

Background: γ-Protocadherin adhesion molecules regulate dendrite arborization by inhibiting focal adhesion kinase (FAK).
Results: Protein kinase C (PKC) phosphorylation of a C-terminal serine disrupts γ-protocadherin inhibition of FAK and reduces dendrite arborization.
Conclusion: The ability of the γ-protocadherins to promote dendrite arborization is regulated by phosphorylation.
Significance: These data provide much-needed mechanistic insight into the regulation of a critical neurodevelopmental adhesion molecule.
Keywords: branching, signaling, phosphorylation, dendrite, cell adhesion, cortex, dendritogenesis, Pcdh, serine/threonine protein kinase
Abstract
The γ-protocadherins (γ-Pcdhs) are a family of 22 adhesion molecules with multiple critical developmental functions, including the proper formation of dendritic arbors by forebrain neurons. The γ-Pcdhs bind to and inhibit focal adhesion kinase (FAK) via a constant C-terminal cytoplasmic domain shared by all 22 proteins. In cortical neurons lacking the γ-Pcdhs, aberrantly high activity of FAK and of PKC disrupts dendrite arborization. Little is known, however, about how γ-Pcdh function is regulated by other factors. Here we show that PKC phosphorylates a serine residue situated within a phospholipid binding motif at the shared γ-Pcdh C terminus. Western blots using a novel phospho-specific antibody against this site suggest that a portion of γ-Pcdh proteins is phosphorylated in the cortex in vivo. We find that PKC phosphorylation disrupts both phospholipid binding and the γ-Pcdh inhibition of (but not binding to) FAK. Introduction of a non-phosphorylatable (S922A) γ-Pcdh construct into wild-type cortical neurons significantly increases dendrite arborization. This same S922A construct can also rescue dendrite arborization defects in γ-Pcdh null neurons cell autonomously. Consistent with these data, introduction of a phosphomimetic (S/D) γ-Pcdh construct or treatment with a PKC activator reduces dendrite arborization in wild-type cortical neurons. Together, these data identify a novel mechanism through which γ-Pcdh control of a signaling pathway important for dendrite arborization is regulated.
Introduction
Perturbations in dendrite arborization have been implicated in a variety of neurodevelopmental and psychiatric diseases (1–3), although much remains unknown about the pathways that specify dendritic development. Recent research has uncovered a role for the protocadherin (Pcdh)2 family of cell adhesion molecules in the regulation of dendrite growth and organization (4–11), and some Pcdhs have been associated with psychiatric disorders (12–14). The “clustered” Pcdhs include ∼60 proteins encoded by the Pcdha, Pcdhb, and Pcdhg gene clusters that are arrayed in tandem at a single genomic site in vertebrates (15, 16). The 22 γ-Pcdhs are of particular interest, as mice lacking the Pcdhg cluster exhibit the strongest phenotypes, including neuronal apoptosis, defective synaptogenesis, disrupted axonal and dendritic arborization, and neonatal death (9–11, 17–22).
Each γ-Pcdh protein is encoded by four exons: one of 22 large “variable” exons, each of which encodes 6 extracellular cadherin repeats, a single transmembrane domain, and an ∼90-amino acid variable cytoplasmic domain plus 3 small “constant” exons that together encode a shared 125-amino acid C terminus. Each neuron expresses a subset of isoforms through a mechanism involving differential transcript initiation from promoters upstream of each variable exon (17, 23, 24). Our studies, confirmed by others, show that γ-Pcdh proteins form cis multimers promiscuously but that these multimers interact strictly homophilically in trans (25, 26). The differential expression of 22 isoforms thus allows, in theory, thousands of distinct adhesive interfaces to be specified by γ-Pcdhs (27, 28).
We recently found that the γ-Pcdhs promote dendritic arborization in cortical neurons by inhibiting a signaling pathway involving FAK, PKC, and the PKC target myristoylated alanine-rich protein kinase C substrate (MARCKS) (9). The C-terminal γ-Pcdh constant domain binds to FAK and prevents its autophosphorylation (29). In Pcdhg mutant cortex, FAK and PKC activation is aberrantly high, and MARCKS is hyperphosphorylated, resulting in disrupted dendrite arborization (9). This defect can be rescued by culturing knock-out neurons with pharmacological inhibitors of FAK or PKC (9). Although these data identify a signaling pathway through which the γ-Pcdhs act, it is not known how γ-Pcdh inhibition of FAK is regulated to allow for proper dendritic arbor elaboration during development.
Here we identify a C-terminal serine (termed Ser-922) within the γ-Pcdh constant domain that is phosphorylated by PKC. We show that Ser-922 is situated within a motif exhibiting binding to membrane phospholipids that is abrogated by PKC phosphorylation. Phosphorylation of Ser-922 also disrupts γ-Pcdh inhibition of FAK without preventing FAK binding. Expression of a non-phosphorylatable (S922A) mutant γ-Pcdh construct can cell-autonomously rescue dendrite arborization defects in knock-out neurons and increase dendrite arborization above normal levels in wild-type neurons. Consistent with these data, expression of a phosphomimetic (S922D) mutant γ-Pcdh construct or treatment with phorbol 12-myristate 13-acetate (PMA), a PKC activator, significantly reduces dendrite arborization in wild-type neurons. Together these data identify phosphorylation by PKC as a key regulatory mechanism modulating the promotion of dendrite arborization by the γ-Pcdhs.
Experimental Procedures
Reagents
PMA, Gö6983, PF-573,228, and prepared phosphoinositides from bovine brain were all from Sigma. Anti-GCP antibodies were generated at Pacific Immunology using the peptide GGNGNKKKSGKKEKK, with the serine phosphorylated. Anti-GCP was affinity-purified against the phosphorylated peptide; the phospho-nonspecific anti-GCN antibody was culled from the flow-through preparation. Additional antibodies include: phospho-(Ser) PKC substrate (Cell Signaling Technology), FAK and phosphorylated FAK (Tyr-397) (Cell Signaling Technology), γ-Pcdh constant domain (N159/5, NeuroMab), HA (Roche Applied Science), GST (Sigma), and GFP (Life Technologies).
In Vitro Phosphorylation Assay
GST-γC proteins (pGEX vector constructs) were purified from isopropyl 1-thio-β-d-galactopyranoside-induced BL21 bacteria using standard methodology and incubated with or without PKC in a 50-μl reaction using 1 μg of protein, 10 μl of 5× reaction buffer, 10 μl of 5× PKC activation buffer (sonicated phosphatidylserine), and 1 μl of PKC (primarily α, β, and γ, with lesser amounts of δ and ζ; all from PepTag Non-Radioactive PKC Assay Kit [Promega]). To remove phosphorylation, samples were incubated with 5 units of Antarctic phosphatase (New England BioLabs).
Micelle Binding Assay
Twelve μm proteins with or without 212 μm sonicated brain phosphoinositides were incubated at room temperature for 5 min before size-exclusion chromatography on an ÄKTA-FPLC using Superdex 75 columns (GE Healthcare). The outflow was monitored continuously by UV light to identify elution peaks.
Co-sedimentation Assay
GST-γC proteins were phosphorylated as above (control non-phosphorylated samples included PKC activation buffer with phosphatidylserine but no PKC enzyme). Proteins (0.05 μg) were diluted in 100 μl and mixed with 250 μg of sonicated micelles of crude brain phosphoinositides for 1 h at 4 °C before ultracentrifugation at 110,000 × g for 1 h at 4 °C (∼55,000 rpm using a Beckman TLA120.1 rotor).
