

Background: P-Rex1 (phosphatidylinositol (3,4,5)-trisphosphate-dependent Rac exchanger-1) activity is correlated with tumorigenesis in cancer.
Results: P-Rex1·Rac1 (Ras-related C3 botulinum toxin substrate-1) crystal structure reveals the molecular mechanism of Rac1 activation.
Conclusion: The P-Rex1·Rac1 interface is critical for Rac1 activation in breast cancer cell lines.
Significance: The study provides a rationale for therapeutic targeting of the P-Rex1·Rac1 interface by describing the structural basis of P-Rex1 activity.
Keywords: breast cancer, crystallography, G protein-coupled receptor (GPCR), guanine nucleotide exchange factor (GEF), Ras-related C3 botulinum toxin substrate 1 (Rac1), receptor tyrosine kinase, Rho (Rho GTPase), P-Rex1, PREX1, PREX2
Abstract
The P-Rex (phosphatidylinositol (3,4,5)-trisphosphate (PIP3)-dependent Rac exchanger) family (P-Rex1 and P-Rex2) of the Rho guanine nucleotide exchange factors (Rho GEFs) activate Rac GTPases to regulate cell migration, invasion, and metastasis in several human cancers. The family is unique among Rho GEFs, as their activity is regulated by the synergistic binding of PIP3 and Gβγ at the plasma membrane. However, the molecular mechanism of this family of multi-domain proteins remains unclear. We report the 1.95 Å crystal structure of the catalytic P-Rex1 DH-PH tandem domain in complex with its cognate GTPase, Rac1 (Ras-related C3 botulinum toxin substrate-1). Mutations in the P-Rex1·Rac1 interface revealed a critical role for this complex in signaling downstream of receptor tyrosine kinases and G protein-coupled receptors. The structural data indicated that the PIP3/Gβγ binding sites are on the opposite surface and markedly removed from the Rac1 interface, supporting a model whereby P-Rex1 binding to PIP3 and/or Gβγ releases inhibitory C-terminal domains to expose the Rac1 binding site.
Introduction
Rho GTPases are small G proteins that regulate cytoskeletal organization, cell-cycle progression, and gene expression, and their deregulation drives tumorigenesis and metastatic dissemination (1, 2). The activation of Rho GTPases, from the inactive GDP-bound state to the active GTP-bound state, is regulated by guanine nucleotide exchange factors (GEFs)7 (3). P-Rex1 (phosphatidylinositol (3,4,5)-trisphosphate (PIP3)-dependent Rac exchanger 1) is a large multidomain GEF that catalyzes nucleotide exchange for the Rac subfamily of Rho GTPases to control cell motility, chemotaxis, and cell morphology (4–7).
Crucially, P-Rex1 overexpression is correlated with tumorigenesis in breast and prostate cancers (8–11), and knockdown of P-Rex1 inhibits cell migration and invasion in melanoma, prostate, and ovarian cancers (8, 12–14). The human PREX1 gene is located on chromosome 20q13; amplification of this region occurs in 8–29% of breast tumors and is associated with poor patient outcomes (10). This region is also frequently deleted or amplified in malignant myeloid diseases, hereditary prostate cancer, pancreatic endocrine tumors, and ovarian cancers (15–18). PREX1 mRNA expression is elevated in the majority of human melanoma cell lines and human melanomas (14) and promotes melanoblast migration via Rac activation. Melanoma-model mice with P-Rex1 ablation are more resistant to metastasis (14). Although P-Rex1 is not detected in the normal breast, the PREX1 gene is amplified or mutated in primary breast tumors, with P-Rex1 positive staining in 58% of all breast cancers. Recent genome sequencing of melanoma and pancreatic cancers has also detected a high frequency of mutation in P-Rex2, a close structural and functional homologue of P-Rex1 (59% sequence identity) (19, 20).
P-Rex1 contains an N-terminal DH-PH tandem domain that forms the catalytic subunit of the protein in vitro (21). C-terminal to the DH-PH domain are two DEP (Disheveled, EGL-10, pleckstrin) domains, two PDZ (postsynapticdensity protein, discs large, zona occludens-1) domains, and a large IP4P (inositol polyphosphate 4-phosphatase) homology domain with no identified phosphatase activity to date (Fig. 1A). Rho GEFs maintain a generally conserved DH-PH tandem domain architecture; however the mode of GTPase binding and activation varies between GEFs. A number of GEFs activate Rho GTPases through interactions with the DH domain alone, whereas several also require molecular contributions from the PH domain and/or regions outside the tandem DH-PH domain (22–26). The difference in contribution of the PH domain to GTPase binding observed between different GEFs is largely due to conformational variability between the relative positioning of the DH and PH domains. This conformational variability is defined primarily by the GEF-specific length and orientation of the final α-helix of the DH domain (27). Therefore, the mechanism of Rho GTPase activation is highly GEF-specific.
FIGURE 1.

Structure of the P-Rex1·Rac1 complex. A, schematic representation of the domain layout of P-Rex1, highlighting the position of the DH (yellow) and PH (purple) domains. Rac1 is shown in teal. B, the structure of P-Rex1-(34–399) bound to Rac1-(1–176) shown schematically. The position of the missing β3-β4 loop within the P-Rex1 PH domain is indicated.
P-Rex proteins are unique within the Rho GEF family, as they function as a key integration point for signals downstream of both receptor tyrosine kinases (RTKs) and G protein-coupled receptors (GPCRs) (28, 29). For example, activation of P-Rex1 by the RTK epidermal growth factor receptor (EGFR) promotes breast cancer cell migration and proliferation (10), whereas P-Rex1 activation by the chemokine GPCR, CXCR4, mediates endothelial cell angiogenesis in vitro, an essential step in tumorigenesis (30). RTK stimulation activates phosphoinositide 3-kinase (PI3K) to phosphorylate PIP2 to PIP3, whereas GPCR activation leads to dissociation of the heterotrimeric G protein into Gα and Gβγ subunits. Both the PIP3 and Gβγ subunits are thought to synergistically activate P-Rex proteins at the plasma membrane (29, 31).
Currently, we have limited molecular insight into how Rho GTPases are activated by P-Rex1 and how P-Rex1 activity is regulated by the coordinated actions of PIP3, the Gβγ subunits, and protein phosphorylation. For instance, P-Rex1 PH domain binding to PIP3 is necessary for activation and membrane localization (21, 31); however the mechanism by which PIP3 binding increases P-Rex1 exchange activity is unresolved. In addition to PIP3 binding, PH domains commonly interact with Gβγ subunits (32–35). However, Gβγ activation of P-Rex1 appears to be a complex process requiring the PH domain, DH domain, and/or additional interactions with the C-terminal DEP, PDZ, and IP4P domains (21, 36). Moreover, intramolecular binding between the C terminus and the DH-PH domain of P-Rex1 can autoregulate GEF activity (21, 36). Finally, distinct patterns of phosphorylation further contribute to the control of P-Rex1 activity (11, 37) and the affinity of P-Rex1 for Gβγ and PIP3 (37, 38).
Herein, we report the 1.95 Å x-ray crystal structure of the P-Rex1 DH-PH domain in complex with its canonical GTPase, Rac1, providing the first structural insight into P-Rex1 activity and regulation. Critically, using structure-guided mutagenesis we have dissected the central role of the P-Rex1 DH domain-Rac1 interface in promoting Rac1 activation both in vitro and downstream of GPCR and RTK signaling in cancer cell lines. Our structural data also indicate that the PIP3/Gβγ binding sites in P-Rex1 are markedly removed and on the opposite surface from the Rac1 interface. Together, these data provide insight into the future therapeutic targeting of P-Rex1 in the treatment of a number of cancers.
Experimental Procedures
Reagents
RaichuEV-Rac1 (39, 40), kindly provided by Prof. Michiyuki Matsuda and Assoc. Prof. Kazuhiro Aoki (Kyoto University, Japan), is contained in the pCAGGS vector (41) provided by Dr. Jun-ichi Miyazaki (Osaka University, Japan). MCF7 cells were from the American Type Culture Collection. The highly metastatic MDA-MB-231 human breast adenocarcinoma cells (42) were a gift from Dr. Zhou Ou (Fudan University Shanghai Cancer Centre, China). P-Rex1 siRNA was from GE Dharmacon (ON-TARGETplus SMARTpool). The monoclonal antibody to P-Rex1 was generated in-house.
Cloning and Mutagenesis
His10-tagged P-Rex1 DH-PH (residues 1–404) was cloned into the BamHI and EcoRI sites of the polyhedron multiple cloning site of pFastBacDual (Invitrogen). Rac1 (residues 1–177) was cloned into the XhoI and KpnI sites of the p10 multiple cloning site within the same vector. For bacterial expression, Rac1 G12V (residues 1–177) was cloned into pGEXTEV, a modified version of pGEX-4T-1 (GE Healthcare) where the thrombin site is replaced with a TEV cleavage site. Full-length HA-tagged P-Rex1 was cloned into the HindIII and XbaI sites of pcDNA3.1(+) (Invitrogen). Mutations were introduced using QuikChange site-directed mutagenesis (Stratagene).
