

Abstract
Background and Purpose
6R-L-erythro-5,6,7,8-tetrahydrobiopterin (BH4) is an essential cofactor for nitric oxide biosynthesis. Substantial clinical evidence indicates that intravenous BH4 restores vascular function in patients. Unfortunately, oral BH4 has limited efficacy. Therefore, orally bioavailable pharmacological activators of endogenous BH4 biosynthesis hold significant therapeutic potential. GTP-cyclohydrolase 1 (GCH1), the rate limiting enzyme in BH4 synthesis, forms a protein complex with GCH1 feedback regulatory protein (GFRP). This complex is subject to allosteric feed-forward activation by L-phenylalanine (L-phe). We investigated the effects of L-phe on the biophysical interactions of GCH1 and GFRP and its potential to alter BH4 levels in vivo.
Experimental Approach
Detailed characterization of GCH1–GFRP protein–protein interactions were performed using surface plasmon resonance (SPR) with or without L-phe. Effects on systemic and vascular BH4 biosynthesis in vivo were investigated following L-phe treatment (100 mg·kg−1, p.o.).
Key Results
GCH1 and GFRP proteins interacted in the absence of known ligands or substrate but the presence of L-phe doubled maximal binding and enhanced binding affinity eightfold. Furthermore, the complex displayed very slow association and dissociation rates. In vivo, L-phe challenge induced a sustained elevation of aortic BH4, an effect absent in GCH1(fl/fl)-Tie2Cre mice.
Conclusions and Implications
Biophysical data indicate that GCH1 and GFRP are constitutively bound. In vivo, data demonstrated that L-phe elevated vascular BH4 in an endothelial GCH1 dependent manner. Pharmacological agents which mimic the allosteric effects of L-phe on the GCH1–GFRP complex have the potential to elevate endothelial BH4 biosynthesis for numerous cardiovascular disorders.
Table of Links
| TARGETS |
|---|
| Enzymes |
| NOS, nitric oxide synthase |
| LIGANDS |
|---|
| GTP |
| L-phe, L-phenylalanine |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
GTP-cyclohydrolase 1 (GCH1) catalyses the committing and rate limiting step in the production of 6R-L-erythro-5,6,7,8-tetrahydrobiopterin (BH4), an essential cofactor for aromatic amino acid hydroxylases (Kaufman, 1963), nitric oxide synthase (NOS) (Tayeh and Marletta, 1989) and alkylglycerol mono-oxygenase (Watschinger et al., 2010). The products of these enzymes have widespread functions (Thöny et al., 2000) and hence GCH1, via its control of BH4 biosynthesis, regulates a number of diverse physiological systems.
Numerous clinical studies have shown that intravascular BH4 administration, at high concentrations, significantly improves endothelial function in patients with a wide range of cardiovascular disorders, by increasing nitric oxide (NO) bioavailability and/or reducing oxidative stress (Heitzer et al., 2000; Higashi et al., 2002; Wyss et al., 2005; Mayahi et al., 2007). Unfortunately, BH4 is very unstable and, when orally administered, has limited efficacy likely due to oxidation during absorption (Cunnington et al., 2012). Therefore, other approaches to directly raise endogenous BH4 biosynthesis hold therapeutic potential – one such approach involves the pharmacological activation of GCH1, within the vascular endothelial cells themselves.
Interestingly, mammals have evolved an endogenous system to dynamically regulate GCH1 activity, whereby the enzyme's activity can be inhibited by BH4 (via end product feedback inhibition), or activated by L-phenylalanine (L-phe). However, this dynamic post-translational regulation only occurs when GCH1 is bound to GCH1 feedback regulatory protein (GFRP) (Harada et al., 1993). In vitro studies have demonstrated that when GCH1 and GFRP are bound, BH4 mediates allosteric feedback inhibition of GCH1, in a non-competitive manner, whilst L-phe can reverse this effect – stimulating GCH1 activity (Harada et al., 1993; Maita et al., 2004). Importantly, this allosteric regulation of GCH1 activity has an absolute requirement for GFRP, as BH4 and L-phe are unable to influence the activity of purified GCH1 protein in isolation (Harada et al., 1993).
These feedback and feed-forward mechanisms ensure that BH4 is kept within a tight physiological range in the body. As BH4 is an essential cofactor for the metabolism of dietary L-phe by phenylalanine hydroxylase, the L-phe mediated feed-forward activation of GCH1 raises endogenous BH4 and ensures that dietary L-phe is adequately metabolized. This is clinically important as L-phe can be neurotoxic above a certain concentration, leading to irreversible mental disability. Indeed, BH4 supplementation is currently used to treat a subset of L-phenylketonuria (PKU) patients who are unable to metabolize L-phe (Heintz et al., 2013). The differential efficacy of oral BH4 supplementation between coronary artery disease patents and those with PKU may be explained by the presence of enhanced oxidative stress, which subsequently leads to oxidative inactivation of BH4 during absorption in the former, but not the latter, patient group (Cunnington et al., 2012).
The presence of a functional GCH1–GFRP complex has been demonstrated in humans, whereby oral administration of L-phe elicits an increase in plasma biopterin (a surrogate marker of BH4) – an effect that is attenuated in patients carrying a loss of function GCH1 mutation (Saunders-Pullman et al., 2004). However, the effects of L-phe on biopterin and, more importantly, BH4 levels in tissues have not been directly determined.
The crystal structure of the GCH1–GFRP complex has been solved, revealing GCH1 as a homodecamer (∼280 kDa) sandwiched between two GFRP homopentamers (∼50 kDa). These crystal structures revealed discrete binding pockets for L-phe and BH4 located at the GCH1–GFRP interface and distinct from the GTP binding site (Maita et al., 2002; 2004,), making these unique and rational drug targets to either enhance or limit BH4 biosynthesis respectively. Upon binding to their respective pockets at the GCH1–GFRP interface, BH4 and L-phe induce conformational changes in the remote GTP substrate binding pocket, impeding or facilitating GTP binding respectively (Maita et al., 2002; 2004,). However, to date, crystallization studies have been limited to a truncated form of mammalian GCH1, lacking the first 42 amino acids due to N-terminal instability (Auerbach et al., 2000; Maita et al., 2002; 2004,). Whilst these studies stated that this N-terminal region did not influence GFRP binding, feedback regulation or GCH1 activity, other studies have contradicted these findings, suggesting that the absence of a complete N-terminal region can alter GCH1 activity and further limits the capacity of GCH1 to bind to GFRP (Swick and Kapatos, 2006; Higgins and Gross, 2011).
We hypothesized that drugs which mimic the allosteric effects of L-phe on the GCH1–GFRP complex have the potential to elevate BH4 within vascular cells and restore endothelial function in numerous cardiovascular disorders, circumventing the limitations of oral BH4 treatment. However, to facilitate a rational drug discovery approach, a greater understanding of the GCH1 : GFRP complex and the potential limitations of the current crystal structures (which used an N-terminal truncation mutant) is required. Furthermore, in vitro and in vivo proof of concept studies validating the GCH1–GFRP axis as a tangible drug target to regulate endothelial BH4 are also lacking.