Constructs
GST constructs diagrammed in Fig. 1 were cloned into the pGEX4 vector (GE Healthcare). Mammalian expression constructs diagrammed in Fig. 1 were cloned into pCMV or pCDNA3.1 vectors modified to include N-terminal HA tags (25).
FIGURE 1.

Identification of a C-terminal serine shared by all 22 γ-Pcdhs as a target for PKC phosphorylation. A, schematic diagram of constructs utilized in these studies. Constant region sequences are shown in red. Primary sequence of the C-terminal lysine rich domain containing Ser-922 is noted. B, NetPhos prediction of phosphorylation sites within the γ-Pcdh constant region; residues are numbered from the start of the 125-amino acid constant region. C, NetPhosK prediction of kinase recognition sites within the γ-Pcdh constant region; residues are numbered from the start of 125 amino acid constant region. Ser-119 here corresponds to Ser-922 in full-length murine γ-Pcdh-A3 utilized in these studies.
Western Blots
Western blots were performed as described previously (25). Briefly, proteins were separated by denaturing SDS-PAGE using TGX precast gels (Bio-Rad) and transferred to nitrocellulose membranes using a TransBlot Turbo transfer system (Bio-Rad). Membranes were blocked with 5% nonfat milk in TBS-Tween (0.1%), primary antibodies were diluted in 5% BSA in TBS-Tween, and blots were incubated overnight at 4 °C followed by washing in TBS-Tween and incubation with HRP-conjugated secondary antibodies for 1–2 h at room temperature. Signal detection utilized either SuperSignal West Pico or Femto ECL reagents (Pierce) and x-ray film. For all Western blots, a wide range of exposures was collected, and multiple exposures were utilized for each quantification. Films were scanned and analyzed in FIJI (NIH Image/J) using the Gel Analyzer module. Graphs shown in the figures represent multiple exposure quantifications from at least three separate experiments as indicated. In all experiments, relationships between treatments were maintained throughout multiple exposures.
Immunoprecipitation
Immunoprecipitation was performed as described previously (25). Briefly, cells were harvested in mild lysis buffer (50 mm Tris-HCl, pH 7.8, 150 mm NaCl, 1% Triton X-100, 10% glycerol, 25 mm NaF, 1× Roche Applied Science MiniComplete Protease Inhibitor), and lysates were centrifuged at 8000 × g for 10 min at 4 °C. Supernatants were rotated overnight at 4 °C with primary antibody (typically ∼0.5–1 μg/ml). Protein A/G-agarose beads (Pierce) were added for 2 h at 4 °C, and complexes were isolated by centrifugation. After several washes of the beads in mild lysis buffer, they were resuspended in 2× Laemmli protein sample buffer and boiled.
Phosphatidylinositol Phosphate (PIP) Strips
PIP strip membranes (Echelon Biosciences) were blocked for 1 h with 3% fatty acid free-BSA in TBS, 0.1% Tween20 (TBST) before incubation with GST-γC proteins for 2 h. They were washed 5 times with TBST and incubated with α-GST for 4 h in 3% fatty acid-free-BSA/TBST, washed, incubated with α-mouse-HRP, and detected with chemiluminescence (Pierce SuperSignal West Pico).
Cortical Neuron Cultures
Cortical neuron cultures were performed as described previously (9) using timed-pregnant C57BL/6 mice (Harlan) or Pcdhgdel homo- and heterozygotes (17) at E18/P0. Briefly, cortices were dissected, and the meninges were removed. Cortices were cut into small chunks and digested in an enzyme solution (papain, 10 units/ml) 2 × 20 min, after which tissue was rinsed with increasing concentrations of trypsin inhibitor followed by plating media (Basal Medium Eagle, 5% FBS, Glutamax (Invitrogen), N2 supplements (Invitrogen), and penicillin/streptomycin). Cells were plated onto German coverglass coated sequentially with poly-l-lysine and laminin or with Matrigel (Corning) at a density of 250,000 cells per coverslip. After 2 h and every 3 days subsequently, media were changed to Neurobasal with Glutamax, B27 supplements (Invitrogen), and penicillin/streptomycin. Transfections with Pcdh constructs and N1-EGFP were performed at 1 day in vitro (DIV) using Lipofectamine 2000 (Invitrogen). Each coverslip was incubated with 0.5 μg of total DNA and 1 μl of Lipofectamine 2000 and mixed in Neurobasal media according to the manufacturer's instructions for 2 h before being changed back to regular complete Neurobasal media. In some experiments, pharmacological inhibitors were added with a media change at 5 DIV: Gö6983 (Tocris Bioscience) at 1 μm, PMA (Sigma) at 100 nm, and PF-228 (Sigma) at 1 μm. Coverslips were fixed at 8 DIV with 4% paraformaldehyde for 15 m at room temperature, rinsed with PBS, and stained with anti-GFP antibody (Invitrogen) as described previously (9, 21). For some biochemistry experiments, neurons were nucleofected using an Amaxa Nucleofector at the time of dissociation using manufacturer's protocols for the Mouse Neuron Nucleofection kit and program G-013. Nucleofected neurons were plated at 500,000 cells per coverslip and lysed for Western blot as detailed above.
Sholl Analysis of Dendrite Complexity
Images of GFP+ transfected neurons were taken using a 20× objective on a Leica TCS SPE microscope. Neurons were traced manually using the Simple Neurite Tracer module in FIJI (NIH Image/J). Sholl analysis used concentric circles spaced either every 5 or 10 μm from the cell body up to 100 μm, and crossings were counted using the Sholl Analysis function of Simple Neurite Tracer. Crossings were graphed in Prism (GraphPad) for display, and the area under the curve was calculated for statistical analysis.
Statistical Analysis
Comparisons between constructs, genotypes, and treatments were performed by one-way analysis of variance using the recommended Tukey post-hoc test (corrected for multiple comparisons) in Prism. When only two conditions were being compared, a two-way t test was utilized. Asterisks in the figures denote the following significance levels: * = p < 0.05; ** = p < 0.01; *** = p < 0.001.
Results
A C-terminal Serine in the γ-Pcdh Constant Domain Is a Target for PKC
Toward identifying the molecular mechanisms by which γ-Pcdhs are regulated, we sought potential phosphorylation sites within the constant domain shared by all 22 isoforms (termed γC; Fig. 1A). One previous study found that clustered Pcdhs (including some γ-Pcdhs) can be phosphorylated by the receptor tyrosine kinase Ret (30); however, the relevant γ-Pcdh tyrosine residue was not identified, and the consequence of Ret phosphorylation for neural development is not known. Because of our previous work implicating the γ-Pcdhs in negatively regulating the serine/threonine kinase PKC (9), we systematically examined predicted Ser/Thr phosphorylation sites within γC. Using NetPhos tools from ExPASy (expasy.org), we generated a list of phosphorylatable residues along with predicted kinases. Of particular interest to us was a serine (referred to here as Ser-922: residue 922 of 928 in the murine γ-Pcdh-A3 protein; residue 119 of 125 in the murine γC domain) with the highest prediction for phosphorylation located at the C terminus (Fig. 1B). Interestingly, PKC was the predicted kinase (Fig. 1C); based on our prior results, we further investigated this residue.