Protein Purification
His10-tagged P-Rex1 DH-PH (residues 1–404) was co-expressed with Rac1 (residues 1–177) in Sf9 cells for 2.5 days following baculovirus infection (pFastBacDual, Bac-to-Bac, Invitrogen). Co-expressed Rac1 was not purified with P-Rex1, as phosphorylation of P-Rex1 significantly affected the yield of the P-Rex1·Rac1 complex purified directly from insect cells. However, co-expression significantly improved P-Rex1 expression in Sf9 cells. Cells were harvested by centrifugation and lysed by sonication in 20 mm Tris, pH 8.0, 500 mm NaCl, 10% (v/v) glycerol, 5 mm β-mercaptoethanol, and 0.02% (w/v) azide. Lysates were cleared by centrifugation, filtered at 0.8 μm, and incubated with Ni-NTA resin (Qiagen) and 20 mm imidazole for 90 min at 4 °C with agitation. The resin was washed with lysis buffer, and P-Rex1 was eluted with 500 mm imidazole in the same buffer.
The His10 tag was removed from P-Rex1 with overnight TEV cleavage during dialysis into 20 mm Tris, pH 8.0, 200 mm NaCl, 10% (v/v) glycerol, 2 mm DTT, and 5 mm MnCl2 and concurrent dephosphorylation with λ-phosphatase. Dephosphorylation of P-Rex1 was necessary to increase the final yield of the purified P-Rex1·Rac1 complex. P-Rex1 was further purified by cation exchange on a ResourceS (GE Healthcare) column with a gradient from 50 mm to 1 m NaCl in 20 mm Tris, pH 8.0, 2 mm DTT, and 5% (v/v) glycerol. P-Rex1-containing fractions were further purified on a HiLoad Superdex 75 16/60 size-exclusion column (GE Healthcare).
GST-Rac1 G12V was expressed overnight in Escherichia coli BL21(DE3) CodonPlus cells by IPTG induction at 18 °C. Cells were harvested by centrifugation and lysed by sonication in 20 mm Tris, pH 8.0, 500 mm NaCl, 2 mm DTT, and 2 mm EDTA. Lysates were cleared by centrifugation, filtered at 0.8 μm, and incubated with glutathione-Sepharose 4B resin (GE Healthcare) for 90 min at 4 °C. The resin was washed with lysis buffer and incubated at 4 °C overnight with TEV added to cleave the GST tag. The protein was eluted from the resin and further purified on a HiLoad Superdex 75 16/60 size-exclusion column (GE Healthcare).
To form the P-Rex1 DH-PH·Rac1 complex for crystallography, purified P-Rex1 and Rac1 were concentrated to 5–10 mg/ml and mixed at a 1:3 (P-Rex1·Rac1) ratio with the further addition of 5 mm EDTA. After a 30-min incubation at room temperature, the complex was purified on a HiLoad Superdex 75 16/60 size-exclusion column (GE Healthcare) equilibrated in 20 mm Tris, pH 8.0, 150 mm NaCl, and 2 mm DTT. Fractions containing the complex were pooled and concentrated to ∼10 mg/ml for crystallography.
Crystallization, Structure Determination, and Refinement
P-Rex1 DH-PH·Rac1 crystals were formed at a 1:1 drop ratio by sitting drop vapor diffusion in 0.1 mm Na-MES, pH 6.5, 0.2 m sodium acetate, and 15% (w/v) PEG 8000. Crystals were cryoprotected in mother liquor containing 20% (v/v) glycerol, and data were collected at the MX2 microfocus beamline at the Australian Synchrotron (43). The structure was determined by molecular replacement in PHENIX (44) within the Phaser (45) module using PDB codes 2DFK and 3SBD (46, 47). The structure was autobuilt in ARP/wARP within the CCP4 suite (48–50). Refinement was carried out using PHENIX Refine (51) with local rebuilding in COOT (52), resulting in a model with an R-factor of 16.23% (Rfree of 18.32%). The structure had a final MolProbity (53) score of 0.70 (100th percentile). Structural alignments were performed with Superpose within the CCP4 suite (54).
N-Methylanthraniloyl (Mant)-GDP Exchange Assay
Nucleotide exchange was measured with Mant-GDP (Invitrogen), a fluorescent GDP analogue. 2 μm Rac1 was equilibrated with 1 μm Mant-GDP in 20 mm Tris, pH 7.4, 50 mm NaCl, 5 mm MgCl2, 1 mm DTT, and 5% (v/v) glycerol for 30 min prior to the addition of P-Rex1 DH-PH to a final concentration of 400 nm. Fluorescence (λex = 360 nm, λem = 440 nm) was measured on a BMG Labtech CLARIOstar microplate reader over a period of 3 h at 25 °C. Traces shown are baseline-subtracted and the mean of at least three independent data sets. GEF activity of mutants was quantified by measuring fluorescence at 60 min after DH-PH addition, relative to the wild type.
Pulldown Analysis
His6-tagged Rac1 was bound to Ni-NTA resin (Qiagen) and incubated with 1 μm untagged P-Rex1 DH-PH wild type or mutants in 20 mm Tris, pH 8.0, 150 mm NaCl, 2 mm DTT, and 20 mm imidazole. Solutions were incubated at 4 °C for 90 min with agitation following which the beads were washed three times in buffer and eluted in buffer containing 500 mm imidazole. Pulldowns were analyzed by SDS-PAGE.
Cell Culture and Transfection
The MCF7 and MDA-MB-231 cell lines were verified by CellBank Australia. MCF7 cells were maintained in Dulbecco's modified Eagle's medium (Invitrogen) with 10% (v/v) FBS, 10 μg/ml insulin, and penicillin/streptomycin. MDA-MB-231 HM cells were maintained in the same medium without insulin. Cells were grown in a humidified atmosphere at 37 °C with 5% CO2.
For verification of endogenous P-Rex1 knockdown using siRNA, cells were seeded at 70% confluency during transfection with Lipofectamine 2000 into 6-well plates in OptiMEM (Invitrogen). Cells were transfected with 10 nm scrambled or P-Rex1 siRNA and 7.2 μl of Lipofectamine 2000/well. The medium was replaced with complete growth medium 6 h after transfection, and protein expression was assessed by Western blotting 72 h after transfection.
For verification of P-Rex1 mutant expression by Western blotting, cells were seeded at 70% confluency during transfection with 25-kDa linear polyethylenimine (PEI) into 6-well plates in complete growth medium. Cells were transfected with 1.6 μg of pcDNA or 1.6 μg of P-Rex1 DNA at a DNA:PEI ratio of 1:4.5 (for MCF7) or 1:6 (for MDA-MB-231). Protein expression was assessed by Western blotting 48 h after transfection.
For FRET experiments, cells were seeded at 70% confluency during transfection with 25-kDa linear PEI into black, optically clear 96-well plates in complete growth medium. MCF7 cells were transfected with 200 ng/well RaichuEV-Rac1 and 100 ng/well pcDNA or P-Rex1 at a 1:4.5 DNA:PEI ratio. MDA-MB-231 cells were transfected with 150 ng/well RaichuEV-Rac1 and 100 ng/well pcDNA or P-Rex1 at a 1:6 DNA:PEI ratio. For P-Rex1 knockdown using siRNA, MCF7 cells were transfected with 100 ng/well RaichuEV-Rac1, 10 nm/well scrambled or P-Rex1 siRNA, and 0.45 μl/well Lipofectamine 2000. FRET was measured following overnight serum restriction (0.5% FBS (v/v) culture medium) at 48 or 72 h (siRNA experiments) following transfection.
Cell Lysate Preparation and Western Blotting
Cells were detached from 6-well plates in PBS and resuspended in 100 μl of lysis buffer/well (50 mm Tris, pH 7.5, 150 mm NaCl, 1% Nonidet P-40, 1 mm benzamidine, 2 mm DTT, 2 mm sodium orthovanadate, 10 mm β-glycerophosphate, 1 mm PMSF, and protease inhibitor mixture). Cells were incubated for 30 min on ice and lysed by passage through a 21-guage needle 10 times. Lysates were centrifuged (200 × g, 4 °C, 5 min), and the supernatant was snap-frozen on dry ice. For Western blotting, lysates were thawed, and total protein concentration measured using a Bradford detection assay. 12 or 14 μg of total protein was run on 8% SDS-PAGE for MCF7 and MDA-MB-231 samples, respectively. Protein was transferred to nitrocellulose membranes by electroblotting in 3-(cyclohexylamino)propanesulfonic acid (CAPS) buffer before membrane blocking in 5% (w/v) milk powder in TBS for 1 h. Blots were incubated overnight at 4 °C with either anti-P-Rex1 or anti-HA (Abcam, ab9110) at 1:1000 in 1% (w/v) milk in TBS. Blots were washed three times with TBS-T (0.05% v/v Tween 20 in TBS) before incubation with anti-rabbit HRP-conjugated antibody (Thermo Scientific, No. 31460) at 1:5000 in 5% (w/v) milk in TBS for 1 h. Blots were then washed three times in TBS-T, and bands were visualized with Amersham Biosciences ECL chemiluminescence reagent (GE Healthcare). Blots were stripped (Millipore ReBlot Plus) and reprobed as described with anti-α-tubulin (Abcam, ab4074, 1 μg/ml) to confirm equal protein loading.
