

Background: Ameloblastin domain redistribution during development serves to pattern mineral microstructure.
Results: Proteasome subunit α type 3 (Psma3) interacts with the C terminus of ameloblastin, and the 20S proteasome can digest ameloblastin.
Conclusion: Ameloblastin-Psma3 interaction facilitates ameloblastin domain separation and redistribution defining the enamel rod boundaries.
Significance: This study investigates the mechanism of enamel microstructural formation at the level of protein-to-protein interaction.
Keywords: biomineralization, extracellular matrix protein, protein-protein interaction, tooth, yeast two-hybrid, 20S proteosome, ameloblastin, enamel microstructure pattern, proteasome subunit α type 3
Abstract
Enamel is a bioceramic tissue composed of thousands of hydroxyapatite crystallites aligned in parallel within boundaries fabricated by a single ameloblast cell. Enamel is the hardest tissue in the vertebrate body; however, it starts development as a self-organizing assembly of matrix proteins that control crystallite habit. Here, we examine ameloblastin, a protein that is initially distributed uniformly across the cell boundary but redistributes to the lateral margins of the extracellular matrix following secretion thus producing cell-defined boundaries within the matrix and the mineral phase. The yeast two-hybrid assay identified that proteasome subunit α type 3 (Psma3) interacts with ameloblastin. Confocal microscopy confirmed Psma3 co-distribution with ameloblastin at the ameloblast secretory end piece. Co-immunoprecipitation assay of mouse ameloblast cell lysates with either ameloblastin or Psma3 antibody identified each reciprocal protein partner. Protein engineering demonstrated that only the ameloblastin C terminus interacts with Psma3. We show that 20S proteasome digestion of ameloblastin in vitro generates an N-terminal cleavage fragment consistent with the in vivo pattern of ameloblastin distribution. These findings suggest a novel pathway participating in control of protein distribution within the extracellular space that serves to regulate the protein-mineral interactions essential to biomineralization.
Introduction
Enamel is the hardest tissue in the vertebrate body. It is formed by biomineralization of a protein extracellular matrix precursor that is ultimately replaced by mineral. Substituted hydroxyapatite (HAP)2 nanocrystallites with scarce residual organic material between them form enamel. The crystallites are aligned parallel along their long axis and tightly packed together, with each bundle formed under the control of a single ameloblast cell. As a consequence of cellular fabrication, the boundaries between cells give rise to a hierarchically integrated microstructure of crystallite bundles, which, in rodents, forms a decussating pattern of woven bioceramic structure (1, 2). This highly organized hierarchical structure provides enamel with its unique material properties of wear resistance, fracture toughness, and in rodents, self-sharpening edges (3–11).
HAP crystallites form within the organic extracellular matrix secreted during tooth development (12). The matrix proteins are synthesized by ameloblast cells and secreted into the extracellular space through the Tomes' process, the distal membrane projection of the ameloblast (13). Once released into the extracellular space, these matrix proteins undergo self-assembly to produce an organized matrix that is competent to guide the initiation, growth, and arrangement of HAP crystallites in the highly patterned rod-interrod microstructure observed in mature enamel (10, 14–17). In addition to protein self-assembly, ameloblast cell-to-matrix interactions also influence the matrix because the Tomes' process is always in contact with the forming enamel matrix (18). The Tomes' process of the ameloblast also determines the boundaries of each corresponding enamel rod, whereas the interrod matrix is deposited among adjacent ameloblasts, thereby unifying the bioceramic tissue into a continuum (Fig. 1), which imparts the enamel's unique material properties (1–4, 19, 20). During enamel formation, the ameloblast circadian rhythm entrains a daily round of protein synthesis followed by a similar period for mineral deposition occurring in the anti-phase of the circadian cycle (21, 22), thereby allowing incremental enamel matrix formation to be coupled to incremental HAP mineral conversion. Proteolytic processing of the enamel matrix proteins starts upon secretion, and removal of the protein matrix is essential to the expansion of the HAP mineral phase (23–28).
FIGURE 1.
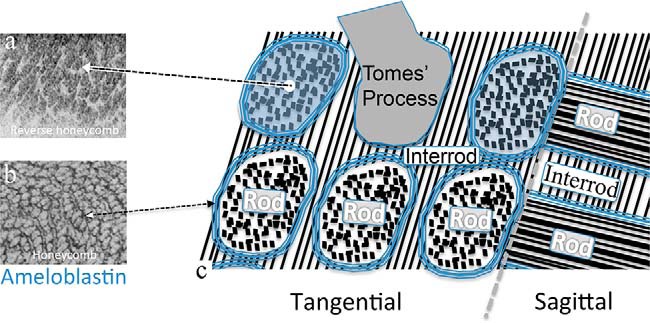
Idealized schematic of cellular fabrication showing the organization of mouse enamel extracellular matrix with HAP crystallite bundles within enamel rod-to-interrod microstructure. a shows developing porcine enamel with newly secreted ameloblastin localized to all of the area of the Tomes' processes (43), but it soon becomes localized exclusively to the perimeter of each enamel rod as a sheath as shown in b (40, 45). c, is a tangential (en face) and sagittal section schematic view of forming mouse enamel. The boundary (sheath) of each enamel rod corresponds to the lateral limit of each ameloblast cell's Tomes' process. The enamel rod is the basic unit of enamel and is created by a single ameloblast cell. Within a rod, the crystallites are packed parallel to one another along their long axis. The interrod crystallites form within the matrix deposited among adjacent ameloblasts, thereby giving rise to the continuity of the highly patterned rod-to-interrod enamel microstructure (22).
To date, two main groups of enamel matrix proteins have been identified. These are the amelogenin proteins and the non-amelogenin proteins, the latter including ameloblastin, enamelin, amelotin, and perhaps other yet-to-be-named proteins (29–34). Ameloblastin (35), also known as amelin (36) or sheathlin (37), is the most abundant of the non-amelogenin enamel matrix proteins. Ameloblastin protein is highly expressed by the secretory-stage ameloblasts and diminishes in abundance during the maturation stage (38–40). Ameloblastin is processed by matrix metalloproteinase 20 (also known as enamelysin or MMP20) (41, 42) immediately upon being secreted into the extracellular space. Remarkably, the ameloblastin cleavage products redistribute into different areas within the enamel rod, producing a pattern. Full-length ameloblastin and its C-terminal cleavage products first accumulate within the newly formed rods, producing a “reverse honeycomb” pattern (Fig. 1) (43–45). In contrast, the N-terminal cleavage products localize around the peripheral boundaries of the ameloblasts to form a “honeycomb” pattern (Fig. 1) (40, 44, 45). Moreover, in mouse models that express a truncated ameloblastin (46–48) or overexpress ameloblastin (49), the resulting enamel shows structural imperfections, with disturbances to the canonical pattern of rod-interrod boundaries. In the truncated ameloblastin animal, rescue of the enamel rod microstructure abnormalities has been achieved with expression of a full-length ameloblastin transgene (10). These observations suggest that the distributions of ameloblastin domains within the forming enamel matrix play important roles in establishing the enamel microstructure comprising the rod-interrod pattern of organization and hence in producing the favorable material properties found in mature enamel.
Based on these observations, we hypothesized that the N-terminal ameloblastin domain undergoes redistribution to the ameloblast cell periphery, thus serving to segregate the forming enamel matrix into individual units (rods) of enamel microstructure. Enamel does not remodel; therefore, correctly forming the matrix through protein self-assembly in the extracellular space is essential to properly forming the mineral phase, which must function for the life of the animal. We hypothesize that ameloblastin redistribution is controlled either by interactions with heretofore-unknown proteins within the matrix or with proteins localized to Tomes' processes, the secretory ends of the ameloblast cells that contact the matrix. The purpose of this investigation was to identify these previously unknown enamel matrix proteins that interact with ameloblastin during amelogenesis and to elucidate their expression and localization in developing mouse enamel.
To identify ameloblastin-interacting proteins, we performed a yeast two-hybrid assay to screen an ameloblast cDNA library using human ameloblastin as the bait. The yeast two-hybrid assay was developed by Fields and Song in 1989 (50) and is based on the fact that the GAL4 transcription factor can be split into two separable domains as follows: a DNA binding domain (BD) and a DNA activating domain (AD) allowing each GAL4 domain to fuse with a query protein to ascertain its interaction(s) with others. Should the two query proteins interact with one another, the two separated transcription factor fragments are brought back into proximity to one another, and the GAL4 factor activates transcription, providing a marker and selection strategy to identify the yeast colony harboring the putative interacting protein partner(s). In our assay, the GAL4 BD is fused to human ameloblastin, and the GAL4 AD is fused to an unknown protein encoded by a cDNA from an ameloblast cDNA library. If the unknown protein interacts with ameloblastin, the two separated transcription factor domains reconstitute the GAL4 factor to activate the reporter/selection genes. The yeast two-hybrid assay has previously been successfully deployed to identify the interacting protein partners for amelogenin and enamelin (15, 51, 52).
