

Abstract
Background and Purpose
Histone hypoacetylation is associated with Parkinson's disease (PD), due possibly to an imbalance in the activities of enzymes responsible for histone (de)acetylation; correction of which may be neuroprotective/neurorestorative. This hypothesis was tested using the anti-epileptic drug sodium valproate, a known histone deacetylase inhibitor (HDACI), utilizing a delayed-start study design in the lactacystin rat model of PD.
Experimental Approach
The irreversible proteasome inhibitor lactacystin was unilaterally injected into the substantia nigra of Sprague–Dawley rats that subsequently received valproate for 28 days starting 7 days after lactacystin lesioning. Longitudinal motor behavioural testing, structural MRI and post-mortem assessment of nigrostriatal integrity were used to track changes in this model of PD and quantify neuroprotection/restoration. Subsequent cellular and molecular analyses were performed to elucidate the mechanisms underlying valproate's effects.
Key Results
Despite producing a distinct pattern of structural re-modelling in the healthy and lactacystin-lesioned brain, delayed-start valproate administration induced dose-dependent neuroprotection/restoration against lactacystin neurotoxicity, characterized by motor deficit alleviation, attenuation of morphological brain changes and restoration of dopaminergic neurons in the substantia nigra. Molecular analyses revealed that valproate alleviated lactacystin-induced histone hypoacetylation and induced up-regulation of brain neurotrophic/neuroprotective factors.
Conclusions and Implications
The histone acetylation and up-regulation of neurotrophic/neuroprotective factors associated with valproate treatment culminate in a neuroprotective and neurorestorative phenotype in this animal model of PD. As valproate induced structural re-modelling of the brain, further research is required to determine whether valproate represents a viable candidate for disease treatment; however, the results suggest that HDACIs could hold potential as disease-modifying agents in PD.
Tables of Links
| TARGETS |
|---|
| HDACs |
| HATs |
| TH (tyrosine hydroxylase) |
| LIGANDS | |
|---|---|
| BDNF | H2O2 |
| D-amphetamine | Hsp70 |
| GDNF | Sodium valproate |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Parkinson's disease (PD) is the most prevalent movement disorder, with cardinal symptoms of rigidity, tremor and bradykinesia, primarily resulting from degeneration of the dopaminergic nigrostriatal pathway (Jenner and Olanow, 2006). The pathological hallmark of the degenerating neurons in PD is intracytoplasmic protein inclusions known as Lewy bodies and Lewy neurites, composed predominantly of a synaptic protein called α-synuclein (αSyn), which are thought to be at least partly responsible for the dopaminergic neuronal cell death observed (Spillantini et al., 1997; Dexter and Jenner, 2013).
In recent years epigenetic mechanisms such as DNA methylation and histone remodelling have become implicated in PD pathogenesis (Ammal Kaidery et al., 2013). Increased expression or the presence of mutated forms of αSyn sequesters DNA methyltransferase 1 to the cytoplasm in PD and the promoter region of the αSyn gene, SNCA, is hypomethylated in both transgenic mice and in sporadic PD (Desplats et al., 2011). Similarly, it has been observed that nuclear αSyn ‘masks’ histone proteins preventing their acetylation, resulting in histone hypoacetylation and subsequent apoptosis (Kontopoulos et al., 2006). In the healthy brain, a carefully controlled balance exists between the activities of the two enzymes that control histone acetylation status: histone acetyltransferases (HATs) and histone deacetylases (HDACs). HATs facilitate the acetylation of histone N-terminal tail lysine residues, causing relaxation of chromatin structure and transcription factor access to DNA. Equally, HDACs reverse this process by removing acetyl groups from histone N-terminal tail lysine residues causing condensation of chromatin structure and transcriptional repression. We and others postulate that this deregulation of the balance of histone acetylation and deacetylation could be rectified with the use of HDAC inhibitors (HDACIs), reducing the neurodegeneration observed as a result of HAT/HDAC misbalance (Hahnen et al., 2008; Kazantsev and Thompson, 2008; Chuang et al., 2009; Dietz and Casaccia, 2010; Harrison and Dexter, 2013).
Sodium valproate (2-propylpentanoic acid), first marketed over 45 years ago for the treatment of epilepsy (Löscher, 2002), is now a commonly prescribed mood stabilizer and anti-convulsant used to control generalized and partial seizures (Perucca, 2002). Subsequently, valproate was discovered to relieve HDAC-dependent transcriptional repression and cause histone hyperacetylation both in vitro and in vivo (Gottlicher et al., 2001; Phiel et al., 2001) by acting as a pan-inhibitor of HDAC classes I and IIa: inhibiting HDAC 1, 2, 3, 4, 5, 7 and 8 with varying potencies (Gurvich et al., 2004). This property of valproate has been shown to reduce neuroinflammation leading to neuroprotection in cell culture models of neuroimmune cell activation and dopaminergic cell death (Peng et al., 2005; Chen et al., 2006; Monti et al., 2007; 2009,; Wu et al., 2008; Kidd and Schneider, 2010). Furthermore, preclinical in vivo studies highlight the neuroprotective potential of valproate in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model (Kidd and Schneider, 2011), and the rotenone (Monti et al., 2010) and 6-hydroxydopamine (6-OHDA) (Monti et al., 2012) rat models of PD when given as a pretreatment. Given its longevity of clinical use, these data suggest valproate is a candidate drug for clinical repositioning in the treatment of PD. Critically however, in previous preclinical neuroprotection studies, valproate was administered as a pretreatment prior to administering the neurotoxin responsible for modelling Parkinsonian nigral degeneration. These studies were also in animal models that induce cell death through mechanisms other than altered protein deposition, the neuropathological hallmark of PD. Hence, this design, although effective for proof-of-concept studies, lacks any clinical translatability or relevance. Likewise the molecular mechanism linking inhibition of HDACs by valproate leading to neuroprotection remains unclear. Current evidence from preclinical studies is therefore insufficient to facilitate translation of these basic findings to the clinic.
In this study, for the first time we have addressed these issues by investigating the neuroprotective effects of delayed-start valproate treatment, initiated 1 week after lesioning the rat nigrostriatal system, using the proteasome inhibitor, lactacystin. Importantly, this model recapitulates the formation of neurotoxic αSyn protein inclusions, which are known to affect epigenetic mechanisms, within the substantia nigra to induce dopaminergic cell death (Vernon et al., 2010a; 2011,; Pienaar et al., 2015). Similarly, this model is associated with a specific reproducible pattern of neuroanatomical changes in the brain overlapping that of late stage PD (Vernon et al., 2010b; 2011,; Duncan et al., 2013). For the first time, we also use a combined phenotyping approach, combining motor behavioural testing, clinically comparable longitudinal structural MRI and post-mortem assessment of the integrity of the rat brain nigrostriatal system to track changes in this model of PD and detect neuroprotection. Subsequent cellular and molecular analyses were also performed to elucidate the mechanisms underlying valproate's neuroprotective effects, including quantification of histone acetylation and expression levels of a number of different neurotrophic factors, apoptotic regulators and genes of interest to PD, previously shown to change upon treatment with HDACIs (Monti et al., 2009).
Methods
Experimental animals
All animal procedures were carried out in accordance with the Home Office Animals (Scientific Procedures) Act, UK, 1986, and were previously approved by the Imperial College Animal Welfare and Ethical Review Board. Presentation of data complies with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath et al., 2010). Male Sprague–Dawley rats (250 ± 10 g, Charles River, Kent, UK) were housed in groups of two or three at 21 ± 1°C on a 12 h light–dark cycle with the relative humidity maintained at 55 ± 10%. Standard rat chow and drinking water were available ad libitum throughout the duration of the study, and were supplemented with standard rat wet diet for 7 days post-surgery.
Animal treatment groups
Five animal treatment groups (Figure 1) underwent serial evaluation of brain structure by MRI and motor behavioural testing. The baseline assessment was performed before lactacystin lesioning of the substantia nigra pars compacta (SNpc) with follow-up assessments at 1, 3 and 5 weeks post-surgery. Animals were injected daily for 28 days with either saline or valproate (200 or 400 mg·kg−1 i.p.) initiated 7 days post-surgery. After the final in vivo assessments, animals were killed and brain tissue harvested for subsequent analysis. An additional group of animals was also stereotaxically lesioned with lactacystin, but killed and brain tissue harvested for subsequent analysis 7 days post-surgery.
Figure 1.
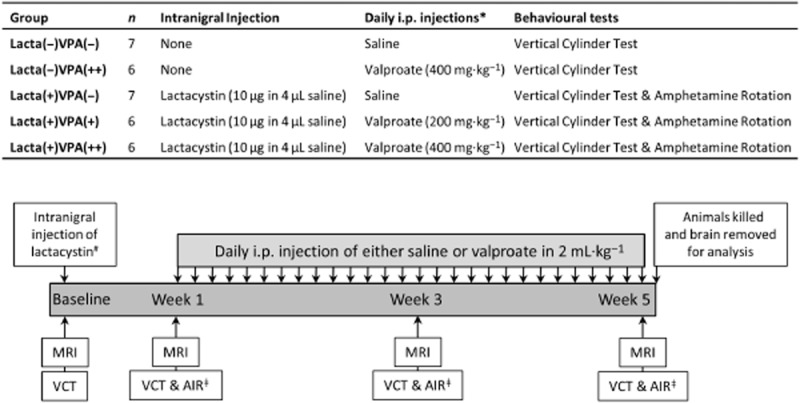
Animal treatment groups and study design. *All daily i.p. injections given as 2 mL·kg−1: saline injections given as 2 mL·kg−1; 400 mg·kg−1 valproate injections given as 2 mL·kg−1 of 200 mg·mL−1 solution of valproate in saline; 200 mg·kg−1 valproate injections given as 2 mL·kg−1 of 100 mg·mL−1 solution of valproate in saline. #Only groups Lacta(+)VPA(−), Lacta(+)VPA(+) and Lacta(+)VPA(++) intranigrally injected with lactacystin. Control groups remained surgically naïve. ǂOnly groups lesioned with lactacystin were tested using the amphetamine-induced rotation test at these time points. Abbreviations: VCT, vertical cylinder test; AIR, amphetamine-induced rotations.