FRET Imaging of Live Cells
Cells were equilibrated in Hanks' balanced salt solution (Invitrogen) at 37 °C. Fluorescence imaging was performed using a high content GE Healthcare IN Cell 2000 analyzer with a FRET module. For emission ratio analysis, cells were excited sequentially using a CFP filter (430/24, 430 ± 12 nm) with emission measured using YFP (535/30) and CFP (470/24) filters and a polychroic filter optimized for the CFP/YFP filter pair (Quad3). Cells were imaged every 1 min, allowing image capture of up to 16 wells/min. Two fields of view per well were captured for the MDA-MB-231 cells due to lower transfection efficiency. Baseline emission ratio images were captured for 4 min. Cells were challenged with an EC80 concentration of isoprenaline (100 nm), SDF1α (30 nm), EGF (10 ng/ml), or vehicle (0.13% w/v BSA in PBS), and images were captured for 20 min. Cells were then stimulated with the positive control (1 μm isoprenaline, 50 ng/ml EGF, 10 μm AlCl3, and 10 mm NaF (55)) for 10 min to generate a maximal increase in Rac1 activity, and positive emission ratio images were captured for 4 min. Data were analyzed within the FIJI distribution of ImageJ (57). Cells with >5% change in F/Fo (FRET ratio relative to baseline for each cell) after stimulation with positive controls were selected for analysis. Data are expressed as the emission ratio relative to the maximal response for each cell (F/Fmax), with the area under the curve calculated using GraphPad Prism. Pseudocolor ratiometric images of the cells were generated as described (58). Data are expressed as the mean ± S.E. of n cells as stated. Statistical significance was determined by two-way ANOVA with Dunnett's multiple comparison test, and p < 0.05 was deemed significant.
Results
Structure of the P-Rex1·Rac1 Complex
To examine the mechanism of P-Rex1-dependent Rac1 activation, we crystalized the P-Rex1 DH-PH domain (residues 1–404) in complex with Rac1. The structure was determined by molecular replacement and refined to 1.95 Å resolution with an Rwork/Rfree of 16.23 and 18.32%, respectively, and excellent geometry (Table 1). The final model comprises P-Rex1-(34–399) bound to Rac1 without its membrane-associated C terminus (residues 1–176) (Fig. 1A). No density was observed for the N-terminal P-Rex1-(1–33) and -(306–322) within the β3-β4 loop of the PH domain, suggesting a high degree of mobility within these regions.
TABLE 1.
Data collection and refinement statistics
| P-Rex1-(1–404)·Rac1-(1–177) | |
|---|---|
| Data collection | |
| PDB ID code | 4YON |
| Beamline | Australian Synchrotron MX2 |
| Oscillation range, ° | 1 |
| Space group | C2221 |
| Cell parameters | a = 94.51 Å, b = 111.25 Å, c = 158.12 Å |
| α = 90°, β = 90°, γ = 90° | |
| Resolution, Åa | 40.56–1.95 (2.00–1.95) |
| Unique reflectionsa | 60,929 (4,244) |
| I/σIa | 10.9 (2.1) |
| Rmeas, %a | 21.7 (139.7) |
| Rpim, %a | 6.3 (40.4) |
| CC½a,b | 99.6 (72.6) |
| Completeness, %a | 100.0 (99.9) |
| Redundancya | 11.7 (11.8) |
| Refinement | |
| Resolution, Å | 40.56–1.95 |
| No. of reflections | 60,883 |
| Rwork/Rfree, % | 16.23/18.32 |
| No. of protein atoms | 4,197 |
| No. of water atoms | 780 |
| r.m.s. deviation from ideal bond length, Å | 0.004 |
| r.m.s. deviation angles, ° | 0.794 |
| Ramachandran statistics, %c | |
| Favored regions | 98.07 |
| Outliers | 0 |
The P-Rex1 DH domain (residues 34–258) is composed of the six main α-helices typical of Dbl family Rho GEFs arranged into an elongated bundle (Fig. 1B). Helices α3 and α4 are further divided into two smaller α-helical segments. The extended helix α6 of the P-Rex1 DH domain bridges both the DH and the PH domains. This arrangement effectively positions the PH domain (residues 259–399) toward the solvent and orients it away from the DH domain. The PH domain forms a seven-β-stranded anti-parallel β-sandwich capped by a C-terminal α-helix. The Rac1 fold, consistent with previously described structures (59), contains one central six-stranded β-sheet surrounded by six α-helices.
The P-Rex1 DH Domain Activates Rac1 with No Direct Molecular Contributions from the PH Domain
Rac1 makes extensive contacts with helices α1, α5, and α6 of the P-Rex1 DH domain, burying a total of 2,578 Å2 (Figs. 1B and 2A). Rac1 makes no molecular contacts with the PH domain (Fig. 2A). The conformational change within two highly conserved regions of GTPases, termed switch 1 and switch 2, facilitates the displacement of both the GDP and a Mg2+ cofactor to allow GTP binding and GTPase activation (59). Of the three highly conserved regions of the P-Rex1 DH domain (termed CR1–3), CR1 and CR3 make extensive contacts with both switch 1 and switch 2 of Rac1 (Fig. 2A). P-Rex1 helix α1 interacts primarily with switch 1 through the highly conserved Glu-56P-Rex1, which forms hydrogen bonds with Tyr-32Rac1 and the main chain amides of Thr-35Rac1 and Val-36Rac1 with excellent density (Figs. 2B and 3, A, B, and D).
FIGURE 2.

The P-Rex1·Rac1 interface. A, the P-Rex1 DH domain helices α1, α5, and α6 interact with the Rac1 switch 1 and 2 regions. Rac1 has been rotated 180° about the y axis relative to P-Rex1 to show the interface between each protein. Interacting residues involved in switch 1 binding (blue) and switch 2 binding (orange) are highlighted as spheres. B, 2Fo − Fc electron density map contoured at 1.0 σ, highlighting interactions between P-Rex1 (gray) and the Rac1 switch 1 region (blue). C, structural alignment of P-Rex1-bound Rac1 with Rac1 bound to GDP and Mg2+ (gray; PDB code 1I4D (64), with an r.m.s. deviation of 1.008 Å over 169 residues). Conformational changes in the Rac1 switch 1 (blue) and switch 2 (orange) regions occur upon P-Rex1 binding, whereas the conformation of the P-loop (green) is unchanged. Residues within the Rac1 switch regions that undergo large conformational changes upon P-Rex1 binding are displayed as sticks.
FIGURE 3.

Structural basis of Rac1 activation by P-Rex1. A, location of the Rac1 switch 1 and switch 2 interaction interfaces within the P-Rex1·Rac1 structure. B, P-Rex1 DH domain residues Glu-56 and Gln-197 interact with the switch 1 region of Rac1. C, P-Rex1 DH domain residues Lys-201, Asn-238, Arg-242, and Glu-245 mediate interactions with the switch 2 region of Rac1. D, multiple sequence alignment of P-Rex1 family members with related GEFs. The P-Rex1 secondary structure is illustrated above the alignment with residues targeted for mutational analysis indicated. E, the rate of in vitro GEF activity of the P-Rex1 DH-PH domain was significantly decreased for E56R, E56A, and Q197A mutants. F, the rate of in vitro GEF activity of the P-Rex1 DH-PH domain was significantly decreased for R242A, K201A, and N238A, but not E245R, mutants. Curves show the average of at least three independent experiments. G, GEF activity at 60 min following P-Rex1 DH-PH addition. H, impaired interactions between wild-type His6-Rac1 and untagged P-Rex1-(1–404) mutants compared with the wild-type control. Error bars indicate means ± S.E. ***, p < 0.001, one-way ANOVA with Sidak's multiple comparison test. The activity of the mutants is expressed relative to the wild-type P-Rex1 DH-PH domain. Binding was assessed by pulldown assays using His6-Rac1 bound to Ni-NTA-agarose beads. Purified untagged wild-type P-Rex1 and the indicated P-Rex1 mutants were added to the pulldown assay at a concentration of 1 μm.
Structural alignment of P-Rex1-bound Rac1 with inactive Rac1-GDP reveals a displacement of Thr-35Rac1 ∼6 Å toward P-Rex1 (Fig. 2C). This large conformational change disrupts the ability of Rac1 to coordinate Mg2+, promoting GDP dissociation and subsequent GTP binding. Also within switch 1, Asn-39Rac1 forms hydrogen bonds with the conserved Gln-197P-Rex1 in CR3 (helix α5) (Fig. 3, B and D). To confirm that these interactions are critical for P-Rex1 activity, we generated P-Rex1 DH-PH domain Q197A and E56A mutants and an additional switch charge E56R mutant. Mutation of Gln-197P-Rex1 decreased activity compared with the wild-type control in an in vitro Mant-GDP exchange assay (Fig. 3, E and G). P-Rex1 DH-PH domain activity was abolished following mutation of Glu-56P-Rex1 in the E56A and E56R mutants (Fig. 3, E and G). As expected, mutation of either Glu-56P-Rex1 or Gln-197P-Rex1 also diminished Rac1 binding compared with wild-type P-Rex1 in vitro (Fig. 3H). Together, these data reveal the functional importance of the conserved Glu-56P-Rex1 and Gln-197P-Rex1 for switch 1 binding and activation of Rac1.