Therefore, in this study, we have quantified GCH1–GFRP protein interactions using surface plasmon resonance (SPR), comparing native/full length (F-GCH1) and truncated GCH1 (T-GCH1) binding to GFRP. Additional studies evaluated the impact of L-phe on GCH1–GFRP interactions. Furthermore, whilst L-phe is known to raise plasma biopterin in human plasma, little is known of the impact of GCH1-GFRP stimulation in vascular cells and tissues. We therefore undertook in vitro studies establishing the effects of L-phe administration on BH4, nitric oxide levels and superoxide anion levels, in cultured endothelial cells. Furthermore, we investigated the in vivo impact of oral L-phe challenge on aortic and systemic biopterin and BH4 levels in both wild-type mice and in mice lacking endothelial GCH1 [GCH1(fl/fl)-Tie2Cre] (Chuaiphichai et al., 2014).
Methods
Cloning and construct formation
Human liver mRNA (AMS Biotechnology UK Ltd: M1234149) was reverse transcribed using the GE Healthcare First Strand cDNA Synthesis Kit. DNA encoding native GFRP, F-GCH1 and T-GCH1 (lacking the first 42 N-terminal amino acids) was amplified from the cDNA by PCR using KOD hot start polymerase (VWR International, Lutterworth, UK). PCR products (GFRP, F-GCH1 and T-GCH1) were individually purified, digested and ligated into the dual expression vector pDuet-1 encoding an N-terminal His6-tag in MCS1, using appropriate restriction enzymes. MCS2 was used for co-expression of non-tagged proteins. All PCR primers encoded a TEV cleavage site for removal of the His6 tag after protein purification (Table 2013). Competent Escherichia coli strains BL21 (DE3) and Rosetta were transformed with ligation mixtures. DNA sequencing authenticated the clones.
Table 1.
Construct details
| Construct | Vector and multiple cloning site | Restriction enzyme ± TEV site | Primers (5′–3′) |
|---|---|---|---|
| T-GCH1 (FWD) | pET-Duet MCS 1 | Sal1-TEV | GATCGTCGACGAAAACCTGTACTTCCA |
| AGGAGAGGCCAAGAGCGCGCAGCCC | |||
| T-GCH1 (REV) | pET-Duet MCS 1 | Not1 | CTGATAGCGGCCGCTCAGCTCCTAATGA |
| GCGTCAGGAA | |||
| F-GCH1 (FWD) | pET-Duet MCS 1 | Sal 1-TEV | GATCGTCGACGAAAACCTGTACTTCCA |
| AGAAATGGAGAGAAGGGCCCTGTGCG | |||
| GGCACCGGCGGAG | |||
| F-GCH1 (RVS) | pET-Duet MCS 1 | Not1 | CTGATAGCGGCCGCTCAGCTCCTAATGA |
| GCGTCAGGAA | |||
| GFRP (FWD) | pET-Duet MCS 1 | Sal1-TEV | GATCGTCGACGAAAACCTGTACTTCCA |
| AGGACCCTACCTGCTCATCAGCACCCA | |||
| GATC | |||
| GFRP (RVS) | pET-Duet MCS 1 | Not1 | CTGATAGCGGCCGCTCACTCCTTGTGCA |
| GACACCACACCAGCGTCTG | |||
| GFRP (FWD) | pET-Duet MCS 2 | Fse1 | CTGATA GGCCGGCC CCC TAC CTG CTC ATC AGC ACC CAG ATC |
| GFRP (RVS) | pET-Duet MCS 2 | Kpn1 | GATC GGTACC TCA CTC CTT GTG CAG ACA CCA CAC CAG CGT CTG |
Each construct was generated by PCR amplification with corresponding oligonucleotide primers, as listed. A TEV site was incorporated for removal of the His6 tag. T-GCH1 (truncated mutant lacking the first N-terminal 42 amino acids).
F-GCH1, native/full length GCH1; GFRP, GCH1 feedback regulatory protein; T-GCH1, truncated GCH1.
Protein expression and purification
Overnight bacterial cultures were induced using 1 mM isopropyl β-D-1-thiogalactopyranoside at 25°C for 12 h. His6-tagged proteins, either alone or bound to non-tagged co-expressed proteins, were purified using Talon cell-thru metal affinity resin (Takara-Bio Europe/Clontech, Saint-Germain-En-Laye, France). For SPR experiments, purified proteins were incubated with TEV protease at 30°C overnight to cleave the His-tag, and then co-incubated with the affinity resin (4 h) to remove the cleaved His-tag. Finally, protein samples were run through a size exclusion chromatography Superdex column (GE Healthcare Ltd, Buckinghamshire, UK).
Proteins were identified by SDS-PAGE and Western blotting using either a polyclonal primary GCH1 anti-peptide antibody (raised against amino acid residues 18–45) (Nandi et al., 2005), a commercial primary GFRP antibody (Santa Cruz Biotechnologies, Santa Cruz, CA) or a His-tag antibody (Abcam, Cambridgeshire, UK). Purified protein samples were combined and concentrated in 100 mM Tris pH 7.8, 100 mM NaCl or 50 mM HEPES pH 7.8, 100 mM KCl.
GCH1 activity (HPLC)
GCH1 activity was assessed by HPLC as previously described (Howells et al., 1986), whereby neopterin content was quantified by isocratic HPLC and fluorescence detection. Quantification of neopterin was carried out by comparison with external standards and was normalized for sample protein content.
GCH1 activity kinetic microplate assay
An established kinetic microplate assay was modified and used to measure GCH1 activity in expressed proteins, in addition to HPLC (Kolinsky and Gross, 2004). This assay measures the accumulation of the intermediate reaction product, dihydroneopterin triphosphate (H2NTP), by monitoring an increase in A340 over time. The assay was set up in a 96-well plate format as follows: 0.25 μM GCH1 protein (T or F) was combined with assay buffer (100 mM Tris–HCl to a final volume of 300 μL) and 100 μM GTP (Thermo Fisher Scientific, Hemel Hempstead, UK). Purified GFRP protein (1 μM) was added in certain experiments. Absorbance (340 nm) was measured using a Spectramax temperature controlled plate reader (Molecular Devices Ltd, Wokingham, UK) at 37°C until the reaction reached saturation. Absorbance units were expressed in mol H2NTP as previously described (Kolinsky and Gross, 2004).
SPR
A Biacore™ T200 was used to conduct SPR experiments. F-GCH1, T-GCH1 or GFRP proteins obtained from single expression vectors were captured on a CM5 sensor chip surface using an anti-His-tag antibody (Biacore His-capture Kit; GE Healthcare Ltd). The sensor chip was activated using a 1:1 mixture of 50 mM N-hydroxysuccinimide and 200 mM of N-ethyl-N′-(diethylaminopropyl) carbodiimide. This was injected across two flow cells simultaneously, with the second flow cell acting as a control surface to identify any non-specific binding. A 50 μg·mL−1 anti-His-tag antibody solution was injected over the experimental flow cell and both experimental and control surfaces were subsequently quenched with 1 M ethanolamine HCl (pH 8.5). His-tagged protein (100 nM) in running buffer (50 mM HEPES pH 7.4, 150 mM NaCl, 3 mM EDTA, and 0.005% (v/v) surfactant P20) was injected across both flow cells, allowing the protein to be captured by the anti-His-tag antibody immobilized on the surface of the experimental flow cell. Experiments were performed with F-GCH1 or T-GCH1 captured on a CM5 surface (with GFRP as the analyte), and reciprocally with GFRP captured on the surface (with either T-GCH1 or F-GCH1 as the analyte). For the analyte proteins, the His-tag was cleaved and the protein incorporated into the running buffer over a range of concentrations (100–2000 nM). Analyte proteins were injected across both flow cells. The surface was regenerated using an injection of 10 mM glycine (pH 1.5) following each analyte cycle. In separate experiments, L-phe (100 μM) or BH4 (20 μM) was introduced along with the analyte protein into the running buffer. Flow rates were adjusted accordingly to enable equilibration (flow rate of 3 μL·min−1 for 7000 s). All SPR assays were conducted at 25°C. Surface density after ligand immobilization was maintained at 1250–1500 RU for all experiments. Dose-dependent association experiments and binding characterization experiments were repeated four times.