We first assessed whether γC was indeed a PKC target in vitro. We generated several GST-tagged constructs, including the wild-type (WT) γC domain, non-phosphorylatable serine 922-to-alanine (S/A) point mutant, and a truncation mutant lacking the last 11 amino acids (ΔC11) (Fig. 1A). To ascertain if PKC is capable of phosphorylating γC, we incubated proteins directly with purified rat brain PKC. Using an antibody specific to phosphorylated serine substrates of PKC, we found that PKC indeed phosphorylated WT γC protein but not S/A or ΔC11 mutants (Fig. 2A), confirming Ser-922 as a target site. To specifically identify Ser-922-phosphorylated γ-Pcdh in vivo, we generated an affinity-purified antibody against peptides containing the 15 C-terminal residues of γC with Ser-922 phosphorylated (anti-GCP). We repeated the in vitro phosphorylation experiment using anti-GCP and obtained results identical to those obtained with anti-phosphoserine (Fig. 2B). Phosphatase treatment of proteins after PKC phosphorylation led to a drastic reduction of signal with the anti-GCP antibody (Fig. 2B). At longer Western blot exposures, weak anti-GCP signal could be observed in unphosphorylated γC but not at all in ΔC11 truncated proteins. Together, these results indicate that the novel anti-GCP strongly recognizes phosphorylated Ser-922.
FIGURE 2.
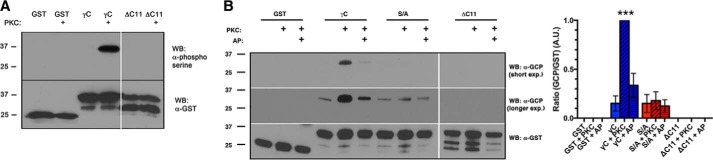
In vitro phosphorylation of γ-Pcdh Ser-922 by PKC. A and B, For in vitro phosphorylation assays the indicated GST fusion proteins were incubated with or without PKC and blots probed with phospho-(Ser) PKC substrate antibody (A) or anti-GCP (B) (two different film exposures are shown). Treatment of phosphorylated γC with Antarctic phosphatase (AP) leads to loss of anti-GCP signal as expected. The graph in B shows quantification of longer exposures from three experiments ± S.E. ***, p < 0.001. A.U., arbitrary units; WB, Western blot.
γ-Pcdhs Are Phosphorylated at Ser-922 in Vivo
To confirm that Ser-922 phosphorylation occurs on intact γ-Pcdh proteins in cells, we generated constructs encoding N-terminally HA-tagged full-length γ-Pcdh-A3 proteins, including A3-WT, A3-S/A, and A3-ΔC15 (Fig. 1A). First, we compared HEK293 lysates from untransfected cells to cells transfected with A3-WT and A3-ΔC15. We found that anti-GCP detected a protein of the expected size in cells expressing A3-WT but not A3-ΔC15 (Fig. 3A, left). An identical result was obtained after immunoprecipitation with anti-HA, confirming that the band detected using anti-GCP is, in fact, A3-WT (Fig. 3A, right) and indicating that a portion of exogenous γ-Pcdh proteins is basally phosphorylated in HEK293 cells. To confirm that anti-GCP detects γ-Pcdhs phosphorylated at Ser-922 by PKC, we exposed transfected cells to PMA, a phorbol-ester PKC activator. We found a robust increase in anti-GCP signal at the expected band size following as little as a 10-min PMA exposure in cells transfected with A3-WT but not with A3-S/A (Fig. 3B). Note that because Fig. 3B shows a short film exposure, no signal is seen without PMA as it is in the longer exposure shown in Fig. 3A; Fig. 3C quantifies the signal from multiple replicates and exposures for comparison. PMA is known to have other targets; thus to ensure that phosphorylation of γ-Pcdhs in response to PMA was due primarily to PKC activation, we exposed A3-WT-transfected HEK cells to PMA ± Gö6983, a potent PKC inhibitor. Pretreatment with Gö6983 significantly reduced PMA-induced A3-WT phosphorylation by ∼60% (Fig. 3D), confirming phosphorylation of γ-Pcdhs at Ser-922 by PKC.
FIGURE 3.

In vivo phosphorylation of γ-Pcdh Ser-922 by PKC. A, analysis of HEK293 cells transfected with A3-WT or A3-ΔC15 shows that anti-GCP recognizes the C-terminal motif (input, left). Immunoprecipitation (IP) for HA tag confirms that the appropriately sized band recognized by anti-GCP is in fact γ-Pcdh-A3 (right). WB, Western blot; UT, untransfected. B, HEK293 cells transfected with A3-WT or A3-S/A and incubated with 648 nm PMA for the indicated times. The anti-GCP signal increases after PMA treatment but not when Ser-922 is mutated to a non-phosphorylatable alanine. The graph shows quantification of three experiments ± S.E. with A3-WT. No significant differences between the three PMA time points were found. A.U., arbitrary units. C, comparison of short and long film exposures of vehicle (DMSO) and PMA-treated HEK293 cells transfected with A3-WT (3 replicates (R)). The graph shows quantification of this blot, confirming a significant increase in anti-GCP signal with PMA treatment. Note that endogenous phosphorylation is observed at longer exposures (A and C). D and E, Gö6983 and PMA exposure in HEK293 (D) and cortical neuron cultures (E). Anti-GCP signal increases with PMA, and this increase is significantly blocked by treatment with PKC inhibitor Gö6983 (1 μm). The graphs show the mean ± S.E. of three experiments. F, anti-GCP (upper blots) detects a portion of endogenous γ-Pcdhs (all γ-Pcdhs were detected with mAb N159/5, lower blots) in embryonic brain and postnatal cerebral cortex. The lack of signal in Emx1-Cre;Pcdh-γfcon3/fcon3 cortically restricted mutants (9) confirms antibody specificity. Although the proportion of phosphorylated γ-Pcdhs trends lower at later postnatal ages, no significant age differences were found (graph shows results of three experiments ± S.E.). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
We next assessed phosphorylation of endogenous γ-Pcdhs in neurons. First, we prepared cortical neuron cultures and treated them acutely at 8 DIV with PMA ± Gö6983. Using anti-GCP we detected a robust increase in γ-Pcdh phosphorylation after PMA treatment compared with the vehicle (DMSO) control that was significantly reduced by Gö6983 (Fig. 3E). Second, we analyzed lysates from embryonic brain and postnatal cortex and detected an appropriately sized band with anti-GCP at all ages (Fig. 3F); importantly, anti-GCP gave no signal in Pcdhg knock-out cortical tissues (Emx1-Cre;Pcdhgfcon3/fcon3; Ref. 9), confirming the specificity of this antibody and suggesting that Ser-922 phosphorylation occurs in vivo. The ratio of anti-GCP signal to that obtained with a non-phospho-specific γ-Pcdh antibody (either anti-GCN or N159/5) did not differ significantly across the ages examined.
A C-terminal Lysine-rich Motif Mediates Phospholipid Binding Abrogated by Ser-922 Phosphorylation
The Ser-922 residue is situated within a lysine-rich motif (-KKKSGKKEKK; Fig. 1A) predicted to be positively charged. We posited that this motif might interact with membrane phospholipids, and, if so, that PKC phosphorylation at Ser-922 might add a negative charge and disrupt such an interaction. To assess whether this motif exhibits lipid binding, we first incubated each GST-tagged γC protein (Fig. 1A) on nitrocellulose membranes spotted with 15 different phosphoinositides (PIP Strips, Echelon Biosciences; Fig. 4A) and detected bound proteins with anti-GST antibody. We detected binding of the intact γC but not any of the truncated constructs or GST alone, to PIP, PIP2, and PIP3 lipids (Fig. 4A).
FIGURE 4.

