

Background: Cone photoreceptors undergo endoplasmic reticulum stress-associated apoptosis in CNG channel deficiency.
Results: Suppressing cGMP/PKG signaling enhances inositol 1,4,5-trisphosphate receptor 1 (IP3R1) phosphorylation and inhibits endoplasmic reticulum stress and cone death.
Conclusion: cGMP/PKG signaling regulates IP3R1 activity and promotes endoplasmic reticulum stress in CNG channel deficiency.
Significance: Understanding of the mechanism(s) of photoreceptor degeneration is essential for therapeutic strategy development.
Keywords: calcium channel, cyclic GMP (cGMP), endoplasmic reticulum stress (ER stress), photoreceptor, protein kinase G (PKG)
Abstract
Photoreceptor cyclic nucleotide-gated (CNG) channels play a pivotal role in phototransduction. Mutations in the cone CNG channel subunits CNGA3 and CNGB3 are associated with achromatopsia and cone dystrophies. We have shown endoplasmic reticulum (ER) stress-associated apoptotic cone death and increased phosphorylation of the ER Ca2+ channel inositol 1,4,5-trisphosphate receptor 1 (IP3R1) in CNG channel-deficient mice. We also presented a remarkable elevation of cGMP and an increased activity of the cGMP-dependent protein kinase (protein kinase G, PKG) in CNG channel deficiency. This work investigated whether cGMP/PKG signaling regulates ER stress and IP3R1 phosphorylation in CNG channel-deficient cones. Treatment with PKG inhibitor and deletion of guanylate cyclase-1 (GC1), the enzyme producing cGMP in cones, were used to suppress cGMP/PKG signaling in cone-dominant Cnga3−/−/Nrl−/− mice. We found that treatment with PKG inhibitor or deletion of GC1 effectively reduced apoptotic cone death, increased expression levels of cone proteins, and decreased activation of Müller glial cells. Furthermore, we observed significantly increased phosphorylation of IP3R1 and reduced ER stress. Our findings demonstrate a role of cGMP/PKG signaling in ER stress and ER Ca2+ channel regulation and provide insights into the mechanism of cone degeneration in CNG channel deficiency.
Introduction
Rod and cone photoreceptor cyclic nucleotide-gated (CNG)3 channels are localized to the plasma membrane of photoreceptor outer segments and play an essential role in phototransduction. In darkness, a portion of the channels are kept open by cGMP, maintaining a steady Na+ and Ca2+ influx. Light induces hydrolysis of cGMP by photoreceptor phosphodiesterase, resulting in closure of the channels and hyperpolarization of the cell (1). The cone CNG channel is a heterotetrameric complex comprised of CNGA3 and CNGB3 subunits, with CNGA3 as an ion-conducting subunit and CNGB3 as a modulator. Mutations in genes encoding CNGA3 and CNGB3 are associated with human cone dystrophies, including achromatopsia, progressive cone dystrophy, and early-onset macular degeneration (2–4). There are about 80 and 40 disease-causing mutations in CNGA3 and CNGB3, respectively, accounting for about 80% of achromatopsia cases (2–4).
Cone loss in patients with achromatopsia and cone dystrophy associated with CNG channel mutations has been well documented by optical coherence tomography (5–7). Impaired cone function, progressive cone degeneration, and apoptotic cone death have also been characterized in Cnga3−/− and Cngb3−/− mice (8–11) and in Cnga3−/−/Nrl−/− and Cngb3−/−/Nrl−/− mice (12, 13). NRL, a rod-specific neural retina leucine zipper transcriptional factor, promotes differentiation of rods, and NRL deficiency produces a cone-only retina (14). Because cones include only 2–3% of the total photoreceptor population in the wild-type mouse retina, the use of the Cnga3−/−/Nrl−/− and Cngb3−/−/Nrl−/− mouse lines enabled us to explore the cellular alterations and biochemical events in CNG channel-deficient cones to understand the mechanism(s) of cone degeneration.
CNG channel-deficient mice display early-onset apoptotic cone death, with cone degeneration evident in the 2nd postnatal week (8, 12). We have shown that apoptotic cone death is associated with endoplasmic reticulum (ER) stress, manifested by elevated ER stress marker proteins, including Grp78/Bip, phospho-eukaryotic initiation factor 2α (phospho-eIF2α), and CCAAT/-enhancer-binding protein homologous protein (CHOP), up-regulated cysteine protease calpains, and increased processing of caspase-12 and caspase-7 (12, 13). We have also shown increased phosphorylation of the ER Ca2+ channel inositol 1,4,5-trisphosphate receptor 1 (IP3R1) (12). Reduced cone death following the treatment with the ER chemical chaperone tauroursodeoxycholic acid (TUDCA) (13) suggests a contribution of ER stress to cone death.