In vitro and vivo studies
Animal welfare and ethical statement
All animals studies described in this paper were conducted following ethics approval and in accordance with the UK Home Office Animals (Scientific Procedures) Act 1986 [Amendment Regulations (SI 2012/3039)]. Experimental design and conduct were undertaken in accord with the ARRIVE guidelines (Kilkenny et al., 2010) and complied with The Basel Declaration and the Concordat on Openness on Animal Research. All techniques used for in vivo studies were as humane as possible. A total of 80 animals were used in the experiments described here.
L-phe challenge in wild-type and GCH1(fl/fl)-Tie2Cre mice
Three groups of mice were used for in vivo studies: (i) male C57BL/6 mice (12–14 weeks old), purchased from a commercial supplier (Harlan Laboratories, Loughborough, UK); (ii) GCH1(fl/fl)-Tie2Cre (KO) mice and (iii) GCH1(fl/fl) mice – hereafter referred to as wild-type littermates – generated by crossing male GCH1(fl/fl)-Tie2cre and female GCH1(fl/fl) mice (Chuaiphichai et al., 2014). All animals were group housed with 12 h light/dark cycle and controlled temperature of 20–22°C and given access to a standard chow diet and water ad libitum. GCH1(fl/fl)-Tie2Cre mice have previously been shown to lack GCH1 in endothelial cells (Chuaiphichai et al., 2014).
L-phe (100 mg·kg−1) in saline was administered orally (by gavage) to all three groups of mice. This dose is equivalent to ∼200 g of beef in a healthy 70 kg adult (Uhe et al., 1997) and matches the dose used in L-phe loading studies in PKU patients (Saunders-Pullman et al., 2004). Mice were killed, by exsangination under isoflourane anaesthesia, after 20 min, 1, 2, 4 or 8 h. Plasma and aortic tissues were collected and snap frozen for subsequent analysis of L-phe, biopterin and BH4 levels. L-phe levels were measured using HPLC detection in plasma and tissue homogenates following sample preparation as described previously (Atherton and Green, 1988). Quantification of L-phe was performed by comparison with external standards (0–250 mmol·L−1) and the lower limit of detection was 2.7 μM. Values were normalized for protein content using a standard Bradford assay.
L-phe challenge in sEnd1 endothelial cells
sEnd-1 cells (a stable murine endothelial cell line) were cultured in DMEM as previously described (Nandi et al., 2008) and used between passages 3 and 7 for all experiments. Human modified oxidized lipoprotein (RP-048 – Source BioScience Life Sciences, Nottingham, UK) (100 μg·mL−1) (Feldmann et al., 2013) was incubated with cells for 2 h to induce oxidative stress (Bowers et al., 2011). L-Phe (500 μM; 0.5 h) or vehicle control was subsequently added to cells, and the impact on nitrite accumulation (a correlate of nitric oxide) in the media and BH4 in cell lysates was assessed.
BH4 measurement
Biopterin and BH4 were measured in cell lysates, tissue and plasma, as previously described using fluorescence and electrochemical detection following sample separation by HPLC (Howells et al., 1986; Starr et al., 2014). Quantification was performed by comparison with external standards after normalizing for sample protein content. All analyses were conducted in a blinded fashion and investigators were unblinded to treatment/genotype following completion of data analysis.
Nitrite measurement
Plasma or tissue homogenates were deproteinated and nitrite content was then quantified using a fluorometric method utilizing 2,3-diaminonaphthalene (Bryan and Grisham, 2007). The amount of nitrite in each sample, expressed as micromolar nitrite·per milligram protein was calculated from a linear calibration curve of known nitrite concentrations (linear range: 0.5–5 μM) and normalized for total amount of protein.
Chemiluminescent measurement of superoxide anion
Superoxide levels were quantified from sEnd1 cells using a Lucigenin chemiluminescence-based assay (Li and Shah, 2001). Briefly, cells were seeded at equal density in a 96-well microplate luminometer (Model Lucy 1, Rosys Anthos, Austria) and pre-treated with human modified oxidized lipoprotein ± L-phe as described above. Media were removed and replaced by 100 μL Krebs solution (119 mM NaCl, 4.7 mM KCl, 1.5 mM MgSO4, 2.5 mM CaCl2, 25 mM NaHCO3, 1.2 mM KH2PO4, 11 mM glucose and 100 μM L-arginine), pH 7.4, and were kept cold on ice. Immediately before recording chemiluminescence, NADPH (final concentration 100 μmol·L−1) and lucigenin, bis-N-methylacridinium nitrate (10 μmol·L−1) was added to tissues and superoxide dismutase (SOD, 200 units·mL−1) was used as a positive control. Light emission was recorded as mean arbitrary light units/cycle over 60 min.
Data analysis
SPR data were analysed using the curve fitting software Origin 7.0 (OriginLab Corporation, Northampton, MA, USA) and Biaevaluation software to determine the kon and koff rate constants and binding parameters, using both first and second order kinetic models. Bmax calculations were normalized for surface density when this differed between experiments. A global fitting approach using the Biaevaluation software was not adequate to fully describe and fit the binding curves. Therefore, individual curve fitting was conducted in order to calculate binding parameters and rate constants (Supporting Information Fig. S2). The representative data shown in the results (Figure 2) were best described using first-order kinetics; hence the values were determined using monophasic fits.
Figure 2.

Surface plasmon resonance sensorgrams and tabulated data for His-tag captured native/full length GCH1 (F-GCH1) or truncated GCH1 (T-GCH1) interacting with GCH1 feedback regulatory protein (GFRP) analyte in the absence and presence of L-phenylalanin (L-phe). (A) Representative sensorgrams comparing F-GCH1–GFRP binding curves (left) and T-GCH1–GFRP binding curves (right). GTP-cyclohydrolase 1 (GCH1) is captured at a surface density of ∼1300 RU via His-capture, and GFRP is introduced in varying concentrations (12.5–400 nM) with tabulated first-order dissociation constants (KD) and on- and off-rate constants (n = 4). (B) Representative sensorgrams for GFRP binding to F-GCH1 in the presence of L-phe. GCH1 immobilized at a surface density of ∼1500 RU via His-capture, and GFRP and L-phe are introduced in varying concentrations (12.5–400 nM), with tabulated first-order dissociation constants (KD) and on- and off-rate constants are tabulated (n = 4). (C) Comparison of binding kinetics; binding profiles for T-GCH1 and F-GCH1 in the presence and absence of ligands with 400 nM GFRP; F-GCH1 + GFRP; T-GCH1 + GFRP; F-GCH1 + GFRP + L-phe.