Rac1 is further anchored in a nucleotide-releasing conformation by extensive contacts between switch 2 and P-Rex1 helices α5 and α6, which buries 850 Å2 of surface area (Fig. 3, A and C). Asn-238P-Rex1 forms hydrogen bonds with Asp-65Rac1 and Arg-66Rac1 (Fig. 3C), and alanine substitution (N238A) completely abolishes both the exchange activity and Rac1 binding (Fig. 3, F and H). Previous studies using sequence conservation reported a “GEF-dead” P-Rex1 N238A/E56A mutation, which disrupts activity both in vitro and in vivo (5, 8, 21). Our data give a structural justification for this GEF-dead mutant, additionally demonstrating that mutation of each residue alone is sufficient to abolish GEF activity in vitro.
Glu-245P-Rex1 was in close enough proximity to form a hydrogen bond and salt bridge with Arg-66Rac1 in switch 2 (Fig. 3C). A switch charge mutation (E245R) had no effect on exchange activity (Fig. 3, F and G). Interestingly however, we detected reduced binding of P-Rex1 E245R to Rac1 (Fig. 3H). Arg-242P-Rex1 and the conserved Lys-201P-Rex1 provided further contacts with the Rac1 switch 2 region via Asp-65Rac1 and Ala-59Rac1, respectively (Fig. 3, C and D). Point mutations of these P-Rex1 residues to alanine (R242A and K201A) diminished GEF activity (Fig. 3, F and G), although they did not completely abrogate Rac1 binding (Fig. 3H).
The Rac1 P-loop (residues 10–18) coordinates the binding to the phosphate groups of the bound nucleotide (27). In the P-Rex1·Rac1 complex, no molecular contacts occur between P-Rex1 and the P-loop, and its relative conformation does not change upon P-Rex1 binding (Fig. 2C). This is consistent with previously published structures in which GEF binding does not promote major conformational change in the nucleotide coordinating Rac1 P-loop (27).
EGFR, CXCR4, and β-Adrenoceptors (βAR) Require P-Rex1 for Rac1 Activation in Breast Cancer Cells
To examine P-Rex1-dependent activation of Rac1 in an intact cell system, we transfected two human breast cancer cell lines with the RaichuEV-Rac1 FRET-based biosensor (39, 40): MCF7 cells, a luminal breast cancer cell line that endogenously expresses P-Rex1 at high levels (Fig. 4D), and a highly metastatic variant of the triple negative breast cancer cell line, MDA-MB-231 (42), that has undetectable levels of endogenous P-Rex1 (10)(Fig. 4F).
FIGURE 4.

EGFR, CXCR4, and βAR require P-Rex1 for Rac1 activation in breast cancer cells. A, Rac1 activity in MCF7 cells following the addition of 100 nm isoprenaline, 30 nm SDF1α, 10 ng/ml EGF, or vehicle (0.13% (w/v) BSA in PBS) (406–526 cells). The FRET response for each cell is expressed relative to the maximal FRET change (F/Fmax). B, representative ratiometric pseudocolor images of MCF7 cells expressing RaichuEV-Rac1 at baseline (0 min) following stimulation with ligands and the maximal FRET response to the positive control (Positive). C, effect of endogenous P-Rex1 knockdown on the Rac1 response in MCF7 cells (26–101 cells). AUC, area under the curve. D, expression of endogenous P-Rex1 in MCF7 cells is knocked down by P-Rex1 siRNA but not by a scrambled siRNA control. E, effect of P-Rex1 overexpression on the Rac1 response in MDA-MB-231 cells (307–484 cells). F, P-Rex1 is not endogenously expressed in MDA-MB-231 cells but can be expressed following transfection. Error bars indicate means ± S.E. ***, p < 0.001 versus vehicle control; ∧∧, p < 0.01; and ∧∧∧, p < 0.001 versus scrambled siRNA controls, two-way ANOVA with Dunnett's multiple comparison test.
Previous studies have shown that in MCF7 cells, stimulation of the chemokine and epidermal growth factor receptors, CXCR4 and EGFR, respectively, leads to an increase in Rac1 activity dependent on P-Rex1 (10, 30). Here, activation of MCF7 cells with 10 ng/ml EGF (EGFR ligand) caused a rapid and sustained increase in Rac1 activity over 20 min (Fig. 4, A and B). In contrast, 30 nm SDF1α (CXCR4 ligand) caused a slow increase in Rac1 activity (Fig. 4, A and B). Previous studies have shown that activation of βAR in HEK293 cells caused a decrease in Rac1 activity, dependent on PKA phosphorylation of P-Rex1 (38). However, in the MCF7 cell line, 100 nm isoprenaline (βAR ligand) caused a slow and sustained increase in Rac1 activity (Fig. 4, A and B).
To confirm that P-Rex1 is required for Rac1 activity in MCF7 cells, we knocked down endogenous P-Rex1 using targeted siRNA (Fig. 4D). Compared with scrambled siRNA controls, P-Rex1 knockdown abolished Rac1 activity in response to all ligands (Fig. 4C). To complement this ‘loss-of-function’ experiment, we performed a ‘gain-of-function’ experiment in the MDA-MB-231 cell line. In MDA-MB-231 cells transfected with empty pcDNA, there was no effect shown of EGF, SDF1α, or isoprenaline on Rac1 activity compared with vehicle control (Fig. 4E). However, following overexpression of P-Rex1 (Fig. 4F), all ligands caused a significant increase in Rac1 activity (Fig. 4E). Taken together, these data highlight the critical role of P-Rex1 in activating Rac1 in MCF7 and MDA-MB-231 breast cancer cells in response to EGF, SDF1α, and isoprenaline.
The P-Rex1·Rac1 Interface Is Critical for Rac1 Activation in Human Breast Cancer Cells
To test the importance of the P-Rex1·Rac1 interaction for Rac1 activity in intact cells, we selected key residues that were critical for Rac1 activity in vitro for mutagenesis in the full-length protein: Glu-56, Gln-197, Lys-201, Asn-238, and Arg-242. These mutants were overexpressed in both MCF7 and MDA-MB-231 cells (Fig. 5, A and B). As expected, overexpression of wild-type P-Rex1 had no effect on the Rac1 response to EGF, SDF1α, or isoprenaline in the MCF7 cell line (Fig. 5I), but it allowed an increase in Rac1 activity in the P-Rex1-deficient MDA-MB-231 cells (Fig. 5J).
FIGURE 5.

The P-Rex1 DH domain activates Rac1 in human breast cancer cells. Transfection with HA-tagged full-length P-Rex1 wild-type (WT) or point mutants induces P-Rex1 overexpression in MCF7 (A) and MDA-MB-231 (B) cells. P-Rex1-dependent Rac1 activity is shown following the addition of vehicle (0.13% (w/v) BSA in PBS), 100 nm isoprenaline, 30 nm SDF1α, or 10 ng/ml EGF in MCF7 cells transfected with wild-type P-Rex1 (347–446 cells) (C), P-Rex1 Q197A (142–206 cells) (D), P-Rex1 R242A (144–287 cells) (E), P-Rex1 K201A (131–155 cells) (F), P-Rex1 R242A/Q197A (94–114 cells) (G), or P-Rex1 E56A/N238A (51–98 cells) (H). I, effect of overexpression of P-Rex1 mutants on Rac1 activity in MCF7 cells (51–556 cells). J, effect of overexpression of P-Rex1 mutants on Rac1 activity in MDA-MB-231 cells (30–484 cells). AUC, area under the curve. Bars/symbols represent means, and error bars = S.E. *, p < 0.05; **, p < 0.01; and ***, p < 0.001 versus vehicle control; ^, p < 0.05; ^^, p < 0.01; and ^^^, p < 0.001 versus wild-type P-Rex1, two-way ANOVA with Dunnett's multiple comparison test.
Overexpression of P-Rex1 Q197A in MCF7 cells abolished Rac1 activity in response to isoprenaline and SDF1α, and significantly decreased the Rac1 response to EGF (Fig. 5, D and I). Interestingly, the temporal profile of the Rac1 response to EGF also changed from sustained to transient with this mutation within the switch 1 interaction interface. This may suggest that within the full-length protein, mutation of Gln-197P-Rex1 destabilizes the P-Rex1·Rac1 interface and/or increases the susceptibility of this complex to negative regulation. There was no Rac1 activity in response to any ligand following expression of P-Rex1 Q197A in MDA-MB-231 cells (Fig. 5J). We also tested two single point mutations within the switch 2 interaction interface (R242A and K201A) (Fig. 5, E and F). Although overexpression of P-Rex1 R242A significantly decreased the Rac1 response to EGF compared with wild-type P-Rex1, all ligands still increased Rac1 activity compared with vehicle control in MCF7 cells (Fig. 5I). In contrast, only isoprenaline caused an increase in Rac1 activity following P-Rex1 R242A expression in MDA-MB-231 cells (Fig. 5J). All Rac1 activity in response to the three ligands was abolished following overexpression of P-Rex1 K201A in both MCF7 and MDA-MB-231 cells (Fig. 5, F, I, and J). Given that there was no additive effect of overexpression of wild-type P-Rex1 in the MCF7 cells, the lack of Rac1 activity following expression of P-Rex1 K201A suggests that transfection of the P-Rex1 mutants has a dominant negative effect over endogenous P-Rex1 in this cell line.