We report here that the proteasome subunit α type 3 (Psma3) interacts with ameloblastin in the yeast two-hybrid assay. Using confocal microscopy, we confirmed the localization of Psma3 to the ameloblast secretory end piece known as Tomes' processes, a physical site where ameloblastin is also present. The interaction of ameloblastin with Psma3 was corroborated by co-immunoprecipitation assay of total mouse ameloblast lysates using either an ameloblastin- or Psma3-specific antibody to identify the reciprocal partner protein. Protein engineering was used to define the C terminus of ameloblastin as the region interacting with Psma3. Finally, we performed in vitro proteasome digestion assays to investigate the potential functional significance of the ameloblastin-Psma3 interaction.
Experimental Procedures
Construction of Plasmids
To construct “bait plasmids” for the expression of human ameloblastin full-length protein (FL, amino acid residues 27–447, GenBankTM accession number AAF73048.1), N-terminal protein domain (Np, amino acid residues 27–222), and C-terminal protein domain (Cp, amino acid residues 223–447) (41, 42, 53, 54), the corresponding cDNAs were prepared without the ameloblastin signal peptide that can contribute to false-positive interactions. The DNA sequence for each of the three proteins was amplified from the human cDNA template (clone sc304427, OriGene Technologies Inc., Rockville, MD), and the PCR products cloned into the pDEST32 vector (ProQuest Two-Hybrid System, Invitrogen) containing the GAL4 BD gene sequence using Gateway subcloning technology, as described previously (55). The inserted cDNA fragments were preserved in their correct reading frame with the GAL4 BD nucleotide sequence. The constructed plasmids were transferred into yeast cells where they expressed the following individual fusion proteins: GAL4 BD-FL; GAL4 BD-Np; and GAL4 BD-Cp.
In this study, the term “prey plasmid” refers to a collection of plasmids containing cDNAs expressed by the mouse enamel organ epithelial cells. To prepare an enamel organ epithelial cDNA library, mandibular first molars were dissected from mouse pups (Swiss Webster) at postnatal day 3 (PN3). The odontogenic epithelia were micro-dissected from the underlying mesenchyme and used to isolate total RNA using an RNeasy Plus mini kit (Qiagen). The RNA sample was used to construct a normalized cDNA library using SMART synthesis techniques that favor full-length cDNAs. The double-stranded cDNAs were ligated in the pDEST22 vectors (ProQuest two-hybrid system, Invitrogen) containing the GAL4 AD gene sequence. The sequence representation of this cDNA library is about 1.75 × 106, and the average insert size is around 2.3 kilobases.
Yeast Two-hybrid Assay
A ProQuest two-hybrid system (Invitrogen) was used to perform the yeast two-hybrid assay, using the MaV203 yeast strain as the host. The MaV203 strain contains three reporter genes (lacZ, HIS3, and URA3), which provide four phenotypes by which to discover and selectively recover the cDNA encoding an unknown interacting protein. Four control plasmids were used to flank positive and negative control protein-to-protein interactions and establish their affinity as follows: pEXP32/Krev1; pEXP22/RalGDS-WT; pEXP22/RalGDS-m1; and pEXp22/RalGDS-m2 (Table 1).
TABLE 1.
Controls used in the yeast two-hybrid assay and phenotypes of reporter genes for each control
Four control plasmids, pEXP32/Krev1, pEXP22/RalGDS-WT, pEXP22/RalGDS-m1, and pEXP22/RalGDS-m2, were used to bracket the affinity of an unknown interacting protein. The positive control is based on the interaction of Krev1 and RalGDS protein. Mutation of the RalGDS protein diminishes (RalGDS-m1) or breaks (RalGDS-m2) the interaction with the Krev1 protein. The backbone of pEXP32/Krev1 is a pDEST32 vector containing the GAL4 binding domain coding gene, whereas the backbone of pEXP22/RalGDS-WT, pEXP22/RalGDS-m1, and pEXP22/RalGDS-m2 is a pDEST22 vector containing the GAL4 activation domain coding gene. The empty vectors pDEST32 and pDEST22 were used as the negative controls for vector self-activation. The plasmid pair of pDEST32-FL and pDEST22 was used to test self-activation of the bait pDEST32-FL. Positive interaction in yeast MaV203 cells activates the transcription of three gene reporters (lacZ, HIS3, and URA3). The lacZ gene expresses β-galactosidase, which generates blue coloration in the X-gal assay. Induction of the HIS3 gene allows the yeast transformants to grow on histidine dropout (null) plates. Induction of the URA3 gene endows the yeast transformants the ability to grow on uracil dropout (null) plates, with growth blocked on 5-FOA-containing plates. The abbreviations used are as follows: β-Gal, β-galactosidase; His−, histidine auxotrophy; Ura−, uracil auxotrophy; 5-FOA, 5-fluoroorotic acid; +, growth; −, no growth.
| Controls |
Purpose | Phenotypes |
||||
|---|---|---|---|---|---|---|
| GAL4 BD plasmid | GAL4 AD plasmid | β-Gal | His− | Ura− | 5-FOA | |
| pEXP32/Krev1 | pEXP22/RalGDS-WT | Strong positive | Blue | + | + | − |
| pEXP32/Krev1 | pEXP22/RalGDS-m1 | Weak positive | Faint blue | + | + | − |
| pEXP32/Krev1 | pEXP22/RalGDS-m2 | Negative | White | − | − | + |
| pDEST32 | pDEST22 | Negative self-activation | White | − | − | + |
| pDEST32-FL | pDEST22 | Test of self-activation | White | − | − | + |
We performed the yeast two-hybrid assay by screening the ameloblast cDNA library using the full-length human ameloblastin as the bait, according to the manufacturer's recommended protocol (Invitrogen). Briefly, the bait plasmid pDEST32-FL and library plasmids were co-transferred into yeast strain MaV203 to provide double-transformed yeast colonies. The double-transformed yeast colonies that grew on auxotrophic plates lacking leucine, tryptophan, and histidine and supplemented with 80 mm 3-amino-1,2,4-triazole (3AT, Sigma) were selected and re-plated on their respective selective plates to confirm the expression of the reporter/selection genes (Table 2). Positive yeast colonies containing ameloblastin-interacting protein candidates were identified according to the phenotypes indicating the activation of the three reporter genes. In addition, auxotrophic plates containing 0.2 weight % 5-fluoroorotic acid (Sigma) were used to eliminate false positives. From these candidates, the corresponding prey plasmids were isolated and used to transform fresh MaV203 yeast cells to confirm the protein interactions in a second round of screening.
TABLE 2.
Assays for testing protein interactions in the yeast two-hybrid system
Four specific assays with the corresponding metabolic selection plates were used to test the induction of three reporter genes in yeast cells. Reporter gene activation provided four unique phenotypes with which to assess the putative protein interactions: (i) blue colony coloration for the X-gal assay; (ii) growth on the Leu−/Trp−/His− with 80 mm 3AT plates; (iii) growth on the Leu−/Trp−/Ura− plates; and (iv) failure to grow on Leu−/Trp− with 0.2 weight % 5-FOA plates. The abbreviations used are as follows: β-Gal, β-galactosidase; Leu−/Trp−/His−, auxotrophic plate lacking leucine, tryptophan, and histidine; Leu−/Trp−Ura−, auxotrophic plate lacking leucine, tryptophan, and uracil; Leu−/Trp−, auxotrophic plate lacking leucine and tryptophan; 5-FOA, 5-fluoroorotic acid.
| Test | Assay | Plates | Phenotypes |
|
|---|---|---|---|---|
| Interaction | No interaction | |||
| lacZ induction (β-Gal activity) | X-gal assay | YPAD containing filter paper | Blue | White |
| HIS3 induction | Histidine auxotrophy | Leu − /Trp − /His − containing 80 mm 3AT | Formed colony | No colony |
| URA3 induction | Uracil auxotrophy | Leu−/Trp−/Ura− | Formed colony | No colony |
| URA3 induction | 5-FOA sensitivity | Leu − /Trp − containing 0.2 weight % 5-FOA | No colony | Formed colony |
For each prey plasmid, the nucleotide sequence was obtained for the cDNA encoding an ameloblastin-interacting protein identified in the yeast two-hybrid assay. The nucleotide sequence for each putative cDNA was analyzed for similarity to previously identified genes using the BLAST algorithm searched against the mouse genome plus transcript database as the comparison reference.