For HPLC based assays, data are presented as mean ± SEM (where n = number of animals). One-way analysis of variance was used to analyse data obtained from HPLC-based assays measuring BH4, biopterin, nitrite and superoxide anion levels in vascular tissues, plasma, cells and media. For the real-time kinetic assays, data are presented as mean ± SEM (where n = number of cell pellets from individually grown cultures). Two-way analysis of variance was used to analyse activity data from kinetic assays. GraphPad Prism 5 (GraphPad Software Inc, La Jolla, CA, USA) was employed to analyse all assay data.
Materials
Bacterial culture reagents, plasmid vectors and competent cells were purchased from Novagen, VWR International. Oligonucleotide primers were synthesized by Sigma-Aldrich (Dorset, UK). All other reagents were purchased from Sigma-Aldrich unless otherwise stated.
Results
Expression and activity of native/full length and T-GCH1 with GFRP
Soluble human recombinant T-GCH1, F-GCH1 and GFRP proteins were successfully expressed individually in BL21 (GCH1) or Rosetta (GFRP) cells (Figure 1A). In dual expression cultures, metal affinity purification of His6T-GCH1 or His6F-GCH1 revealed that GFRP was able to bind to both forms of GCH1 and could be co-isolated (Figure 1B).
Figure 1.

Expression and activity of human recombinant native/full length GCH1 (F-GCH1), truncated GCH1 (T-GCH1) and GTP cyclohydrolase 1 (GCH1) feedback regulatory protein (GFRP). (A) F-GCH1 expression in uninduced (lane 1) and IPTG induced (lane 2) cells. T-GCH1 expression from IPTG induced (lanes 3 and 4) cells. GFRP expression in uninduced (lane 5) and IPTG (isopropyl β-D-1-thiogalactopyranoside) induced (lanes 6 and 7) cells. Products were resolved on a 4–15% SDS-PAGE gradient gel. (B) Elution of purified proteins from dual expression cultures: His-T-GCH1 and GFRP bands are observed at ∼25 and ∼10 kDa, respectively; His-F-GCH1–GFRP bands observed at ∼28 and ∼10 kDa respectively. (C) (i): Immunoreactive bands for GFRP at ∼10 kDa were observed using a commercially available GFRP antibody; (ii) immunoreactive bands for both GFRP and T-GCH1 using a commercially available His-tag antibody were observed at ∼10 and ∼24 kDa, respectively; (iii) immunoreactive band at ∼28 kDa for F-GCH1 using an N-terminal GCH1 antibody; no immunoreactive bands for T-GCH1. (D) Native 5% PAGE gels run prior to surface plasmon resonance (SPR) analysis. Predominant bands highlighted for F-GCH1, T-GCH1 and GFRP as indicated, from two independent experiments. (E) Activity of purified F-GCH1 and T-GCH1 was quantified by HPLC detecting neopterin production; mouse liver homogenate and empty vector control were used for comparison (n = 6, mean ± SEM). *P < 0.05, ***P < 0.001, significantly different from control. (F) GCH1 activity measured by microplate assay in the presence of 250 nM GCH1, 1 μM GFRP and 100 μM GTP (n = 10, mean ± SEM). ***P < 0.01, significantly different from F-GCH1 without GFRP).
Western blotting using specific antibodies revealed immunoreactive bands for GFRP and all His-tagged proteins (Figure 1C, i and ii). The GCH1 antibody (which recognizes an N-terminal epitope) correctly identified native but not T-GCH1 (Figure 1C, iii), confirming an intact N-terminal region on native GCH1 and demonstrating that the two forms of GCH1 could be readily distinguished from one another.
Activity assays and native PAGE gels run immediately before SPR analysis, demonstrating that proteins were predominantly in an oligomeric form (Figure 1D–F) (Maita et al., 2001). Consistent with previous reports (Higgins and Gross, 2011), F-GCH1 displayed lower levels of activity compared with T-GCH1 (Figure 1E), an effect that was reversed when F-GCH1 was co-incubated with GFRP (Figure 1F)
Protein–protein interactions between native or T-GCH1 with GFRP
Representative binding profiles illustrate differences between GFRP binding to F-GCH1 (Figure 2A, left) or T-GCH1 (Figure 2A, right). Binding parameters and determined kon and koff values are listed in the associated table. Binding was observed between both forms of GCH1 with GFRP, in the absence of additional L-phe, BH4 or GTP. The analysis yielded KD values of 8 nM for F-GCH1 : GFRP and 17 nM for T-GCH1 : GFRP in the absence of ligands (tabulated in Figure 2A).
Effects of L-phe on F-GCH1–GFRP protein–protein interactions
L-phe changed both the association and dissociation rate constants with both forms of GCH1 and GFRP resulting in an eight-fold increase in binding affinity for F-GCH1–GFRP interactions: 1 nM KD, and 10-fold increase in binding affinity for T-GCH1–GFRP interactions: 1.7 nM KD. Representative sensorgrams for F-GCH1–GFRP + L-Phe (Figure 2B); T-GCH1–GFRP + L-Phe (Supporting Information Fig. S1) and corresponding values for F-GCH1 and T-GCH1 are summarized in the associated table (Figure 2B). In addition to these findings, a clear, two-fold rise in maximal binding (Bmax) was observed in F-GCH1–GFRP interactions in the presence of L-phe. A comparison of SPR derived data and binding kinetics are summarized in Figure 2C
The effect of BH4 (20 μM) on GCH1–GFRP interactions was also investigated, revealing increased binding affinity in its presence but no distinguishable differences between T-GCH1–GFRP and F-GCH1–GFRP interactions (Supporting Information Table S1).
Effects of L-phe challenge on Ox-LDL pre-treated endothelial cells
Addition of human modified oxidized lipoprotein to endothelial cells led to a significant elevation of superoxide anion (Figure 3A), reduction in nitrite (Figure 3B) and reduction in BH4 (Figure 3C) compared to baseline and consistent with published observations (Bowers et al., 2011). Application of L-phe (500 μM, 30 min) restored superoxide, nitrite and BH4 towards baseline levels (Figure 3A–C).
Figure 3.
