

Abstract
Background and Purpose
Ectonucleotide pyrophosphatase/PDE1 (NPP1) is an ectoenzyme, which plays a role in several disorders including calcific aortic valve disease (CAVD). So far, compounds that have been developed as inhibitors of NPP1 lack potency and specificity. Quinazoline-4-piperidine sulfamides (QPS) have been described as potent inhibitors of NPP1. However, their mode of inhibition as well as their selectivity and capacity to modify biological processes have not been investigated.
Experimental Approach
In the present series of experiments, we have evaluated the efficacy of two derivatives, QPS1-2, in inhibiting human NPP1, and we have evaluated the effect of the most potent derivative (QPS1) on other ectonucleotidases as well as on the ability of this compound to prevent phosphate-induced mineralization of human primary aortic valve interstitial cells (VICs).
Key Results
The QPS1 derivative is a potent (Ki 59.3 ± 5.4 nM) and selective non-competitive inhibitor of human NPP1. Moreover, QPS1 also significantly inhibited the K121Q NPP1 gene variant (Ki 59.2 ± 14.5 nM), which is prevalent in the general population. QPS1 did not significantly alter the activity of other nucleotide metabolizing ectoenzymes expressed at the cell surface, namely NPP3, NTPDases (1–3), ecto-5′-nucleotidase and ALP. Importantly, QPS1 in the low micromolar range (≤10 μM) prevented phosphate-induced mineralization of VICs and lowered the rise of osteogenic genes as expected for NPP1 inhibition.
Conclusions and Implications
We have provided evidence that QPS1 is a potent and selective non-competitive inhibitor of NPP1 and that it prevented pathological mineralization in a cellular model.
Table of Links
| TARGETS |
|---|
| Enzymes |
| NT5E/CD73, ecto-5′-nucleotidase |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013)
Introduction
The ectonucleotide pyrophosphatase/PDE-1 (NPP1/PC-1) is involved in different disorders including diabetes, osteoarthritis and atherosclerosis (Goding et al., 2003). We recently found that NPP1 promotes the mineralization of the aortic valve and that it is highly expressed during the development of calcific aortic valve disease (CAVD; Cote et al., 2012b). It is worth emphasizing that NPP1 is an enzyme that hydrolyses nucleotides such as ATP and produces AMP and pyrophosphate (PPi), the latter being a potent inhibitor of ectopic mineralization. On one hand, knockout mice for NPP1 show ectopic mineralization of tendons (Okawa et al., 1998). On the other hand, when NPP1 is overexpressed, it promotes pathological mineralization of chondrocytes and valve interstitial cells (VICs) of the aortic valve (Johnson et al., 2001a; Cote et al., 2012a). The mechanism by which NPP1 promotes pathologic mineralization is twofold. First, pathological mineralization is often accompanied by the expression of alkaline phosphatase (ALP), which uses PPi to produce the pro-mineralizing phosphate (Pi; Towler, 2005). Hence, a high level of NPP1 and ALP will promote a build-up of Pi, which triggers calcification of soft tissues. Second, a high level of NPP1 contributes to the depletion of the extracellular level of ATP, which reduces purinergic signalling. In turn, a decreased purinergic signalling triggers apoptosis-mediated mineralization.
The ectonucleotidase family of enzymes encompasses the ectonucleoside triphosphate diphosphohydrolases (NTPDases), which catalyse the conversion of ATP into ADP and also ADP into AMP. Ecto-5′-nucleotidase (NT5E/CD73) promotes the conversion of AMP into adenosine, whereas alkaline phosphatase (ALP/TNAP) uses a wide variety of substrates (Goding, 2000). So far, the compounds that have been used to probe the role of different ectonucleotidases lack specificity and potency. For instance, ARL67156 (6-N, N-Diethyl-D-β, γ-dibromomethylene ATP trisodium salt) inhibits NPP1 but also significantly affects other ectonucleotidases (Levesque et al., 2007). Quinazoline-4-piperidine sulfamides (QPS) have been described as potent inhibitors of NPP1 (Patel et al., 2009). However, it is presently unknown whether these compounds are specific and have biological activities in cell culture models. Furthermore, the mode of inhibition has not been described. In this work, we have evaluated QPS compounds as inhibitors of NPP1 and also determined whether they also affect NPP3, NTPDase1-3, NT5E and ALP. Furthermore, we evaluated the capacity of QPS1 to inhibit in vitro phosphate-induced mineralization of human primary VICs.
Methods
Cell transfection with vectors
Cos-7 cells were seeded in 10 cm cell culture dishes. At 80–90% confluence, cells were transected with 10 μg of NPP1, NPP3, NTPDase1-3, CD73, ALP human cDNA. ENPP1 ORF clone incorporated into the vector pCMV6-AC-GFP was purchased from Origene (Rockville, MD, USA). Vectors for NPP3, NTPDase1, 2, 3, CD73 and ALP were described previously (Kaczmarek et al., 1996; Jin-Hua et al., 1997; Smith and Kirley, 1998; Chiang and Knowles, 2008; Lecka et al., 2010). The transfection was done using NanoJuice transfection reagent from Novagen (Darmstadt, Germany). After 24 h, cells were harvested for enzymatic activities.