PKC phosphorylation of Ser-922 regulates lipid binding of a C-terminal γ-Pcdh lysine-rich motif. A, PIP Strip (Echelon Biosciences) membranes spotted with phospholipids incubated with the indicated GST fusion proteins and detected with anti-GST antibody. Only proteins with an intact, Ser-922-containing lysine-rich domain exhibited binding to phosphoinositides. PE, phosphatidylethanolamine; PA, phosphatidic acid; PC, phosphatidylcholine; PS, phosphatidylserine; Ptdins, phosphatidylinositol; LPA, lysophosphatidic acid; LPC, lysophosphatidyl choline; S1P, sphingosine-1-phosphate. B, micelle binding assay. Shown are absorbance traces from size exclusion chromatography of the indicated GST fusion proteins with or without phosphoinositide micelles. The addition of micelles results in a major shift to larger sizes with intact γC protein but not ΔC11. C and D, co-sedimentation assay. Indicated GST fusion proteins were incubated with crude phosphoinositide micelles before ultracentrifugation to separate the lipids and lipid-associated proteins from the soluble supernatant. Phosphorylation with PKC shifts γC, but not the S/A mutant, from pellet (P) to supernatant (S), indicating reduced phospholipid binding. mAU, milliabsorbance units; WB, Western blot. D shows results from three experiments ± S.E. S/A pellet localization is significantly higher than that of γC. **, p < 0.01.
We next used a micelle-binding assay (31) to confirm interaction with membrane lipids. Proteins were incubated alone or in the presence of micelles prepared from sonicated crude bovine brain phosphoinositide lipids. Preparations were separated by size-exclusion chromatography; in this assay binding is detected as a shift to an earlier elution fraction in protein + micelle samples compared with protein alone. The γC protein, but not the ΔC11 truncation lacking the lysine-rich motif or GST alone, exhibited a strong elution shift, confirming that the constant domain interacts with brain phospholipids (Fig. 4B).
We next sought to address whether PKC phosphorylation of Ser-922 might disrupt membrane lipid binding by the lysine-rich motif. This was not possible using the assays described above: phosphatidylserine is an essential cofactor for the PKC in vitro phosphorylation reaction, and its presence led to an unacceptably high background. We thus turned to a membrane lipid co-sedimentation assay. The γC and S/A mutant GST fusion proteins with or without PKC in vitro phosphorylation were incubated with sonicated crude brain phosphoinositides. The samples were then subjected to ultracentrifugation; unbound proteins remain in the supernatant, whereas lipid-associated proteins come down in the pellet.
We found that γC was enriched in the pellet compared with GST but that this enrichment was lost upon phosphorylation (Fig. 4, C and D). Similar to the native γC protein, the S/A mutant protein was found primarily in the lipid pellet (actually almost entirely so) but in contrast was insensitive to PKC phosphorylation, with no redistribution to the supernatant observed (Fig. 4, C and D). These data clearly indicate that Ser-922 phosphorylation by PKC disrupts the membrane phospholipid binding properties of the C-terminal motif, suggesting a novel mechanism by which PKC may regulate γ-Pcdh functions.
Phosphorylation of Ser-922 Disrupts γ-Pcdh Inhibition of, but Not Binding to, FAK
The γ-Pcdhs bind to FAK via the γC domain and inhibit its activation by autophosphorylation at tyrosine 397 (29). We previously presented evidence that γ-Pcdh inhibition of FAK is important for tempering PKC phosphorylation of MARCKS, thus allowing normal development of dendritic arbors in cortical neurons in vivo and in vitro (9). We hypothesized that PKC phosphorylation of Ser-922 might act in turn to regulate the ability of γ-Pcdh to inhibit FAK. To investigate this, we cotransfected cells with A3-WT or A3-S/A along with a construct encoding GFP-tagged FAK, immunoprecipitated A3 with α-HA, and examined the amount and Tyr-397 phosphorylation state of bound FAK. We found, consistent with Chen et al. (29), that very little FAK that came down with γ-Pcdhs, in this case A3-WT, was phosphorylated (Fig. 5A). After a brief treatment with PMA (30 min, which we confirmed did not result in indirect phosphorylation of FAK itself; see Input lanes, Fig. 5A), the proportion of γ-Pcdh-associated FAK that was phosphorylated was doubled (Fig. 5, A and B). PMA had no effect on the phosphorylation state of FAK associated with the A3-S/A point mutant, confirming that PKC phosphorylation of Ser-922 was the cause of this reduction in FAK inhibition (Fig. 5, A and B).
FIGURE 5.

Phosphorylation of Ser-922 disrupts γ-Pcdh inhibition of, but not binding to, FAK. A, lysates of HEK293 cells co-transfected with HA-tagged γ-Pcdh A3-WT or A3-S/A and GFP-FAK immunoprecipitated with anti-HA to pull down γ-Pcdh-associated FAK. PMA treatment did not disrupt the ability of A3-WT to bind FAK but did result in a higher proportion of A3-associated FAK being phosphorylated (PFAK). PMA treatment has no effect on activation state of A3-S/A-associated FAK, confirming the role of PKC phosphorylation of Ser-922. IP, immunoprecipitation; WB, Western blot. B and C, quantification of three experiments performed as in A. The ratio of PFAK to total FAK associated with A3 was calculated and normalized to the no-PMA A3-WT ratio (B). Total amount of FAK pulled down with A3-WT, but not A3-S/A, actually increased after PMA treatment (C). D–F, experiments were similar to those shown in A–C but compared A3-WT to A3-ΔC15. Loss of the C-terminal lipid binding domain led to more FAK being pulled down, similar to the effect of PMA treatment on A3-WT, although as expected ΔC15 is insensitive to PMA as it lacks Ser-922 (D and F). The ratio of PFAK/FAK in ΔC15 co-immunoprecipitations trended higher, but due to high variability in these experiments this difference was not statistically significant (D and E). Graphs show the means ± S.E. of three (A–C) or 6 (D–F) experiments. *, p < 0.05; **, p < 0.01.
Interestingly, PMA treatment did not disrupt the ability of A3-WT to associate with FAK; in fact, when cells were exposed to PMA, we found a significant increase in the total amount of FAK pulled down (Fig. 5, A and C). Again, A3-S/A was insensitive to PMA in this regard (Fig. 5, A and C). Importantly, in all analyses of γ-Pcdh-associated FAK, we normalized phospho-FAK levels to total FAK levels (Fig. 5B). Thus, even though PMA treatment did increase the total amount of FAK associated with A3-WT, the relative proportion of phospho-FAK was still doubled (Fig. 5B). We conclude that phosphorylation of γ-Pcdhs at Ser-922 by PKC did not prevent them from binding to FAK (in fact, it may enhance such binding) but does reduce their ability to inhibit FAK activation.
FAK activation depends on its FERM domain binding to PIP2, which opens up its structure, exposing its kinase domain and allowing autophosphorylation (32, 33). It could be that the γ-Pcdh lipid binding motif competes locally with FAK for binding to phospholipids, which might reduce the activation of γ-Pcdh-associated FAK. To address this possibility, we performed similar experiments using A3-ΔC15 truncation mutants. As expected (due to the loss of Ser-922), PMA treatment did not change the binding of FAK to A3-ΔC15 nor did it affect the phosphorylation state of the FAK pulled down with A3-ΔC15 (Fig. 5, D–F). Interestingly, A3-ΔC15 exhibited significantly enhanced binding to FAK similar to that observed when A3-WT protein was phosphorylated (Fig. 5, D and F). Although the proportion of A3-ΔC15-associated FAK that was phosphorylated was elevated compared with A3-WT (Fig. 5, D and E), results for this truncation construct were variable enough that this result did not reach statistical significance. Thus, from the existing data we cannot conclude that γC phospholipid binding is critical for FAK inhibition by the γ-Pcdhs.