Immunolocalization of Proteins
Hemi-mandibles dissected from PN3 mouse pups (Swiss Webster) were fixed with 4% paraformaldehyde in PBS overnight at 4 °C. Tissues were decalcified with osmotically balanced 10% EDTA (pH 7.4) for 3 days and embedded in paraffin for sectioning. Sagittal and tangential sections of 5 μm in thickness were prepared for immunofluorescence. Immunostaining procedures followed a published protocol (56). In brief, sections were blocked with 5% BSA/PBS containing 0.1% Tween 20 for 1 h before incubation with primary antibody. Polyclonal primary antibodies were used to detect ameloblastin (M-300, sc-50534, Santa Cruz Biotechnology, Dallas, TX) and Psma3 (A-17, sc-54707, Santa Cruz Biotechnology). The locations of the primary antibody immunoglobulins were detected by Alexa Fluor 594 (Invitrogen) or FITC-conjugated (Santa Cruz Biotechnology) secondary antibodies. Nuclei were stained with DAPI in mounting medium (Vector Laboratories, Burlingame, CA). Tissue sections incubated with secondary antibody alone were used as negative controls. Fluorescent microscopy (Leica DMI3000 B, Leica Microsystems, Buffalo Grove, IL) and confocal microscopy (Leica TCS SP5 II, Leica Microsystems) were used to record immunofluorescent signal. To visualize the Tomes' processes more readily, sections tangential to the ameloblast long axis from PN3 mouse mandibular incisors were used to assess co-localization of ameloblastin and putative ameloblastin-interacting proteins. Three-dimensional reconstructions of a Z-stack of adjacent images were created using Leica software (version 2.7.3.9723, Leica Microsystems).
Co-immunoprecipitation and Western Blot Analysis
Enamel organ epithelia were dissected from PN3 mouse mandibular incisors and then solubilized into lysis buffer containing 20 mm Tris (pH 7.4), 150 mm NaCl, and 1% Triton X-100. Protein concentration was determined using a Pierce BCA protein assay kit (Thermo Scientific, Rockford, IL). The procedure for co-immunoprecipitation (57) and analysis by Western blot (58) has been described and was used without modification. Cell lysates were subject to immunoprecipitation with an anti-ameloblastin antibody (N-18, sc-33100, Santa Cruz Biotechnology) or an anti-Psma3 antibody (A-17); the immunoprecipitant was resolved to size by PAGE and analyzed by Western blotting with anti-Psma3 antibody (EPR5455, ab109532, Abcam, Cambridge, MA) or anti-ameloblastin antibody (M-300) accordingly. The negative control consisted of identical lysate incubated with precipitation beads (Santa Cruz Biotechnology) without the primary antibody.
Interactions with Yeast-synthesized Fusion Proteins
The three human ameloblastin bait plasmids (pDEST32-FL, pDEST32-Np, and pDEST32-Cp) were separately transformed into yeast MaV203 cells and cultured on selection media (Leu− plate). Western blot analysis was used to confirm the expression of ameloblastin fusion proteins in yeast using a GAL4 BD-specific antibody (15–6E10A7, ab135397, Abcam, Cambridge, MA). The prey plasmid pDEST22-Psma3 was co-transformed into MaV203 cells and cultured on selection medium (Trp− plate). Total yeast proteins were recovered as a lysate as described previously (59) and subjected to co-immunoprecipitation. Anti-ameloblastin primary antibody (M-300) was used to recover the interacting proteins onto precipitation beads (Santa Cruz Biotechnology), and Western blot analysis was used to determine the binding domain of the Psma3 with ameloblastin by anti-Psma3 antibody (A-17). Lysate samples incubated with precipitation beads (Santa Cruz Biotechnology) but without primary antibody were used as the negative controls.
In Vitro Proteolysis
Recombinant human ameloblastin (rhAMBN) (Abnova, Taiwan) was incubated with purified 20S proteasome (Enzo Life Sciences, Farmingdale, NY) at a proteasome-to-substrate molar ratio of 1:1 in 50 mm Tris-HCl buffer (pH 7.0) at 37 °C. After 1 or 3 h, the reactions were stopped by boiling in SDS loading buffer and analyzed by Western blotting with an ameloblastin N-terminal antibody (N-18, Santa Cruz Biotechnology) and a C-terminal antibody (ab116347, Abcam). Proteasome digestion of proline-rich homeodomain (PRH) protein was performed using the same procedure to confirm the enzymatic activity of the purified 20S proteasome (60). The PRH proteins and anti-PRH mouse polyclonal antibody were kind gifts provided by Dr. P.-S. Jayaraman (University of Bristol, Bristol, UK). Densitometry of PAGE-resolved protein bands was obtained by optical scanning, and the relative intensity of the bands was quantified using ImageJ densitometry (rsb.info.nih.gov).
To test the inhibition of proteolytic activity, 50 μm epoxomicin (Enzo Life Sciences), a proteasome-specific inhibitor, was added to the mixture of rhAMBN and 20S proteasome. The reactions were carried out at 37 °C and terminated after 3 h. Proteolysis was analyzed by Western blotting with the two ameloblastin-specific antibodies described above. Incubation of rhAMBN with 20S proteasome in the absence of epoxomicin at 37 °C for 3 h was used as the control.
Results
Psma3 Interacts with Ameloblastin
Using a yeast two-hybrid assay, we screened 6.4 × 106 colonies from a mouse ameloblast cDNA library using full-length human ameloblastin protein as bait, which yielded 43 colonies that grew on auxotrophic plates, indicating a protein-to-protein interaction. These 43 candidates were recovered and inoculated onto selective auxotrophic plates to corroborate the interaction and to eliminate false-positive reactions. Secondary screening eliminated 36 candidates as false positives but identified seven colonies that met all of the criteria for protein-to-protein interaction between the ameloblastin and its interacting partner protein as follows: (i) blue colony coloration for the X-gal assay; (ii) growth on the Leu−/Trp−/Ura− plates; (iii) growth on the Leu−/Trp−/His− with 80 mm 3AT plates; and (iv) failure to grow on Leu−/Trp− with 0.2 weight % 5-fluoroorotic acid plates. Nucleotide sequencing revealed that each of the seven candidates was preserved in the same reading frame as the GAL4 AD, supporting the notion that the encoded protein could interact with ameloblastin. The BLAST algorithm used for comparison of the cDNA nucleotide sequences of the seven candidates revealed that all of the candidates encoded the Psma3 as follows: four clones encoding full-length Psma3 (255 amino acids) and the remaining three encoding various lengths of the N-terminal segment of Psma3, with the most truncated protein being 216 amino acids.
Distribution of Ameloblastin in the Developing Tooth
We used a rabbit polyclonal antibody (M-300, Santa Cruz Biotechnology) to examine the distribution of ameloblastin in the developing PN3 mouse mandibular incisors by immunofluorescence. Sagittal sections (Fig. 2, a–f) revealed ameloblastin expression to be limited to the ameloblast cell layer (Fig. 2d). The intensity of immunoreactivity increased in the area corresponding to the secretory stage ameloblast cells (Fig. 2e) relative to the pre-secretory stage (Fig. 2f). Intense immunostaining was found in the distal membrane projections of secretory ameloblast cells, an area of membrane specialization in ameloblasts known as Tomes' processes (Fig. 2e). Moreover, on tangential sections (Fig. 2, g–k) that favor imaging en face visualization of the Tomes' processes (Fig. 1), immunostaining revealed ameloblastin distribution in a decussating pattern, corresponding to the positional pattern of Tomes' processes and most easily visualized in three-dimensional reconstruction of a confocal Z-stack of images (Fig. 2, j and k). This decussating staining pattern corresponds precisely to the arrangement of enamel rods observed in mature mouse enamel, suggesting the ameloblastin was localized to the secretory surface of a rod with little ameloblastin appearing in the interrod matrix shared among adjacent ameloblasts. Negative controls showed an absence of fluorochrome indicating no specific immunolocalization of the target antigen.
FIGURE 2.

Histology of mouse incisors and immunolocalization of ameloblastin in incisors. Sagittal (a–f) and tangential (g–k) sections were prepared from PN3 mouse mandibles. a is a schematic diagram of a tissue section of the jaw cut on the sagittal plane. b and c are hematoxylin-eosin-stained sagittal sections showing a mandibular incisor with c being a higher magnification of the boxed area in b, and arrows identifying Tomes' processes. d–f show representative immunofluorescent staining of ameloblastin (red fluorochrome) in sagittal sections with DAPI staining used for nuclear localization. Immunoreactivity showed increasing intensity in the area corresponding to the secretory stage ameloblast cells (e) relative to the pre-secretory stage cells (f). e and f are higher magnification views of the corresponding boxed areas in d. Arrows in e indicate intense immunostaining in the Tomes' processes. g is a schematic diagram of a tissue section of the jaw cut on the tangential section. h and i are hematoxylin-eosin-stained tangential sections showing a mandibular incisor with i being a higher magnification view of the boxed area in h. j and k are three-dimensional reconstructions of confocal Z-stack images from tangential sections, in which the immunodetection signal reveals ameloblastin antigen distributed across each Tomes' process (arrows), forming a decussating pattern of staining, which corresponds to the rod arrangement in mature mouse enamel. k is a higher magnification view of the area boxed in i. The abbreviations used are as follows: Am, ameloblast; EM, enamel matrix; D, dentine; pD, pre-dentine; P, pulp; Od, odontoblast; TP, Tomes' process.