For the preparation of protein extracts, transfected cells were washed three times with Tris-saline buffer at 4°C, collected by scraping in the harvesting buffer containing 95 mM NaCl, 0.1 mM PMSF, 45 mM Tris at pH 7.5 and 10 μg·mL−1 aprotinin and sonicated. Nucleus and cellular debris were discarded by centrifugation at 300× g for 10 min at 4°C and the supernatant was collected and stored at −80°C until used for the activity assays. Protein concentration was estimated by the Bradford microplate assay using bovine serum albumin as a standard.
Enzymic assays
NPPs
Evaluation of the effect of QPS1 or QPS2 on human NPP1, NPP1 K121Q and NPP3 activity was carried out with para-nitrophenyl thymidine 5′-monophosphate (pnp-TMP). The reactions were carried out at 37°C in 0.2 mL of the following incubation mixture, 1 mM CaCl2, 140 mM NaCl, 5 mM KCl and 50 mM Tris, pH 8.5, with or without QPS1 or QPS2 (50, 100, 500 and 1000 nM). Reaction was initiated by adding human NPP1, NPP1 K121Q or NPP3 cell extracts to the pre-incubated reaction mixture containing pnp-TMP (25, 50, 100 and 200 μM). The production of paranitrophenol was measured at 410 nm, 60 min after the initiation of the reaction. Results were normalized for protein content. The type of inhibition was determined by competition assay and the level of inhibition was reported as percent inhibition. Ki was calculated by plotting the data of independent experiments using Sigma Plot 12.3 (Systat Software Inc., CA, USA). Results were presented using Dixon plots.
NTPDases
Activity was measured in 96-well plate in 0.2 mL of incubation medium (5 mM CaCl2 and 80 mM Tris, pH 7.4) at 37°C with or without QPS1 (50, 100, 500 and 1000 nM). The reaction was initiated by adding NTPDase protein extracts to the pre-incubated reaction mixture containing 100 μM ATP (Sigma Aldrich, Oakville, ON, Canada) and stopped after 15 min with 50 μL of malachite green reagent. The released inorganic phosphate (Pi) was measured at 630 nm (Baykov et al., 1988). Results were normalized to protein contents and reported as percent changes.
NT5E
Activity was measured at 37°C for 15 min in 0.2 mL of incubation solution (5 mM CaCl2 and 80 mM Tris, pH 7.4) containing AMP (25, 50, 100 and 200 μM) with or without QPS1 (50, 100, 500 and 1000 nM). Reaction was started by the addition of NT5E cell extracts and stopped with 50 μL malachite green reagent after 15 min. Released inorganic phosphate was measured as described above for NTPDases. Results were normalized to protein contents and reported as percent changes.
ALP
Activity was assayed using p-nitrophenyl phosphate as a substrate (PNPP; Sigma, Oakville, ON, Canada). Samples were incubated in the presence of substrate for 30 min at 37°C. The ALP activity was then measured by absorbance reading at 410 nm. The assay was carried out in triplicates. Results were normalized to protein contents and reported as percent changes.
VICs isolation and in vitro analyses of calcification
The protocol for human tissue samples was approved by the local ethical committee and informed consent was obtained from the donors. Human VICs were isolated from non-mineralized aortic leaflets, by collagenase digestion. Cells were isolated from non-calcified aortic valves obtained during heart transplant procedures. Patients with a history of rheumatic disease, endocarditis and inflammatory diseases were excluded. Aortic valves with sclerosis/stenosis or moderate to severe regurgitation (grade >2) were excluded.
To promote calcification, cells were incubated for 7 days with a pro-calcifying medium containing: DMEM + 5% FBS, 10−7 M insulin, 50 μg·mL−1 ascorbic acid and NaH2PO4 at 2 mM. In some experiments, QPS1 (0.1–10 μM; dissolved in DMSO) was added as specified. The calcium content was determined by the Arsenazo III method (Synermed, Monterey Park, CA, USA), which relies on the specific reaction of Arsenazo III with calcium to produce a blue complex. The results were measured at 650 nm on the Roche Diagnostics Modular P800 Elecsys (Roche Diagnostics, Laval, QC, Canada). This reaction is specific to calcium. Magnesium is prevented from forming a complex with the reactive. The results were normalized for protein contents and reported as percent changes.
Alizarin red staining
To visualize the calcium formation, cells were stained with 2% Alizarin red solution. Alizarin red solution was prepared by dissolving 2 g of Alizarin red (Sigma, Oakville, ON, Canada) in 100 mL distilled water, well mixed and pH was adjusted to 4.1–4.3 with 0.1% NH4OH. The solution was filtered before use. Human VICs were incubated for 7 days with pro-calcifying media as stated above to promote calcification. The medium was changed every 2 days. After 7 days, cells were washed once with PBS and fixed with 4% formaldehyde (Sigma, Oakville, ON, Canada) for 30 min, and then washed once with distilled water. Filtered 2% alizarin red solution was added to the cells for 2–3 min and washed with distilled water four times.
Detection of apoptosis
Apoptosis was documented in mouse VIC culture by the caspase 3/7 detection reagent (Invitrogen, Toronto, ON, Canada) according to manufacturer's instructions. After treatment with or without QPS1 (0.1–10 μM), cells were labelled with 2 μM CellEventTM (Life Technologies, Burlington, ON, Canada) caspase-3/7 green detection reagent in complete medium for 30 min at 37°C, 5% CO2 in the dark. Cells were fixed in 3.7% formaldehyde for 20 min at room temperature, then treated with 50 mM ammonium chloride for 15 min at room temperature and samples were mounted in DAPI containing mounting media (Sigma, Oakville, ON, Canada). Caspase-3/7 stained cells were counted over total population using an Olympus IX81 inverted microscope.