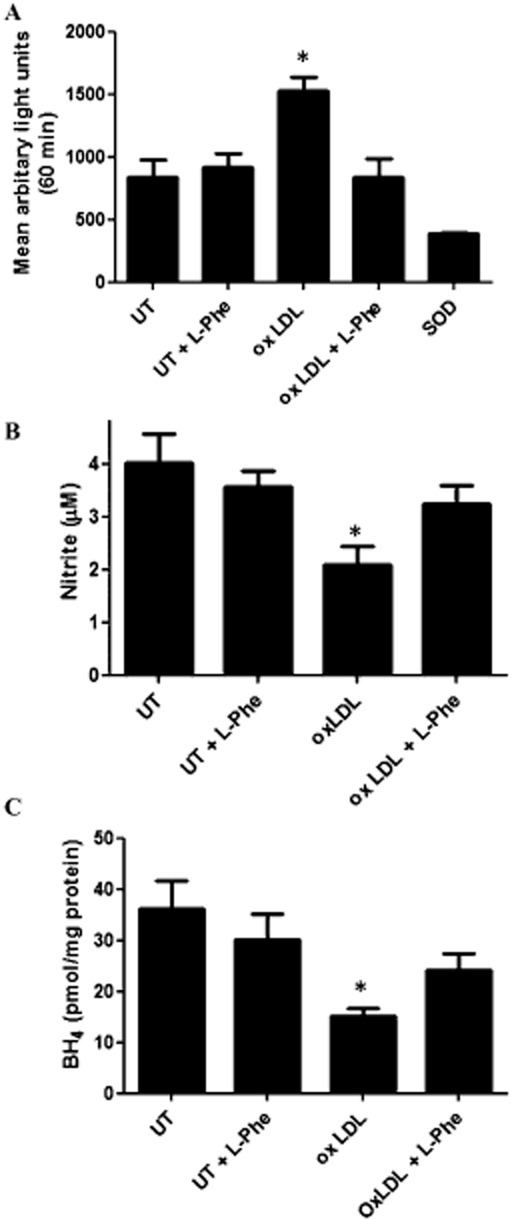
Effects of L-phenylalanine (L-phe) challenge on superoxide anion, nitrite and 6R-L-erythro-5,6,7,8-tetrahydrobiopterin (BH4) levels in cultured endothelial (sEnd1) cells in the presence and absence of human modified oxidized lipoprotein. (A) Cellular superoxide anion concentration as quantified by mean arbitrary lights units (lucigenin chemiluminescence) using superoxide dismutase as positive control, (B) nitrite accumulation in media, (C) cellular BH4 concentration, in sEnd 1 cells untreated (UT) or treated with Human modified oxidized lipoprotein (100 μM, 2 h) in the absence or presence of L-phe (500 μM, 0.5 h); n = 4–9, mean ± SEM. *P < 0.05, significantly different from untreated control).
Effects of oral L-phe challenge on biopterin and BH4 levels in vivo
Oral L-phe challenge (100 mg·kg−1 bolus) in C57BL/6 mice led to a peak plasma L-phe concentration at 20 min which normalized to baseline by 4 h (Figure 4A). Similarly, the peak increase in plasma biopterin was rapid and normalized by 8 h (Figure 4B). In contrast, biopterin in aortic tissue followed a different profile, showing a more gradual and continual rise over the 8 h period (Figure 4C). Importantly, the functionally important pterin, BH4 (which behaves as an NOS cofactor and has vaso-protective properties), was also significantly elevated in aorta 4 h after administration of L-phe, normalizing by 8 h (Figure 4D). Finally, whilst administration of L-phe to GCH1 wild-type littermates stimulated biopterin/BH4 production in a similar manner to that observed in commercially purchased C57BL/6 mice (Figure 4B, E and F), L-phe had no significant effect in mice lacking endothelial GCH1 [GCH1(fl/fl)-Tie2Cre] (Figure 4E and F). These data suggest that L-phe stimulates endothelial GCH1, leading to a rise in BH4 in the aorta.
Figure 4.
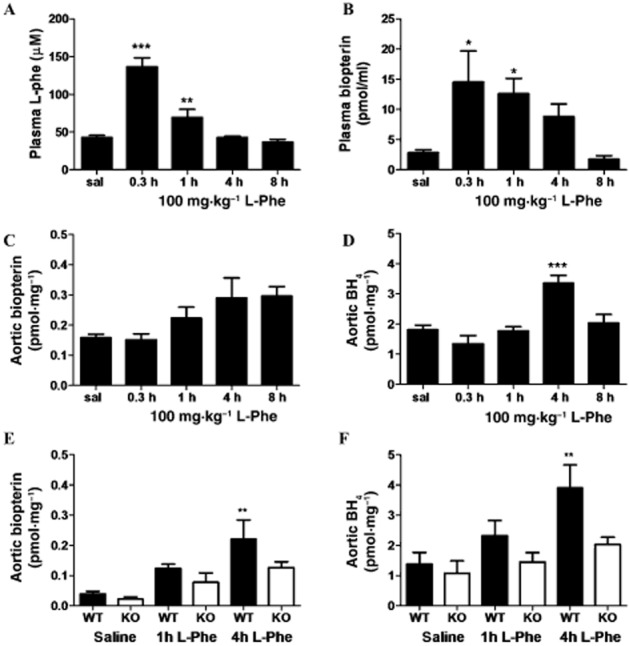
Effects of oral L-phe challenge on systemic and vascular biopterin levels in wild-type mice and GCH1(fl/fl)-Tie2Cre (KO) mice. (A) Plasma L-phenylalanine, (B) plasma total biopterins, (C) aortic total biopterins and (D) aortic 6R-L-erythro-5,6,7,8-tetrahydrobiopterin (BH4) levels detected by HPLC, over an 8 h time course, following 100 mg·kg−1 oral L-phe challenge in wild-type mice. n = 6–12 for plasma and n = 4–8 aorta, mean ± SEM. ***P < 0.001, **P < 0.01, *P < 0.05: significantly different from saline control. (E) Aortic total biopterins and (F) aortic BH4 levels in GCH1(fl/fl)-Tie2Cre (KO) mice and wild-type littermates, 1 and 4 h after 100 mg·kg−1 oral L-phe challenge. n = 6–8, mean ± SEM. **P < 0.01, significantly different from WT saline.
Discussion and conclusion
Protein interactions of native and truncated GCH1 with GFRP
Published structural studies of the GCH1–GFRP complex have used a truncated form of mammalian GCH1, lacking a large portion of the N-terminal region, suggesting that this region had no influence on either activity or binding to GFRP (Auerbach et al., 2000; Maita et al., 2002; 2004,). This suggestion has, however, been challenged, as the N-terminal region has been shown to modulate GCH1 activity and to be essential for GFRP binding (Swick and Kapatos, 2006; Higgins and Gross, 2011). Furthermore, previous studies have suggested that the known ligands, L-phe and BH4, or substrate, GTP, are an essential requirement for GCH1–GFRP binding (Harada et al., 1993; Yoneyama and Hatakeyama, 1998).