Finally, we assessed whether combined mutations within the switch 1 and switch 2 regions would impair P-Rex1 activation of Rac1. Expression of the P-Rex1 R242A/Q197A double mutant abolished Rac1 activity in response to EGF, SDF1α, and isoprenaline in both cell types (Fig. 5, G, I, and J), highlighting the importance of switch 1 and switch 2 for P-Rex1-dependent Rac1 activation. Similarly, and in agreement with previous studies, overexpression of the GEF-dead P-Rex1 (N238A/E56A) abolished Rac1 activation in response to EGF, SDF1α, and isoprenaline in both the MCF7 and MDA-MB-231 cells (Fig. 5, H, I, and J). Together, these data demonstrate the critical role of key interactions between the P-Rex1 DH domain and Rac1 in integrating the signaling of GPCRs and RTKs in human breast cancer cells.
The PIP3 and Gβγ Binding Sites Are on the Opposite Side of P-Rex1 from the Rac1 Interface
The P-Rex1 PH domain is necessary for PIP3 binding and coordinates P-Rex1 membrane recruitment and exchange activity (21, 31). PIP3 binding therefore plays a crucial role in P-Rex1 regulation. The electrostatic surface potential of the P-Rex1 DH-PH domain structure reveals a positively charged surface pocket situated within the PH domain that could coordinate the negatively charged PIP3 head group (Fig. 6B). The P-Rex1 PH domain aligns closely with the PIP3-bound PH domain of CENTA1, with an r.m.s. deviation of 1.82 Å over 86 residues (Fig. 6A). In this alignment, PIP3 sits between the β-sheets of the P-Rex1 PH domain β-sandwich within this positively charged pocket (Fig. 6, A and B).
FIGURE 6.

Structural insights into P-Rex1·Gβγ and P-Rex1·PIP3 binding. A, alignment of the PH domain of P-Rex1 with the GRK2 PH·Gβγ complex (green; PDB code 1OMW (34), with an r.m.s. deviation of 2.046 Å across 93 residues) positions the Gβγ heterodimer on the opposite side of P-Rex1 from Rac1. In the alignment, Gβγ contacts both the P-Rex1 DH and PH domains. Inset, shows PIP3 modeled by structural alignment of the P-Rex1 PH domain with the CENTA1 PH·PIP3 complex (purple; PDB code 3LJU (65), with an r.m.s. deviation of 1.815 Å over 86 residues). B, electrostatic surface of P-Rex1 in the same orientation as shown in A, highlighting a highly negatively charged surface patch on P-Rex1 at the site of Gβγ binding and a highly positively charged pocket, where modeling places the negatively charged PIP3 head group. C and D, the membrane-interacting regions of each component of the P-Rex1·Rac1·Gβγ tetramer are located on the same face of the complex (C), including the C terminus of Rac1 extending from Val-176 (orange), the PIP3-binding pocket of the P-Rex1 PH domain, and the C-terminal Cys-68 of Gγ (blue) (D). PIP3 (green) shown as modeled in A. Val-176 of Rac1 and Cys-68 of Gγ are highlighted as spheres.
In addition to PIP3 binding, the binding of Gβγ subunits increases P-Rex1 activity and facilitates the efficient recruitment of P-Rex1 to the plasma membrane (21, 29, 31). PH domains frequently interact with Gβγ subunits (32–35), and the P-Rex1 PH domain is required for Gβγ-mediated membrane recruitment (31). Importantly, alignment of the PH domains of P-Rex1 and GRK2 within the P-Rex1·Rac1 and GRK2·Gβγ complexes positions Gβγ into a highly negatively charged surface patch of the P-Rex1 structure with few steric clashes (Fig. 6, A and B). In this model, the Gβγ heterodimer contacts both the DH and PH domains via helices α3 and α6 of the DH domain and sheet β4 and helix α7 of the PH domain (Fig. 6A). Interestingly, the Gβγ heterodimer sits on the opposite side of P-Rex1 from Rac1 and does not make any direct interactions with the GTPase. The C-terminal Cys-68 of Gγ undergoes prenylation to enable attachment to the cell membrane. This model is compatible with membrane recruitment of the three proteins, as Cys-68 of Gγ aligns on the same side of the tetramer as the membrane-associated C terminus of Rac1 and the proposed PIP3 binding site of the P-Rex1 PH domain (Fig. 6, C and D).
Discussion
The P-Rex family of Rho GEFs is rapidly emerging as critical effectors of cell migration, invasion, and metastasis in several human cancers including melanoma and prostate and breast cancer (8, 10, 14). Herein, we have undertaken the first investigation into the structural basis for P-Rex1 activity. The P-Rex1·Rac1 crystal structure highlights the critical role of the P-Rex1 DH domain in coordinating Rac1 activation. P-Rex1 DH domain helices α1, α5, and α6 coordinate interactions with the nucleotide-binding regions of Rac1 with no direct molecular contributions from the PH domain. P-Rex1 binding induces a large conformational change in Rac1, principally localized to the switch 1 region. This conformational change disrupts Mg2+ and GDP binding, thereby facilitating GDP displacement and subsequent GTP loading. Critically, the disruption of key residues within this interface abolishes both P-Rex1 exchange activity in vitro and Rac1 signaling downstream of RTKs and GPCRs in breast cancer cell lines.
We observed significant decreases in P-Rex1 in vitro activity upon alanine substitution of either Arg-242P-Rex1 or Gln-197P-Rex1 that correlated with reduced signaling in both the MCF7 and MDA-MB-231 cancer cell lines. Importantly, the combined mutation of these two residues in P-Rex1 completely abolished Rac1 signaling in breast cancer cells. Of the two mutations, Gln-197P-Rex1 is highly conserved among Rho GEFs and makes analogous interactions with Asn-39Rac1 in the Tiam1·Rac1 and Vav1·Rac1 complexes (25, 60). Indeed, an equivalent mutation of the conserved glutamine residue in Vav1 significantly abrogates Rac1 activation in HEK293 cells (60).
Lys-201P-Rex1 forms a key hydrogen bond to Ala-59Rac1, effectively positioning the Rac1 residue in proximity to sterically inhibit efficient Mg2+ association. This interaction is highly conserved, and has previously been shown to be critical for the activation of a number of related Rho GEFs (27). Accordingly, removal of the stabilizing hydrogen bond by mutation of Lys-201P-Rex1 reduced the exchange activity in vitro and completely abolished Rac1 activity in both cancer cell lines. However, we observed only a minor reduction in Rac1 binding in the Lys-201P-Rex1 mutant in pulldown assays. Together, these data indicate that Lys-201P-Rex1 does not greatly contribute to Rac1 binding, although it plays a critical role in Rac1 activation by promoting GDP dissociation. This important role of Lys-201P-Rex1 in Rac1 activation is further highlighted upon comparison with the single Arg-242P-Rex1 and Gln-197P-Rex1 mutants, which displayed a similar exchange activity in vitro but had less of an inhibitory effect in the cellular environment.
Interestingly, we observed reduced binding of P-Rex1 E245R to Rac1 in pulldown assays without a concomitant reduction in in vitro exchange activity. Although the cause of this disparity is unclear, these data may suggest that the Glu-245P-Rex1 interaction with Arg-66Rac1 predominantly supports complex association and has little direct role in modulating Rac1 nucleotide exchange. Additionally, the mutation of Glu-56P-Rex1 and Asn-238P-Rex1, previously identified in the GEF-dead P-Rex1 mutant (21), abrogated activity both in vitro and in the two breast cancer cell lines. The important role of Glu-56P-Rex1 in stabilizing the Rac1 switch 1 region is illustrated by its high level of conservation. Indeed, numerous studies of related Rho GEFs show a comparable reduction in activity upon mutation of the equivalent glutamic acid residue (27, 60).
Taken together, these data substantiate the P-Rex1·Rac1 interface as a potential therapeutic target in the treatment of cancer. Rho GTPases, such as Rac1, are typically viewed as difficult targets for small molecule inhibitors owing to their globular fold and lack of suitable hydrophobic surface pockets (61). Further, although mutation of Ras GTPases is commonly found as a driver of cancer, oncogenic dysregulation of Rho family GTPases occurs primarily through the aberrant expression of their upstream regulators (62). As such, a number of studies have begun to focus on Rho GEF inhibition as an alternative mechanism to provide tissue-specific inhibition of Rho GTPase activity (62, 63). Within the P-Rex1·Rac1 structure, the Rac1 switch regions interface with surface grooves of P-Rex1 formed by the α-helices of the DH domain. As clearly demonstrated by the P-Rex1 point mutations in intact cancer cell lines, interference with the P-Rex1·Rac1 interface represents a potential mechanism to inhibit Rac1 activation in vivo. Future studies will be aimed at the development of small molecules targeting these surface grooves on P-Rex1 to ascertain whether this is a viable approach to inhibiting Rac1 activation in cancer.