Quantitative real-time PCR
RNA was extracted from cells during in vitro experiments. Total RNA was isolated with RNeasy micro kit from Qiagen (Mississauga, ON, Canada). The RNA extraction protocol was performed according to the manufacturer's instructions. The quality of total RNA was monitored by capillary electrophoresis (Experion, Biorad, Mississauga, ON, Canada). One μg of RNA was reverse transcribed using the Quantitec Reverse Transcription Kit from Qiagen. Quantitative real-time PCR (q-PCR) was performed with Quantitec SYBR Green PCR kit from Qiagen on the Rotor-Gene 6000 system (Corbett Robotics Inc, San Francisco, CA, USA). Primers for runx2, osteopontin, osteocalcin, ALP and COL1A1 were obtained from Qiagen (Mississauga, ON, Canada). Cycling conditions were as follows: 2 min denaturation at 94°C, and 40 amplification cycles consisting of 15 s at 94°C and 1 min at 60°C. The expression of HPRT was used as a reference gene to normalize the results.
ATP measurements
Cells were incubated for 7 days with a pro-calcifying medium containing: DMEM + 5% FBS, 10−7 M insulin, 50 mg·mL−1 ascorbic acid and NaH2PO4 at 2 mM. In some experiments, QPS1 was added as specified. At day 7, the medium was changed for a phenol free medium and ATP level was determined after an incubation of 1 h. The level of ATP released by cells in supernatants was measured using the ATP SL luminescent kit from BioThema (Cedarlane, ON, Canada) according to manufacturer's instructions.
Data analyses
For the comparisons of groups, the results were expressed as means ± SEM. For continuous data, values were compared between groups with Student's t-test or anova when two or more than two groups were compared respectively. Post hoc Tukey analyses were done when the P value of the anova was <0.05. A P value < 0.05 was considered as statistically significant. Statistical analyses were performed with a commercially available software package JMP IN 8.1 (SAS Institute Inc, Cary, NC, USA).
Materials
DMEM and fetal bovine serum (FBS) were purchased from Invitrogen (Grand Island, NY, USA). QPS1 and QPS2 were synthesized (see Supporting Information for details). Para-nitrophenyl thymidine 5′-monophosphate (pnp-TMP), AMP, ATP and phosphatase substrate were purchased from Sigma Aldrich (Oakville, ON, Canada).
Results
QPS are potent and selective inhibitors of human NPP1
QPS have been shown to be potent inhibitors of NPP1. However, their mode of inhibition as well as their specificity over other members of the ectonucleotidase family is unknown. Two QPS compounds (QPS1, QPS2) were synthesized (>95% purity estimated by 1H NMR; Supporting Information) and used in enzyme kinetic assays (Figure 1). QPS1 inhibited NPP1 (Panova < 0.0001) in a concentration-dependent manner (Figure 2A and B). The maximal rate of inhibition was 84% with 1000 nM QPS1. The inhibition rate did not vary significantly when increasing the concentration of substrate, suggesting that QPS1 is a non-competitive inhibitor of NPP1. Analyses of NPP1 activity in the Dixon plot indicated that QPS1 inhibited NPP1 with a Ki of 59.3 ± 5.4 nM (Figure 2C and G). Inhibited and uninhibited NPP1 had different intercepts on the Y-axis, confirming a non-competitive inhibition (Figure 2C). We next documented that QPS2 is less potent than QPS1. The maximal inhibition rate of NPP1 activity achieved with 1000 nM QPS2 was 77% (Panova < 0.0006; Figure 2D and E). The Ki obtained for QPS2 was 110 ± 23 nM (Figure 2F and G).
Figure 1.
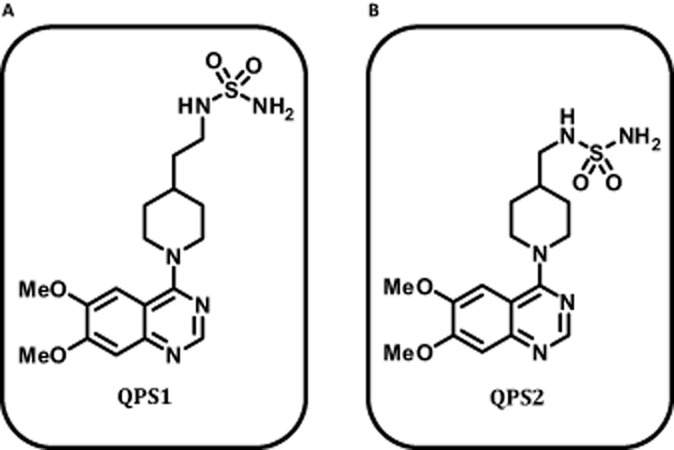
Molecular structure of NPP1 inhibitors QPS1 (A) and QPS2 (B).
Figure 2.

Inhibition of NPP1 by QPS1 (A and B; n = 5) and Dixon plot representation (C). Inhibition of NPP1 by QPS2 (D and E; n = 4) and Dixon plot representation (F). G) Characteristics of inhibition by QPS1 and QPS2. S: substrate. Data are mean ± SEM. *P < 0.05 compared with control (no QPS); #P < 0.05 compared with 50 nM dose; ‡P < 0.05 compared with 100 nM dose.