In contrast to these observations, using SPR, we have shown that T-GCH1 and F-GCH1 are able to bind to GFRP with nanomolar affinity, in the absence of known ligands and substrate. Furthermore, whilst the N-terminal region modestly enhanced the affinity of interaction with GFRP, high affinity interactions were still observed between T-GCH1 and GFRP. This ability of both native and truncated forms of GCH1 to interact with GFRP was independently confirmed using a GCH1–GFRP dual expression plasmid, where untagged GFRP was co-isolated with both His6-F-GCH1 and His6-T-GCH1. These findings are inconsistent with yeast two hybrid studies, in which N-terminal deletion diminished GCH1–GFRP interactions by 80% (Swick and Kapatos, 2006) or where the truncated enzyme displayed a relative inability to engage in higher ordered complexes with His6-GFRP (Higgins and Gross, 2011). The reasons for this discrepancy probably reflect differences in protein expression methodologies and the enhanced sensitivity of SPR to detect protein–protein interactions. A noticeable difference in maximal binding (Bmax) between F-GCH1–GFRP and TGCH1–GFRP was, however, detected, suggesting altered stoichiometry between the native and truncated complexes – a finding that requires further investigation but beyond the scope of the current study. Furthermore, absence of the N-terminal region conferred higher measured activity consistent with published observations (Higgins and Gross, 2011). It has been suggested that the N-terminal region may exert an auto-inhibitory effect leading to the observed activity changes mediated via a direct peptide bond, rather than a transient obstruction of the active site by a mobile N-terminal region (Higgins and Gross, 2011). Furthermore, instability of the full-length enzyme during purification has been reported by several groups (Yim and Brown, 1976; Auerbach et al., 2000), and it may equally be feasible that the enhanced activity of T-GCH1 can be, in part, attributed to greater T-GCH1 stability compared to F-GCH1 following purification. Together, our in vitro and biophysical data indicate that the GCH1 N-terminal region is not essential for GFRP binding, but that GCH1–GFRP binding kinetics and activity are altered when the N-terminal region is deleted – indicating a functionally important role for this region.
Effects of L-phe on GCH1–GFRP interactions
The interactions between GFRP and both forms of GCH1 were subsequently quantified in the presence of the allosteric effector molecule, L-phe. Whilst both forms of GCH1 were able to bind to GFRP in its absence, L-phe changed both the association and dissociation rate constants between both forms of GCH1 and GFRP, resulting in an 8- to 10-fold increase in binding affinity – an effect that was mimicked by another allosteric modulator, BH4. The increase in binding affinity induced by L-phe between the truncated and native forms of GCH1 with GFRP was indistinguishable. These changes in binding affinity are consistent with the structural changes reported in the stimulatory crystal structure of the rodent T-GCH1–GFRP complex (Maita et al., 2002).
For the physiologically expressed form (F-GCH1) a clear two-fold rise in maximal binding (Bmax) values was also observed upon the addition of L-phe, indicating a different stoichiometry to that previously suggested (Maita et al., 2002). This unexpected observation is, however, consistent with a previous report that native GCH1 binding to GFRP generates a very high molecular weight band which exceeds the size that would have been predicted for a GCH1 homodecamer bound to a GFRP pentamer (Higgins and Gross, 2011). However, further studies are required to fully understand this observation.
We thus demonstrate that SPR can readily detect the changes in GCH1–GFRP protein interactions, induced by small effector molecules such as L-phe. The very slow dissociation rates between GCH1 and GFRP observed using SPR indicate that within a physiological context, the two proteins would likely remain tightly complexed to one another.
L-phe challenge on BH4, nitric oxide and superoxide anion levels in vitro
We used an in vitro model of endothelial dysfunction and eNOS ‘uncoupling’ (Xie et al., 2012) to establish the effects of L-phe on BH4 and nitrite levels. Consistent with previous reports, incubation of endothelial cells with human oxidized lipoprotein led to a significant elevation of superoxide anion levels and concomitant reduction in nitrite and BH4. Incubation with L-phe reversed this effect, indicating that stimulation of endothelial GCH1 by L-phe analogues has the potential to reverse endothelial dysfunction.
L-phe challenge on biopterin and BH4 levels in vivo
A single 100 mg·kg−1 L-phe oral dose led to a significant increase in plasma and aortic biopterin/BH4 levels, in all wild-type mice – an effect that was not observed in aortas from GCH1(fl/fl)-Tie2Cre mice. Based on published evidence and data obtained in this study, we conclude that the raised biopterin/BH4 levels following oral L-phe administration occur via activation of the GCH1 : GFRP complex in endothelial cells (Harada et al., 1993; Saunders-Pullman et al., 2004).
Interestingly, aortic BH4 was still detectable in mice lacking endothelial GCH1, suggesting that cell types such as smooth muscle or adventitial fibroblasts may also generate BH4. Our findings are in agreement with the initial characterization of these genetically modified mice, which demonstrated that endothelial GCH1 gene deletion or endothelial denudation in wild-type mice reduced, but did not abolish, aortic BH4 levels (Chuaiphichai et al., 2014). Whilst we were unable to show a statistically significant difference in basal BH4 levels within aortic tissue between GCH1(fl/fl)-Tie2Cre mice and WT littermates, we did observe a trend reduction in the baseline state – the differences observed between this study and that of Chuaiphichai et al. (2014) are likely to reflect subtle differences in tissue dissection and preparation.
Importantly, in the present study, L-phe administration did not stimulate a significant rise in aortic BH4 in GCH1(fl/fl)-Tie2Cre mice, indicating that the GCH1–GFRP complex is primarily located in endothelial cells. As such, novel therapies activating the GCH1–GFRP axis are most likely to target this cell type. Indeed, the vascular endothelium is believed to be the primary source of BH4 (d'Uscio and Katusic, 2006), and previous studies have demonstrated that changes in GCH1–GFRP interactions are a critical regulator of BH4 and NO biosynthesis in endothelial cells, in response to laminar shear stress (Li et al., 2010).
The profiles of plasma L-phe and biopterin in mice mirrored that previously observed in humans challenged with oral L-phe (Saunders-Pullman et al., 2004), indicating that this is a suitable and clinically translatable model with which to investigate the GCH1–GFRP axis.
The observation that L-phe challenge stimulated aortic BH4 levels for at least 4 h is encouraging, as it suggests that the activation of the GCH1–GFRP complex elicits a sustained elevation of vaso-protective BH4 in target vascular tissues. These in vitro and in vivo findings thus provide mechanistic evidence to support published functional studies that have shown that L-phe administration restores endothelial function and attenuates the observed hypertension induced by administration of the GCH1 inhibitor di-amino-hydroxypyrimidine (Mitchell et al., 2004).
In conclusion, we have undertaken a detailed analysis of GCH1 and GFRP using complementary in vitro biophysical analysis with in vitro and in vivo murine studies. We have successfully expressed soluble human GCH1 and GFRP and, for the first time, quantified the binding rate constants between GCH1 and GFRP, using SPR. We have also demonstrated that the N-terminal region of GCH1 is not essential for GFRP to interact, but that deletion of this region alters the binding kinetics between the two proteins. Whilst GCH1 and GFRP were able to bind in the absence of known ligands, the presence of L-phe substantially elevated the maximal binding and the affinity of interaction – suggesting that, in an in vivo system (where ligands and substrate would be circulating), the two proteins would display high affinity interactions. Indeed, the rapid rise in plasma biopterin (observed within 20 min) coupled with slow GCH1–GFRP dissociation rates (obtained by SPR) supports the view that GCH1 and GFRP are likely to be constitutively bound in vivo, rather than binding in response to an acute elevation of circulating L-phe, following dietary intake.
Our biophysical and in vivo data suggest that the L-phe binding pockets on the GCH1–GFRP complex represent a rational drug target to raise vascular BH4, for the treatment of endothelial dysfunction. It is important to note that L-phe itself is not a feasible chronic therapeutic intervention due to its diverse physiological functions and role in catecholamine biosynthesis. However, given the marked allosteric changes induced by L-phe, low MW small molecule mimetics that alter interactions between GCH1 and GFRP in a similar manner have the potential to regulate intracellular BH4 for therapeutic purposes. Indeed, the sustained effect on aortic BH4 levels following a single oral L-phe challenge is highly encouraging from a therapeutic standpoint as this could elevate endothelial NO levels and/or limit damaging reactive oxygen species, circumventing the limited bioavailability/efficacy of oral BH4. Such an agent would have use in a wide spectrum of cardiovascular diseases, underpinned by reduced NO bioavailability and/or enhanced oxidative stress.