Stereotaxic lesioning of the SNpc with lactacystin
The left SNpc was stereotaxically lesioned using the irreversible proteasome inhibitor, lactacystin (BML-PI104, Enzo Life Sciences, Exeter, UK), as previously described (Vernon et al., 2010a). Briefly, isoflurane (05260-05, IsoFlo®, Abbott Laboratories, Maidenhead, UK) anaesthetized animals were then positioned in a stereotaxic frame (Kopf Instruments, Tujunga, CA, USA) in the horizontal skull position with the incisor bar positioned 3.3 mm below the interaural line. Buprenorphine (QN02AE01, Alstoe Animal Health, York, UK) was injected i.m. to provide analgesia while bupivacaine (PL-20910/0008, Taro Pharmaceuticals, Ossining, NY, USA) was utilized to produce local anaesthesia in the scalp before a midline incision was made to expose the skull where bregma was identified (Paxinos and Watson, 2009). A small burr hole was made in the skull above the SNpc and 10 μg of lactacystin (2.5 μg·μL−1 in sterile (0.9%) saline, 4 μL in total) was stereotaxically administered via a Hamilton syringe to the SNpc: anteroposterior, −5.2 mm, mediolateral, +2.5 mm and ventral to dura, −7.6 mm (Paxinos and Watson, 2009). The lactacystin was injected at a rate of 1 μL·min−1, the needle was left in situ for 3 min before being retracted. Post-surgery fluid replacement (glucosaline, 5 mL of 0.18% NaCl, 4% glucose, given i.p. before surgery) was administered and animals were left to recover in a heated recovery chamber.
Behavioural testing
Vertical cylinder test
To assess asymmetry in forelimb motor function, the vertical cylinder test was employed similar to that previously published (Schallert et al., 2000). Briefly, animals were placed into a Perspex cylinder (200 mm in diameter by 300 mm in height) and rearing behaviour was recorded with a video camera for either 10 complete rears or 3 min. Forelimb movements were examined using frame-by-frame analysis of video recordings. The percentage of contralateral forelimb use was calculated as:
 |
Amphetamine-induced rotation test
Rotational asymmetry was assessed using the amphetamine-induced rotation test similar to that previously published (Ungerstedt and Arbuthnott, 1970). Briefly, animals were administered 5 mg·kg−1 amphetamine (i.p., D-amphetamine sulphate (A0922, Sigma, Poole, UK) in sterile saline at 5 mg and placed in a clear circular test arena of dimensions 400 mm in diameter by 360 mm in height (Circling Bowl, Harvard Apparatus, Holliston, MA, USA) for 30 min to acclimatize. After this time behaviour was recorded for 30 min and the number of contraversive and ipsiversive rotations were counted in bins of 5 min. The net number of ipsiversive rotations per 5 min bin was calculated as:
MRI
T2-weighted (T2W) MRIs were acquired similar to that previously described (Vernon et al., 2010a; 2011,) using a 4.7 Tesla DirectDrive horizontal small bore MRI scanner (Varian, Palo Alto, CA, USA) and a separate 72 mm quadrature birdcage head RF coil (M2M Imaging, Cleveland, OH, USA) linked to a LINUS-based control console running VnmrJ acquisition software (v2.3, Varian). Briefly, animals were anaesthetized by inhalation of isoflurane vaporized into O2 in an anaesthetic chamber. The animal was then positioned into an MRI-compatible polytetrafluoroethylene stereotaxic head holder and bed (M2M Imaging) and maintained under anaesthesia using isoflurane. Depth of anaesthesia was monitored using a respiratory balloon (SA Instruments, Stoney Brook, NY, USA) placed under the animal's chest and body temperature was monitored and maintained using a rectal probe and heated fan (SA Instruments) respectively. T2W images were acquired using a multi-echo, multi-slice spin-echo pulse sequence, with the following scan parameters: field of view = 35 × 35 mm; matrix = 192 × 192; TR = 5155.2 ms; TE = 10, 20, 30, 40, 50, 60, 70, 80, 90, 100 ms; four averages, scan duration 1 h 5 min 59 s. Fifty contiguous 500 μm thick coronal slices with an in plane resolution of 256 × 256 μm were acquired such that the entire brain of each animal was covered. Once scanning was completed, animals were removed from the magnet bore to a separate holding room and placed in a heated recovery chamber. Following full recovery from anaesthesia, animals were then returned to their home cages.
MRI analysis
Post-acquisition, images corresponding to the 10 TE times used were summed using the ‘Z Project’ function in ImageJ (v1.4, National Institutes of Health, Bethesda, MD, USA) to give the 50 T2W images representing 50 contiguous 500 μm thick coronal slices of the rat brain.
For regional volumetric analysis of MRIs, structures were delineated manually using ImageJ software by a single rater blinded to animal treatment. Six brain regions (whole brain, lateral ventricles, corpus striatum, hippocampus, midbrain and cerebellum) were delineated based on anatomical landmarks previously published (Vernon et al., 2010a; 2011,) and with reference to the rat brain stereotaxic map (Paxinos and Watson, 2009) (Supporting Information Figure S1). Volumes were then calculated by multiplying the sum of the areas for a given structure on all slices by the slice thickness (500 μm). Brain volumes were then expressed as percentage change from baseline.
To perform an unbiased whole brain analysis of apparent volumetric differences between groups, an automated image-processing pipeline for tensor-based morphometry (TBM) was used (Crum et al., 2013a; Rattray et al., 2013). Briefly, a single, well-positioned good-quality control animal (surgically naïve, saline treated) scan was chosen as an initial reference for rigid (6 degrees of freedom) and rigid + scaling (9 degrees of freedom) registration using a robust population approach (Crum et al., 2013b). Then, a template image was constructed as the mean of the registered control group (surgically naïve, saline treated) scans and used as the reference in subsequent processing. High-dimensional non-rigid registration was applied to each scan to warp it onto the control template, and thereby obtain maps of apparent local volume difference for each scan, encoded as the Jacobian determinant at each voxel (Crum et al., 2013a). Non-parametric t-tests at each voxel were used to detect differences in the Jacobian determinants between each study group and the control group and thereby infer differences in volume across groups (Bullmore et al., 1999). Significance values were corrected for multiple comparisons using the false discovery rate with q = 0.05 (Genovese et al., 2002).
Tissue collection and preparation
At the end of the study period, animals were killed, decapitated and the brain removed from the skull. Using a rodent brain matrix, each brain was cut coronally at the level of the infundibular stem to produce forebrain and hindbrain blocks. Frontal brain tissue was dissected out, snap frozen on dry ice and stored at −80°C for protein and mRNA extraction. The hindbrain was fixed in 4% paraformaldehyde in PBS (pH 7.4) for 72 h before being cryoprotected in 30% sucrose in PBS until the tissue was observed to have sunk. Blocks were then snap frozen in isopentane pre-chilled on dry ice and stored at −80°C for subsequent sectioning.
Immunohistochemistry
A number of 30 μm thick coronal sections were collected throughout the extent of the SNpc onto SuperFrost® Plus slides (HECH2409/1, VWR International, Lutterworth, UK) using a cryostat (Bright Instruments, Huntingdon, UK). Slides were then stored at −80°C until analysis. Immunohistochemistry for TH, the rate-limiting enzyme in monoamine synthesis was utilized as the cellular marker for dopaminergic neurons in the SNpc, and cresyl violet was used as a counterstain for the Nissl body of all neurons. For this the avidin–biotin complex (ABC)/peroxidase method of immunohistochemistry with a cresyl violet counterstain was performed similar to that previously published by our group (Pienaar et al., 2015). Briefly, endogenous peroxidase activity was blocked by incubation in 0.3% H2O2 in methanol for 45 min before sections were rehydrated in a descending series of alcohol washes followed by PBS containing 0.1% Triton X-100 (PBS-T). Non-specific binding was blocked by incubation with 20% normal goat serum in PBS-T for 1 h at room temperature before incubating with the primary antibody (rabbit polyclonal anti-TH, AB152, Millipore, Billerica, MA, USA) at 1:1000 in PBS-T for 24 h at room temperature. Sections were then washed in PBS-T and incubated in the secondary antibody (biotinylated goat anti-rabbit secondary antibody, BA-1000, Vector Labs, Peterborough, UK) at 1:200 in PBS-T for 1 h at room temperature. Sections were then washed again and incubated in ABC (Vectastain Elite ABC Kit, PK-6200, Vector Labs) for a further 1 h at room temperature before being washed thoroughly in tris-buffered saline (TBS) (pH 8.4), and staining visualized with 3, 3′-diaminobenzidine. Sections were then washed in H2O before being counterstained using cresyl violet (0.1% in dH2O) for 2 min and mounted.
Stereological cell quantification
Stereological quantification of the number of TH-positive (TH+) and Nissl-positive (Nissl+) cells were made in the entire SNpc. For this, a computer-based stereology software system (ImagePro, MediaCybernetics, Rockville, MD, USA) attached to a Nikon Eclipse E8 – microscope (Nikon Instruments, Surrey, UK) and JVC (London, UK) 3CCD camera was used. The optical fractionator method of stereology was used as previously published (West et al., 1991). Briefly, for each section the SNpc was delineated manually with relation to previously published boundaries (Carman et al., 1991), to create an area of interest (AOI). The software system then created counting frames (140 × 160 μm) which fell within the AOI using the uniform random sampling method. The total area of the counting frames relative to the area of the AOI gives the area sampling fraction. The height of the optical dissector, which was measured by taking an average of three random points across the section using a Heidenhain microcator (Hedenhain, Traunreut, Germany), relative to the section thickness gives the height sampling fraction. The section sampling fraction was 1/6 as every sixth section throughout the SNpc was analysed. To avoid edge effects, when counting TH+ and Nissl+ cells within the counting frames, ‘acceptance’ and ‘forbidden’ lines were used. Total cell estimates were calculated as follows, where n equals the number of positive cells counted:
Protein and mRNA extraction and quantification
Thirty milligrams of frontal brain tissue were homogenized in QIAzol® Lysis Reagent (79306, Qiagen, Crawley, UK) using a tissue homogenizer (Ultra-Turrax T18, IKA, Staufen, Germany). mRNA and protein was extracted from homogenized brain tissue using the RNeasy® Plus Universal Mini Kit (73404, Qiagen) as per the manufacturer's instructions. Isolated RNA was quantified spectrophotometrically using a NanoDrop ND-1000 spectrophotometer (Thermo Fischer Scientific, Waltham, MA, USA) and RNA purity verified by an average A260/280 ratio of 1.99 (range 1.97–2.01). The quantity of isolated protein was determined using the 96-well variant of the Bradford Assay (B6916, Sigma): colour change was determined using a 96-well plate reader (VersaMax Microplate Reader, Molecular Devices, CA, USA) at A595. mRNA and protein were stored at −80 and −20°C, respectively, until further analysis.