Previous studies have indicated that the PH domain is required for full P-Rex1 activity; however the molecular basis for this function is unclear (21, 31). It has been suggested that the PH domain may interact directly with Rac1, as observed in the Rho GEF GTPase structures of Dbs-Cdc42, LARG-RhoA, and PDZ-Rho GEF-RhoA (22–24). However, here we saw no evidence for direct molecular contacts between the P-Rex1 PH domain and Rac1. Instead, the extended α6-helix of the P-Rex1 DH domain effectively oriented the PH domain away from Rac1. As such, we propose that the PH domain instead plays an indirect role in P-Rex1 activity by regulating membrane localization and/or by releasing autoinhibitory interactions with the C-terminal domains of P-Rex1 following PIP3 binding.
As a consequence of the orientation of the P-Rex1 PH domain relative to the DH domain, both the modeled PIP3 and Gβγ binding sites are positioned away from the GTPase interaction interface. This suggests that both PIP3 and Gβγ subunits may control P-Rex1 activity via a unique regulatory mechanism rather than via direct stabilization of GTPase binding. Indeed, previous studies have demonstrated that the C-terminal P-Rex1 domains can autoinhibit activity, presumably by interacting with the DH-PH domain (21, 36). This led the authors to postulate that key regulatory processes, including P-Rex1 phosphorylation patterns and plasma membrane recruitment by Gβγ and/or PIP3 binding, may activate P-Rex1 by relieving this C-terminal autoinhibition (21, 31, 36, 37). Taken together with these studies, our data begin to build a structural context for the unique synergistic regulation of the P-Rex family members by PIP3 and Gβγ. Within the cytoplasm, the P-Rex1 C-terminal domains sterically inhibit activity via interactions with the DH-PH domain (21, 36) (Fig. 7). Upon membrane localization, PIP3 and/or Gβγ compete for DH-PH domain binding, releasing the autoinhibitory C-terminal domains and allowing GTPase activation by the DH domain.
FIGURE 7.
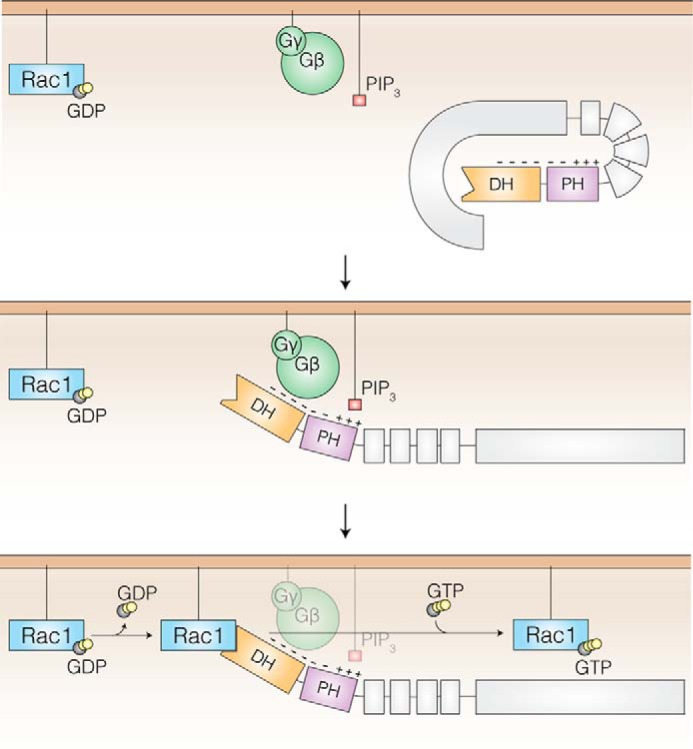
Model of Gβγ and PIP3-dependent P-Rex1 GEF activity. Low basal P-Rex1 activity is maintained through inhibitory interactions between the DH-PH tandem domain and C-terminal domains (gray) that block the GTPase binding site (DH domain, orange). At the plasma membrane, PIP3 binds via a positively charged pocket within the PH domain (purple), and Gβγ subunits bind to a negatively charged patch that spans the DH and PH domains. The binding of PIP3 and Gβγ subunits releases the C-terminal domains and exposes the Rac1 binding interface on the opposite side of P-Rex1. This allows Rac1 nucleotide exchange via the P-Rex1 DH domain.
Phosphorylation of P-Rex1 likely adds another layer of complexity to this regulatory process. A number of studies have demonstrated that P-Rex1 activity is carefully coordinated by the phosphorylation state of the protein (11, 37, 38). Interestingly, phosphorylation of P-Rex1 residues Ser-313 and Ser-319 within the β3-β4 loop negatively regulates Rac1 activation in breast cancer cells (11). The conserved P-Rex1 β3-β4 loop (residues 306–322) within the PH domain (Fig. 1B) could not be resolved in the crystal structure, suggesting that this region of the protein is highly mobile. The orientation of the residues on either side of the β3-β4 loop in the structure suggests that the loop is solvent-exposed and faces toward the DH domain·Rac1 interface adjacent to helix α6. It is possible that upon phosphorylation, the β3-β4 loop may interact with the surface of the DH domain to sterically inhibit Rac1 binding.
In conclusion, our findings provide the first high-resolution molecular description of P-Rex1 activity, highlighting the critical role of the DH domain in enabling GDP displacement and GTP loading. The mutations in the P-Rex1·Rac1 interface reveal a critical role of this complex in Rac1 activation downstream of RTKs and GPCRs. The PIP3/Gβγ binding sites are on the opposite surface and markedly removed from the Rac1 interface, supporting a model whereby P-Rex1 binding to PIP3 and/or Gβγ releases inhibitory C-terminal domains to expose the Rac1 binding site. Lastly, these data uncover the P-Rex1·Rac1 interface as a potential therapeutic target in the treatment of cancer.
Author Contributions
C. M. L., M. L. H., L. M. O., H. J. L., and A. M. E. performed the research; C. M. L., M. L. H., and A. M. E. analyzed the data; C. M. L., M. L. H., C. A. M., J. C. W., and A. M. E. designed the research; and C. M. L. and A. M. E. wrote the manuscript, which was drafted by all of the authors.
Acknowledgments
We thank Cindy K. Pon for assistance. Crystallographic data were collected at the MX2 beamline at the Australian Synchrotron, Victoria, Australia.
This research was supported by a Strategic Grant from the Faculty of Medicine, Nursing, and Health Sciences (to A. M. E.) and Project Grants APP1062230 (to A. M. E. and J. C. W.) and APP1044737 (to C. A. M.) from the National Health and Medical Research Council of Australia (NHMRC). The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 4YON) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- GEF
- guanine nucleotide exchange factor
- PIP3
- phosphatidylinositol (3,4,5)-trisphosphate
- P-Rex1
- PIP3-dependent Rac exchanger 1
- PEI
- polyethylenimine
- RTK
- receptor tyrosine kinase
- GPCR
- G protein-coupled receptor
- EGFR
- EGF receptor
- Mant
- N-methylanthraniloyl
- TEV
- tobacco etch virus
- Ni-NTA
- nickel-nitrilotriacetic acid
- CAPS
- 3-(cyclohexylamino)propanesulfonic acid
- CFP
- cyan fluorescent protein
- ANOVA
- analysis of variance
- βAR
- β-adrenoceptor
- r.m.s.
- root mean square
- PDB
- Protein Data Bank
- DH
- Dbl homology
- PH
- pleckstrin homology.