Expression of Psma3 Protein in the Developing Tooth
To analyze the expression and localization of Psma3 in the developing tooth, we again performed imaging experiments on sagittal (Fig. 3, a–e) and tangential sections (Fig. 3, f–j) of mouse mandibular incisors at PN3. Using a goat polyclonal antibody (A-17, Santa Cruz Biotechnology), Psma3 protein was detected in the cytoplasm of ameloblasts and in Tomes' processes (Fig. 3d). Tomes' processes showed robust immunostaining (Fig. 3e). To better observe the localization and distribution of Psma3, we used confocal microscopy to perform three-dimensional reconstruction of a Z-stack of tissue sections cut tangential to the ameloblast long axis (en face) (Fig. 3, i and j). The results showed a distribution pattern for Psma3 localized to the Tomes' processes, which was identical to the location observed for ameloblastin distribution in the same region of forming enamel. The immunostaining for Psma3 revealed a decussating pattern localized to the matrix that corresponds to Tomes' processes, a pattern consistent with the position of enamel rods in mature mouse enamel. The interrod matrix, representing the shared secretory products of adjacent ameloblasts, revealed little Psma3 staining. Negative controls showed no specific immunolocalization of the target antigen.
FIGURE 3.

Histology of mouse incisors and immunolocalization of Psma3 in incisors. Sagittal (a–e) and tangential (f–j) sections were prepared from PN3 mouse mandibles. a is a schematic diagram of a sagittal section through the jaw. b and c are hematoxylin-eosin-stained sagittal sections showing a mandibular incisor with c being a higher magnification view of the approximate area in b shown by the black arrow. Arrows in c identify the Tomes' processes. d and e show representative immunofluorescent staining for Psma3 (green fluorochrome) in a sagittal section with DAPI staining used for nuclear localization. Psma3 was detected in the cytoplasm of ameloblasts and Tomes' processes. The higher magnification image in e shows that intense immunoreactivity was observed in the distal membrane projections of the secretory ameloblasts, i.e. Tomes' processes (arrows). f is a schematic diagram of the plane from a tangential section of the jaw. g and h are a hematoxylin-eosin-stained tangential section showing a mandibular incisor, with h being a higher magnification view of the boxed area in g. i and j are three-dimensional reconstructions of confocal Z-stack images of immunostaining of Psma3 from tangential section. j is a higher magnification view of the approximate area boxed in i. In these reconstruction images, Psma3 was detected and localized to each of the Tomes' processes (arrows), producing a decussating pattern consistent with the arrangement of rods in mature mouse enamel. The abbreviations used are as follows: Am, ameloblast; EM, enamel matrix; D, dentine; pD, pre-dentine; Od, odontoblast; TP, Tomes' process.
Co-localization of Ameloblastin and Psma3 in the Developing Tooth
Immunofluorescent staining was performed to examine the co-localization of ameloblastin and Psma3 using polyclonal antibodies M-300 and A-17, respectively, on tangential sections (for orientation see Figs. 2, g–i, and 3, f–h) of mouse mandibular incisors at PN3. At the resolution of the confocal microscope, immunostaining results indicated that ameloblastin and Psma3 co-localized in the cytoplasm of secretory ameloblasts and the Tomes' processes, with intense immunostaining observed in Tomes' processes (Fig. 4). The co-localized immunostaining pattern identified for Tomes' processes revealed a decussating organization of alternating rows of ameloblasts, a pattern that corresponds to the rod arrangement in mature mouse enamel, where one ameloblast is responsible for creation of one enamel rod. Negative controls showed no specific immunolocalization of the target antigen.
FIGURE 4.

Confocal micrographs reveal co-localization of ameloblastin and Psma3 in Tomes' processes. Immunofluorescent staining performed on tangential sections of PN3 mouse mandibular incisors shows that ameloblastin (red fluorophore, a) and Psma3 (green fluorophore, b) co-localized to the Tomes' processes of ameloblasts. The co-localization of ameloblastin and Psma3 is revealed in a merged image as a yellow fluorescence signal (d). a, b, and d are three-dimensional reconstructions of confocal Z-stack images of the 10-μm-thick immunostained section. c is the bright field view of the stained section showing the Tomes' processes and their arrangement. a–d, arrows point to identical Tomes' processes in each panel. The co-localized immunostaining of ameloblastin and Psma3 in Tomes' processes (d) revealed a decussating pattern of protein localization corresponding to alternating rows of ameloblasts, a pattern that is consistent with the rod arrangement in mature mouse enamel.
Interaction between Ameloblastin and Psma3 in Vitro
To independently corroborate the interaction between ameloblastin and Psma3, a co-immunoprecipitation assay was performed using a lysate prepared from PN3 mouse incisor enamel organ epithelium. In this assay, neither Psma3 nor ameloblastin protein was overexpressed; rather, native physiological expression levels were assayed. Psma3 protein was co-immunoprecipitated with ameloblastin by a polyclonal anti-ameloblastin antibody (N-18) and detected by Western blot analysis using a monoclonal anti-Psma3 antibody (EPR5455) (Fig. 5a). The same experimental procedure, reciprocally performed, detected the ameloblastin protein in an immunocomplex containing Psma3. In the reciprocal experiment, ameloblastin was co-immunoprecipitated with Psma3 by a Psma3-specific antibody (A-17) and detected by an anti-ameloblastin antibody (M-300) in Western blot analysis (Fig. 5b). Taken together, these data confirm Psma3's interaction with ameloblastin at the protein level in ameloblast cell lysate expressing physiologically relevant levels of Psma3 and ameloblastin.
FIGURE 5.

Co-immunoprecipitation analysis of ameloblastin and Psma3. Ameloblast cell lysates prepared from PN3 mouse incisor enamel organ epithelium were subjected to immunoprecipitation (a and b). Total cell lysate was used as the positive control. Cell lysate incubated with Protein A/G Plus-agarose beads served as the negative control. a shows that Psma3 was co-immunoprecipitated with ameloblastin by an ameloblastin-specific antibody (N-18, Santa Cruz Biotechnology) and probed by a Psma3 antibody (EPR5455, Abcam) in Western blot. b shows that ameloblastin was reciprocally co-immunoprecipitated with Psma3 by a Psma3-specific antibody (A-17, Santa Cruz Biotechnology) and probed by an ameloblastin antibody (M-300, Santa Cruz Biotechnology) in Western blot. Ameloblastin fusion protein was expressed in yeast cells, and cell lysates were prepared for an immunoprecipitation assay (c and d). c, Western blot detection of GAL4 BD-ameloblastin fusion protein in yeast cell lysate using GAL4 BD protein-specific antibody (15–6E10A7, Abcam). Total cell lysate prepared from untransformed yeast was used as the negative control (lane 1); asterisks indicate the corresponding bands for the GAL4 BD protein (lane 2), GAL4 BD-FL fusion protein (lane 3), GAL4 BD-Np fusion protein (lane 4), and GAL4 BD-Cp fusion (lane 5). Molecular mass markers (in kDa) are given at left. d, co-immunoprecipitation assay using cell lysates (shown in c) prepared from transformed yeast and used to detect the Psma3-binding site on human ameloblastin (AMBN). Lane 1, co-IP AMBN FL with Psma3; lane 2, negative control (precipitation beads alone) of lane 1; lane 3, co-IP AMBN Np with Psma3; lane 4, negative control (precipitation beads alone) of lane 3; lane 5, co-IP AMBN Cp with Psma3; lane 6, negative control (precipitation beads alone) of lane 5. Psma3 GAL4 AD fusion protein (arrowhead) was co-immunoprecipitated with either the AMBN FL or the Cp fusion proteins, showing the interaction is confined to the ameloblastin C terminus. The abbreviations used are as follows: IP, immunoprecipitated; WB, Western blot; co-IP, co-immunoprecipitation; Ab., antibody.
Binding Site of Psma3 to Ameloblastin Maps to the C-terminal Domain
MaV203 yeast cells were separately transformed with the expression plasmids pDEST32, pDEST32-FL, pDEST32-Np, and pDEST32-Cp. Total yeast proteins were recovered and subjected to Western blot for detection of ameloblastin GAL4-BD fusion proteins (Fig. 5c). The GAL4 BD protein alone was detected as an 18-kDa protein (Fig. 5c, lane 2); the GAL4 BD-FL fusion protein (561 amino acids) was recognized as a band of ∼65 kDa (Fig. 5c, lane 3); and the GAL4 BD-Np fusion protein (351 amino acids) was recognized as a band of ∼22 kDa (Fig. 5c, lane 4), the GAL4 BD-Cp fusion protein (372 amino acids) as a band of close to 24 kDa as a monomer, and ∼53-kDa as a dimer (Fig. 5c, lane 5). These data confirmed that the yeast transformants expressed the ameloblastin FL, Np, and Cp fusion proteins.