Expression of Non-phosphorylatable γ-Pcdh Increases Dendrite Arborization by Reducing FAK Activation
Both FAK and PKC are negative regulators of dendrite arborization (9, 34–38). Because PKC phosphorylation of Ser-922 reduces γ-Pcdh inhibition of FAK (Fig. 5), we asked whether introducing the non-phosphorylatable A3-S/A into cortical neurons might increase dendrite arborization. We co-transfected cortical neuron cultures at 1 DIV with A3-WT, A3-S/A, A3-ΔC15, or empty vector plus GFP at low efficiency (∼1–2%) using lipofection. At 8 DIV, cultures were fixed, and Sholl analysis of dendrite arbor complexity was performed on tracings from micrographs of GFP+ neurons (Fig. 6 shows representative micrographs, whereas Fig. 8 shows representative tracings, to illustrate methodology).
FIGURE 6.

Neurons expressing a non-phosphorylatable γ-Pcdh exhibit increased dendrite arborization. A and B, representative cortical neuron micrographs from cultures transfected at low efficiency with an empty vector, A3-WT, A3-ΔC15, or A3-S/A plus GFP to allow for tracing. Cultures were treated at 5 DIV with the FAK inhibitor PF-228 (B) or vehicle alone (DMSO; A) and analyzed at 8 DIV. C, Sholl analysis curves demonstrate increased dendritic complexity of neurons expressing A3-S/A compared with A3-WT or empty vector. No decrease in arborization was observed in neurons expressing A3-ΔC15, suggesting that lipid binding of the γC domain is not critical to regulate FAK. All neurons treated with PF-228 (C, dotted lines) exhibit similarly high dendritic complexity regardless of transfection conditions. Expression of A3-S/A did not result in a further increase in arborization, consistent with its effect being to increase the level of FAK inhibition, which is maximized by PF-228 treatment. D, area under the curve graphs of Sholl analysis data in C. E, Western blots of lysate from neurons nucleofected with A3-WT or A3-S/A, probed with the indicated antibodies. F, FAK activation, measured as the normalized PFAK/FAK ratio, is significantly reduced in neurons expressing A3-S/A (left axis). This is not due to higher expression of this construct (right axis). The graph shows results from three separate cultures ± S.E., n = 54–60 neurons per condition. **, p < 0.01. The bar in A is 100 μm.
FIGURE 8.

Phosphorylation of Ser-922 by PKC decreases dendrite arborization. A–D, representative tracings of cortical neuron dendritic arbors from cultures transfected at low efficiency with an empty vector (Control), A3-WT, A3-S/A, or A3-S/D plus GFP and treated for 3 days with vehicle only (DMSO), Gö6983, or PMA. E–G, Sholl analysis curves of all transfection conditions treated with vehicle (E, DMSO), Gö6983 (F, Gö), or PMA (G). H, area under the curve representation of data shown in E–G, compared with control or A3-WT-expressing neurons, neurons expressing A3-S/A exhibit increased dendrite complexity, whereas those expressing A3-S/D exhibit reduced dendrite complexity (E and H). PKC inhibition (Gö6983 treatment) did not further increase arborization in A3-S/A neurons, in contrast to all other conditions, where PKC inhibition elevates (control, A3-WT) or rescues (A3-S/D) arborization (F and H). Because PMA is a broad activator of PKC, which is known to inhibit dendrite arborization downstream of γ-Pcdhs, PMA treatment reduced arborization to similarly low levels in all neurons examined (G and H). Treatment of neurons expressing A3-S/D with PF-228 rescued arborization to control levels (H). Graphs show results from three separate cultures ± S.E., 20–60 neurons per condition. *, p < 0.05; **, p < 0.01; ***, p < 0.001. The bar in A is 100 μm. PF, PF-573,228.
Neurons expressing A3-S/A did indeed exhibit a significant increase in dendrite arborization compared with control and A3-WT conditions (Fig. 6, A, C, and D). Phosphorylation of Ser-922 reduces phospholipid binding at the γ-Pcdh C terminus (Fig. 4, C and D); thus it was possible that A3-S/A promoted dendrite arborization simply due to unhampered lipid binding. This did not seem to be the case, however, as transfection of neurons with A3-ΔC15 (lacking the entire lipid binding motif) did not reduce dendrite arborization compared with control (Fig. 6, A, C, and D). Intriguingly, transfection of Pcdhgdel null mutant neurons (9, 17) with A3-S/A, but not with A3-WT, was able to rescue their defective dendrite arborization to levels significantly above that of control neurons (Fig. 7).
FIGURE 7.

Expression of a non-phosphorylatable γ-Pcdh cell autonomously rescues arborization defects in Pcdhg-null neurons. A and B, Sholl analysis curves of wild type (A) and Pcdhgdel/del null mutant (knock-out) neurons (B) transfected with empty vector, A3-WT, or A3-S/A plus GFP. C, area under the curve graph of data in A and B. As observed in our prior work (9), knock-out (KO) neurons exhibit significantly reduced dendrite arborization, which is rescued to above control wild-type levels by transfection with A3-S/A but not with A3-WT. As observed in parallel experiments (Fig. 6), transfection of wild-type neurons with A3-S/A, but not with A3-WT, significantly increases dendrite complexity. Graphs show results from 3–4 animals per genotype ± S.E., 36–57 neurons per condition. *, p < 0.05; ***, p < 0.001.
The fact that A3-S/A, but not A3-WT, can cell-autonomously promote dendrite arborization suggests that a critical control point is the level of Ser-922 phosphorylation and, by extension, efficiency of FAK inhibition. Indeed, we found that FAK activation (as measured by phosphorylated FAK/FAK ratio) was significantly decreased in neurons expressing A3-S/A compared with those expressing A3-WT (Fig. 6, E and F, left axis). Importantly, expression levels of A3-WT and A3-S/A were identical in nucleofected neurons (Fig. 6, E and F, right axis), demonstrating that this difference is not simply due to differential expression of the A3-S/A construct.
Exposure of neurons to a specific FAK inhibitor (PF-228) substantially increased dendrite arborization in all transfection conditions to the same level of elevated complexity (Fig. 6, B–D); that is, PF-228 effects were not additive with those of A3-S/A expression. This implies A3-S/A and PF-228 act in the same pathway to inhibit FAK, as we do not see a further potentiation of arborization in neurons expressing A3-S/A versus other constructs exposed to PF-228. Arbor complexity when FAK was strongly inhibited by PF-228 exceeded that seen with A3-S/A transfection alone (Fig. 6D). Consistent with our prior work (9), this indicates that γ-Pcdhs are a significant, but clearly not the only, upstream regulator of FAK activation relevant to dendrite arborization.
Phosphorylation of Ser-922 Negatively Regulates Dendrite Arborization
PKC phosphorylation of Ser-922 reduces the ability of γ-Pcdh to inhibit FAK activation (Fig. 5), and consistent with this, neurons expressing A3-S/A exhibit decreased FAK activation and increased dendrite arborization (Fig. 6). This suggests that γ-Pcdh phosphorylation state per se should, in part, determine dendrite complexity. We addressed this by assessing arborization in neurons 1) treated with a pharmacological activator or inhibitor of PKC and 2) transfected with a predicted phosphomimetic γ-Pcdh construct, A3-S/D.
As expected, in control neurons (transfected with empty vector + GFP), treatment with Gö6983 significantly increased dendrite arborization compared with vehicle controls (Fig. 8, panels A and E and panels F and H). Identical results were obtained in neurons transfected with A3-WT (Fig. 8 panels B and E and panels F and H), consistent with its inability to cell autonomously increase arborization (Fig. 6D). As in previous experiments (Fig. 6D), expression of A3-S/A resulted in significantly increased dendrite arborization to levels as high as those obtained with PKC inhibition by Gö6983 (Fig. 8, C, E, and H). Treatment of A3-S/A-expressing neurons with Gö6983 did not further elevate arborization (Fig. 8, C, F, and H), confirming that γ-Pcdh phosphorylation is one mechanism through which PKC can inhibit dendrite arborization. Consistent with this, vehicle-treated neurons expressing the predicted phosphomimetic A3-S/D construct exhibited significantly decreased arborization (Fig. 8, D, E, and H).