Western blot analysis
Laemmli sample buffer (S3401, Sigma) was added to 10 μg of extracted protein sample and denatured by incubating at 95°C for 15 min. Samples were loaded onto a 1 mm thick hand-cast 15% tris-glycine gel and proteins were separated by electrophoresis (65 mA for 40 min). Proteins were transferred onto a methanol-soaked PVDF membrane with a pore size of 0.45 μm using semi-dry transfer (20 V for 45 min). Membranes were then equilibrated in TBS containing 0.2% Tween-20 (TBS-T), before being blocked in 5% non-fat milk in TBS-T for 1 h at room temperature. Membrane was washed in TBS-T again before being incubated in primary antibodies against histone protein H3 acetylated on lysine 9 (rabbit anti-AcH3-Lys9, H9286, Sigma, 1:10 000) and mouse anti-β-actin antibody (1:20 000, Ab6276, Abcam, Cambridge, UK) for 1 h at RT. Membranes were then washed again and incubated in HRP-conjugated secondary antibodies [either goat anti-rabbit (1:10 000) and horse anti-mouse (1:10 000) for AcH3-Lys9 or β-actin, respectively, both Vector Labs] for 1 h at RT. Membranes were washed again in TBS-T and developed using chemiluminescence (170–5060, Clarity Western ECL Substrate, Bio-Rad, Hemel Hempstead, UK). Bands were quantified using densitometry analysis software (ImageJ, v1.4).
Quantitative real-time PCR (qRT-PCR)
For cDNA synthesis, 500 ng of total RNA from each sample was reverse transcribed according to the manufacturer's instructions using the QuantiTect® reverse transcription kit (205310, Qiagen) with integrated removal of genomic DNA contamination. The reactions were stored at −20°C until further use. qRT-PCR experiments were performed using a Mx3000P™ real-time PCR system with MxPro software (v4.10, Stratagene, La Jolla, CA, USA) and the Brilliant® II QPCR master mix with low ROX (600806, Agilent Technologies UK Ltd, Edinburgh, UK). For each gene of interest in each sample, 20 μL reactions were set up in triplicate, and run in duplex with a novel reference gene {XPNPEP1 [X-prolyl aminopeptidase (aminopeptidase P) 1; Durrenberger et al., 2012 ]}, with each reaction containing 10 μL of 2 × Brilliant® II QPCR master mix, 7 μL of RNase-free water, 1 μL template cDNA, 2 μL (1 μL gene of interest + 1 μL reference gene) of 10 × PrimeTime™ qPCR assays (Integrated DNA Technology, Coralville, IA, USA; Supporting Information Table S1). Reactions were carried out with the following cycling protocol: 95°C for 10 min, then 60 cycles with a three-step programme (95°C for 30 s, 55°C for 30 s and 72°C for 30 s). Fluorescence data collection was performed during the annealing step. A negative control containing no cDNA template was also run in each plate. Similarly an inter-plate calibrator, created by pooling control cDNA samples, was also run in each plate. Relative gene expression was determined using the 2−ΔΔCT method normalizing to the expression of the novel reference gene (Durrenberger et al., 2012) and the appropriate control group.
Statistical analysis
All data are presented as mean ± SEM. Two-way (repeated measures) anova with Bonferroni post-tests were used for analysis of vertical cylinder test, amphetamine-induced rotation test and MRI manual segmentation analysis datasets. An unpaired t-test was used to compare the baseline and week 1 forelimb use of lactacystin-lesioned animals in vertical cylinder test data. Paired t-tests were used to compare stereological cell counts in the ipsilateral and contralateral hemispheres of animal brains. A one-way anova with Bonferroni post-tests were used to compare cell loss percentages calculated from stereological cell counts. A two-way anova with Bonferroni post-tests were used to compare qRT-PCR data. A one-way anova with Bonferroni post-tests were used to compare Western blot data. All statistical tests were performed using GraphPad Prism (v5.0 for Windows, GraphPad Software, San Diego, CA, USA).
Results
Valproate attenuates behavioural motor deficits caused by lactacystin
The vertical cylinder test and amphetamine-induced rotations were utilized to assess for motor asymmetry in the animals' movement due to unilateral injection of lactacystin into the SNpc.
Vertical cylinder test
At baseline there was equal use of both the left and right forelimbs in all animals (Figure 2A). However, 1 week after surgery there was a significant reduction in the contralateral forelimb use of lactacystin-lesioned animals compared with non-lesioned animals. Contralateral forelimb use of saline-treated animals continued to decline with time. However, the continued decline in motor deficits due to lactacystin toxicity from week 1 was halted in animals treated with the lower dose of valproate (200 mg·kg−1). Whereas in animals treated with the highest dose of valproate (400 mg·kg−1), the continued decline in motor deficits due to lactacystin toxicity from week 1 was totally reversed with the animals failing to show any deficit after 28 days of treatment.
Figure 2.

Valproate attenuates behavioural motor deficits caused by lactacystin. (A) Vertical cylinder test outcomes show that once animals begin treatment with valproate at week 1 the lactacystin-induced reduction in percentage contralateral forelimb use is reversed in a dose-dependent manner. (B and C) Amphetamine-induced rotation test outcomes show that animals treated with valproate perform dose-dependently fewer rotations at each of the examined time points after starting treatment with valproate at week 1. Statistical significance denoted with either asterisks: (*P < 0.05) or letters where P < 0.05 for each comparison (see text for degrees of significance): aSignificantly different from group Lacta(−)VPA(−); bSignificantly different from group Lacta(−)VPA(++); cSignificantly different from group Lacta(+)VPA(−); dSignificantly different from group Lacta(+)VPA(+). Data are presented as mean ± SEM. n = 6–7 per group. Abbreviations: Lacta(−), lactacystin naïve; Lacta(+), lactacystin lesioned; VPA(−), treated with vehicle; VPA(+), treated with 200 mg·kg−1·day−1 valproate; VPA(++), treated with 400 mg·kg−1·day−1 valproate.
Amphetamine-induced rotations
Ipsilateral rotations following an amphetamine challenge were observed at 1 week in all lactacystin-lesioned animals (Figure 2B). Saline-treated lactacystin-lesioned animals performed increasing numbers of rotations over the further two time points examined (AUC produced by plotting number of rotations vs. time, Figure 2C). However, animals treated with the lowest dose of valproate performed fewer rotations than saline-treated animals after 28 days of valproate treatment, whereas animals treated with the highest dose of valproate (400 mg·kg−1) performed fewer rotations than the saline-treated group after both 14 and 28 days of valproate treatment.
MRI reveals dose-dependent attenuation of lactacystin-induced volumetric changes by valproate
In vivo rat brain MRIs were acquired longitudinally to follow the neuropathological progression of the lactacystin PD model as previously described (Vernon et al., 2010a; 2011,). A combination of manual segmentation analysis and automated, unbiased TBM were used to quantify neuropathological changes observed as a result of lactacystin lesioning and determine the effects of valproate treatment upon this progression.
Manual segmentation analysis
No significant difference was observed in baseline brain volume between groups (data not shown); however, to account for individual variations in brain size, data are presented as percentage change from baseline. In line with rat growth and consistent with MRI finding in rats of comparable age to those used in this study, there was a steady increase in whole brain volume in surgically naïve saline-treated animals over the 5-week study (Figure 3A). Lactacystin-lesioned animals treated with saline and lesioned animals treated with the lower dose of valproate, however, both exhibited a greater increase in brain volume compared with control animals. Whereas animal groups treated with valproate at its higher dose, both lactacystin lesioned and surgically naïve, exhibited a discernible shallower increase in whole brain volume over the 5 weeks of treatment.
Figure 3.
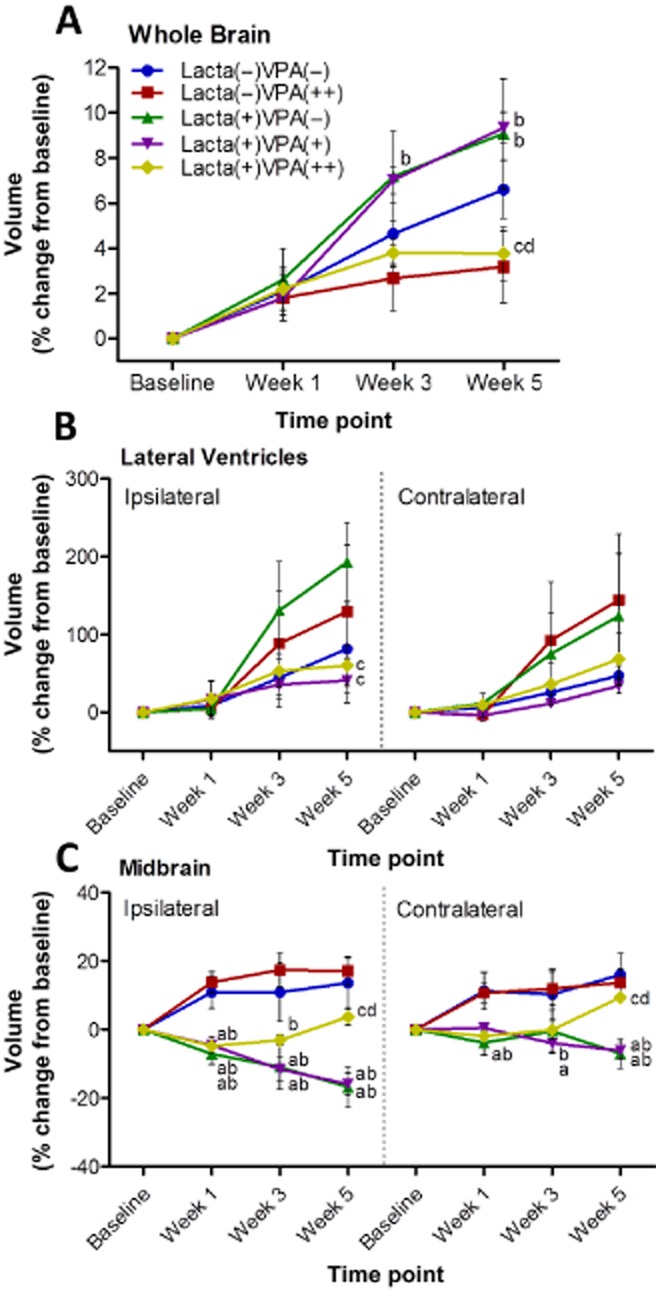
Manual segmentation analysis of MRI reveals dose-dependent attenuation of lactacystin-induced volumetric changes by valproate. Administration of valproate dose-dependently attenuated volumetric changes observed in the (B) lateral ventricles and (C) the midbrain as a result of lactacystin lesioning, as ascertained through manual segmentation analysis of rat brain MRI. Similarly, administration of valproate and/or lactacystin had marked effects on (A) whole brain volume. Statistical significance denoted with letters where P < 0.05 for each comparison (see text for degrees of significance): aSignificantly different from group Lacta(−)VPA(−); bSignificantly different from group Lacta(−)VPA(++); cSignificantly different from group Lacta(+)VPA(−); dSignificantly different from group Lacta(+)VPA(+). Data are presented as mean ± SEM. n = 6–7 per group. Abbreviations: Lacta(−), lactacystin naïve; Lacta(+), lactacystin lesioned; VPA(−), treated with vehicle; VPA(+), treated with 200 mg·kg−1·day−1 valproate; VPA(++), treated with 400 mg·kg−1·day−1 valproate. See also Supporting Information Figure S2.