To investigate the interaction site of Psma3 and ameloblastin, lysate prepared from yeast cells expressing Psma3 fusion protein was separately mixed with lysate prepared from yeast cells expressing ameloblastin-FL, -Np, or -Cp fusion protein (Fig. 5c). The resulting protein mixtures were used for co-immunoprecipitation analysis (Fig. 5d). The results showed that Psma3 was co-immunoprecipitated with both ameloblastin-FL and -Cp using an anti-ameloblastin antibody (M-300). The GAL4 AD-Psma3 fusion protein was detected as a protein band of ∼50 kDa in the immunoprecipitated complex by an anti-Psma3 antibody (A-17) in Western blot analysis. These data indicated that the binding site of Psma3 was restricted to the C-terminal domain of the ameloblastin protein.
Proteolysis of Ameloblastin by 20S Proteasome in Vitro
To investigate the biological consequence of the ameloblastin-Psma3 interaction, we examined whether ameloblastin is a substrate for proteasome activity. An in vitro digestion assay was set up between rhAMBN and purified 20S proteasome. In the presence of the 20S proteasome, the densitometrically measured intensity of full-length ameloblastin protein detected by both anti-N-terminal and anti-C-terminal ameloblastin antibody decreased after 1 h of incubation and still further after 3 h of incubation time (Fig. 6a). Moreover, digestion with the 20S proteasome resulted in generating a smaller molecular weight ameloblastin fragment that was detected exclusively by the N-terminal specific antibody (Fig. 6a, lane 8). We verified activity of the 20S proteasome using PRH proteins, a substrate known to be degraded by 20S proteasome (Fig. 6b). The ability of the 20S proteasome to digest ameloblastin was blocked by the proteasome-specific inhibitor epoxomicin (Fig. 6c). In the presence of epoxomicin, the mass of the rhAMBN protein was not significantly changed after incubation with 20S proteasome at 37 °C for up to 3 h.
FIGURE 6.

Digestion of ameloblastin with 20S proteasome in vitro. a, Western blot with the ameloblastin C-terminal antibody (ab116347, Abcam) (lanes 1–4) or with the N-terminal antibody (N-18, Santa Cruz Biotechnology) (lanes 5–8). Lanes 1 and 5, recombinant human ameloblastin (rhAMBN) alone in assay buffer (50 mm Tris buffer (pH 7.0)) and the band at ∼100 kDa corresponds to the intact rhAMBN protein; lanes 2 and 6, purified 20S proteasome alone in assay buffer and no specific band was detected by either of the ameloblastin antibodies; lanes 3 and 7, rhAMBN incubated with purified 20S proteasome in assay buffer at 37 °C for 1 h with decreased intensity of the full-length rhAMBN band (∼100 kDa); lanes 4 and 8, rhAMBN incubated with purified 20S proteasome in assay buffer at 37 °C for 3 h, and the intensity of the full-length rhAMBN band (∼100 kDa) diminished further. Furthermore, with 20S digestion, a band corresponding to a cleavage product (∼43 kDa) was detected only by the N-terminal antibody (lane 8). Equal amount of rhAMBN was used in each lane. b, positive control of recombinant PRH demonstrated that the purified 20S proteasome used in the digestion assay retained its proteolytic activity. Equal amount of PRH was used in each lane. c, inhibition of proteasome digestion of ameloblastin by epoxomicin. Western blot with ameloblastin antibody specific to the C terminus (ab116347) (lanes 1–3) or the N terminus (N-18) (lanes 4–6) was used to detect ameloblastin with or without 50 μm epoxomicin. In the presence of epoxomicin and the 20S proteasome, the quantity of ameloblastin protein was not reduced dramatically (lanes 2 and 5). In contrast, without epoxomicin, incubation of rhAMBN with 20S resulted in diminution of ameloblastin protein quantity (lanes 3 and 6) with the release of a smaller molecular weight fragment (lane 6). Equal amounts of rhAMBN were used in each lane. Densitometry tracing of 20S proteasome digestion for rhAMBN and PRH is shown as a percentage of the total substrate mass used in each lane.
Discussion
Enamel is known for its unique mechanical properties that integrate hardness with remarkable resistance to fracture (3, 4, 6–10). These unique properties are dependent on the hierarchical structure of enamel imparted by the protein matrix precursor. Enamel forms in the extracellular space with the replacement of enamel matrix proteins by carbonated HAP crystallites. To provide optimal material properties required for fracture toughness (3, 19, 61), the crystallites within the rods are woven together with the interrod crystallites to form a continuum. The enamel rod is the smallest divisible repeating structural element of enamel, and each rod is fabricated by a single ameloblast cell that synthesizes and secretes enamel matrix proteins that guide their replacement by the mineral phase. During enamel formation, protein self-assembly and protein-to-protein interaction serve to organize the matrix proteins that in turn control HAP crystal habit and patterning. To date, numerous studies have described the essential roles of enamel matrix proteins, including amelogenin, ameloblastin, and enamelin in the assembly and interactions that occur during enamel biomineralization (14–16, 20, 39, 46, 51, 52, 62–64). In particular, ameloblastin was reported to serve as an interfacial anchor between the ameloblast and the extracellular matrix (46, 65). Moreover, ameloblastin proteolytic fragments become enriched along the cell perimeter that defines the lateral boundary of each enamel rod (40, 45). Here, we extend our studies to investigate ameloblastin-protein interactions hypothesized to facilitate redistribution of ameloblastin fragments responsible for protein patterning within the matrix that establishes the lateral boundaries of each enamel rod fabricated by an ameloblast cell.
In this work, Psma3 was identified as interacting with ameloblastin in the developing mouse tooth. We used human ameloblastin protein as the bait in a yeast two-hybrid assay to screen a mouse ameloblast cDNA library. The amino acid sequence for ameloblastin is well conserved among diverse vertebrate species, with human and mouse ameloblastin sharing extensive homology at the amino acid level (53). Because of limitations in obtaining developing human teeth as a source of mRNAs, we generated an ameloblast cDNA library using postnatal day 3 mouse molars. At this developmental stage, most ameloblasts are in the secretory stage, and the expression of matrix proteins is robust.
The yeast two-hybrid assay is a mature molecular tool for detecting protein-protein interactions. However, false-positive interactions remain a problem for this assay, especially in large scale screening such as the cDNA library screening employed here. To considerably decrease the rate of false-positive interactions, we used a stringent yeast strain coupled with stringent selection conditions and added a high concentration of 3AT (80 mm) to the triple auxotrophic plate (Leu−/Trp−/His−) for yeast colony analysis. After secondary screening, seven of 43 candidate colonies were identified as containing ameloblastin-interacting proteins. We picked these seven colonies based on their interactions being stronger than the positive control interaction of Krev1 with mutant RalGDS-m1, while excluding candidates that contained more weakly interacting proteins. DNA nucleotide sequence analysis showed that each of the seven candidates encoded the Psma3 protein. The most truncated Psma3 protein comprised only N-terminal residues 1–216, supporting the interpretation that the N-terminal 216-amino acid fragment is sufficient for Psma3 interaction with ameloblastin.
Proteasome subunit α type 3 is one of the components of proteasome core structure. The proteasome is more widely known as an important intracellular device for protein degradation. Organized as a cylindrical complex containing a core structure and two “cap” subunits, the core is composed of four stacked rings with the two outer rings composed of seven α subunits and the two inner rings composed of seven β subunits. The outer rings are responsible for maintaining a pore structure, and the inner rings contain catalytic sites where protein degradation occurs (66). To date, seven different α subunits and 10 different β subunits have been reported in mammals (67). In the mouse, Psma3 is 255 amino acids in length and has no isoform (NCBI accession number NP_035314.3). The proteasome is widely acknowledged to localize in the nucleus and cytoplasm in eukaryotes (68). However, several recent investigations reported that a biologically active proteasome core is present in extracellular space, including the alveolar space (69, 70), blood plasma (71), and cerebrospinal fluid (72). These data suggest that cells secrete/shed proteasome and/or proteasome subunits into the extracellular space, wherein the proteasome components may retain function. In this study, the immunofluorescence data revealed that the distribution pattern of Psma3 on Tomes' processes is very similar to the pattern of ameloblastin, suggesting they co-localize and therefore could interact there. Furthermore, the results from co-immunoprecipitation analysis demonstrated that Psma3 interacted with ameloblastin and that the interaction is sufficiently stable that either protein can pull down the other partner. These findings lead to the interpretation that Psma3 may interact with ameloblastin at the Tomes' process or may be secreted into extracellular space and interact with the newly secreted ameloblastin in the enamel extracellular matrix.