Because QPS1 was the more potent inhibitor, the next series of experiments was carried out with this compound. We tested QPS1 on the K121Q NPP1 variant, which is expressed in ∼ 15% of the population and associated with diabetes. Similar to the results obtained with wild-type NPP1, QPS1 provided a strong inhibition of the K121Q NPP1 variant. The maximal rate of inhibition on NPP1 activity was 77% (Panova = 0.0035; Figure 3A and B) with a Ki of 59.2 ± 14.5 nM (Figure 3C and D). Taken together, these findings indicate that QPS1 is a potent inhibitor of NPP1 including its common variant K121Q.
Figure 3.

Inhibition of NPP1 K121Q by QPS1 (A and B; n = 5), Dixon plot representation (C) and characteristics of inhibition (D). Data are mean ± SEM. *P < 0.05 compared with control (no QPS).
Specificity of QPS1
The specificity of QPS1 inhibition was tested on the most important ectonucleotidases. Among the NPP family of enzymes, NPP2 has a poor pyrophosphatase activity and has instead a lysophospholipase D activity. On the other hand, NPP3 has a pyrophosphatase activity. Hence, we next tested QPS1 on NPP3. In these experiments, QPS1 did not significantly inhibit NPP3 with a maximal inhibition rate of 7% on enzyme-specific activity (Figure 4A). Ecto-5′-nucleotidase (NT5E; CD73) converts AMP into adenosine. We next verified the activity of ecto-5′-nucleotidase by using the malachite green technique and AMP as a substrate. In this series of experiments, QPS1 did not significantly affect ecto-5′-nucleotidase activity, with a maximal inhibition rate of 5% (Figure 4B). We next tested the activity of QPS1 on human NTPDases, which catalyse the hydrolysis of ATP into ADP and also ADP into AMP. Enzyme activity of NTPDases was tested with the malachite green technique with ATP as a substrate. In experiments with human NTPDase1,-2,-3, QPS1 did not produce a significant inhibition, with a maximal inhibition rate of 12% for NTPDase2 (Figure 4C). ALP or tissue non-specific ALP (TNAP) is a phosphatase with a wide spectrum of substrates. We used the p-nitrophenyl phosphate (PNPP) in cells transfected with a human vector encoding ALP. QPS1 yielded non-significant inhibition on ALP activity with a maximal inhibition rate of 6% (Figure 4C). Hence, QPS1 was specific for NPP1 and did not significantly affect NPP3, NTPDases (1–3), ALP and NT5E.
Figure 4.

Percentage enzyme inhibition by QPS1 for NPP3 (A; n = 4) with pnp-TMP as the substrate, NT5E (B; n = 5) with AMP as the substrate, NTPDases1-3 (C; n = 5) (ATP as the substrate) and ALP (C; n = 4) with PNPP as the substrate. Data are mean ± SEM. Panova = NS.
Blockade of NPP1 prevents calcification of isolated VICs in response to phosphate
We previously showed that NPP1 is highly expressed in CAVD and that it promotes the mineralization of VICs. Hence, we used QPS1 in isolated human VICs exposed to a mineralizing medium (phosphate-containing medium) to document the biological activity of QPS1. Human primary VICs were isolated from seven patients who had undergone heart transplantation (Table 2005). A treatment of VICs with the mineralizing medium increased the level of calcium measured in cell culture after 7 days. A treatment of VIC cultures with QPS1 reduced in a dose-dependent manner the amount of calcium measured biochemically and normalized to the protein content (Panova < 0.0001; Figure 5A). These findings were corroborated by using alizarin red staining of cell cultures, which showed that QPS prevented the mineralization of VIC cultures (Figure 5B). Phosphate-induced mineralization relies on apoptosis and osteoblastic transition of cells. Phosphate induces the expression of NPP1, which contributes to depleting the extracellular level of ATP. In turn, a lower purinergic signalling in VICs promotes the mineralization through osteogenic and apoptotic mechanisms (Cote et al., 2012b). After 7 days of culture, we measured the level of ATP in VIC supernatants. We found in osteogenic medium (containing phosphate) that the level of extracellular ATP was decreased, whereas a treatment with QPS1 (0.5 μM) significantly increased the level of nucleotides (Panova < 0.0001; Figure 5C). We next measured the level of apoptosis in VIC cultures treated with the phosphate-containing medium by using the caspase 3/7 activity assay. Although treatment with phosphate-containing medium increased apoptosis by a factor of 3.1, QPS1 negated this effect (Panova = 0.0003; Figure 5D). We next measured the level of osteoblastic genes following stimulation with the mineralizing medium with or without QPS1 at 10 μM. After a treatment with phosphate, the following osteoblastic genes were increased: runx2, osteopontin and osteocalcin (Panova < 0.03; Figure 5E). The mRNA encoding for ALP was not significantly modulated by a phosphate treatment (Figure 5E). QPS1 prevented the rise of runx2, osteopontin and osteocalcin, whereas it did not affect the expression of ALP (Figure 5E). In addition, phosphate-induced expression of collagen α-1 chain type 1 (COL1A1) was also reduced by QPS1 (Panova = 0.0001; Figure 5F). These findings indicate that QPS1 has biological activities in the low micromolar range and prevents pathological mineralization/fibrosis in an in vitro model.
Figure 5.