References
- 1. Jaffe A. B., Hall A. (2005) Rho GTPases: biochemistry and biology. Annu. Rev. Cell Dev. Biol. 21, 247–269 [DOI] [PubMed] [Google Scholar]
- 2. Aspenström P., Ruusala A., Pacholsky D. (2007) Taking Rho GTPases to the next level: the cellular functions of atypical Rho GTPases. Exp. Cell Res. 313, 3673–3679 [DOI] [PubMed] [Google Scholar]
- 3. Schmidt A., Hall A. (2002) Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev. 16, 1587–1609 [DOI] [PubMed] [Google Scholar]
- 4. Herter J. M., Rossaint J., Block H., Welch H., Zarbock A. (2013) Integrin activation by P-Rex1 is required for selectin-mediated slow leukocyte rolling and intravascular crawling. Blood 121, 2301–2310 [DOI] [PubMed] [Google Scholar]
- 5. Waters J. E., Astle M. V., Ooms L. M., Balamatsias D., Gurung R., Mitchell C. A. (2008) P-Rex1: a multidomain protein that regulates neurite differentiation. J. Cell Sci. 121, 2892–2903 [DOI] [PubMed] [Google Scholar]
- 6. Welch H. C., Condliffe A. M., Milne L. J., Ferguson G. J., Hill K., Webb L. M., Okkenhaug K., Coadwell W. J., Andrews S. R., Thelen M., Jones G. E., Hawkins P. T., Stephens L. R. (2005) P-Rex1 regulates neutrophil function. Curr. Biol. 15, 1867–1873 [DOI] [PubMed] [Google Scholar]
- 7. Yoshizawa M., Kawauchi T., Sone M., Nishimura Y. V., Terao M., Chihama K., Nabeshima Y., Hoshino M. (2005) Involvement of a Rac activator, P-Rex1, in neurotrophin-derived signaling and neuronal migration. J. Neurosci. 25, 4406–4419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Qin J., Xie Y., Wang B., Hoshino M., Wolff D. W., Zhao J., Scofield M. A., Dowd F. J., Lin M. F., Tu Y. (2009) Upregulation of PIP3-dependent Rac exchanger 1 (P-Rex1) promotes prostate cancer metastasis. Oncogene. 28, 1853–1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wong C. Y., Wuriyanghan H., Xie Y., Lin M. F., Abel P. W., Tu Y. (2011) Epigenetic regulation of phosphatidylinositol 3,4,5-triphosphate-dependent Rac exchanger 1 gene expression in prostate cancer cells. J. Biol. Chem. 286, 25813–25822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sosa M. S., Lopez-Haber C., Yang C., Wang H., Lemmon M. A., Busillo J. M., Luo J., Benovic J. L., Klein-Szanto A., Yagi H., Gutkind J. S., Parsons R. E., Kazanietz M. G. (2010) Identification of the Rac-GEF P-Rex1 as an essential mediator of ErbB signaling in breast cancer. Mol. Cell 40, 877–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Montero J. C., Seoane S., Ocaña A., Pandiella A. (2011) P-Rex1 participates in Neuregulin-ErbB signal transduction and its expression correlates with patient outcome in breast cancer. Oncogene 30, 1059–1071 [DOI] [PubMed] [Google Scholar]
- 12. Kim E. K., Yun S. J., Ha J. M., Kim Y. W., Jin I. H., Yun J., Shin H. K., Song S. H., Kim J. H., Lee J. S., Kim C. D., Bae S. S. (2011) Selective activation of Akt1 by mammalian target of rapamycin complex 2 regulates cancer cell migration, invasion, and metastasis. Oncogene 30, 2954–2963 [DOI] [PubMed] [Google Scholar]
- 13. Campbell A. D., Lawn S., McGarry L. C., Welch H. C., Ozanne B. W., Norman J. C. (2013) P-Rex1 cooperates with PDGFRβ to drive cellular migration in 3D microenvironments. PLoS One 8, e53982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lindsay C. R., Lawn S., Campbell A. D., Faller W. J., Rambow F., Mort R. L., Timpson P., Li A., Cammareri P., Ridgway R. A., Morton J. P., Doyle B., Hegarty S., Rafferty M., Murphy I. G., McDermott E. W., Sheahan K., Pedone K., Finn A. J., Groben P. A., Thomas N. E., Hao H., Carson C., Norman J. C., Machesky L. M., Gallagher W. M., Jackson I. J., Van Kempen L., Beermann F., Der C., Larue L., Welch H. C., Ozanne B. W., Sansom O. J. (2011) P-Rex1 is required for efficient melanoblast migration and melanoma metastasis. Nat. Commun. 2, 555–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Asimakopoulos F. A., Green A. R. (1996) Deletions of chromosome 20q and the pathogenesis of myeloproliferative disorders. Br. J. Haematol. 95, 219–226 [DOI] [PubMed] [Google Scholar]
- 16. Berry R., Schroeder J. J., French A. J., McDonnell S. K., Peterson B. J., Cunningham J. M., Thibodeau S. N., Schaid D. J. (2000) Evidence for a prostate cancer-susceptibility locus on chromosome 20. Am. J. Hum. Genet. 67, 82–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stumpf E., Aalto Y., Höög A., Kjellman M., Otonkoski T., Knuutila S., Andersson L. C. (2000) Chromosomal alterations in human pancreatic endocrine tumors. Genes Chromosomes Cancer 29, 83–87 [DOI] [PubMed] [Google Scholar]
- 18. Larramendy M. L., Lushnikova T., Björkqvist A. M., Wistuba I. I., Virmani A. K., Shivapurkar N., Gazdar A. F., Knuutila S. (2000) Comparative genomic hybridization reveals complex genetic changes in primary breast cancer tumors and their cell lines. Cancer Genet. Cytogenet. 119, 132–138 [DOI] [PubMed] [Google Scholar]
- 19. Berger M. F., Hodis E., Heffernan T. P., Deribe Y. L., Lawrence M. S., Protopopov A., Ivanova E., Watson I. R., Nickerson E., Ghosh P., Zhang H., Zeid R., Ren X., Cibulskis K., Sivachenko A. Y., Wagle N., Sucker A., Sougnez C., Onofrio R., Ambrogio L., Auclair D., Fennell T., Carter S. L., Drier Y., Stojanov P., Singer M. A., Voet D., Jing R., Saksena G., Barretina J., Ramos A. H., Pugh T. J., Stransky N., Parkin M., Winckler W., Mahan S., Ardlie K., Baldwin J., Wargo J., Schadendorf D., Meyerson M., Gabriel S. B., Golub T. R., Wagner S. N., Lander E. S., Getz G., Chin L., Garraway L. A. (2012) Melanoma genome sequencing reveals frequent PREX2 mutations. Nature 485, 502–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Waddell N., Pajic M., Patch A. M., Chang D. K., Kassahn K. S., Bailey P., Johns A. L., Miller D., Nones K., Quek K., Quinn M. C., Robertson A. J., Fadlullah M. Z., Bruxner T. J., Christ A. N., Harliwong I., Idrisoglu S., Manning S., Nourse C., Nourbakhsh E., Wani S., Wilson P. J., Markham E., Cloonan N., Anderson M. J., Fink J. L., Holmes O., Kazakoff S. H., Leonard C., Newell F., Poudel B., Song S., Taylor D., Waddell N., Wood S., Xu Q., Wu J., Pinese M., Cowley M. J., Lee H. C., Jones M. D., Nagrial A. M., Humphris J., Chantrill L. A., Chin V., Steinmann A. M., Mawson A., Humphrey E. S., Colvin E. K., Chou A., Scarlett C. J., Pinho A. V., Giry-Laterriere M., Rooman I., Samra J. S., Kench J. G., Pettitt J. A., Merrett N. D., Toon C., Epari K., Nguyen N. Q., Barbour A., Zeps N., Jamieson N. B., Graham J. S., Niclou S. P., Bjerkvig R., Grützmann R., Aust D., Hruban R. H., Maitra A., Iacobuzio-Donahue C. A., Wolfgang C. L., Morgan R. A., Lawlor R. T., Corbo V., Bassi C., Falconi M., Zamboni G., Tortora G., Tempero M. A., Gill A. J., Eshleman J. R., Pilarsky C., Scarpa A., Musgrove E. A., Pearson J. V., Biankin A. V., Grimmond S. M., et al. (2015) Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 518, 495–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hill K., Krugmann S., Andrews S. R., Coadwell W. J., Finan P., Welch H. C., Hawkins P. T., Stephens L. R. (2005) Regulation of P-Rex1 by phosphatidylinositol (3,4,5)-trisphosphate and Gβγ subunits. J. Biol. Chem. 280, 4166–4173 [DOI] [PubMed] [Google Scholar]
- 22. Rossman K. L., Worthylake D. K., Snyder J. T., Siderovski D. P., Campbell S. L., Sondek J. (2002) A crystallographic view of interactions between Dbs and Cdc42: PH domain-assisted guanine nucleotide exchange. EMBO J. 21, 1315–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kristelly R., Gao G., Tesmer J. J. (2004) Structural determinants of RhoA binding and nucleotide exchange in leukemia-associated Rho guanine-nucleotide exchange factor. J. Biol. Chem. 279, 47352–47362 [DOI] [PubMed] [Google Scholar]
- 24. Derewenda U., Oleksy A., Stevenson A. S., Korczynska J., Dauter Z., Somlyo A. P., Otlewski J., Somlyo A. V., Derewenda Z. S. (2004) The crystal structure of RhoA in complex with the DH/PH fragment of PDZRhoGEF, an activator of the Ca(2+) sensitization pathway in smooth muscle. Structure 12, 1955–1965 [DOI] [PubMed] [Google Scholar]
- 25. Worthylake D. K., Rossman K. L., Sondek J. (2000) Crystal structure of Rac1 in complex with the guanine nucleotide exchange region of Tiam1. Nature 408, 682–688 [DOI] [PubMed] [Google Scholar]
- 26. Snyder J. T., Worthylake D. K., Rossman K. L., Betts L., Pruitt W. M., Siderovski D. P., Der C. J., Sondek J. (2002) Structural basis for the selective activation of Rho GTPases by Dbl exchange factors. Nat. Struct. Biol. 9, 468–475 [DOI] [PubMed] [Google Scholar]
- 27. Rossman K. L., Der C. J., Sondek J. (2005) GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 6, 167–180 [DOI] [PubMed] [Google Scholar]