In addition to ER stress, CNG channel deficiency is associated with elevation of cellular cGMP levels. Retinal cGMP levels in Cnga3−/−/Nrl−/− mice sharply increased at postnatal day 8 (P8), peaked around P10–15, remained high through P30–60, and returned to near control levels at P90 (15). The cGMP elevation pattern correlated with apoptotic cone death (8, 12). Using Cnga3−/−/Gucy2e−/− mice lacking retinal guanylyl cyclase 1 (retGC1), an enzyme responsible for biosynthesis of cGMP in photoreceptors, we showed that cGMP accumulation contributed to cone death in the absence of CNG channels. Cone density and expression levels of cone proteins were significantly increased in Cnga3−/−/Gucy2e−/− mice, compared with Cnga3−/− mice (15). We also showed that the activity and expression levels of cGMP-dependent protein kinase (protein kinase G, PKG) were significantly increased, suggesting a potential role of cGMP/PKG signaling in cone death.
In this study, we investigated whether cGMP/PKG signaling contributes to ER stress and regulates IP3R1 phosphorylation in CNG channel-deficient cones. Treatment with PKG inhibitors and deletion of retGC1 effectively reduced apoptotic cone death and Müller glial cell activation and increased expression levels of cone proteins in CNG channel-deficient mice. Furthermore, inhibition of cGMP/PKG signaling significantly increased phosphorylation of IP3R1 and reduced ER stress. Findings from this work support a cGMP/PKG-regulated, IP3R1-associated ER stress/apoptosis in CNG channel-deficient cones.
Experimental Procedures
Mice, Antibodies, and Other Materials
The Cnga3−/− (9), Nrl−/− (14), Gucy2e−/− (16), and Cnga3−/−/Nrl−/− (12, 13) mouse lines were generated as described previously. Cnga3−/−/Nrl−/−/Gucy2e−/− line was generated by cross-breeding. All mice were maintained under cyclic light (12-h light-dark) conditions. During the light cycle, cage illumination was ∼7 foot-candles. All animal maintenance and experiments were approved by the local Institutional Animal Care and Use Committee (University of Oklahoma Health Sciences Center, Oklahoma City, OK) and conformed to the guidelines on the care and use of animals adopted by the Society for Neuroscience and the Association for Research in Vision and Ophthalmology (Rockville, MD).
Primary antibodies used in this study are listed in Table 1. Horseradish peroxidase (HRP)-conjugated anti-rabbit and anti-mouse secondary antibodies were purchased from Kirkegaard & Perry Laboratories Inc. (Gaithersburg, MD). Fluorescent goat anti-rabbit and goat anti-mouse antibodies were obtained from Invitrogen. All other reagents were purchased from Sigma, Bio-Rad, and Invitrogen.
TABLE 1.
List of antibodies used in this study
| Antibody | Provider | Catalog no. | Dilutions used in immunoblotting (IB) or immunofluorescence (IF) labeling |
|---|---|---|---|
| M-opsin | Dr. Cheryl Craft, Keck School of Medicine, University of Southern California | 1:2000 (IB) | |
| S-opsin | Dr. Muna Naash, University of Oklahoma Health Sciences Center | 1:1000 (IB) | |
| Cone arrestin | Dr. Cheryl Craft, Keck School of Medicine | 1:2000 (IB) | |
| GADD 153 (CHOP-10) | Santa Cruz Biotechnology Inc., Santa Cruz, CA | sc-575 | 1:100 (IB) |
| Phospho-CREB | sc-7978 | 1:500 (IF) | |
| Phospho-eIF2α | Cell Signaling Technology, Beverly, MA | 3398 | 1:500 (IB) |
| Phospho-IP3R | 3760 | 1:250 (IB) | |
| Caspase-7 | 9492 | 1:250 (IB) | |
| Histone 3 (H3) | 4499 | 1:2000 (IB) | |
| ATF-6 | Active Motif, Carlsbad, CA | 40,962 | 1:250 (IB) |
| GFAP | Dako Denmark A/S, Glostrup, Denmark | Z0334 | 1:500 (IF) |
| β-Actin | ab-6276 | 1:2000 (IB) | |
| Phospho-IRE1α | Abcam, Inc., Cambridge, MA | ab-48187 | 1:100 (IB) |
PKG Inhibitor Treatment
We used two PKG inhibitors, KT5823 (Sigma) and (Rp)-8-Br-cGMPS (Santa Cruz Biotechnology, Dallas, TX), in this study (17–19). Starting at P7, Cnga3−/−/Nrl−/− mice received KT5823 (1.0 μmol/kg body weight/day, i.p.) or (Rp)-8-Br-cGMPS (5.0 μmol/kg body weight/day, i.p.) or vehicle for 8 days for the apoptosis and ER stress study, or for 20 days for the cone protein expression level study. Retinas and eyes were collected at the end of the experiments for analysis of PKG activity, TUNEL labeling, caspase-7 cleavage, phospho-eIF2α levels, and expression levels of cone proteins.