(A) Concentration-dependent inhibition of PO4-induced VICs mineralization by QPS1 (n = 7). (B) Alizarin red staining of VIC cultures. (C) Percentage of ATP in VIC supernatants (n = 4). (D) QPS1 prevented PO4-induced apoptosis of VICs as evaluated with the caspase3/7 assay (n = 4). (E and F) QPS1 (10 μM) significantly reduced the rise of osteogenic/fibrotic markers following a treatment of VICs with PO4 (n = 3). Data are mean ± SEM. In A and D, *P < 0.05 compared with control (CTL), #P < 0.05 compared with CTL with QPS, ‡P < 0.05 compared with PO4; in panels E and F, *P < 0.05 compared with control (CTL), #P < 0.05 compared with PO4 without QPS.
Table 1.
Clinical characteristics of patients
| Aortic valves for VIC isolation (n = 7) | |
|---|---|
| Age | 45 ± 3 |
| Male (%) | 71 |
| Smoking (%) | 29 |
| Hypertension (%) | 29 |
| Diabetes (%) | 14 |
| BMI (kg·m−2) | 30.2 ± 2.10 |
| Triglycerides (mmol·L−1) | 1.40 ± 0.34 |
| LDL (mmol·L−1) | 2.36 ± 0.68 |
| HDL (mmol·L−1) | 1.39 ± 0.11 |
| Creatinine (μmol·L−1) | 90.3 ± 9.55 |
Values are mean ± SEM or %.
BMI, body mass index.
Discussion
In this work, we have shown that two QPS compounds are specific and potent non-competitive inhibitors of human NPP1. Specifically, we found that QPS1 did not significantly inhibit NPP3, NTPDases (1–3), ecto-5′-nucleotidase and ALP. Furthermore, we found that QPS1 prevented phosphate-induced mineralization of human VICs. QPS1 was efficient, in the low micromolar range, to prevent phosphate-induced apoptosis of VICs. Also, the rise of osteoblastic gene markers induced by phosphate-containing medium was prevented by QPS1. Taken together, these findings indicate that QPS1 prevents the pathological mineralization of VICs by specifically inhibiting NPP1.
Inhibitors of NPP1
ARL67156, a non-hydrolysable competitive inhibitor of ectonucleotidases, is widely used in cell experiments. A report showed that ARL67156 inhibits NPP1 with a Ki of 12 ± 3 μM (Levesque et al., 2007). The effect of ARL67156 is relatively non-specific and impacts significantly on the activity of NTPDase1 and NTPDase3 with Ki of 11 ± 3 μM and 18 ± 4 μM respectively. Also, derivatives of oxadiazole-2(3h)-thione have been described as inhibitors of NPP1 (Khan et al., 2009). However, the potency of these compounds is low with Ki ∼ 100 μM. More recently, novel diadenosine polyphosphonate derivatives have been described as inhibitors of NPP1 with Ki varying between 9 and 51 μM (Eliahu et al., 2010). Diadenosine polyphosphonate compounds also significantly inhibited NPP2 and NPP3. In the present series of experiments, we documented that QPS1 and QPS2 were potent inhibitors of NPP1 with Ki of 0.06 μM (59.3 nM) and 0.11 μM (110.4 nM) respectively. Patel et al. (2009) reported Ki of 0.036 μM and 0.061 μM, respectively, for QPS1 and QPS2, which is similar to the present findings. Hence, the present study confirmed the potency of QPS derivatives as significant inhibitors of NPP1.
Specificity of QPS1
The first report on QPS derivatives neither addressed the specificity of this class of compounds on other ectonucleotidases, nor their efficacy in a biological system such as in cell culture models. In the present study, we underlined that QPS1 did not significantly impact on NPP3, NTPDases (1–3), ecto-5′-nucleotidase and ALP. It is worth emphasizing that it is, to our knowledge, the first description of a compound that is both a potent and a specific inhibitor of NPP1. Therefore, QPS1 could be used in biological assays where different ectonucleotidases are expressed to help to discriminate between the role of NPP1 and that of other ectonucleotidases. In this regard, different ectonucleotidases are expressed by cell populations and exert a powerful control over purinergic signalling and several biological functions (Mathieu, 2012).
QPS1 inhibits pathological mineralization of isolated human cells
NPP1 has both anti and pro-mineralizing properties. Mice with deletion of NPP1 develop extensive mineralization of tendons and soft tissues owing to the absence of PPi (Okawa et al., 1998). On the other hand, the expression of NPP1 is increased considerably in human pathological specimens of CAVD. We documented that overexpression of NPP1 promotes the mineralization of VICs by depleting the level of extracellular ATP. In turn, a low level of ATP triggers apoptosis of cell cultures, whereby mineralization is promoted. Human VICs express NPP1, NPP2, ecto-5′-nucleotidase and ALP (Cote et al., 2012b). In this work, the use of QPS1, a potent and selective inhibitor of NPP1, prevented both apoptosis and mineralization of VIC cultures induced by phosphate. Phosphate-induced expression of osteogenic genes relies on the sodium/phosphate co-transporter Pit1 (SLC20A1), which is highly expressed during ectopic vascular/valvular mineralization (El Husseini et al., 2013). However, the molecular mechanism by which intracellular phosphate mediates gene expression is still unknown. QPS1 prevented the rise of several osteogenic markers induced by phosphate, indicating that QPS1 has strong anti-mineralizing properties on VICs. Whether QPS can also inhibit the mineralization of vascular smooth muscle cells remains to be explored. It should be stressed that expression of osteogenic genes is increased in human specimens of CAVD (Bosse et al., 2009). Furthermore, phosphate is a strong promoter of the expression of several osteoblastic genes (El Husseini et al., 2013). Therefore, the present findings suggest that QPS1 has strong biological activities in the low micromolar range (≤10 μM), which prevents both apoptosis and osteoblastic transition of VICs. Notably, COL1A1 was the most up-regulated gene following exposure of VICs to phosphate. QPS1 significantly decreased the rise of COL1A1 in VICs, suggesting that this derivative may prevent fibrotic processes. It is worthwhile to highlight that the expression of genes related to fibrosis is elevated in human CAVD tissues (Chen and Simmons, 2011).