In surgically naïve animals, the volume of the lateral ventricles increased comparably in both hemispheres over the 5 week study (Figure 3B). However, lactacystin-lesioned animals subsequently treated with saline exhibited a greater increase in ventricular volume, a change that was more pronounced in the lesioned hemisphere. In contrast, lactacystin-lesioned animals treated with valproate (both 200 and 400 mg·kg−1) did not exhibit these increases in ventricular volume over time.
One week after lesioning surgery, prior to starting vehicle/drug treatment, the ipsilateral midbrain volume of all lactacystin-lesioned animal groups was significantly lower than the volume of both surgically naïve groups (Figure 3C). The ipsilateral midbrain volume in lactacystin-lesioned saline and low-dose (200 mg·kg−1) valproate-treated animals continued to decline over the further two time points examined, remaining significantly different from the volume change in surgically naïve groups at both week 3 and week 5 (P < 0.001 in all comparisons). In contrast, lactacystin-lesioned animals treated with the higher dose (400 mg·kg−1) of valproate displayed a reversal of the decrease in ipsilateral midbrain volume seen at week 1 in the following two time points: reaching a significant difference from saline and low dose (200 mg·kg−1) treated animals at week 5. Identical albeit more subtle changes were similarly observed in the contralateral hemisphere of the midbrain.
No significant differences were observed in corpus striatum volume in either the ipsilateral or contralateral hemispheres. However, a number of trends in the ipsilateral hemisphere exist subtly mimicking those changes observed in the midbrain (Supporting Information Figure S2). Similarly, no discernible changes in volume of the either ipsilateral or contralateral hippocampus, or the cerebellum were observed (Supporting Information Figure S2).
TBM analysis
To confirm and extend our manual segmentation observations, we applied TBM analysis to MRIs acquired from all groups 5 weeks post-surgery (Figure 4). After correcting for global differences in brain volume (9 dof registration) to search for relative differences in brain volume, several distinct anatomical patterns were observed across treatment groups (all data shown are corrected for multiple comparison over voxels using the false discovery rate with q < 0.05).
Figure 4.
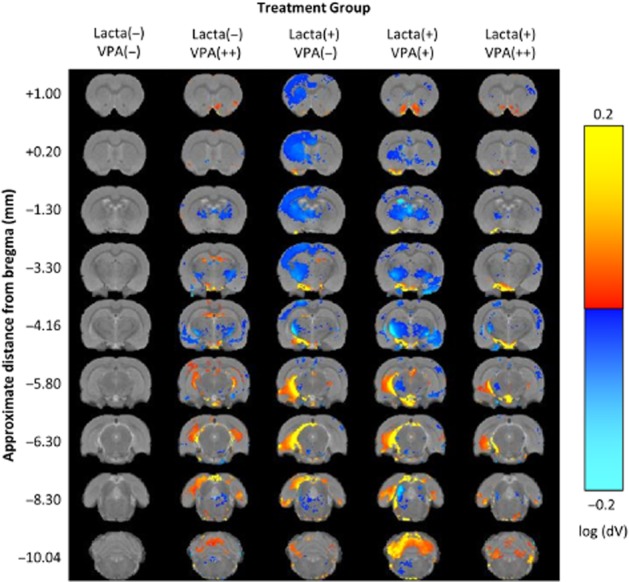
Tensor-based morphometry validates findings from manual segmentation analyses of rat brain MRI. Regions of significant volume difference relative to whole brain for each group compared with Lacta(−)VPA(−) at week 5 are shown. Positive differences indicate where each group has volume increases compared with Lacta(−)VPA(−) and negative differences indicate where each group has volume decreases compared with Lacta(−)VPA(−). Results shown are significant after correction for multiple comparisons across voxels using the false discovery rate with q = 0.05. n = 6–7 per group. Abbreviations: Lacta(−), lactacystin naïve; Lacta(+), lactacystin lesioned; VPA(−), treated with vehicle; VPA(+), treated with 200 mg·kg−1·day−1 valproate; VPA(++), treated with 400 mg·kg−1·day−1 valproate.
Surgically naïve animals treated with valproate showed bilateral clusters of contracted voxels in the globus pallidus, internal capsule, third ventricle, ventromedial thalamic nuclei, perirhinal cortex, amygdala and brainstem. Conversely, clusters of expanded voxels were detected in the hippocampus (dorsal and ventral regions), dorsal entorhinal cortex, external capsule and the third and fourth cerebellar lobules.
In lactacystin-lesioned animals treated with saline, we observed widespread significant contraction of cortical voxels in the ipsilateral (lesioned) hemisphere of the brain. These included the cingulate, motor, somatosensory and parietal cortex sub-fields. Sub-cortically, widespread clusters of contracted voxels were observed in the ipsilateral striatum, globus pallidus, thalamus, ventral midbrain and brainstem nuclei. Some of these clusters extended across the midline into the contralateral (non-lesioned) hemisphere. No significantly contracted voxels were observed in the hippocampus. These data are consistent with prior observations in this model (Vernon et al., 2011). Clusters of significantly expanded voxels were also observed in the ipsilateral hemisphere. Primarily, this reflected an increase in CSF signal accompanying deformation of the ventral midbrain. Significantly, expanded voxels were however also seen in the cerebellar white matter and dorsolateral entorhinal cortex.
In lactacystin-lesioned animals treated with low (200 mg·kg−1) and high doses (400 mg·kg−1) of valproate a more complex pattern of anatomical changes were detected. Valproate treatment dose dependently reversed cortical atrophy in the cingulate, motor, sensorimotor and parietal cortices in the ipsilateral (lesioned) hemisphere. Sub-cortically, valproate treatment also dose dependently reversed atrophy of the ventromedial thalamus, ventral midbrain and expansion of CSF space. These effects were more marked at the higher dose tested (400 mg·kg−1). Drug-specific effects of valproate were however also present, which make interpretation of the results difficult. Indeed, low-dose (200 mg·kg−1) valproate treatment resulted in bilateral clusters of contracted voxels in the ventromedial thalamus, globus pallidus, internal capsule and third ventricle. These patterns are consistent with the effects observed in surgically naïve animals treated with valproate alone, suggesting a drug-specific pattern. Interestingly, these patterns were however absent in lactacystin-lesioned animals given a high dose of valproate (400 mg·kg−1), perhaps suggestive of dose-dependent drug × disease pathology interactions.
Valproate treatment causes dose-dependent protection/restoration of dopaminergic neurons in the SNpc in lactacystin-lesioned animals
Post-study, animals were culled and hind brain tissue collected for immunohistochemical staining and stereological quantification of dopaminergic neurons (TH+) in the SNpc (Figures 5 and 6). Non-lesioned animals treated with either saline or valproate (400 mg·kg−1) did not show any interhemispheric loss of TH+ neurons in the SNpc. However, animals lesioned with lactacystin and culled 7 days post-surgery, the time point when valproate administration was initiated, exhibited a marked interhemispheric loss of TH+ neurons due to the intranigral injection of lactacystin. The loss of TH+ neurons continued to increase with time, rising to −64.24 ± 11.34% in the lactacystin-lesioned animals treated with saline for 28 days, demonstrating further neurodegeneration after week 1. In contrast, valproate administration in lactacystin-lesioned animals resulted in a dose-dependent neuroprotective/restorative effect. When administered at its higher dose (400 mg·kg−1) for 28 days it afforded near complete protection/restoration of SNpc TH+ neurons against the toxic effects of lactacystin. Similarly, administration of the lower dose (200 mg·kg−1) of valproate resulted in only a partial protection of the TH+ neurons against lactacystin toxicity. All changes observed in SNpc TH+ cell number were reproduced in the numbers of Nissl+ cell number, indicative of TH+ neuronal cell death rather than loss of the TH enzyme expression in dying neurons.
Figure 5.

Valproate treatment causes dose-dependent protection/restoration of dopaminergic neurons in the SNpc in lactacystin-lesioned animals. Stereologically estimated (A) TH+ and (B) Nissl+ neuron numbers in the SNpc of rats suggest a dose-dependent neuroprotective/restorative effect of valproate in this lactacystin rat model of Parkinson's disease. This is exemplified by the percentage interhemispheric loss of TH+ (C) and Nissl+ (D) neurons calculated between hemispheres of the SNpc. Statistical significance indicated with asterisks: *P < 0.05; **P < 0.01, ***P < 0.001. Data are presented as mean ± SEM. n = 6–7 per group. Abbreviations: C, contralateral; I, ipsilateral; Lacta(−), lactacystin naïve; Lacta(+), lactacystin lesioned; VPA(−), treated with vehicle; VPA(+), treated with 200 mg·kg−1·day−1 valproate; VPA(++), treated with 400 mg·kg−1·day−1 valproate.
Figure 6.