Acknowledgments
We would like to thank Dr. Mark Crabtree, Division of Cardiovascular Medicine, University of Oxford, for access to the HPLC detection systems for neopterin and biopterin measurements.
The work was funded by British Heart Foundation PG/09/073 and RG/12/5/29576, The Royal Society RG120151 and PhD studentship funding from the Ministry of Higher Education, Saudi Arabia.
Glossary
- BH4
6R-L-erythro-5,6,7,8-tetrahydrobiopterin
- Bmax
maximal binding
- F-GCH1
native/full length GCH1
- GCH1
GTP cyclohydrolase 1
- GFRP
GCH1 feedback regulatory protein
- KD
equilibrium dissociation constant
- koff
dissociation rate constant
- kon
association rate constant
- L-phe
L-phenylalanine
- NOS
nitric oxide synthase
- PKU
phenylketonurea
- SPR
surface plasmon resonance
- T-GCH1
truncated GCH1
Author contributions
M. N. designed the research study. D. H., A. S. and L. H. performed the research. E. M. and K. M. C. provided access to and advice on GCH1(fl/fl)-Tie2Cre mice. P. R. B. directed all cloning and protein expression studies. B. J. S. and J. M. M. directed and contributed to analysis of all SPR data. D. H. and M. N. wrote the manuscript. All authors contributed to editorial changes in the manuscript.
Conflict of interest
The authors declare no conflict of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 Binding profile for truncated GCH1 (T-GCH1) with GCH1 feedback regulatory protein (GFRP) in the presence of L-phenylalanine (L-phe). T-GCH1 is immobilized at a surface density of ∼1500 RU via His-capture, and GFRP and L-phe are introduced in varying concentrations (400–25 nM) (n = 4 for each experiment at varying immobilization patterns and surface densities). Representative sensorgrams are shown.
Figure S2 SPR data analysis by Origin software, curve fittings and residuals.
Table S1 Summary of the binding kinetics for both native/full-length GCH1 (F-GCH1) and GCH1 feedback regulatory protein (GFRP) as well as truncated GCH1 (T-GCH1) and GFRP in the presence and absence of BH.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, Peters JA, Harmar AJ CGTP Collaborators. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atherton ND, Green A. HPLC measurement of phenylalanine in plasma. Clin Chem. 1988;34:2241–2244. [PubMed] [Google Scholar]
- Auerbach G, Herrmann A, Bracher A, Bader G, Gütlich M, Fischer M, et al. Zinc plays a key role in human and bacterial GTP cyclohydrolase I. PNAS. 2000;97:13567–13572. doi: 10.1073/pnas.240463497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers MC, Hargrove LA, Kelly KA, Wu G, Meininger CJ. Tetrahydrobiopterin attenuates superoxide-induced reduction in nitric oxide. Front Biosci (Schol Ed) 2011;3:1263–1272. doi: 10.2741/s224. [DOI] [PubMed] [Google Scholar]
- Bryan NS, Grisham MB. Methods to detect nitric oxide and its metabolites in biological samples. Free Radic Biol Med. 2007;43:645–657. doi: 10.1016/j.freeradbiomed.2007.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuaiphichai S, McNeill E, Douglas G, Crabtree MJ, Bendall JK, Hale AB, et al. Cell-autonomous role of endothelial GTP cyclohydrolase 1 and tetrahydrobiopterin in blood pressure regulation. Hypertension. 2014;64:530–540. doi: 10.1161/HYPERTENSIONAHA.114.03089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunnington C, Van Assche T, Shirodaria C, Kylintireas I, Lindsay AC, Lee JM, et al. Systemic and vascular oxidation limits efficacy of oral tetrahydrobiopterin treatment in patients with coronary artery disease. Circulation. 2012;125:1356–1366. doi: 10.1161/CIRCULATIONAHA.111.038919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Uscio LV, Katusic ZS. Increased vascular biosynthesis of tetrahydrobiopterin in apolipoprotein E-deficient mice. Am J Physiol Heart Circ Physiol. 2006;290:H2466–H2471. doi: 10.1152/ajpheart.00366.2005. [DOI] [PubMed] [Google Scholar]
- Feldmann R, Geikowski A, Weidner C, Witzke A, Kodelja V, Schwarz T, et al. Foam cell specific LXRalpha ligand. PLoS ONE. 2013;8:e57311. doi: 10.1371/journal.pone.0057311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada T, Kagamiyama H, Hatakeyama K. Feedback regulation mechanisms for the control of GTP cyclohydrolase I activity. Science. 1993;260:1507–1510. doi: 10.1126/science.8502995. [DOI] [PubMed] [Google Scholar]
- Heintz C, Cotton RGH, Blau N. Tetrahydrobiopterin, its mode of action on phenylalanine hydroxylase, and importance of genotypes for pharmacological therapy of phenylketonuria. Hum Mutat. 2013;34:927–936. doi: 10.1002/humu.22320. [DOI] [PubMed] [Google Scholar]
- Heitzer T, Brockhoff C, Mayer B, Warnholtz A, Mollnau H, Henne S, et al. Tetrahydrobiopterin improves endothelium-dependent vasodilation in chronic smokers: evidence for a dysfunctional nitric oxide synthase. Circ Res. 2000;86:e36–e41. doi: 10.1161/01.res.86.2.e36. [DOI] [PubMed] [Google Scholar]
- Higashi Y, Sasaki S, Nakagawa K, Fukuda Y, Matsuura H, Oshima T, et al. Tetrahydrobiopterin enhances forearm vascular response to acetylcholine in both normotensive and hypertensive individuals. Am J Hypertens. 2002;15:326–332. doi: 10.1016/s0895-7061(01)02317-2. [DOI] [PubMed] [Google Scholar]
- Higgins CE, Gross SS. The N-terminal peptide of mammalian GTP cyclohydrolase is an autoinhibitory control element and contributes to binding the allosteric regulatory protein GFRP. J Biol Chem. 2011;286:11919–11928. doi: 10.1074/jbc.M110.196204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howells DW, Smith I, Hyland K. Estimation of tetrahydrobiopterin and other pterins in cerebrospinal fluid using reversed-phase high-performance liquid chromatography with electrochemical and fluorescence detection. J Chromatogr B Biomed Sci Appl. 1986;381:285–294. doi: 10.1016/s0378-4347(00)83594-x. [DOI] [PubMed] [Google Scholar]
- Kaufman S. The structure of the phenylalanine-hydroxylation cofactor. Proc Natl Acad Sci U S A. 1963;50:1085–1093. doi: 10.1073/pnas.50.6.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. J Gene Med. 2010;12:561–563. doi: 10.1002/jgm.1473. [DOI] [PubMed] [Google Scholar]
- Kolinsky MA, Gross SS. The mechanism of potent GTP cyclohydrolase I inhibition by 2,4-diamino-6-hydroxypyrimidine: requirement of the GTP cyclohydrolase I feedback regulatory protein. J Biol Chem. 2004;279:40677–40682. doi: 10.1074/jbc.M405370200. [DOI] [PubMed] [Google Scholar]