In this study, we generated the ameloblastin N-terminal domain and C-terminal domain by separating the full-length protein between Arg222 and Leu223 according to the previous experimental evidence (42, 54). We found that Psma3 co-immunoprecipitated with both full-length and C-terminal ameloblastin protein, indicating that the binding site for Psma3 maps to the C-terminal domain of ameloblastin. Using confocal analysis of immunostained ameloblasts, we observed that the rod matrix demonstrated abundant levels of Psma3 and ameloblastin, whereas the interrod matrix was relatively depleted of both ameloblastin and Psma3. These data support the interpretation that Psma3 interacts with the C-terminal domain of ameloblastin at or near Tomes' processes during enamel formation.
Several studies have reported that binding to Psma3 results in protein degradation by proteasome in a ubiquitin-independent pathway (73–76). Moreover, investigators found that proteins binding to Psma3 can undergo proteasome cleavage to form truncated fragments that retain biological activity (60). Therefore, we hypothesized that the binding of Psma3 to ameloblastin may assist the proteolytic processing of the C-terminal domain, leaving the N-terminal domain to appear enriched along the perimeter of the rod.
In the proteolysis studies, we showed that ameloblastin is digested by the 20S proteasome in vitro, which generates a smaller fragment that was detected only by the antibody to ameloblastin N terminus. This proteolytic activity was inhibited by addition of the proteasome-specific inhibitor epoxomicin. Thus, it is possible that ameloblastin-Psma3 interaction facilitates proteasome cleavage of ameloblastin resulting in the N-terminal domain being separated from the C-terminal domain. The liberated N-terminal products could accumulate around the enamel rod-to-interrod boundaries with degradation of the C-terminal domain (40, 45). Redistribution of the ameloblastin N-terminal domain may be facilitated through its recently described self-assembly domain defined by N-terminal amino acid residues 36–72 (77). The C-terminal domain was previously reported to distribute evenly throughout the newly formed immature enamel rods, becoming less abundant at later developmental stages of enamel formation (43, 45, 78). We suggest the ameloblastin C-terminal domain may be degraded by the proteasome or may dissociate from Psma3 and be digested by other matrix enzymes, such as MMP20 (41). These data suggest that the interaction between Psma3 and ameloblastin could facilitate separation and redistribution of ameloblastin fragments in the forming enamel matrix.
The localization of ameloblastin N-terminal domains at the rod perimeter (e.g. the interrod) provides patterning to the matrix where it can play a role in establishing the enamel microstructure by defining the rod and interrod boundaries that contribute the required material properties for a lifetime of masticatory function when fully mineralized. Defined rod boundaries are essential to the creation of a partly interrupted oriented continuum, greatly increasing the toughness and plasticity of enamel over its fundamental constituent, crystalline hydroxyapatite (3). Although the underlying molecular mechanism is still obscure, our study offers the possible interpretation that ameloblastin-Psma3 interaction facilitates the redistribution of ameloblastin domains, resulting in segregation of the rod and the interrod regions to form the highly patterned enamel structure. Ultimately, further studies will improve understanding of the unique structure-function relationship imparted by protein-to-protein interactions and reflected in the final mineral phase, and thus it may contribute to translational application of enamel regeneration.
Author Contributions
S. G. carried out the experiments, interpreted data, wrote the manuscript, revised it, and critically evaluated it; S. N. W. interpreted data, wrote the manuscript, and critically evaluated it; M. L. P. interpreted data, wrote the manuscript, revised it, and critically evaluated it; M. L. S. helped to carry out experiments, interpreted data, wrote the manuscript, revised it, and critically evaluated it.
Acknowledgments
We thank Professor Staale Petter Lyngstadaas for stimulating discussion as this project emerged. We thank Dr. Yaping Lei, Dr. Xin Wen, and Dr. Bridget Samuels for their technical assistance throughout this study. We also thank Dr. P.-S. Jayaraman (University of Bristol, Bristol, United Kingdom) for the kind gifts of PRH proteins and anti-PRH mouse polyclonal antibody.
Note Added in Proof
Figs. 1 and 3 and the text describing these data have been revised since this article was published on June 12, 2015, as a Paper in Press in order to clarify the presentation. These changes do not affect the interpretation of the results or the conclusions.
This work was supported, in whole or in part, by National Institutes of Health Grants DE13045 (to M. L. S.) and DE06988 (to M. L. S.) from USPHS and NIDCR. The authors declare that they have no conflicts of interest with the contents of this article.
- HAP
- hydroxyapatite
- 3AT
- 3-amino-1,2,4-triazole
- BD
- binding domain
- AD
- activating domain
- PRH
- proline-rich homeodomain
- PN3
- postnatal day 3
- rh
- recombinant human.
References
- 1. Boyde A. (1987) A 3-D model of enamel development at the scale of one inch to the micron. Adv. Dent. Res. 1, 135–140 [DOI] [PubMed] [Google Scholar]
- 2. Warshawsky H., Smith C. E. (1971) A three-dimensional reconstruction of the rods in rat maxillary incisor enamel. Anat. Rec. 169, 585–591 [DOI] [PubMed] [Google Scholar]
- 3. White S. N., Luo W., Paine M. L., Fong H., Sarikaya M., Snead M. L. (2001) Biological organization of hydroxyapatite crystallites into a fibrous continuum toughens and controls anisotropy in human enamel. J. Dent. Res. 80, 321–326 [DOI] [PubMed] [Google Scholar]
- 4. Fong H., White S. N., Paine M. L., Luo W., Snead M. L., Sarikaya M. (2003) Enamel structure properties controlled by engineered proteins in transgenic mice. J. Bone Miner. Res. 18, 2052–2059 [DOI] [PubMed] [Google Scholar]
- 5. White S. N., Miklus V. G., Chang P. P., Caputo A. A., Fong H., Sarikaya M., Luo W., Paine M. L., Snead M. L. (2005) Controlled failure mechanisms toughen the dentino-enamel junction zone. J. Prosthet. Dent. 94, 330–335 [DOI] [PubMed] [Google Scholar]
- 6. Imbeni V., Kruzic J. J., Marshall G. W., Marshall S. J., Ritchie R. O. (2005) The dentin-enamel junction and the fracture of human teeth. Nat. Mater. 4, 229–232 [DOI] [PubMed] [Google Scholar]
- 7. Simmer J. P., Papagerakis P., Smith C. E., Fisher D. C., Rountrey A. N., Zheng L., Hu J. C. (2010) Regulation of dental enamel shape and hardness. J. Dent. Res. 89, 1024–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Smith C. E., Chong D. L., Bartlett J. D., Margolis H. C. (2005) Mineral acquisition rates in developing enamel on maxillary and mandibular incisors of rats and mice: implications to extracellular acid loading as apatite crystals mature. J. Bone Miner. Res. 20, 240–249 [DOI] [PubMed] [Google Scholar]
- 9. Baldassarri M., Margolis H. C., Beniash E. (2008) Compositional determinants of mechanical properties of enamel. J. Dent. Res. 87, 645–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chun Y. H., Lu Y., Hu Y., Krebsbach P. H., Yamada Y., Hu J. C., Simmer J. P. (2010) Transgenic rescue of enamel phenotype in Ambn null mice. J. Dent. Res. 89, 1414–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Boyde A. (1997) Microstructure of enamel. Ciba Found. Symp. 205, 18–27 [DOI] [PubMed] [Google Scholar]
- 12. Robinson C., Briggs H. D., Atkinson P. J. (1981) Histology of enamel organ and chemical composition of adjacent enamel in rat incisors. Calcif. Tissue Int. 33, 513–520 [DOI] [PubMed] [Google Scholar]