Clinical implications
NPP1 is suspected of playing a role in several disorders. As previously described, expression of NPP1 is elevated during the development of CAVD. Moreover, a high level of NPP1 has been shown to promote the pathological mineralization of meniscal cells. Blocking NPP1 has been suggested as a means to prevent the formation of calcium pyrophosphate dehydrate crystals and as such could be used in the treatment of chondrocalcinosis and osteoarthritis (Johnson et al., 2001b). In apoE−/− NPP1−/− and apoE−/− NPP1+/− mice, the level of vascular atherosclerosis is reduced. Notably, vascular mineralization was not significantly increased in haploinsufficient apoE−/− NPP1+/− compared with ApoE−/− mice. Hence, NPP1 promotes atherosclerosis and a certain amount is required to prevent ectopic vascular mineralization (Nitschke et al., 2011). Also, studies have stressed that overexpression of NPP1 could play a role in the pathogenesis of diabetes. In this regard, overexpression of NPP1 in the liver promotes insulin resistance. Expression of NPP1 is increased in fibroblast of patients with type 2 diabetes (T2D; Maddux et al., 1995). It is suspected that interaction of NPP1 with the insulin receptor could impede insulin signalling. However, whether enzyme activity of NPP1 is necessary to hinder insulin signalling is still a matter of debate (Grupe et al., 1995; Chin et al., 2009). The K121Q variant of NPP1, prevalent in the population, has been associated in different studies with T2D (Abate et al., 2005). Considering the prevalence of this gene variant in the general population, we tested the QPS1 compound on the K121Q NPP1 variant. Of interest, we documented that QPS1 was efficient at inhibiting the variant K121Q with a Ki of 0.06 μM (59.2 nM). Hence, the development of novel inhibitors for NPP1, which are specific and potent, is desperately needed to probe the role of NPP1 in different disorders. As a result, this study may have important implications as it underlines that QPS1 may be used, at least in vitro, to investigate the pathogenic role of NPP1.
Limitations
Though potent and selective, the QPS derivatives have not been validated in vivo. The in vivo kinetics and resistance to hydrolysis of QPS derivatives remain to be investigated. Some of the QPS derivatives bind to the hERG potassium channel and thus may induce prolongation of the QT interval (Patel et al., 2009). QPS1 is a potent ligand of hERG, whereas QPS2 has the best ratio of NPP1 inhibition to hERG binding. Hence, further development of these compounds will require voltage clamp experiments to assess properly this class of compounds on hERG function. Also, their transmembrane kinetics and their effects on other types of intracellular enzymes remain to be established. Nonetheless, the present series of investigations have cast some light on this novel class of NPP1 inhibitors. We have identified that QPS1 is potent, selective and efficient in a cellular model to inhibit pathological mineralization.
Conclusion
In this study, we have provided evidence that QPS compounds are a novel class of non-competitive inhibitors of NPP1. Furthermore, we found that QPS1 is selective and prevents phosphate-induced apoptosis and mineralization of human VICs. Moreover, we identified that this selective inhibitor negated the osteogenic transition of VICs induced by mineralizing medium, thus highlighting its potential role to prevent ectopic mineralization.
Acknowledgments
This work was supported by grants from the Heart and Stroke Foundation of Canada (HSFC) (P. M.), the Canadian Institutes of Health Research (CIHR) (P. M.) (MOP245048) and the Fonds de recherche du Québec – Nature et technologies (FRQNT) (P. M.). P. M. and J. S. are recipients of a scholarship from the Fonds de recherche du Québec – Santé (FRQS). The Canada Research Chair Program (J. F. P.), the Natural Sciences and Engineering Research Council of Canada (J. F. P.), the Canada Foundation for Innovation (J. F. P.; P. M.), PROTEO and Université Laval are also acknowledged.
Glossary
- ALP/TNAP
alkaline phosphatase
- CAVD
calcific aortic valve disease
- NPP1/PC-1
ectonucleotide pyrophosphatase/PDE-1
- NT5E/CD73
ecto-5′-nucleotidase
- NTPDases
ectonucleotide triphosphate diphosphohydrolases
- QPS
quinazoline-4-piperidine sulfamides
- VICs
valve interstitial cells
Author contributions
E. E. S., M. C. B. and A. M. conducted in vitro experiments on enzyme kinetics and in cell culture. E. E. S. performed data analyses. E. F. and S. D. synthesized compounds used in this study. J. F. P. supervised synthesis of chemical compounds. X. B., J. S. and P. L. critically reviewed data. P. M. supervised in vitro experiments and wrote the manuscript. All the authors critically reviewed the manuscript.
Competing interests
P. Mathieu has patent applications for the use of ectonucleotidase inhibitors and purinergic agonists in the treatment of CAVD.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 1H NMR spectrum of compound 2.
Figure S2 13C NMR spectrum of compound 2.