- Li JM, Shah AM. Differential NADPH- versus NADH-dependent superoxide production by phagocyte-type endothelial cell NADPH oxidase. Cardiovasc Res. 2001;52:477–486. doi: 10.1016/s0008-6363(01)00407-2. [DOI] [PubMed] [Google Scholar]
- Li L, Rezvan A, Salerno JC, Husain A, Kwon K, Jo H, et al. GTP cyclohydrolase I phosphorylation and interaction with GTP cyclohydrolase feedback regulatory protein provide novel regulation of endothelial tetrahydrobiopterin and nitric oxide. Circ Res. 2010;106:328–336. doi: 10.1161/CIRCRESAHA.109.210658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maita N, Okada K, Hirotsu S, Hatakeyama K, Hakoshima T. Preparation and crystallization of the stimulatory and inhibitory complexes of GTP cyclohydrolase I and its feedback regulatory protein GFRP. Acta Crystallogr D Biol Crystallogr. 2001;57(Pt 8):1153–1156. doi: 10.1107/s0907444901005558. [DOI] [PubMed] [Google Scholar]
- Maita N, Okada K, Hatakeyama K, Hakoshima T. Crystal structure of the stimulatory complex of GTP cyclohydrolase I and its feedback regulatory protein GFRP. PNAS. 2002;99:1212–1217. doi: 10.1073/pnas.022646999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maita N, Hatakeyama K, Okada K, Hakoshima T. Structural basis of biopterin-induced inhibition of GTP cyclohydrolase I by GFRP, its feedback regulatory protein. J Biol Chem. 2004;279:51534–51540. doi: 10.1074/jbc.M409440200. [DOI] [PubMed] [Google Scholar]
- Mayahi L, Heales S, Owen D, Casas JP, Harris J, MacAllister RJ, et al. (6R)-5,6,7,8-tetrahydro-L-biopterin and its stereoisomer prevent ischemia reperfusion injury in human forearm. Arterioscler Thromb Vasc Biol. 2007;27:1334–1339. doi: 10.1161/ATVBAHA.107.142257. [DOI] [PubMed] [Google Scholar]
- Mitchell BM, Dorrance AM, Webb RC. Phenylalanine improves dilation and blood pressure in GTP cyclohydrolase inhibition-induced hypertensive rats. J Cardiovasc Pharmacol. 2004;43:758–763. doi: 10.1097/00005344-200406000-00004. [DOI] [PubMed] [Google Scholar]
- Nandi M, Miller A, Stidwill R, Jacques TS, Lam AAJ, Haworth S, et al. Pulmonary hypertension in a GTP-cyclohydrolase 1-deficient mouse. Circulation. 2005;111:2086–2090. doi: 10.1161/01.CIR.0000163268.32638.F4. [DOI] [PubMed] [Google Scholar]
- Nandi M, Kelly P, Vallance P, Leiper J. Over-expression of GTP-cyclohydrolase 1 feedback regulatory protein attenuates LPS and cytokine-stimulated nitric oxide production. Vasc Med. 2008;13:29–36. doi: 10.1177/1358863X07085916. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 2014;42(Database Issue):D1098–1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders-Pullman R, Blau N, Hyland K, Zschocke J, Nygaard T, Raymond D, et al. Phenylalanine loading as a diagnostic test for DRD: interpreting the utility of the test. Mol Genet Metab. 2004;83:207–212. doi: 10.1016/j.ymgme.2004.07.010. [DOI] [PubMed] [Google Scholar]
- Starr A, Sand CA, Heikal L, Kelly PD, Spina D, Crabtree M, et al. Overexpression of GTP cyclohydrolase 1 feedback regulatory protein is protective in a murine model of septic shock. Shock. 2014;42:432–439. doi: 10.1097/SHK.0000000000000235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swick L, Kapatos G. A yeast 2-hybrid analysis of human GTP cyclohydrolase I protein interactions. J Neurochem. 2006;97:1447–1455. doi: 10.1111/j.1471-4159.2006.03836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tayeh MA, Marletta MA. Macrophage oxidation of L-arginine to nitric oxide, nitrite, and nitrate. Tetrahydrobiopterin is required as a cofactor. J Biol Chem. 1989;264:19654–19658. [PubMed] [Google Scholar]
- Thöny B, Auerbach G, Blau N. Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochem J. 2000;347:1–16. [PMC free article] [PubMed] [Google Scholar]
- Uhe AM, O'Dea K, Collier GR. Amino acid levels following beef protein and amino acid supplement in male subjects. Asia Pac J Clin Nutr. 1997;6:219–223. [PubMed] [Google Scholar]
- Watschinger K, Keller MA, Golderer G, Hermann M, Maglione M, Sarg B, et al. Identification of the gene encoding alkylglycerol monooxygenase defines a third class of tetrahydrobiopterin-dependent enzymes. Proc Natl Acad Sci U S A. 2010;107:13672–13677. doi: 10.1073/pnas.1002404107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss C, Koepfli P, Namdar M, Siegrist P, Luscher T, Camici P, et al. Tetrahydrobiopterin restores impaired coronary microvascular dysfunction in hypercholesterolaemia. Eur J Nucl Med Mol Imaging. 2005;32:84–91. doi: 10.1007/s00259-004-1621-y. [DOI] [PubMed] [Google Scholar]
- Xie L, Liu Z, Lu H, Zhang W, Mi Q, Li X, et al. Pyridoxine inhibits endothelial NOS uncoupling induced by oxidized low-density lipoprotein via the PKCalpha signalling pathway in human umbilical vein endothelial cells. Br J Pharmacol. 2012;165:754–764. doi: 10.1111/j.1476-5381.2011.01607.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim JJ, Brown GM. Characteristics of guanosine triphosphate cyclohydrolase I purified from Escherichia coli. J Biol Chem. 1976;251:5087–5094. [PubMed] [Google Scholar]
- Yoneyama T, Hatakeyama K. Decameric GTP cyclohydrolase I forms complexes with two pentameric GTP cyclohydrolase I feedback regulatory proteins in the presence of phenylalanine or of a combination of tetrahydrobiopterin and GTP. J Biol Chem. 1998;273:20102–20108. doi: 10.1074/jbc.273.32.20102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Binding profile for truncated GCH1 (T-GCH1) with GCH1 feedback regulatory protein (GFRP) in the presence of L-phenylalanine (L-phe). T-GCH1 is immobilized at a surface density of ∼1500 RU via His-capture, and GFRP and L-phe are introduced in varying concentrations (400–25 nM) (n = 4 for each experiment at varying immobilization patterns and surface densities). Representative sensorgrams are shown.
Figure S2 SPR data analysis by Origin software, curve fittings and residuals.
Table S1 Summary of the binding kinetics for both native/full-length GCH1 (F-GCH1) and GCH1 feedback regulatory protein (GFRP) as well as truncated GCH1 (T-GCH1) and GFRP in the presence and absence of BH.