- 28. Donald S., Hill K., Lecureuil C., Barnouin R., Krugmann S., John Coadwell W., Andrews S. R., Walker S. A., Hawkins P. T., Stephens L. R., Welch H. C. (2004) P-Rex2, a new guanine-nucleotide exchange factor for Rac. FEBS Letters 572, 172–176 [DOI] [PubMed] [Google Scholar]
- 29. Welch H. C., Coadwell W. J., Ellson C. D., Ferguson G. J., Andrews S. R., Erdjument-Bromage H., Tempst P., Hawkins P. T., Stephens L. R. (2002) P-Rex1, a PtdIns(3,4,5)P3- and Gβγ-regulated guanine-nucleotide exchange factor for Rac. Cell 108, 809–821 [DOI] [PubMed] [Google Scholar]
- 30. Carretero-Ortega J., Walsh C. T., Hernández-García R., Reyes-Cruz G., Brown J. H., Vázquez-Prado J. (2010) Phosphatidylinositol 3,4,5-triphosphate-dependent Rac exchanger 1 (P-Rex-1), a guanine nucleotide exchange factor for Rac, mediates angiogenic responses to stromal cell-derived factor-1/chemokine stromal cell derived factor-1 (SDF-1/CXCL-12) linked to Rac activation, endothelial cell migration, and in vitro angiogenesis. Mol. Pharmacol. 77, 435–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Barber M. A., Donald S., Thelen S., Anderson K. E., Thelen M., Welch H. C. (2007) Membrane translocation of P-Rex1 is mediated by G protein beta subunits and phosphoinositide 3-kinase. J. Biol. Chem. 282, 29967–29976 [DOI] [PubMed] [Google Scholar]
- 32. Wang D. S., Shaw R., Winkelmann J. C., Shaw G. (1994) Binding of PH domains of β-adrenergic receptor kinase and β-spectrin to WD40/β-transducin repeat containing regions of the β-subunit of trimeric G-proteins. Biochem. Biophys. Res. Commun. 203, 29–35 [DOI] [PubMed] [Google Scholar]
- 33. Wang T., Dowal L., El-Maghrabi M. R., Rebecchi M., Scarlata S. (2000) The pleckstrin homology domain of phospholipase C-β2 links the binding of Gβγ to activation of the catalytic core. J. Biol. Chem. 275, 7466–7469 [DOI] [PubMed] [Google Scholar]
- 34. Lodowski D. T., Pitcher J. A., Capel W. D., Lefkowitz R. J., Tesmer J. J. (2003) Keeping G proteins at bay: a complex between G protein-coupled receptor kinase 2 and Gβγ. Science 300, 1256–1262 [DOI] [PubMed] [Google Scholar]
- 35. Jamora C., Yamanouye N., Van Lint J., Laudenslager J., Vandenheede J. R., Faulkner D. J., Malhotra V. (1999) Gβγ-mediated regulation of Golgi organization is through the direct activation of protein kinase D. Cell 98, 59–68 [DOI] [PubMed] [Google Scholar]
- 36. Urano D., Nakata A., Mizuno N., Tago K., Itoh H. (2008) Domain-domain interaction of P-Rex1 is essential for the activation and inhibition by G protein βγ subunits and PKA. Cell. Signal. 20, 1545–1554 [DOI] [PubMed] [Google Scholar]
- 37. Barber M. A., Hendrickx A., Beullens M., Ceulemans H., Oxley D., Thelen S., Thelen M., Bollen M., Welch H. C. (2012) The guanine-nucleotide-exchange factor P-Rex1 is activated by protein phosphatase 1α. Biochem. J. 443, 173–183 [DOI] [PubMed] [Google Scholar]
- 38. Mayeenuddin L. H., Garrison J. C. (2006) Phosphorylation of P-Rex1 by the cyclic AMP-dependent protein kinase inhibits the phosphatidylinositiol (3,4,5)-trisphosphate and Gβγ-mediated regulation of its activity. J. Biol. Chem. 281, 1921–1928 [DOI] [PubMed] [Google Scholar]
- 39. Itoh R. E., Kurokawa K., Ohba Y., Yoshizaki H., Mochizuki N., Matsuda M. (2002) Activation of Rac and Cdc42 video imaged by fluorescent resonance energy transfer-based single-molecule probes in the membrane of living cells. Mol. Cell Biol. 22, 6582–6591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Komatsu N., Aoki K., Yamada M., Yukinaga H., Fujita Y., Kamioka Y., Matsuda M. (2011) Development of an optimized backbone of FRET biosensors for kinases and GTPases. Mol. Biol. Cell 22, 4647–4656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Niwa H., Yamamura K., Miyazaki J. (1991) Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108, 193–199 [DOI] [PubMed] [Google Scholar]
- 42. Chang X. Z., Li D. Q., Hou Y. F., Wu J., Lu J. S., Di G. H., Jin W., Ou Z. L., Shen Z. Z., Shao Z. M. (2008) Identification of the functional role of AF1Q in the progression of breast cancer. Breast Cancer Res. Treat. 111, 65–78 [DOI] [PubMed] [Google Scholar]
- 43. McPhillips T. M., McPhillips S. E., Chiu H. J., Cohen A. E., Deacon A. M., Ellis P. J., Garman E., Gonzalez A., Sauter N. K., Phizackerley R. P., Soltis S. M., Kuhn P. (2002) Blu-Ice and the distributed control system: software for data acquisition and instrument control at macromolecular crystallography beamlines. J. Synchrotron Radiat. 9, 401–406 [DOI] [PubMed] [Google Scholar]
- 44. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xiang S., Kim E. Y., Connelly J. J., Nassar N., Kirsch J., Winking J., Schwarz G., Schindelin H. (2006) The crystal structure of Cdc42 in complex with collybistin II, a gephyrin-interacting guanine nucleotide exchange factor. J. Mol. Biol. 359, 35–46 [DOI] [PubMed] [Google Scholar]
- 47. Krauthammer M., Kong Y., Ha B. H., Evans P., Bacchiocchi A., McCusker J. P., Cheng E., Davis M. J., Goh G., Choi M., Ariyan S., Narayan D., Dutton-Regester K., Capatana A., Holman E. C., Bosenberg M., Sznol M., Kluger H. M., Brash D. E., Stern D. F., Materin M. A., Lo R. S., Mane S., Ma S., Kidd K. K., Hayward N. K., Lifton R. P., Schlessinger J., Boggon T. J., Halaban R. (2012) Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 44, 1006–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Langer G., Cohen S. X., Lamzin V. S., Perrakis A. (2008) Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Echols N., Headd J. J., Hung L. W., Jain S., Kapral G. J., Grosse Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R. D., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2011) The Phenix software for automated determination of macromolecular structures. Methods 55, 94–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 53. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Krissinel E., Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 [DOI] [PubMed] [Google Scholar]
- 55. Sternweis P. C., Gilman A. G. (1982) Aluminum: a requirement for activation of the regulatory component of adenylate cyclase by fluoride. Proc. Natl. Acad. Sci. U.S.A. 79, 4888–4891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Deleted in proof.
- 57. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J. Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., Cardona A. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kardash E., Bandemer J., Raz E. (2011) Imaging protein activity in live embryos using fluorescence resonance energy transfer biosensors. Nat. Protoc. 6, 1835–1846 [DOI] [PubMed] [Google Scholar]
- 59. Vetter I. R., Wittinghofer A. (2001) The guanine nucleotide-binding switch in three dimensions. Science 294, 1299–1304 [DOI] [PubMed] [Google Scholar]
- 60. Chrencik J. E., Brooun A., Zhang H., Mathews I. I., Hura G. L., Foster S. A., Perry J. J., Streiff M., Ramage P., Widmer H., Bokoch G. M., Tainer J. A., Weckbecker G., Kuhn P. (2008) Structural basis of guanine nucleotide exchange mediated by the T-cell essential Vav1. J. Mol. Biol. 380, 828–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Verdine G. L., Walensky L. D. (2007) The challenge of drugging undruggable targets in cancer: lessons learned from targeting BCL-2 family members. Clin. Cancer Res. 13, 7264–7270 [DOI] [PubMed] [Google Scholar]
- 62. Vigil D., Cherfils J., Rossman K. L., Der C. J. (2010) Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat. Rev. Cancer 10, 842–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nassar N., Cancelas J., Zheng J., Williams D. A., Zheng Y. (2006) Structure-function based design of small molecule inhibitors targeting Rho family GTPases. Curr. Top. Med. Chem. 6, 1109–1116 [DOI] [PubMed] [Google Scholar]
- 64. Tarricone C., Xiao B., Justin N., Walker P. A., Rittinger K., Gamblin S. J., Smerdon S. J. (2001) The structural basis of Arfaptin-mediated cross-talk between Rac and Arf signalling pathways. Nature 411, 215–219 [DOI] [PubMed] [Google Scholar]
- 65. Tong Y., Tempel W., Wang H., Yamada K., Shen L., Senisterra G. A., MacKenzie F., Chishti A. H., Park H. W. (2010) Phosphorylation-independent dual-site binding of the FHA domain of KIF13 mediates phosphoinositide transport via centaurin alpha1. Proc. Natl. Acad. Sci. U.S.A. 107, 20346–20351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Karplus P. A., Diederichs K. (2012) Linking crystallographic model and data quality. Science 336, 1030–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]