Figure S3 1H NMR spectrum of compound 4.
Figure S4 13C NMR spectrum of compound 4.
Figure S5 1H NMR spectrum of compound QPS1.
Figure S6 13C NMR spectrum of compound QPS1.
Figure S7 1H NMR spectrum of compound 6.
Figure S8 13C NMR spectrum of compound 6.
Figure S9 1H NMR spectrum of compound 7.
Figure S10 13C NMR spectrum of compound 7.
Figure S11 1H NMR spectrum of compound 8.
Figure S12 13C NMR spectrum of compound 8.
Figure S13 1H NMR spectrum of compound 9.
Figure S14 13C NMR spectrum of compound 9.
Figure S15 1H NMR spectrum of compound QPS2.
Figure S16 13C NMR spectrum of compound QPS2.
Scheme 1 Synthesis of QPS1a.
Scheme 2 Synthesis of QPS2a.
References
- Abate N, Chandalia M, Satija P, Adams-Huet B, Grundy SM, Sandeep S, et al. ENPP1/PC-1 K121Q polymorphism and genetic susceptibility to type 2 diabetes. Diabetes. 2005;54:1207–1213. doi: 10.2337/diabetes.54.4.1207. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. CGTP Collaborators. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baykov AA, Evtushenko OA, Avaeva SM. A malachite green procedure for orthophosphate determination and its use in alkaline phosphatase-based enzyme immunoassay. Anal Biochem. 1988;171:266–270. doi: 10.1016/0003-2697(88)90484-8. [DOI] [PubMed] [Google Scholar]
- Bosse Y, Miqdad A, Fournier D, Pepin A, Pibarot P, Mathieu P. Refining molecular pathways leading to calcific aortic valve stenosis by studying gene expression profile of normal and calcified stenotic human aortic valves. Circ Cardiovasc Genet. 2009;2:489–498. doi: 10.1161/CIRCGENETICS.108.820795. [DOI] [PubMed] [Google Scholar]
- Chen JH, Simmons CA. Cell-matrix interactions in the pathobiology of calcific aortic valve disease: critical roles for matricellular, matricrine, and matrix mechanics cues. Circ Res. 2011;108:1510–1524. doi: 10.1161/CIRCRESAHA.110.234237. [DOI] [PubMed] [Google Scholar]
- Chiang WC, Knowles AF. Transmembrane domain interactions affect the stability of the extracellular domain of the human NTPDase 2. Arch Biochem Biophys. 2008;472:89–99. doi: 10.1016/j.abb.2008.02.011. [DOI] [PubMed] [Google Scholar]
- Chin CN, Dallas-Yang Q, Liu F, Ho T, Ellsworth K, Fischer P, et al. Evidence that inhibition of insulin receptor signaling activity by PC-1/ENPP1 is dependent on its enzyme activity. Eur J Pharmacol. 2009;606:17–24. doi: 10.1016/j.ejphar.2009.01.016. [DOI] [PubMed] [Google Scholar]
- Cote N, El Husseini D, Pepin A, Bouvet C, Gilbert LA, Audet A, et al. Inhibition of ectonucleotidase with ARL67156 prevents the development of calcific aortic valve disease in warfarin-treated rats. Eur J Pharmacol. 2012a;689:139–146. doi: 10.1016/j.ejphar.2012.05.016. [DOI] [PubMed] [Google Scholar]
- Cote N, El Husseini D, Pepin A, Guauque-Olarte S, Ducharme V, Bouchard-Cannon P, et al. ATP acts as a survival signal and prevents the mineralization of aortic valve. J Mol Cell Cardiol. 2012b;52:1191–1202. doi: 10.1016/j.yjmcc.2012.02.003. [DOI] [PubMed] [Google Scholar]
- El Husseini D, Boulanger MC, Fournier D, Mahmut A, Bosse Y, Pibarot P, et al. High expression of the Pi-transporter SLC20A1/Pit1 in calcific aortic valve disease promotes mineralization through regulation of Akt-1. PLoS ONE. 2013;8:e53393. doi: 10.1371/journal.pone.0053393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliahu S, Lecka J, Reiser G, Haas M, Bigonnesse F, Levesque SA, et al. Diadenosine 5′,5″-(boranated) polyphosphonate analogues as selective nucleotide pyrophosphatase/phosphodiesterase inhibitors. J Med Chem. 2010;53:8485–8497. doi: 10.1021/jm100597c. [DOI] [PubMed] [Google Scholar]
- Goding JW. Ecto-enzymes: physiology meets pathology. J Leukoc Biol. 2000;67:285–311. doi: 10.1002/jlb.67.3.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goding JW, Grobben B, Slegers H. Physiological and pathophysiological functions of the ecto-nucleotide pyrophosphatase/phosphodiesterase family. Biochim Biophys Acta. 2003;1638:1–19. doi: 10.1016/s0925-4439(03)00058-9. [DOI] [PubMed] [Google Scholar]
- Grupe A, Alleman J, Goldfine ID, Sadick M, Stewart TA. Inhibition of insulin receptor phosphorylation by PC-1 is not mediated by the hydrolysis of adenosine triphosphate or the generation of adenosine. J Biol Chem. 1995;270:22085–22088. doi: 10.1074/jbc.270.38.22085. [DOI] [PubMed] [Google Scholar]