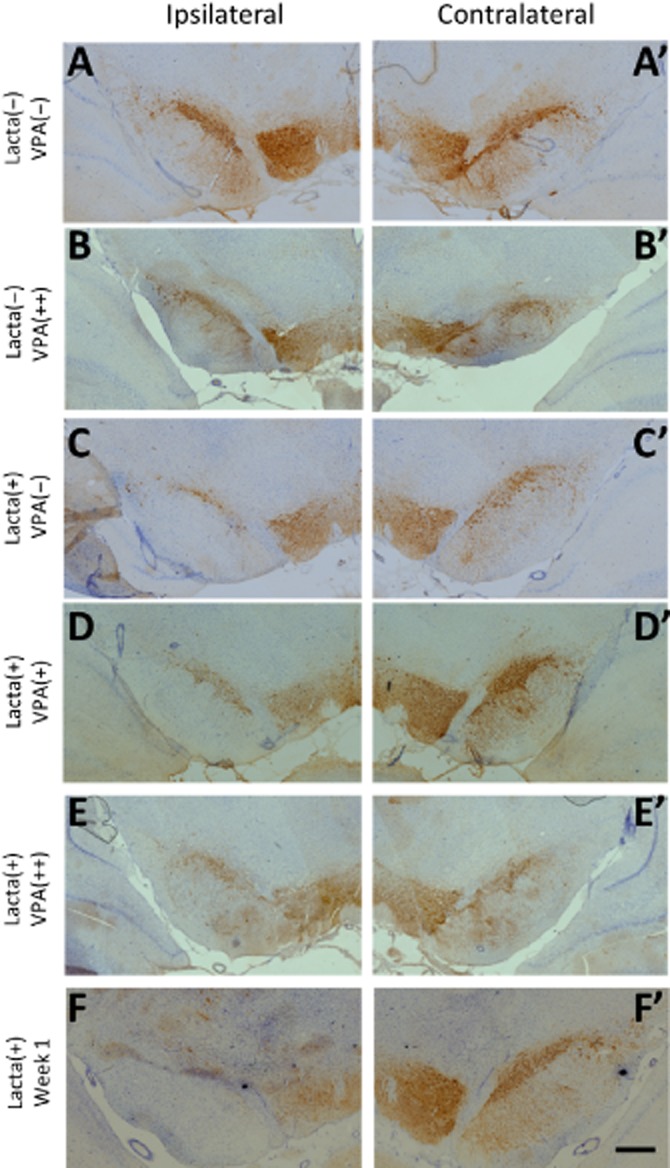
Neuroprotection/restoration in the SNpc of lactacystin-lesioned animals. Representative examples of the TH and Nissl-stained ipsilateral (A–F) and contralateral (A′–F′) SNpc of rats in each of the five treatment groups and group of animals killed before drug treatment. Scale bar equal to 500 μm. Abbreviations: Lacta(−), lactacystin naïve; Lacta(+), lactacystin lesioned; VPA(−), treated with vehicle; VPA(+), treated with 200 mg·kg−1·day−1 valproate; VPA(++), treated with 400 mg·kg−1·day−1 valproate.
Valproate dose-dependently attenuated lactacystin-induced reduction in histone acetylation
Upon removal of brain tissue at the end of the study, the frontal brain was snap frozen for subsequent quantification of histone acetylation through quantification of AcH3-Lys9 using Western blot analysis (Figure 7). Significantly, less AcH3-Lys9 was observed in both hemispheres of lactacystin-lesioned animals treated with saline compared with non-lesioned animals. This effect was dose-dependently attenuated in both hemispheres of animals treated with valproate, reaching significance from saline-treated animals in the ipsilateral hemisphere (ipsilateral hemisphere, saline-treated animals, 0.02 ± 0.03 vs. animals treated with 400 mg·kg−1 valproate, 0.84 ± 0.08, P < 0.05). No difference in the amount of AcH3-Lys9 in either hemisphere was observed in non-lesioned animals treated with valproate compared with control.
Figure 7.
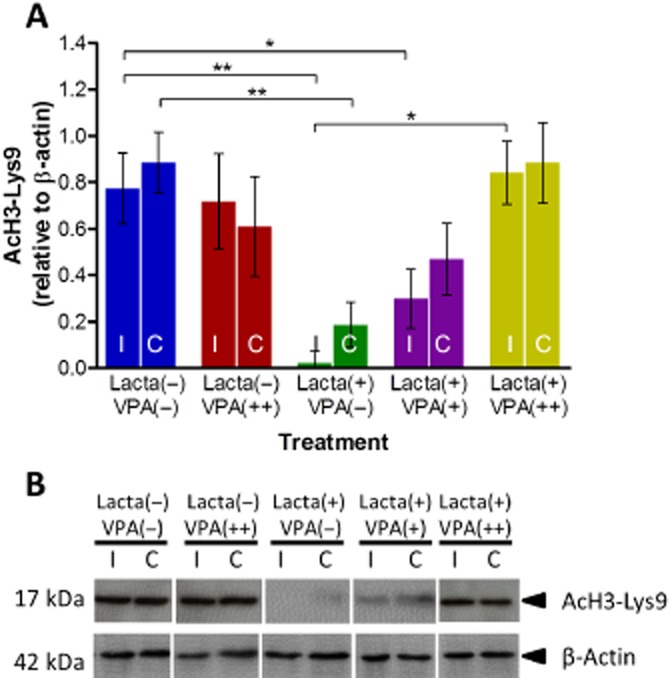
Valproate attenuates lactacystin-induced reduction in frontal brain histone acetylation. Administration of systemic valproate dose-dependently reversed the reduction in histone H3-lysine 9 acetylation caused by lactacystin. (A) Densitometry analysis of the AcH3-Lys9 band relative to the β-actin band used as a loading control. (B) Representative blot of data presented in (A). Statistical significance indicated with asterisks: *P < 0.05; **P < 0.01. Data are presented as mean ± SEM. n = 6–7 per group. Abbreviations: C, contralateral; I, ipsilateral; Lacta(−), lactacystin naïve; Lacta(+), lactacystin lesioned; VPA(−), treated with vehicle; VPA(+), treated with 200 mg·kg−1·day−1 valproate; VPA(++), treated with 400 mg·kg−1·day−1 valproate.
Valproate up-regulates expression of neurotrophic growth factors and neuroprotective proteins
In non-lesioned animals, administration of valproate induced a marked up-regulation of brain-derived neurotrophic factor (BDNF) (Figure 8A). Valproate did not alter the expression of the other genes examined in these surgically naïve animals. However, in lactacystin-lesioned animals valproate significantly and dose-dependently up-regulated the expression of BDNF, glial-derived neurotrophic factor (GDNF) and the anti-apoptotic factor Bcl2, in the frontal brain hemisphere contralateral to the lesion. Most notably, GDNF expression was greater in the contralateral hemisphere of valproate compared with saline-treated animals (Figure 8B). Similarly, expression of BDNF and Bcl2 was elevated in a dose-dependent manner in the frontal brain hemisphere contralateral to the lesion; however, this only reached significance at the higher dose of valproate. Similar trends of an up-regulation of gene expression of native αSyn and heat shock protein 70 upon treatment with valproate were observed, but these did not reach significance.
Figure 8.
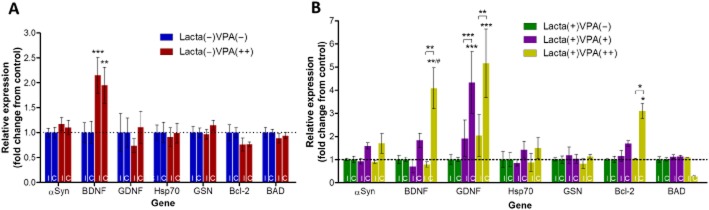
Valproate up-regulates expression of neuroprotective and neurotrophic growth factor mRNA in the frontal brain. (A) Administration of systemic valproate alone, in surgically naïve rats up-regulated bilateral expression of BDNF. (B) In lactacystin-lesioned animals valproate dose-dependently up-regulated unilateral expression of αSyn, BDNF, GDNF, heat shock protein 70 (Hsp70) and Bcl2, as well as reducing the expression of BAD when given at its highest dose. Statistical significance indicated with asterisks and hashes: *P < 0.05, **P < 0.01, ***P < 0.001 compared with the same hemisphere of saline-treated group; #P < 0.05, ##P < 0.01, ###P < 0.001 compared with the same hemisphere of Lacta(+)VPA(+). Data are presented as mean ± SEM. n = 6–7 per group. Abbreviations: C, contralateral; I, ipsilateral; Lacta(−), lactacystin naïve; Lacta(+), lactacystin lesioned; VPA(−), treated with vehicle; VPA(+), treated with 200 mg·kg−1·day−1 valproate; VPA(++), treated with 400 mg·kg−1·day−1 valproate. See also Supporting Information Table S1.
Discussion
Valproate dose-dependently afforded neuroprotection and neurorestoration of nigrostriatal dopaminergic neurons in the lactacystin animal model of PD, as evidenced by an attenuation of motor behavioural deficits, longitudinal MRI brain volume changes and quantification of the dopaminergic neurons within the SNpc. Molecular analyses indicate that valproate's neuroprotective effects may be mediated through epigenetic changes via inhibition of histone deacetylation, chromatin remodelling and up-regulation of neurotrophic and neuroprotective genes, culminating in the observed neuroprotective and neurorestorative effect.