- 13. Tomes J. (1849) On the structure of the dental tissues of the order Rodentia. Phil. Trans. R. Soc. 140, 529–567 [Google Scholar]
- 14. Fincham A. G., Moradian-Oldak J., Diekwisch T. G., Lyaruu D. M., Wright J. T., Bringas P., Jr., Slavkin H. C. (1995) Evidence for amelogenin “nanospheres” as functional components of secretory-stage enamel matrix. J. Struct. Biol. 115, 50–59 [DOI] [PubMed] [Google Scholar]
- 15. Paine M. L., Snead M. L. (1997) Protein interactions during assembly of the enamel organic extracellular matrix. J. Bone Miner. Res. 12, 221–227 [DOI] [PubMed] [Google Scholar]
- 16. Bartlett J. D., Ganss B., Goldberg M., Moradian-Oldak J., Paine M. L., Snead M. L., Wen X., White S. N., Zhou Y. L. (2006) 3. Protein-protein interactions of the developing enamel matrix. Curr. Top. Dev. Biol. 74, 57–115 [DOI] [PubMed] [Google Scholar]
- 17. Gibson C. W., Yuan Z. A., Hall B., Longenecker G., Chen E., Thyagarajan T., Sreenath T., Wright J. T., Decker S., Piddington R., Harrison G., Kulkarni A. B. (2001) Amelogenin-deficient mice display an amelogenesis imperfecta phenotype. J. Biol. Chem. 276, 31871–31875 [DOI] [PubMed] [Google Scholar]
- 18. Smith C. E., Nanci A. (1995) Overview of morphological changes in enamel organ cells associated with major events in amelogenesis. Int. J. Dev. Biol. 39, 153–161 [PubMed] [Google Scholar]
- 19. Zhu D., Paine M. L., Luo W., Bringas P., Jr., Snead M. L. (2006) Altering biomineralization by protein design. J. Biol. Chem. 281, 21173–21182 [DOI] [PubMed] [Google Scholar]
- 20. Paine M. L., Zhu D. H., Luo W., Bringas P., Jr., Goldberg M., White S. N., Lei Y. P., Sarikaya M., Fong H. K., Snead M. L. (2000) Enamel biomineralization defects result from alterations to amelogenin self-assembly. J. Struct. Biol. 132, 191–200 [DOI] [PubMed] [Google Scholar]
- 21. Lacruz R. S., Hacia J. G., Bromage T. G., Boyde A., Lei Y., Xu Y., Miller J. D., Paine M. L., Snead M. L. (2012) The circadian clock modulates enamel development. J. Biol. Rhythms 27, 237–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boyde A. (1979) Carbonate concentration, crystal centers, core dissolution, caries, cross striations, circadian rhythms, and compositional contrast in the SEM. J. Dent. Res. 58, 981–983 [DOI] [PubMed] [Google Scholar]
- 23. Simmer J. P., Fincham A. G. (1995) Molecular mechanisms of dental enamel formation. Crit. Rev. Oral Biol. Med. 6, 84–108 [DOI] [PubMed] [Google Scholar]
- 24. Bartlett J. D., Beniash E., Lee D. H., Smith C. E. (2004) Decreased mineral content in MMP-20 null mouse enamel is prominent during the maturation stage. J. Dent. Res. 83, 909–913 [DOI] [PubMed] [Google Scholar]
- 25. Lacruz R. S., Smith C. E., Kurtz I., Hubbard M. J., Paine M. L. (2013) New paradigms on the transport functions of maturation-stage ameloblasts. J. Dent. Res. 92, 122–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Smith C. E. (1998) Cellular and chemical events during enamel maturation. Crit. Rev. Oral Biol. Med. 9, 128–161 [DOI] [PubMed] [Google Scholar]
- 27. Paine M. L., Snead M. L., Wang H. J., Abuladze N., Pushkin A., Liu W., Kao L. Y., Wall S. M., Kim Y. H., Kurtz I. (2008) Role of NBCe1 and AE2 in secretory ameloblasts. J. Dent. Res. 87, 391–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lacruz R. S., Smith C. E., Chen Y. B., Hubbard M. J., Hacia J. G., Paine M. L. (2011) Gene-expression analysis of early- and late-maturation-stage rat enamel organ. Eur. J. Oral Sci. 119, 149–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Eastoe J. E. (1979) Enamel protein chemistry–past, present and future. J. Dent. Res. 58, 753–764 [DOI] [PubMed] [Google Scholar]
- 30. Termine J. D., Torchia D. A., Conn K. M. (1979) Enamel matrix: structural proteins. J. Dent. Res. 58, 773–781 [DOI] [PubMed] [Google Scholar]
- 31. Smith C. E., Chen W. Y., Issid M., Fazel A. (1995) Enamel matrix protein turnover during amelogenesis: basic biochemical properties of short-lived sulfated enamel proteins. Calcif. Tissue Int. 57, 133–144 [DOI] [PubMed] [Google Scholar]
- 32. Fincham A. G., Moradian-Oldak J., Simmer J. P. (1999) The structural biology of the developing dental enamel matrix. J. Struct. Biol. 126, 270–299 [DOI] [PubMed] [Google Scholar]
- 33. Iwasaki K., Bajenova E., Somogyi-Ganss E., Miller M., Nguyen V., Nourkeyhani H., Gao Y., Wendel M., Ganss B. (2005) Amelotin–a novel secreted, ameloblast-specific protein. J. Dent. Res. 84, 1127–1132 [DOI] [PubMed] [Google Scholar]
- 34. Moffatt P., Smith C. E., St-Arnaud R., Simmons D., Wright J. T., Nanci A. (2006) Cloning of rat amelotin and localization of the protein to the basal lamina of maturation stage ameloblasts and junctional epithelium. Biochem. J. 399, 37–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Snead M. L. (1996) Enamel biology logodaedaly: getting to the root of the problem, or “who's on first”. J. Bone Miner. Res. 11, 899–904 [DOI] [PubMed] [Google Scholar]
- 36. Fong C. D., Slaby I., Hammarström L. (1996) Amelin: an enamel-related protein, transcribed in the cells of epithelial root sheath. J. Bone Miner. Res. 11, 892–898 [DOI] [PubMed] [Google Scholar]
- 37. Hu C. C., Fukae M., Uchida T., Qian Q., Zhang C. H., Ryu O. H., Tanabe T., Yamakoshi Y., Murakami C., Dohi N., Shimizu M., Simmer J. P. (1997) Sheathlin: cloning, cDNA/polypeptide sequences, and immunolocalization of porcine enamel sheath proteins. J. Dent. Res. 76, 648–657 [DOI] [PubMed] [Google Scholar]
- 38. Krebsbach P. H., Lee S. K., Matsuki Y., Kozak C. A., Yamada K. M., Yamada Y. (1996) Full-length sequence, localization, and chromosomal mapping of ameloblastin. A novel tooth-specific gene. J. Biol. Chem. 271, 4431–4435 [DOI] [PubMed] [Google Scholar]
- 39. Fukumoto S., Yamada Y. (2005) Review: extracellular matrix regulates tooth morphogenesis. Connect. Tissue Res. 46, 220–226 [DOI] [PubMed] [Google Scholar]
- 40. Uchida T., Tanabe T., Fukae M., Shimizu M., Yamada M., Miake K., Kobayashi S. (1991) Immunochemical and immunohistochemical studies, using antisera against porcine 25-kDa amelogenin, 89-kDa enamelin, and the 13–17-kDa nonamelogenins, on immature enamel of the pig and rat. Histochemistry 96, 129–138 [DOI] [PubMed] [Google Scholar]
- 41. Iwata T., Yamakoshi Y., Hu J. C., Ishikawa I., Bartlett J. D., Krebsbach P. H., Simmer J. P. (2007) Processing of ameloblastin by MMP-20. J. Dent. Res. 86, 153–157 [DOI] [PubMed] [Google Scholar]
- 42. Chun Y. H., Yamakoshi Y., Yamakoshi F., Fukae M., Hu J. C., Bartlett J. D., Simmer J. P. (2010) Cleavage site specificity of MMP-20 for secretory-stage ameloblastin. J. Dent. Res. 89, 785–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Murakami C., Dohi N., Fukae M., Tanabe T., Yamakoshi Y., Wakida K., Satoda T., Takahashi O., Shimizu M., Ryu O. H., Simmer J. P., Uchida T. (1997) Immunochemical and immunohistochemical study of the 27- and 29-kDa calcium-binding proteins and related proteins in the porcine tooth germ. Histochem. Cell Biol. 107, 485–494 [DOI] [PubMed] [Google Scholar]
- 44. Nanci A., Zalzal S., Lavoie P., Kunikata M., Chen W., Krebsbach P. H., Yamada Y., Hammarström L., Simmer J. P., Fincham A. G., Snead M. L., Smith C. E. (1998) Comparative immunochemical analyses of the developmental expression and distribution of ameloblastin and amelogenin in rat incisors. J. Histochem. Cytochem. 46, 911–934 [DOI] [PubMed] [Google Scholar]
- 45. Hu J. C., Yamakoshi Y. (2003) Enamelin and autosomal-dominant amelogenesis imperfecta. Crit. Rev. Oral Biol. Med. 14, 387–398 [DOI] [PubMed] [Google Scholar]
