels. t0 = day 0, t4 = day 4. As we observed slightly different contractile response within our control groups, an own control for each single experimental treatment was set. These individual controls were set as 100% and the results of parallel conducted treatment studies were normalized to this internal control. When, for example, the control group produced a 70% contraction and a treatment a 35% contraction, then the data are presented as 100% contraction for control and 50% contraction for the treatment group.