MRI studies using toxin-based models of PD have largely been focused on alterations in T2 water 1H relaxation time and 1H MR spectroscopy, whereas potential morphological changes have been overlooked. Recently, however, a method of using MRI to non-invasively monitor the morphological progression and nigrostriatal neuropathology in this proteasome inhibitor rat model of PD has been established (Vernon et al., 2010a; 2011,). To the best of our knowledge, the current study is the first to use this method in conjunction with a candidate neuroprotective drug to longitudinally assess its efficacy in an animal model of PD. Manual segmentation analysis was performed to examine the temporal progression of morphological changes in selected brain regions. In line with previously published data following nigrostriatal neuropathology induced as a result of SNpc lactacystin (Vernon et al., 2010a; 2011,), we observed a marked reduction in the volume of the ipsilateral midbrain in the weeks following lactacystin lesioning. Similarly, we observe an increase in the volume of the lateral ventricles; more pronounced in the ipsilateral as opposed to the contralateral hemisphere. Both of these changes were dose dependently attenuated by delayed-start valproate treatment (200 and 400 mg·kg−1·day−1). Automated, unbiased TBM analysis (Vernon et al., 2011; Crum et al., 2013b) confirms and extends the manual segmentation data. Indeed, TBM highlights that chronic valproate treatment by itself induces a specific pattern of structural re-modelling (expansion and contraction) in the healthy brain. This is interesting in light of recent observations for other psychotropic drugs, including lithium, a mood stabilizer (Vernon et al., 2012; 2013,). Similarly, TBM data confirm lactacystin lesioning by itself is associated with a specific reproducible pattern of neuroanatomical changes in the brain, detectable by MRI (Vernon et al., 2011), modelling features of advanced PD, but also atypical forms of PD, such as progressive supranuclear palsy and multiple system atrophy (Vernon et al., 2010b; Duncan et al., 2013). Thus, our analysis pipeline is robust and reproducible enough to detect the same patterns in data generated and acquired in different academic centres. Together, the data suggest a complex pattern of drug × disease interactions potentially driving the anatomical effects observed herein. On the one hand, TBM identifies a clear neuroprotective effect of valproate, namely prevention of brain atrophy induced by lactacystin lesioning, entirely consistent with the manual segmentation results. On the other hand, valproate itself induces both brain atrophy and growth in topographically distinct brain regions, reinforcing the importance of examining drug effects in both normal and diseased animals. Nevertheless, our unbiased, automated approach re-enforces our manual observations. That is, we are able to detect a dose-dependent neuroprotective effect of valproate on lactacystin-induced brain atrophy, which was associated with both cellular and functional neuroprotection and neurorestoration and improvements in motor behaviour. The combination of longitudinal in vivo MRI and automated TBM, a clinically comparable technology, therefore has great potential for preclinical assessment of drugs with disease-modifying potential in preclinical models of PD, and other neurological disorders, which may speed the translation of basic findings to the clinic.
The data described here importantly illustrate the neuroprotective effects of valproate when administered post-lesion in the lactacystin rat model of PD are consistent with previous studies in which valproate was administered prior to the neurotoxin in other preclinical PD models. Monti et al. (2010) observed that dietary pretreatment resulted in a significant preservation of the SNpc TH+ dopaminergic neurons in rats subchronically administered rotenone. They also observed an attenuation of the loss of striatal dopamine and consistent with in vitro findings from their group (Monti et al., 2007). They observed an increase in native αSyn expression in the SNpc and striatum in line with a reduction in mono-ubiquitinated αSyn and its nuclear translocation following valproate administration. αSyn is thought to result in histone hypoacetylation through its ‘masking’ of histone proteins (Kontopoulos et al., 2006). Therefore, consistent with the lack of αSyn nuclear translocation, the group also observe an increase in histone acetylation upon valproate treatment. Monti et al. (2012) also demonstrated that dietary pretreatment with valproate resulted in protection of the dopaminergic neuronal terminals in the striatum as well as the dopaminergic cell bodies in the SNpc against striatally administered 6-OHDA. Similarly, Kidd and Schneider (2011) demonstrated the neuroprotective effects of valproate, at both the SNpc dopaminergic cell body and striatal dopamine level, when administered systemically prior to the administration of the mitochondrial toxin MPTP to mice. Importantly, in all of these studies valproate was administered prior to the toxin and hence they do not mirror the clinical setting in which a neuroprotective agent will currently be used: by the time the cardinal motor symptoms of PD become evident, 60–70% of neurons in the SNpc and ∼80% of striatal dopamine have already been depleted (Riederer and Wuketich, 1976). Conversely, in this important study we have for the first time shown valproate to be both neuroprotective and neurorestorative when administered 1 week after toxin administration in a ‘delayed-start design’, when the neurodegenerative process has been initiated and we can already observe clinical behavioural and MRI changes in the animal model. Stereological quantification of dopaminergic neurons in animals culled 1 week post lactacystin lesion, that is, the point at which valproate treatment was started, indicate that the vast majority of nigral degeneration occurs in these first 7 days. Importantly, however, despite the delayed start in treatment, less neurodegeneration of nigral dopaminergic neurons was observed after 4 weeks of valproate treatment, indicating that valproate is not only acting neuroprotectively towards dopaminergic nigral neurons but is also acting neurorestoratively. Appropriately, Nissl+ neuronal degeneration was markedly less than that of TH+ cells in animals culled prior to valproate treatment. It could be possible then that valproate in the current study is acting to rescue unhealthy and/or dying neurons that have lost TH expression in the cell body and therefore have reduced function, yet retain their TH+ projections, aiding their functional recovery, translating to performance recovery in behavioural tests such as the vertical cylinder test.
Valproate has also been shown to partially protect against motor deficits in animal models of traumatic brain injury (Dash et al., 2010), spinal cord injury (Lee et al., 2012) and stroke (Kim et al., 2007) yet to our knowledge the present study is the first to examine the ability of valproate to relieve motor symptoms in an animal model of PD. We demonstrate that delayed valproate treatment dose dependently reverses lactacystin-induced reduction in the use of the forelimb contralateral to the lesion, and also attenuates the number of rotations performed upon amphetamine challenge. Interestingly, using a battery of behavioural tests, Castro et al. (2012) also demonstrated that valproate pretreatment prevented the development of early non-motor symptoms of PD, for example, cognitive and emotional deficits, in animals nasally administered MPTP (Prediger et al., 2011). Authors demonstrated that this was accompanied by a significant preservation of olfactory bulb and striatal dopamine content in MPTP-treated animals (Castro et al., 2012). This suggests that valproate is also neuroprotective to other neuronal systems that is particularly important since the neurodegenerative process in PD is not merely confined to the dopaminergic nigrostriatal system (Braak et al., 2003). Our MRI findings are consistent with this: lactacystin-induced atrophy in numerous extra-nigral brain regions such as the cingulate, motor, sensorimotor and parietal cortices are ameliorated by valproate.
The misbalance between HATs and HDACs that are thought to lead to histone hypoacetylation in neurodegeneration was first noted by Rouaux et al. (2003). Since then, histone hypoacetylation has become heavily implicated in neurodegenerative diseases such as PD: it being demonstrated that αSyn accumulation actively promotes histone hypoacetylation both in vitro in SH-SY5Y cells and in vivo in drosophila, both overexpressing αSyn (Kontopoulos et al., 2006). Lactacystin dopaminergic neurotoxicity is associated with the aggregation of αSyn to form inclusion bodies in the SNpc, a finding that has been extensively verified since (McNaught et al., 2002; Zhu et al., 2007; Niu et al., 2009; Vernon et al., 2011). αSyn is thought to result in histone hypoacetylation through its ‘masking’ of histone proteins (Kontopoulos et al., 2006). Therefore, in line with these previous observations of the effect of αSyn on histone acetylation, in the current study we observed a reduction in AcH3-Lys9, in the brains of lactacystin-lesioned animals, used as a surrogate marker for histone deacetylation in the brains of these animals. Importantly, valproate treatment was observed to dose dependently attenuate this histone hypoacetylation, in parallel with the cellular neuroprotective and restorative effect of the drug shown through stereological cell quantification of SNpc dopaminergic neurons. Valproate is a somewhat promiscuous drug: affecting glutamatergic and GABAergic transmission in the brain, and modulating ionic channels such as the voltage-gated Na+ and T-type Ca2+ channels. However, the dose-dependent attenuation of histone hypoacetylation induced by valproate in line with the extent of neuroprotection/restoration observed is suggestive that valproate's inhibition of HDACs is at least partly responsible for the effects observed.
Addition of an acetyl group to histone lysine residues neutralizes the positive charge of the residue and hence reduces the electrostatic interaction between the lysine in the histone tail and the negatively charged phosphate group on DNA. This disrupts the inter- and intra-nucleosomal interactions between the histone and DNA and hence relaxes the structure of the chromatin allowing transcription factor access. Inhibition of HDACs and histone acetylation in the brain has therefore been shown to be associated with transcriptional up-regulation of numerous factors that are thought to contribute to the neuroprotective/restorative effects observed by valproate (Monti et al., 2009). Similarly, we have observed that the neurotrophic factors BDNF and GDNF are significantly up-regulated upon valproate treatment, confirming the studies by Wu et al. (2008) who demonstrated that an astrocytic cell line treated with valproate displays a time-dependent increase in expression of both BDNF and GDNF; an effect that translated to neuroprotection in midbrain neuronal cultures in medium transfer experiments. Additionally, Wu and colleagues demonstrated that the GDNF promoter-associated histone H3 is significantly hyperacetylated when astrocytes are treated with valproate. Importantly, GDNF was the only one of the neuroprotective and neurotrophic factors measured to be up-regulated in both the ipsilateral and contralateral hemispheres, highlighting the possible role of astrocytes in the neurorestorative mechanism of valproate observed here. The magnitude of up-regulation of GDNF expression from control was far greater in the contralateral hemisphere in the current study; however, it is important to note that a twofold up-regulation of GDNF was also observed in the ipsilateral hemisphere despite being on a background of severe neurodegeneration. Given the body of evidence in favour of the neuroprotective and neurorestorative effects of GDNF in vivo (Allen et al., 2013; Kordower and Bjorklund, 2013; Hegarty et al., 2014), the up-regulation observed here in the ipsilateral hemisphere may well contribute to the neuroprotective and neuroregenerative phenotype observed in this model. We also showed in this animal study for the first time that gene expression of the anti-apoptotic molecule BCL2 is up-regulated upon valproate treatment, and the pro-apoptotic molecule BAD is down-regulated. This is in agreement with previous data from Kidd and Schneider (2010) in which dopaminergic cells treated with valproate in vitro displayed a reduction in MPP+-induced activation of caspase-3 indicative of apoptosis inhibition. The findings from the current study therefore suggest that valproate's mechanism of histone acetylation mediated neuroprotection/restoration is a multifaceted and complex affair, thought to be mainly mediated by the effects of GDNF. Such a mechanism may therefore hold therapeutic potential against a complex disorder such as PD.
The doses of valproate administered to rats in this study translate to human equivalent doses of 64 and 32 mg·kg−1·day−1 (400 and 200 mg·kg−1·day−1 rat dose, respectively, as rats metabolize valproate quicker) [as calculated using FDA (2005) guidelines]. These doses are far greater than the usual therapeutic maintenance dose of valproate used for the treatment of epilepsy in humans (1000–2000 mg·day−1) and hence may result in increased incidence of side effects such as extrapyramidal effects, tremor, sleepiness, cognitive problems etc. if administered to humans (Britain BMAatRPSoG, 2009). Obviously, such side effects would aggravate PD symptomatology; hence, further research is required to determine if valproate itself, at the doses examined here, represents a candidate for repositioning for PD. However, the current study acts as proof of principle that delayed-start treatment with a HDACI is capable of producing a neuroprotective phenotype in this animal model of PD.