- 46. Fukumoto S., Kiba T., Hall B., Iehara N., Nakamura T., Longenecker G., Krebsbach P. H., Nanci A., Kulkarni A. B., Yamada Y. (2004) Ameloblastin is a cell adhesion molecule required for maintaining the differentiation state of ameloblasts. J. Cell Biol. 167, 973–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wazen R. M., Moffatt P., Zalzal S. F., Yamada Y., Nanci A. (2009) A mouse model expressing a truncated form of ameloblastin exhibits dental and junctional epithelium defects. Matrix Biol. 28, 292–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Smith C. E., Wazen R., Hu Y., Zalzal S. F., Nanci A., Simmer J. P., Hu J. C. (2009) Consequences for enamel development and mineralization resulting from loss of function of ameloblastin or enamelin. Eur. J. Oral Sci. 117, 485–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Paine M. L., Wang H. J., Luo W., Krebsbach P. H., Snead M. L. (2003) A transgenic animal model resembling amelogenesis imperfecta related to ameloblastin overexpression. J. Biol. Chem. 278, 19447–19452 [DOI] [PubMed] [Google Scholar]
- 50. Fields S., Song O. (1989) A novel genetic system to detect protein-protein interactions. Nature 340, 245–246 [DOI] [PubMed] [Google Scholar]
- 51. Wang H., Tannukit S., Zhu D., Snead M. L., Paine M. L. (2005) Enamel matrix protein interactions. J. Bone Miner. Res. 20, 1032–1040 [DOI] [PubMed] [Google Scholar]
- 52. Paine C. T., Paine M. L., Snead M. L. (1998) Identification of tuftelin- and amelogenin-interacting proteins using the yeast two-hybrid system. Connect. Tissue Res. 38, 257–267 [DOI] [PubMed] [Google Scholar]
- 53. Vymetal J., Slabý I., Spahr A., Vondrásek J., Lyngstadaas S. P. (2008) Bioinformatic analysis and molecular modelling of human ameloblastin suggest a two-domain intrinsically unstructured calcium-binding protein. Eur. J. Oral Sci. 116, 124–134 [DOI] [PubMed] [Google Scholar]
- 54. Wald T., Bednárová L., Osička R., Pachl P., Sulc M., Lyngstadaas S. P., Slaby I., Vondrášek J. (2011) Biophysical characterization of recombinant human ameloblastin. Eur. J. Oral Sci. 119, 261–269 [DOI] [PubMed] [Google Scholar]
- 55. Landy A. (1989) Dynamic, structural, and regulatory aspects of λ site-specific recombination. Annu. Rev. Biochem. 58, 913–949 [DOI] [PubMed] [Google Scholar]
- 56. Ausubel F. M., Brent R., Kingston R. E., Moore D. D., Seidman J. G., Smith J. A., Struhl K. (eds) (2008) Current Protocols Molecular Biology, pp. 14.6.1–14.6.23, John Wiley and Sons, Inc., New York [Google Scholar]
- 57. Zhou Y. L., Lei Y., Snead M. L. (2000) Functional antagonism between Msx2 and CCAAT/enhancer-binding protein α in regulating the mouse amelogenin gene expression is mediated by protein-protein interaction. J. Biol. Chem. 275, 29066–29075 [DOI] [PubMed] [Google Scholar]
- 58. Zhou Y. L., Snead M. L. (2000) Identification of CCAAT/enhancer-binding protein α as a transactivator of the mouse amelogenin gene. J. Biol. Chem. 275, 12273–12280 [DOI] [PubMed] [Google Scholar]
- 59. Li B., Fields S. (1993) Identification of mutations in p53 that affect its binding to SV40 large T antigen by using the yeast two-hybrid system. FASEB J. 7, 957–963 [DOI] [PubMed] [Google Scholar]
- 60. Bess K. L., Swingler T. E., Rivett A. J., Gaston K., Jayaraman P. S. (2003) The transcriptional repressor protein PRH interacts with the proteasome. Biochem. J. 374, 667–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Paine M. L., White S. N., Luo W., Fong H., Sarikaya M., Snead M. L. (2001) Regulated gene expression dictates enamel structure and tooth function. Matrix Biol. 20, 273–292 [DOI] [PubMed] [Google Scholar]
- 62. Du C., Falini G., Fermani S., Abbott C., Moradian-Oldak J. (2005) Supramolecular assembly of amelogenin nanospheres into birefringent microribbons. Science 307, 1450–1454 [DOI] [PubMed] [Google Scholar]
- 63. Fang P. A., Conway J. F., Margolis H. C., Simmer J. P., Beniash E. (2011) Hierarchical self-assembly of amelogenin and the regulation of biomineralization at the nanoscale. Proc. Natl. Acad. Sci. U.S.A. 108, 14097–14102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Moradian-Oldak J., Paine M. L., Lei Y. P., Fincham A. G., Snead M. L. (2000) Self-assembly properties of recombinant engineered amelogenin proteins analyzed by dynamic light scattering and atomic force microscopy. J. Struct. Biol. 131, 27–37 [DOI] [PubMed] [Google Scholar]
- 65. Beyeler M., Schild C., Lutz R., Chiquet M., Trueb B. (2010) Identification of a fibronectin interaction site in the extracellular matrix protein ameloblastin. Exp. Cell Res. 316, 1202–1212 [DOI] [PubMed] [Google Scholar]
- 66. Coux O., Tanaka K., Goldberg A. L. (1996) Structure and functions of the 20S and 26S proteasomes. Annu. Rev. Biochem. 65, 801–847 [DOI] [PubMed] [Google Scholar]
- 67. Elenich L. A., Nandi D., Kent A. E., McCluskey T. S., Cruz M., Iyer M. N., Woodward E. C., Conn C. W., Ochoa A. L., Ginsburg D. B., Monaco J. J. (1999) The complete primary structure of mouse 20S proteasomes. Immunogenetics 49, 835–842 [DOI] [PubMed] [Google Scholar]
- 68. Peters J. M., Franke W. W., Kleinschmidt J. A. (1994) Distinct 19 S and 20 S subcomplexes of the 26 S proteasome and their distribution in the nucleus and the cytoplasm. J. Biol. Chem. 269, 7709–7718 [PubMed] [Google Scholar]
- 69. Sixt S. U., Beiderlinden M., Jennissen H. P., Peters J. (2007) Extracellular proteasome in the human alveolar space: a new housekeeping enzyme? Am. J. Physiol. Lung Cell. Mol. Physiol. 292, L1280–L1288 [DOI] [PubMed] [Google Scholar]
- 70. Sixt S. U., Peters J. (2010) Extracellular alveolar proteasome: possible role in lung injury and repair. Proc. Am. Thorac. Soc. 7, 91–96 [DOI] [PubMed] [Google Scholar]
- 71. Sixt S. U., Dahlmann B. (2008) Extracellular, circulating proteasomes and ubiquitin–incidence and relevance. Biochim. Biophys. Acta 1782, 817–823 [DOI] [PubMed] [Google Scholar]
- 72. Mueller O., Anlasik T., Wiedemann J., Thomassen J., Wohlschlaeger J., Hagel V., Keyvani K., Schwieger I., Dahlmann B., Sure U., Sixt S. U. (2012) Circulating extracellular proteasome in the cerebrospinal fluid: a study on concentration and proteolytic activity. J. Mol. Neurosci. 46, 509–515 [DOI] [PubMed] [Google Scholar]
- 73. Shi Z., Li Z., Li Z. J., Cheng K., Du Y., Fu H., Khuri F. R. (2015) Cables1 controls p21/Cip1 protein stability by antagonizing proteasome subunit α type 3. Oncogene 34, 2538–2545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Shu F., Guo S., Dang Y., Qi M., Zhou G., Guo Z., Zhang Y., Wu C., Zhao S., Yu L. (2003) Human aurora-B binds to a proteasome α-subunit HC8 and undergoes degradation in a proteasome-dependent manner. Mol. Cell. Biochem. 254, 157–162 [DOI] [PubMed] [Google Scholar]
- 75. Bae M. H., Jeong C. H., Kim S. H., Bae M. K., Jeong J. W., Ahn M. Y., Bae S. K., Kim N. D., Kim C. W., Kim K. R., Kim K. W. (2002) Regulation of Egr-1 by association with the proteasome component C8. Biochim. Biophys. Acta 1592, 163–167 [DOI] [PubMed] [Google Scholar]
- 76. Boelens W. C., Croes Y., de Jong W. W. (2001) Interaction between αB-crystallin and the human 20S proteasomal subunit C8/α7. Biochim. Biophys. Acta 1544, 311–319 [DOI] [PubMed] [Google Scholar]
- 77. Wald T., Osickova A., Sulc M., Benada O., Semeradtova A., Rezabkova L., Veverka V., Bednarova L., Maly J., Macek P., Sebo P., Slaby I., Vondrasek J., Osicka R. (2013) Intrinsically disordered enamel matrix protein ameloblastin forms ribbon-like supramolecular structures via an N-terminal segment encoded by exon 5. J. Biol. Chem. 288, 22333–22345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Uchida T., Murakami C., Dohi N., Wakida K., Satoda T., Takahashi O. (1997) Synthesis, secretion, degradation, and fate of ameloblastin during the matrix formation stage of the rat incisor as shown by immunocytochemistry and immunochemistry using region-specific antibodies. J. Histochem. Cytochem. 45, 1329–1340 [DOI] [PubMed] [Google Scholar]