- Jin-Hua P, Goding JW, Nakamura H, Sano K. Molecular cloning and chromosomal localization of PD-Ibeta (PDNP3), a new member of the human phosphodiesterase I genes. Genomics. 1997;45:412–415. doi: 10.1006/geno.1997.4949. [DOI] [PubMed] [Google Scholar]
- Johnson K, Hashimoto S, Lotz M, Pritzker K, Goding J, Terkeltaub R. Up-regulated expression of the phosphodiesterase nucleotide pyrophosphatase family member PC-1 is a marker and pathogenic factor for knee meniscal cartilage matrix calcification. Arthritis Rheum. 2001a;44:1071–1081. doi: 10.1002/1529-0131(200105)44:5<1071::AID-ANR187>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Johnson K, Pritzker K, Goding J, Terkeltaub R. The nucleoside triphosphate pyrophosphohydrolase isozyme PC-1 directly promotes cartilage calcification through chondrocyte apoptosis and increased calcium precipitation by mineralizing vesicles. J Rheumatol. 2001b;28:2681–2691. [PubMed] [Google Scholar]
- Kaczmarek E, Koziak K, Sevigny J, Siegel JB, Anrather J, Beaudoin AR, et al. Identification and characterization of CD39/vascular ATP diphosphohydrolase. J Biol Chem. 1996;271:33116–33122. doi: 10.1074/jbc.271.51.33116. [DOI] [PubMed] [Google Scholar]
- Khan KM, Fatima N, Rasheed M, Jalil S, Ambreen N, Perveen S, et al. 1,3,4-Oxadiazole-2(3H)-thione and its analogues: a new class of non-competitive nucleotide pyrophosphatases/phosphodiesterases 1 inhibitors. Bioorg Med Chem. 2009;17:7816–7822. doi: 10.1016/j.bmc.2009.09.011. [DOI] [PubMed] [Google Scholar]
- Lecka J, Rana MS, Sevigny J. Inhibition of vascular ectonucleotidase activities by the pro-drugs ticlopidine and clopidogrel favours platelet aggregation. Br J Pharmacol. 2010;161:1150–1160. doi: 10.1111/j.1476-5381.2010.00951.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levesque SA, Lavoie EG, Lecka J, Bigonnesse F, Sevigny J. Specificity of the ecto-ATPase inhibitor ARL 67156 on human and mouse ectonucleotidases. Br J Pharmacol. 2007;152:141–150. doi: 10.1038/sj.bjp.0707361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddux BA, Sbraccia P, Kumakura S, Sasson S, Youngren J, Fisher A, et al. Membrane glycoprotein PC-1 and insulin resistance in non-insulin-dependent diabetes mellitus. Nature. 1995;373:448–451. doi: 10.1038/373448a0. [DOI] [PubMed] [Google Scholar]
- Mathieu P. Pharmacology of ectonucleotidases: relevance for the treatment of cardiovascular disorders. Eur J Pharmacol. 2012;696:1–4. doi: 10.1016/j.ejphar.2012.09.003. [DOI] [PubMed] [Google Scholar]
- Nitschke Y, Weissen-Plenz G, Terkeltaub R, Rutsch F. Npp1 promotes atherosclerosis in ApoE knockout mice. J Cell Mol Med. 2011;15:2273–2283. doi: 10.1111/j.1582-4934.2011.01327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okawa A, Nakamura I, Goto S, Moriya H, Nakamura Y, Ikegawa S. Mutation in Npps in a mouse model of ossification of the posterior longitudinal ligament of the spine. Nat Genet. 1998;19:271–273. doi: 10.1038/956. [DOI] [PubMed] [Google Scholar]
- Patel SD, Habeski WM, Cheng AC, de la Cruz E, Loh C, Kablaoui NM. Quinazolin-4-piperidin-4-methyl sulfamide PC-1 inhibitors: alleviating hERG interactions through structure based design. Bioorg Med Chem Lett. 2009;19:3339–3343. doi: 10.1016/j.bmcl.2009.04.006. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TM, Kirley TL. Cloning, sequencing, and expression of a human brain ecto-apyrase related to both the ecto-ATPases and CD39 ecto-apyrases1. Biochim Biophys Acta. 1998;1386:65–78. doi: 10.1016/s0167-4838(98)00063-6. [DOI] [PubMed] [Google Scholar]
- Towler DA. Inorganic pyrophosphate: a paracrine regulator of vascular calcification and smooth muscle phenotype. Arterioscler Thromb Vasc Biol. 2005;25:651–654. doi: 10.1161/01.ATV.0000158943.79580.9d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 1H NMR spectrum of compound 2.
Figure S2 13C NMR spectrum of compound 2.
Figure S3 1H NMR spectrum of compound 4.
Figure S4 13C NMR spectrum of compound 4.
Figure S5 1H NMR spectrum of compound QPS1.
Figure S6 13C NMR spectrum of compound QPS1.
Figure S7 1H NMR spectrum of compound 6.
Figure S8 13C NMR spectrum of compound 6.
Figure S9 1H NMR spectrum of compound 7.
Figure S10 13C NMR spectrum of compound 7.
Figure S11 1H NMR spectrum of compound 8.
Figure S12 13C NMR spectrum of compound 8.
Figure S13 1H NMR spectrum of compound 9.
Figure S14 13C NMR spectrum of compound 9.
Figure S15 1H NMR spectrum of compound QPS2.
Figure S16 13C NMR spectrum of compound QPS2.
Scheme 1 Synthesis of QPS1a.
Scheme 2 Synthesis of QPS2a.