In addition to its effects on γ-Pcdh function shown here, PKC is well known to be a negative downstream regulator of dendrite arborization (9, 34, 35, 38, 39). Consistent with this, broad activation of PKC by PMA reduced arborization to low levels regardless of which construct was expressed (Fig. 8, A–D, G, and H). Arbor complexity in PMA-treated control neurons was nearly identical to that of untreated neurons expressing A3-S/D; treatment of A3-S/D-expressing neurons with PMA did not further decrease complexity (Fig. 8, D, G, and H). FAK inhibition with PF-228 rescued arborization to control levels in neurons expressing A3-S/D, consistent with FAK acting downstream of the γ-Pcdhs (Fig. 8H). Together, these data strongly indicate that γ-Pcdh Ser-922 phosphorylation state and PKC act in the same pathway to regulate dendrite arborization in developing neurons.
Discussion
This work expands our understanding of the role γ-Pcdhs play in dendrite arborization by identifying a novel molecular mechanism by which they are regulated. Together, the present data combined with our prior work (9) support the following model (Fig. 9). The γ-Pcdhs bind to FAK and inhibit FAK Tyr-397 phosphorylation and activation through γC (29), which leads to reduced activity of PKC and allows its target MARCKS to remain unphosphorylated, promoting dendrite arborization (9). PKC phosphorylation of γ-Pcdhs at Ser-922 reduces their ability to inhibit FAK (Fig. 5). In this way PKC may act to balance the pathway, preventing overly exuberant branching of dendrites, such as is seen in the A3-S/A-transfected or PF-228-treated cortical neurons (Fig. 6).
FIGURE 9.

Model of how γ-Pcdh constant domain phosphorylation by PKC affects dendrite arborization. γ-Pcdhs exist in cis multimers at the neuronal membrane (25). The γ-Pcdh constant C terminus binds to and inhibits FAK (29), which leads to reduced PKC activity (9) (left side, see the heat map legend). This allows the PKC target MARCKS to remain hypophosphorylated and to promote dendrite arborization via interaction with actin filaments and the membrane (9). When γ-Pcdh phosphorylation at Ser-922 (A3 isoform; equivalent residue numbers in other isoforms) by PKC (circled P) is increased (right side), their ability to inhibit FAK is reduced, which leads to higher PKC activity, hyperphosphorylated MARCKS, and reduced dendrite arborization (such as that seen in Pcdhg mutant neurons; Ref. 9, right side, see heat map legend). PKC phosphorylation of γ-Pcdhs may be mediated by distinct PKC isozymes or subcellular pools rather than simply as a direct negative feedback loop. This may be important for keeping the signaling pathway balanced and allowing for transient, developmentally related increases and decreases in dendrite arborization.
Importantly, the PKC that acts upon γ-Pcdhs may not be from the same pool or be the same isozyme of PKC that is downstream of γ-Pcdh-FAK signaling. If it were, we might expect a runaway signaling feedback loop, which clearly did not occur. There are 12 PKC isozymes exhibiting differential subcellular localization through interaction with distinct anchoring proteins (40). Although we have shown that at least three isozymes, PKCα, -δ, and -γ, are hyperactive in Pcdhg mutant cortex (9), it is unclear if others are involved nor do we know which isozymes may phosphorylate γ-Pcdhs in vivo. Of course, many signaling pathways may converge onto PKC to affect dendrite arborization; γ-Pcdhs are unlikely to be the only major player. In support of this, although Gö6983 treatment did not enhance branching further in neurons expressing A3-S/A, PMA did reduce branching to low levels in these neurons. In any case, PKC is known to regulate dendrite arborization in multiple neuronal subtypes (34, 35, 38, 39); given the widespread neural expression of the γ-Pcdhs (9, 17, 21), our identification of Ser-922 as a PKC target may thus have broad relevance to PKC signaling and dendrite development throughout the brain.
In addition to our identification of Ser-922 as a site for PKC phosphorylation, we show that this residue resides in a lysine-rich domain that can mediate binding to membrane phospholipids. As discussed above, FAK activation depends on PIP2 binding, which allows its autophosphorylation (32, 33). It seems plausible that the γ-Pcdh lipid binding motif competes locally with FAK for binding to phospholipids. When PKC phosphorylates Ser-922, γC loses its ability to bind to phospholipids (Fig. 4, C and D), and this has the potential to prevent inhibition of associated FAK. However, a γ-Pcdh lacking the lipid binding motif (A3-ΔC15) did not significantly fail to inhibit FAK (Fig. 5, D and E) nor did transfection of neurons with such a construct reduce dendrite arborization (Fig. 6), as might have been expected. In other studies, a portion of γ-Pcdh proteins was found to be associated with membrane vesicles of the endolysosome system in non-neuronal cells, and their overexpression can increase the formation of membrane tubules (41, 42). However, it has been shown that trafficking within this system is regulated by the variable cytoplasmic domain (42), not the constant domain in which Ser-922 resides, so the lipid binding we demonstrate here is unlikely to relate directly to these studies. Although the location of Ser-922 within the lipid binding domain makes it difficult to fully dissociate the effects of PKC phosphorylation from those of phospholipid binding, our data suggest that the phosphorylation event itself is the critical regulatory point.
Intriguingly, in the co-sedimentation assay (Fig. 4, C and D), the S/A mutant protein was almost entirely found in the lipid-bound pellet; this could indicate that a small change to the structure of γC in this mutant actually potentiated lipid binding. Similarly, phosphorylated A3-WT protein and truncated A3-ΔC15 protein pulled down increased amounts of FAK in co-immunoprecipitations (Fig. 5), again suggesting that small structural changes may affect γ-Pcdh function. Nothing is known about the structure of γC, although its C terminus is predicted by online protein structure programs to be an intrinsically disordered region. It is increasingly clear that intrinsically disordered regions are critical modulators of cell signaling pathways participating in the assembly of dynamic protein complexes, apparently due to their malleable structure (43, 44). Although beyond the scope of this study, our identification of a lipid binding motif at the end of the C terminus provides an interesting question as to the function, if any, of the motif, and whether PKC phosphorylation-induced abrogation of lipid binding has any physiological role in γ-Pcdh signaling.
A key remaining question is how the balance of this pathway might be regulated spatially within the growing neuron. It is unclear as yet whether γ-Pcdh inhibition of the FAK/PKC pathway is global or is localized, for example, near branch points within dendrites. The cytoplasmic domain is important for the trafficking of exogenous, GFP-tagged γ-Pcdhs within dendrites (45, 46). It is thus plausible that Ser-922 phosphorylation could regulate the trafficking of γ-Pcdhs to dendritic branch points, allowing their inhibition of FAK to be localized. Using HA antibodies, we saw no gross changes in the localization of A3-S/A versus A3-WT in neurons, but unfortunately, our novel anti-GCP antibody is not effective for immunohistochemistry. Other existing antibodies similarly do not stain endogenous proteins well enough for high resolution analyses in neurons. Future efforts thus should be aimed at developing methods to track phosphorylated versus non-phosphorylated endogenous γ-Pcdhs in growing neurons.