In conclusion, utilizing a clinically relevant drug testing platform this study clearly demonstrates that the HDACI valproate is dose-dependently neuroprotective and neurorestorative in the lactacystin rodent model of PD when administered chronically starting 7 days after the toxin administration when behavioural and MRI deficits are already evident and a significant deficit in dopaminergic neurons is observed. These effects of valproate are associated with a reversal of histone hypoacetylation and an up-regulation of neuroprotective and neurotrophic factors. This therefore supports the potential neuroprotective benefits of HDAC inhibition in PD and advocates further investigation of which specific HDAC isoforms are responsible for the neuroprotective effects of valproate in lieu of a clinically translatable HDACI treatment strategy for PD.
Acknowledgments
I. F. H. was supported by a UK Medical Research Council PhD studentship. W. R. C. acknowledges support from the King's College London Centre of Excellence in Medical Engineering funded by the Wellcome Trust and EPSRC (WT 088641/Z/09/Z). A. C. V. acknowledges support from the Medical Research Council (G1002198), the Psychiatry Research Trust (McGregor 97), the Royal Society (RG130610), King's Health Partners and the Guy's and St. Thomas' Charitable Trust (R140805).
Glossary
- 6-OHDA
6-hydroxydopamine
- αSyn
α-synuclein
- AcH3-Lys9
histone protein H3 acetylated on lysine 9
- BDNF
brain-derived neurotrophic factor
- GDNF
glial-derived neurotrophic factor
- HATs
histone acetyltransferases
- HDACIs
histone deacetylase inhibitors
- HDACs
histone deacetylases
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- PD
Parkinson's disease
- qRT-PCR
quantitative real-time PCR
- SNpc
substantia nigra pars compacta
- TBM
tensor-based morphometry
Author contributions
I. F. H. contributed to the design of the study, performed all experiments and analyses, and wrote the manuscript. W. R. C. performed the tensor-based morphometry and contributed to the manuscript critique. A. C. V. contributed to the interpretation of data and contributed to the manuscript critique. D. T. D. conceived and helped design the study, and contributed to the manuscript critique.
Conflict of interest
Authors declare that there are no conflicts of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 Anatomical landmarks and criteria used for manual segmentation analysis of MRI. Six brain structures delineated manually with reference to the anatomical landmarks: whole brain, lateral ventricles, corpus striatum, midbrain, hippocampus and cerebellum.30
Figure S2 Supplemental data from manual segmentation analysis of MRI. Changes in volume observed in the (B) corpus striatum and (C) the hippocampus as a result of lactacystin lesioning/valproate treatment. Similarly administration of valproate and/or lactacystin have marked effects on the (A) cerebellum volume as ascertained through manual segmentation analysis of rat brain MRI. Statistical significance denoted with letters where P < 0.05 for each comparison (see text for degrees of significance): aSignificantly different from group Lacta(−)VPA(−); bSignificantly different from group Lacta(−)VPA(++); cSignificantly different from group Lacta(+)VPA(−); dSignificantly different from group Lacta(+)VPA(+). Data are presented as mean ± SEM. n = 6–7 per group. Abbreviations: Lacta(−), lactacystin naïve; Lacta(+), lactacystin lesioned; VPA(−), treated with vehicle; VPA(+), treated with 200 mg·kg−1·day−1 valproate; VPA(++), treated with 400 mg·kg−1·day−1 valproate.
Table S1 Probe and primer sequences of PrimeTime™ qPCR assays used. All PrimeTime™ qPCR assays were obtained from integrated DNA technology (Coralville, IA, USA) and contained 2.5 nM of probe, 5 nM of primer 1 and 5 nM of primer 2. Abbreviations: A, adenine; C, cytosine; G, guanine; T, thymine; HEX™, hexachlorofluorescein; IABkFQ, Iowa Black® FQ.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: enzymes. Br J Pharmacol. 2013;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen SJ, Watson JJ, Shoemark DK, Barua NU, Patel NK. GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol Ther. 2013;138:155–175. doi: 10.1016/j.pharmthera.2013.01.004. [DOI] [PubMed] [Google Scholar]
- Ammal Kaidery N, Tarannum S, Thomas B. Epigenetic landscape of Parkinson's disease: emerging role in disease mechanisms and therapeutic modalities. Neurother. 2013;10:698–708. doi: 10.1007/s13311-013-0211-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Tredici KD, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Britain BMAatRPSoG. British National Formulary. 58 edn. London: BMJ Publishing Group; 2009. [Google Scholar]
- Bullmore ET, Suckling J, Overmeyer S, Rabe-Hesketh S, Taylor E, Brammer MJ. Global, voxel, and cluster tests, by theory and permutation, for a difference between two groups of structural MR images of the brain. IEEE Trans Med Imaging. 1999;18:32–42. doi: 10.1109/42.750253. [DOI] [PubMed] [Google Scholar]
- Carman LS, Gage FH, Shults CW. Partial lesion of the substantia nigra: relation between extent of lesion and rotational behavior. Brain Res. 1991;553:275–283. doi: 10.1016/0006-8993(91)90835-j. [DOI] [PubMed] [Google Scholar]
- Castro AA, Ghisoni K, Latini A, Quevedo J, Tasca CI, Prediger RDS. Lithium and valproate prevent olfactory discrimination and short-term memory impairments in the intranasal 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) rat model of Parkinson's disease. Behav Brain Res. 2012;229:208–215. doi: 10.1016/j.bbr.2012.01.016. [DOI] [PubMed] [Google Scholar]
- Chen PS, Peng GS, Li G, Yang S, Wu X, Wang CC, et al. Valproate protects dopaminergic neurons in midbrain neuron/glia cultures by stimulating the release of neurotrophic factors from astrocytes. Mol Psychiatry. 2006;11:1116–1125. doi: 10.1038/sj.mp.4001893. [DOI] [PubMed] [Google Scholar]
- Chuang D-M, Leng Y, Marinova Z, Kim H-J, Chiu C-T. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 2009;32:591–601. doi: 10.1016/j.tins.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crum WR, Giampietro VP, Smith EJ, Gorenkova N, Stroemer RP, Modo M. A comparison of automated anatomical–behavioural mapping methods in a rodent model of stroke. J Neurosci Methods. 2013a;218:170–183. doi: 10.1016/j.jneumeth.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crum WR, Modo M, Vernon AC, Barker GJ, Williams SCR. Registration of challenging pre-clinical brain images. J Neurosci Methods. 2013b;216:62–77. doi: 10.1016/j.jneumeth.2013.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash PK, Orsi SA, Zhang M, Grill RJ, Pati S, Zhao J, et al. Valproate administered after traumatic brain injury provides neuroprotection and improves cognitive function in rats. PLoS ONE. 2010;5:e11383. doi: 10.1371/journal.pone.0011383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desplats P, Spencer B, Coffee E, Patel P, Michael S, Patrick C, et al. a-Synuclein sequesters dnmt1 from the nucleus: a novel mechanism for epigenetic alterations in Lewy body diseases. J Biol Chem. 2011;286:9031–9037. doi: 10.1074/jbc.C110.212589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dexter DT, Jenner P. Parkinson disease: from pathology to molecular disease mechanisms. Free Radic Biol Med. 2013;62:132–144. doi: 10.1016/j.freeradbiomed.2013.01.018. [DOI] [PubMed] [Google Scholar]
- Dietz KC, Casaccia P. HDAC inhibitors and neurodegeneration: at the edge between protection and damage. Pharmacol Res. 2010;62:11–17. doi: 10.1016/j.phrs.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan GW, Firbank MJ, O'Brien JT, Burn DJ. Magnetic resonance imaging: a biomarker for cognitive impairment in Parkinson's disease? Mov Disord. 2013;28:425–438. doi: 10.1002/mds.25352. [DOI] [PubMed] [Google Scholar]
- Durrenberger PF, Fernando FS, Magliozzi R, Kashefi SN, Bonnert TP, Ferrer I, et al. Selection of novel reference genes for use in the human central nervous system: a BrainNet Europe Study. Acta Neuropathol. 2012;124:893–903. doi: 10.1007/s00401-012-1027-z. [DOI] [PubMed] [Google Scholar]
- FDA. Guidance for Industry: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. Silver Spring, MD: U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER); 2005. [Google Scholar]
- Genovese CR, Lazar NA, Nichols T. Thresholding of statistical maps in functional neuroimaging using the false discovery rate. Neuroimage. 2002;15:870–878. doi: 10.1006/nimg.2001.1037. [DOI] [PubMed] [Google Scholar]
- Gottlicher M, Minucci S, Zhu P, Kramer OH, Schimpf A, Giavara S, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001;20:6969–6978. doi: 10.1093/emboj/20.24.6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurvich N, Tsygankova OM, Meinkoth JL, Klein PS. Histone deacetylase is a target of valproic acid-mediated cellular differentiation. Cancer Res. 2004;64:1079–1086. doi: 10.1158/0008-5472.can-03-0799. [DOI] [PubMed] [Google Scholar]
- Hahnen E, Hauke J, Tränkle C, Eyüpoglu IY, Wirth B, Blümcke I. Histone deacetylase inhibitors: possible implications for neurodegenerative disorders. Expert Opin Investig Drugs. 2008;17:169–184. doi: 10.1517/13543784.17.2.169. [DOI] [PubMed] [Google Scholar]
- Harrison IF, Dexter DT. Epigenetic targeting of histone deacetylase: therapeutic potential in Parkinson's disease? Pharmacol Ther. 2013;140:34–52. doi: 10.1016/j.pharmthera.2013.05.010. [DOI] [PubMed] [Google Scholar]
- Hegarty SV, O'Keeffe GW, Sullivan AM. Neurotrophic factors: from neurodevelopmental regulators to novel therapies for Parkinson's disease. Neural Regen Res. 2014;9:1708–1711. doi: 10.4103/1673-5374.143410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner P, Olanow CW. The pathogenesis of cell death in Parkinson's disease. Neurology. 2006;66(10 Suppl 4):S24–S36. doi: 10.1212/wnl.66.10_suppl_4.s24. [DOI] [PubMed] [Google Scholar]