Finally, it is of interest that the increase in dendrite arborization observed after expression of A3-S/A in wild-type neurons (Fig. 6) and the rescue of dendritic defects in γ-Pcdh knock-out neurons by A3-S/A was cell-autonomous. Although the γ-Pcdhs can clearly mediate adhesive homophilic interactions in trans, between two cells (25) there is evidence that at least in neurons with planar dendritic arbors such as Purkinje cells and starburst amacrine cells, the γ-Pcdhs mediate repulsive self-avoidance between processes of the same cell (8). The reduction in dendrite arborization we previously observed in Pcdhg mutant cortical neurons (9) was not accompanied by any obvious increase in dendrite self-crossing or fasciculation, suggesting that the role of γ-Pcdhs may vary depending on neuronal subtype, possibly due to differences in cis binding partners. In the present experiments neurons were transfected using a low efficiency (only ∼1–2%) lipofection method. Thus, transfection with a particular γ-Pcdh (here, A3) would not be expected to increase trans homophilic interactions between neurons, as few surrounding untransfected neurons are likely to contain endogenous A3 due to the stochastic, differential expression of Pcdhg isoforms (23, 28).
In this regard it is interesting that transfection of neurons with A3-WT neither increased nor decreased dendrite arborization in wild-type neurons nor could it rescue defects in knock-out neurons. If γ-Pcdh self-avoidance is important in cortical dendrite arborization, we might have expected this manipulation to increase such repulsive signaling and have an effect cell autonomously. Given that expression of A3-S/A did increase arborization cell autonomously, we suggest the possibility that homophilic interaction between neurons with matching γ-Pcdh repertoires might reduce Ser-922 phosphorylation levels, possibly through a conformational change that renders the site inaccessible to PKC. If so, then introduction of a non-phosphorylatable S/A point mutant γ-Pcdh would be expected to have the cell-autonomous effect observed here. We know that γ-Pcdhs are phosphorylated throughout development (Fig. 3F); although there was no significant overall difference in anti-GCP signal across ages, it is possible that phosphorylation is locally dynamically controlled as neurons extend, retract, and stabilize dendrite branches. Thus, it is plausible that γ-Pcdhs are phosphorylated at a high ratio locally as an initial “default” state in immature dendrites. This, according to our model, would reduce γ-Pcdh inhibition of FAK, helping to maintain immature dendrites before the postnatal increase in dendritic arborization. Then, as further dendritic growth occurs and dendritically localized γ-Pcdhs interact with other γ-Pcdhs presented in the environment, such as other neurons or astrocytes, a change in signaling could occur. Homophilic trans-interaction could recruit new binding partners, resulting in blockage of γ-Pcdh phosphorylation or even direct phosphatase activity. A key goal for the future will be to determine the relationship between homophilic interaction (either in trans or between a neuron's own dendritic branches) and Ser-922 phosphorylation in the regulation of FAK activity and dendrite arborization in developing neurons.
Author Contributions
A. B. K., D. S., and J. A. W. conceived the study, A. B. K. and J. A. W. performed the experiments, and A. B. K., D. S., and J. A. W. wrote the paper.
This work was supported, in whole or in part, by National Institutes of Health Grant R01NS055272 (to J. A. W.). The authors declare that they have no conflicts of interest with the contents of this article.
- Pcdh
- protocadherin
- FAK
- focal adhesion kinase
- MARCKS
- myristoylated alanine-rich C-kinase substrate
- γC
- γ-constant
- GCP
- γ-constant phosphorylated
- PMA
- phorbol 12-myristate 13-acetate
- DIV
- day(s) in vitro
- Emx1
- empty spiracles homeobox 1
- Cre
- Cre recombinase
- PIP
- phosphatidylinositol phosphate
- S/A
- S922A.
References
- 1. Lin Y. C., Koleske A. J. (2010) Mechanisms of synapse and dendrite maintenance and their disruption in psychiatric and neurodegenerative disorders. Annu. Rev. Neurosci. 33, 349–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jan Y. N., Jan L. Y. (2010) Branching out: mechanisms of dendritic arborization. Nat. Rev. Neurosci. 11, 316–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Koleske A. J. (2013) Molecular mechanisms of dendrite stability. Nat. Rev. Neurosci. 14, 536–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Piper M., Dwivedy A., Leung L., Bradley R. S., Holt C. E. (2008) NF-protocadherin and TAF1 regulate retinal axon initiation and elongation in vivo. J. Neurosci. 28, 100–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Deans M. R., Krol A., Abraira V. E., Copley C. O., Tucker A. F., Goodrich L. V. (2011) Control of neuronal morphology by the atypical cadherin Fat3. Neuron 71, 820–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shima Y., Kengaku M., Hirano T., Takeichi M., Uemura T. (2004) Regulation of dendritic maintenance and growth by a mammalian 7-pass transmembrane cadherin. Dev. Cell 7, 205–216 [DOI] [PubMed] [Google Scholar]
- 7. Shima Y., Kawaguchi S. Y., Kosaka K., Nakayama M., Hoshino M., Nabeshima Y., Hirano T., Uemura T. (2007) Opposing roles in neurite growth control by two seven-pass transmembrane cadherins. Nat. Neurosci. 10, 963–969 [DOI] [PubMed] [Google Scholar]
- 8. Lefebvre J. L., Kostadinov D., Chen W. V., Maniatis T., Sanes J. R. (2012) Protocadherins mediate dendritic self-avoidance in the mammalian nervous system. Nature 488, 517–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Garrett A. M., Schreiner D., Lobas M. A., Weiner J. A. (2012) γ-Protocadherins control cortical dendrite arborization by regulating the activity of a FAK/PKC/MARCKS signaling pathway. Neuron 74, 269–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Suo L., Lu H., Ying G., Capecchi M. R., Wu Q. (2012) Protocadherin clusters and cell adhesion kinase regulate dendrite complexity through Rho GTPase. J. Mol. Cell Biol. 4, 362–376 [DOI] [PubMed] [Google Scholar]
- 11. Ledderose J., Dieter S., Schwarz M. K. (2013) Maturation of postnatally generated olfactory bulb granule cells depends on functional γ-protocadherin expression. Sci. Rep. 3, 1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lachman H. M., Petruolo O. A., Pedrosa E., Novak T., Nolan K., Stopkova P. (2008) Analysis of protocadherin alpha gene deletion variant in bipolar disorder and schizophrenia. Psychiatr. Genet. 18, 110–115 [DOI] [PubMed] [Google Scholar]
- 13. Pedrosa E., Stefanescu R., Margolis B., Petruolo O., Lo Y., Nolan K., Novak T., Stopkova P., Lachman H. M. (2008) Analysis of protocadherin alpha gene enhancer polymorphism in bipolar disorder and schizophrenia. Schizophr. Res. 102, 210–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Anitha A., Thanseem I., Nakamura K., Yamada K., Iwayama Y., Toyota T., Iwata Y., Suzuki K., Sugiyama T., Tsujii M., Yoshikawa T., Mori N. (2013) Protocadherin α (PCDHA) as a novel susceptibility gene for autism. J. Psychiatry Neurosci. 38, 192–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wu Q., Maniatis T. (1999) A striking organization of a large family of human neural cadherin-like cell adhesion genes. Cell 97, 779–790 [DOI] [PubMed] [Google Scholar]
- 16. Wu Q. (2001) Comparative DNA sequence analysis of mouse and human protocadherin gene clusters. Genome Res. 11, 389–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang X., Weiner J. A., Levi S., Craig A. M., Bradley A., Sanes J. R. (2002) γ-Protocadherins are required for survival of spinal interneurons. Neuron 36, 843–854 [DOI] [PubMed] [Google Scholar]
- 18. Weiner J. A., Wang X., Tapia J. C., Sanes J. R. (2005) γ protocadherins are required for synaptic development in the spinal cord. Proc. Natl. Acad. Sci. U.S.A. 102, 8–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Prasad T., Wang X., Gray P. A., Weiner J. A. (2008) A differential developmental pattern of spinal interneuron apoptosis during synaptogenesis: insights from genetic analyses of the protocadherin-γ gene cluster. Development 135, 4153–4164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lefebvre J. L., Zhang Y., Meister M., Wang X., Sanes J. R. (2008) γ-Protocadherins regulate neuronal survival but are dispensable for circuit formation in retina. Development 135, 4141–4151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Garrett A. M., Weiner J. A. (2009) Control of CNS synapse development by γ-protocadherin-mediated astrocyte-neuron contact. J. Neurosci. 29, 11723–11731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Prasad T., Weiner J. A. (2011) Direct and indirect regulation of spinal cord ia afferent terminal formation by the γ-protocadherins. Front. Mol. Neurosci. 4, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kaneko R., Kato H., Kawamura Y., Esumi S., Hirayama T., Hirabayashi T., Yagi T. (2006) Allelic gene regulation of Pcdh-α and Pcdh-γ clusters involving both monoallelic and biallelic expression in single Purkinje cells. J. Biol. Chem. 281, 30551–30560 [DOI] [PubMed] [Google Scholar]
- 24. Zou C., Huang W., Ying G., Wu Q. (2007) Sequence analysis and expression mapping of the rat clustered protocadherin gene repertoires. Neuroscience 144, 579–603 [DOI] [PubMed] [Google Scholar]
- 25. Schreiner D., Weiner J. A. (2010) Combinatorial homophilic interaction between γ-protocadherin multimers greatly expands the molecular diversity of cell adhesion. Proc. Natl. Acad. Sci. U.S.A. 107, 14893–14898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thu C. A., Chen W. V., Rubinstein R., Chevee M., Wolcott H. N., Felsovalyi K. O., Tapia J. C., Shapiro L., Honig B., Maniatis T. (2014) Single-cell identity generated by combinatorial homophilic interactions between α, β, and γ protocadherins. Cell 158, 1045–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zipursky S. L., Sanes J. R. (2010) Chemoaffinity revisited: dscams, protocadherins, and neural circuit assembly. Cell 143, 343–353 [DOI] [PubMed] [Google Scholar]
- 28. Yagi T. (2012) Molecular codes for neuronal individuality and cell assembly in the brain. Front. Mol. Neurosci. 5, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen J., Lu Y., Meng S., Han M. H., Lin C., Wang X. (2009) α- and γ-protocadherins negatively regulate PYK2. J. Biol. Chem. 284, 2880–2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schalm S. S., Ballif B. A., Buchanan S. M., Phillips G. R., Maniatis T. (2010) Phosphorylation of protocadherin proteins by the receptor tyrosine kinase Ret. Proc. Natl. Acad. Sci. U.S.A. 107, 13894–13899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yu F. X., Sun H. Q., Janmey P. A., Yin H. L. (1992) Identification of a polyphosphoinositide-binding sequence in an actin monomer-binding domain of gelsolin. J. Biol. Chem. 267, 14616–14621 [PubMed] [Google Scholar]
- 32. Frame M. C., Patel H., Serrels B., Lietha D., Eck M. J. (2010) The FERM domain: organizing the structure and function of FAK. Nat. Rev. Mol. Cell Biol. 11, 802–814 [DOI] [PubMed] [Google Scholar]
- 33. Goñi G. M., Epifano C., Boskovic J., Camacho-Artacho M., Zhou J., Bronowska A., Martín M. T., Eck M. J., Kremer L., Gräter F., Gervasio F. L., Perez-Moreno M., Lietha D. (2014) Phosphatidylinositol 4,5-bisphosphate triggers activation of focal adhesion kinase by inducing clustering and conformational changes. Proc. Natl. Acad. Sci. U.S.A. 111, E3177–E3186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Metzger F., Kapfhammer J. P. (2000) Protein kinase C activity modulates dendritic differentiation of rat Purkinje cells in cerebellar slice cultures. Eur. J. Neurosci. 12, 1993–2005 [DOI] [PubMed] [Google Scholar]
- 35. Schrenk K., Kapfhammer J. P., Metzger F. (2002) Altered dendritic development of cerebellar Purkinje cells in slice cultures from protein kinase Cγ-deficient mice. Neuroscience 110, 675–689 [DOI] [PubMed] [Google Scholar]
- 36. Beggs H. E., Schahin-Reed D., Zang K., Goebbels S., Nave K. A., Gorski J., Jones K. R., Sretavan D., Reichardt L. F. (2003) FAK deficiency in cells contributing to the basal lamina results in cortical abnormalities resembling congenital muscular dystrophies. Neuron 40, 501–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rico B., Beggs H. E., Schahin-Reed D., Kimes N., Schmidt A., Reichardt L. F. (2004) Control of axonal branching and synapse formation by focal adhesion kinase. Nat. Neurosci. 7, 1059–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gundlfinger A., Kapfhammer J. P., Kruse F., Leitges M., Metzger F. (2003) Different regulation of Purkinje cell dendritic development in cerebellar slice cultures by protein kinase Cα and -β. J. Neurobiol. 57, 95–109 [DOI] [PubMed] [Google Scholar]
- 39. Pilpel Y., Segal M. (2004) Activation of PKC induces rapid morphological plasticity in dendrites of hippocampal neurons via Rac and Rho-dependent mechanisms. Eur. J. Neurosci. 19, 3151–3164 [DOI] [PubMed] [Google Scholar]
- 40. Hoque M., Rentero C., Cairns R., Tebar F., Enrich C., Grewal T. (2014) Annexins: scaffolds modulating PKC localization and signaling. Cell. Signal. 26, 1213–1225 [DOI] [PubMed] [Google Scholar]
- 41. Hanson H. H., Kang S., Fernández-Monreal M., Oung T., Yildirim M., Lee R., Suyama K., Hazan R. B., Phillips G. R. (2010) LC3-dependent intracellular membrane tubules induced by γ-protocadherins A3 and B2: a role for intralumenal interactions. J. Biol. Chem. 285, 20982–20992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. O'Leary R., Reilly J. E., Hanson H. H., Kang S., Lou N., Phillips G. R. (2011) A variable cytoplasmic domain segment is necessary for γ-protocadherin trafficking and tubulation in the endosome/lysosome pathway. Mol. Biol. Cell 22, 4362–4372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. van der Lee R., Buljan M., Lang B., Weatheritt R. J., Daughdrill G. W., Dunker A. K., Fuxreiter M., Gough J., Gsponer J., Jones D. T., Kim P. M., Kriwacki R. W., Oldfield C. J., Pappu R. V., Tompa P., Uversky V. N., Wright P. E., Babu M. M. (2014) Classification of intrinsically disordered regions and proteins. Chem. Rev. 114, 6589–6631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wright P. E., Dyson H. J. (2015) Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 16, 18–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fernández-Monreal M., Kang S., Phillips G. R. (2009) γ-protocadherin homophilic interaction and intracellular trafficking is controlled by the cytoplasmic domain in neurons. Mol. Cell Neurosci. 40, 344–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fernández-Monreal M., Oung T., Hanson H. H., O'Leary R., Janssen W. G., Dolios G., Wang R., Phillips G. R. (2010) γ-Protocadherins are enriched and transported in specialized vesicles associated with the secretory pathway in neurons. Eur. J. Neurosci. 32, 921–931 [DOI] [PMC free article] [PubMed] [Google Scholar]