- Kazantsev AG, Thompson LM. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov. 2008;7:854–868. doi: 10.1038/nrd2681. [DOI] [PubMed] [Google Scholar]
- Kidd SK, Schneider JS. Protection of dopaminergic cells from MPP+-mediated toxicity by histone deacetylase inhibition. Brain Res. 2010;1354:172–178. doi: 10.1016/j.brainres.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd SK, Schneider JS. Protective effects of valproic acid on the nigrostriatal dopamine system in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson's disease. Neuroscience. 2011;194:189–194. doi: 10.1016/j.neuroscience.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8:e1000412. doi: 10.1371/journal.pbio.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Rowe M, Ren M, Hong J-S, Chen P-S, Chuang D-M. Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action. J Pharmacol Exp Ther. 2007;321:892–901. doi: 10.1124/jpet.107.120188. [DOI] [PubMed] [Google Scholar]
- Kontopoulos E, Parvin JD, Feany MB. a-Synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum Mol Genet. 2006;15:3012–3023. doi: 10.1093/hmg/ddl243. [DOI] [PubMed] [Google Scholar]
- Kordower JH, Bjorklund A. Trophic factor gene therapy for Parkinson's disease. Mov Disord. 2013;28:96–109. doi: 10.1002/mds.25344. [DOI] [PubMed] [Google Scholar]
- Lee JY, Kim HS, Choi HY, Oh TH, Ju BG, Yune TY. Valproic acid attenuates blood–spinal cord barrier disruption by inhibiting matrix metalloprotease-9 activity and improves functional recovery after spinal cord injury. J Neurochem. 2012;121:818–829. doi: 10.1111/j.1471-4159.2012.07731.x. [DOI] [PubMed] [Google Scholar]
- Löscher W. Basic pharmacology of valproate: a review after 35 years of clinical use for the treatment of epilepsy. CNS Drugs. 2002;16:669–694. doi: 10.2165/00023210-200216100-00003. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNaught KSP, Bjorklund LM, Belizaire R, Isacson O, Jenner P, Olanow CW. Proteasome inhibition causes nigral degeneration with inclusion bodies in rats. Neuroreport. 2002;13:1437–1441. doi: 10.1097/00001756-200208070-00018. [DOI] [PubMed] [Google Scholar]
- Monti B, Polazzi E, Batti L, Crochemore C, Virgili M, Contestabile A. Alpha-synuclein protects cerebellar granule neurons against 6-hydroxydopamine-induced death. J Neurochem. 2007;103:518–530. doi: 10.1111/j.1471-4159.2007.04778.x. [DOI] [PubMed] [Google Scholar]
- Monti B, Polazzi E, Contestabile A. Biochemical, molecular and epigenetic mechanisms of valproic acid neuroprotection. Curr Mol Pharmacol. 2009;2:95–109. doi: 10.2174/1874467210902010095. [DOI] [PubMed] [Google Scholar]
- Monti B, Gatta V, Piretti F, Raffaelli S, Virgili M, Contestabile A. Valproic acid is neuroprotective in the rotenone rat model of Parkinson's disease: involvement of α-synuclein. Neurotox Res. 2010;17:130–141. doi: 10.1007/s12640-009-9090-5. [DOI] [PubMed] [Google Scholar]
- Monti B, Mercatelli D, Contestabile A. Valproic acid neuroprotection in 6-OHDA lesioned rat, a model for parkinson's disease. HOAJ Biol. 2012;1:4. [Google Scholar]
- Niu C, Mei J, Pan Q, Fu X. Nigral degeneration with inclusion body formation and behavioral changes in rats after proteasomal inhibition. Stereotact Funct Neurosurg. 2009;87:69–81. doi: 10.1159/000202972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl. Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 6th edn. New York: Academic Press; 2009. [Google Scholar]
- Peng G-S, Li G, Tzeng N-S, Chen P-S, Chuang D-M, Hsu Y-D, et al. Valproate pretreatment protects dopaminergic neurons from LPS-induced neurotoxicity in rat primary midbrain cultures: role of microglia. Brain Res Mol Brain Res. 2005;134:162–169. doi: 10.1016/j.molbrainres.2004.10.021. [DOI] [PubMed] [Google Scholar]
- Perucca E. Pharmacological and therapeutic properties of valproate: a summary after 35 years of clinical experience. CNS Drugs. 2002;16:695–714. doi: 10.2165/00023210-200216100-00004. [DOI] [PubMed] [Google Scholar]
- Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem. 2001;276:36734–36741. doi: 10.1074/jbc.M101287200. [DOI] [PubMed] [Google Scholar]
- Pienaar IS, Harrison IF, Elson JL, Bury A, Woll P, Simon AK, et al. An animal model mimicking pedunculopontine nucleus cholinergic degeneration in Parkinson's disease. Brain Struct Funct. 2015;220:479–500. doi: 10.1007/s00429-013-0669-5. [DOI] [PubMed] [Google Scholar]
- Prediger R, Aguiar AJ, Moreira E, Matheus F, Castro A, Walz R, et al. The intranasal administration of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): a new rodent model to test palliative and neuroprotective agents for Parkinson's disease. Curr Pharm Des. 2011;17:489–507. doi: 10.2174/138161211795164095. [DOI] [PubMed] [Google Scholar]
- Rattray I, Smith EJ, Crum WR, Walker TA, Gale R, Bates GP, et al. Correlations of behavioral deficits with brain pathology assessed through longitudinal MRI and histopathology in the R6/1 mouse model of Huntington's disease. PLoS ONE. 2013;8:e60012. doi: 10.1371/journal.pone.0060012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riederer P, Wuketich S. Time course of nigrostriatal degeneration in parkinson's disease. A detailed study of influential factors in human brain amine analysis. J Neural Transm. 1976;38:277–301. doi: 10.1007/BF01249445. [DOI] [PubMed] [Google Scholar]
- Rouaux C, Jokic N, Mbebi C, Boutillier S, Loeffler J-P, Boutillier A-L. Critical loss of CBP/p300 histone acetylase activity by caspase-6 during neurodegeneration. EMBO J. 2003;22:6537–6549. doi: 10.1093/emboj/cdg615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schallert T, Fleming SM, Leasure JL, Tillerson JL, Bland ST. CNS plasticity and assessment of forelimb sensorimotor outcome in unilateral rat models of stroke, cortical ablation, parkinsonism and spinal cord injury. Neuropharmacology. 2000;39:777–787. doi: 10.1016/s0028-3908(00)00005-8. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM-Y, Trojanowski JQ, Jakes R, Goedert M. a-Synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Ungerstedt U, Arbuthnott GW. Quantitative recording of rotational behavior in rats after 6-hydroxy-dopamine lesions of the nigrostriatal dopamine system. Brain Res. 1970;24:485–493. doi: 10.1016/0006-8993(70)90187-3. [DOI] [PubMed] [Google Scholar]
- Vernon A, Johansson S, Modo M. Non-invasive evaluation of nigrostriatal neuropathology in a proteasome inhibitor rodent model of Parkinson's disease. BMC Neurosci. 2010a;11:1. doi: 10.1186/1471-2202-11-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernon AC, Ballard C, Modo M. Neuroimaging for Lewy body disease: is the in vivo molecular imaging of α-synuclein neuropathology required and feasible? Brain Res Rev. 2010b;65:28–55. doi: 10.1016/j.brainresrev.2010.05.006. [DOI] [PubMed] [Google Scholar]
- Vernon AC, Crum WR, Johansson SM, Modo M. Evolution of extra-nigral damage predicts behavioural deficits in a rat proteasome inhibitor model of Parkinson's disease. PLoS ONE. 2011;6:e17269. doi: 10.1371/journal.pone.0017269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernon AC, Natesan S, Crum WR, Cooper JD, Modo M, Williams SCR, et al. Contrasting effects of haloperidol and lithium on rodent brain structure: a magnetic resonance imaging study with postmortem confirmation. Biol Psychiatry. 2012;71:855–863. doi: 10.1016/j.biopsych.2011.12.004. [DOI] [PubMed] [Google Scholar]
- Vernon AC, Crum WR, Lerch JP, Chege W, Natesan S, Modo M, et al. Reduced cortical volume and elevated astrocyte density in rats chronically treated with antipsychotic drugs – linking magnetic resonance imaging findings to cellular pathology. Biol Psychiatry. 2013;75:982–990. doi: 10.1016/j.biopsych.2013.09.012. [DOI] [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJG. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- Wu X, Chen PS, Dallas S, Wilson B, Block ML, Wang C-C, et al. Histone deacetylase inhibitors up-regulate astrocyte GDNF and BDNF gene transcription and protect dopaminergic neurons. Int J Neuropsychopharmacol. 2008;11:1123–1134. doi: 10.1017/S1461145708009024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Xie W, Pan T, Xu P, Fridkin M, Zheng H, et al. Prevention and restoration of lactacystin-induced nigrostriatal dopamine neuron degeneration by novel brain-permeable iron chelators. FASEB J. 2007;21:3835–3844. doi: 10.1096/fj.07-8386com. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Anatomical landmarks and criteria used for manual segmentation analysis of MRI. Six brain structures delineated manually with reference to the anatomical landmarks: whole brain, lateral ventricles, corpus striatum, midbrain, hippocampus and cerebellum.30
Figure S2 Supplemental data from manual segmentation analysis of MRI. Changes in volume observed in the (B) corpus striatum and (C) the hippocampus as a result of lactacystin lesioning/valproate treatment. Similarly administration of valproate and/or lactacystin have marked effects on the (A) cerebellum volume as ascertained through manual segmentation analysis of rat brain MRI. Statistical significance denoted with letters where P < 0.05 for each comparison (see text for degrees of significance): aSignificantly different from group Lacta(−)VPA(−); bSignificantly different from group Lacta(−)VPA(++); cSignificantly different from group Lacta(+)VPA(−); dSignificantly different from group Lacta(+)VPA(+). Data are presented as mean ± SEM. n = 6–7 per group. Abbreviations: Lacta(−), lactacystin naïve; Lacta(+), lactacystin lesioned; VPA(−), treated with vehicle; VPA(+), treated with 200 mg·kg−1·day−1 valproate; VPA(++), treated with 400 mg·kg−1·day−1 valproate.
Table S1 Probe and primer sequences of PrimeTime™ qPCR assays used. All PrimeTime™ qPCR assays were obtained from integrated DNA technology (Coralville, IA, USA) and contained 2.5 nM of probe, 5 nM of primer 1 and 5 nM of primer 2. Abbreviations: A, adenine; C, cytosine; G, guanine; T, thymine; HEX™, hexachlorofluorescein; IABkFQ, Iowa Black® FQ.