PKG Activity Assay
PKG activity in the retinal lysate was assayed using the CycLex cGMP-dependent protein kinase assay kit (purchased from MBL International, Woburn, MA) as we described previously (15). This immune colorimetric assay analyzes PKG activity by measuring the levels of phosphorylated PKG substrate, which is phosphorylated by PKG family members PKGI and PKGII. The phosphorylated substrate is detected by the phospho-specific monoclonal antibody 10H11 that recognizes the phosphothreonine 68/119 residues on the substrate. Briefly, retinal proteins (100 μg) in kinase buffer (200 μl) were added to the assay plates coated with recombinant PKG substrate containing threonine 68/119 residues. The plates were incubated in the presence of Mg2+ and ATP for 30 min at 30 °C to phosphorylate the substrate. After washing, 100 μl of the HRP-conjugated, phosphospecific antibody was added to each well, and samples were incubated for 1 h at room temperature. The wells were washed and substrate reagent was added, leading to a color change from colorless to blue catalyzed by HRP. The reaction was stopped with stop solution, which changes the color from blue to yellow, and the absorbance at 450 nm was measured with a SpectraMax 190 microplate spectrophotometer (Molecular Devices). Each reaction was performed in duplicate. Results are an average of three to four independent experiments using retinas prepared from five to six mice.
Eye Preparation, Immunofluorescence Labeling, and Confocal Microscopy
We prepared mouse eye cross-sections for immunohistochemical analysis as described previously (11). Briefly, euthanasia of mice was performed by CO2 asphyxiation, and mouse eyes were enucleated and fixed with Prefer (Anatech Ltd., Battle Creek, MI) for 25–30 min at room temperature. The superior portion of the cornea was marked with a green dye for orientation before enucleation. Fixed eyes were then transferred in 70% ethanol and stored at 4 °C until the eyes were processed and embedded in paraffin. We prepared 5-μm-thick paraffin sections passing vertically through the retina along the vertical meridian passing through the optic nerve head using a Leica microtome (Leica Biosystems, Buffalo Grove, IL). Immunofluorescence labeling was performed as described previously (11). Briefly, eye sections were blocked with PBS containing 5% BSA and 0.5% Triton X-100 for 1 h at room temperature. Antigen retrieval was performed by incubating tissues in 10 mm sodium citrate buffer, pH 6.0, for 30 min in a 70 °C water bath. Primary antibody incubation was performed at room temperature for 2 h (see Table 1 for antibody dilutions). Following fluorescence-conjugated secondary antibody incubation and rinses, slides were mounted and coverslipped. Fluorescent signals were imaged using an Olympus FV1000 confocal laser scanning microscope (Olympus, Melville, NY) and FluoView imaging software (Olympus, Melville, NY). Fluorescence intensity of GFAP in the retinal sections was analyzed as described previously (20, 21). Data were analyzed and graphed using GraphPad Prism software (GraphPad software, San Diego, CA).
TUNEL Assay
The terminal deoxynucleotidyltransferase dUTP nick end-labeling (TUNEL) was performed to evaluate photoreceptor apoptotic death as described previously (12). We used paraffin-embedded retinal sections and the In Situ Cell Death Fluorescein Detection kit (Roche Diagnostics) in this analysis. Immunohistochemical labeling was imaged using an Olympus FV1000 confocal laser scanning microscope. The total TUNEL-positive cells in the outer nuclear layer that passed through the optical nerve were counted and averaged from four sections from one eye. The averages from at least four eyes were obtained. Data were analyzed and graphed using GraphPad Prism® software (GraphPad Software, San Diego).
cGMP ELISA
cGMP level in the retinal lysate was measured by ELISA using the cyclic GMP complete kit (Assay Designs, Farmingdale, NY) as we described previously (15). Briefly, dissected retinas were homogenized in 0.1 m HCl. The acidic supernatants were used, and the assays were performed per the manufacturer's instructions. We used a SpectraMax 190 microplate spectrophotometer (Molecular Devices, CA) to measure the absorbance at 405 nm. Each reaction was performed in duplicate. Results are an average of three to four independent experiments using retinas prepared from five to eight mice.
Retinal Protein Preparation, SDS-PAGE, and Western Blot Analysis
Protein SDS-PAGE and Western blotting were performed as described previously (12). Briefly, retinas were homogenized in homogenization buffer A (20 mm HEPES-NaOH, pH 7.4, 5 mm EDTA, 320 mm sucrose, containing protease and phosphatase inhibitor mixture (catalog no. 04906387001, Roche Applied Science)), and the homogenates were centrifuged at 1000 × g for 10 min at 4 °C. The resulting supernatant and pellet were subjected to extraction of cytosolic/membrane proteins and nuclear protein, respectively. To separate membrane protein from cytosolic protein, the supernatant was centrifuged at 16,000 × g for 30 min at 4 °C, and the resulting pellet was used as membrane fraction. The nuclei protein was extracted by resuspending the pellet in buffer B (20 mm HEPES-NaOH, pH 7.4, 5 mm EDTA, 320 mm sucrose, containing protease and phosphatase inhibitor mixture as described above) and sonicating for 10 s at medium speed using an ultrasonic cell disruptor (Masonix, model XL2000) twice, allowing a 30-s recovery between disruptions, followed by incubation on ice for 1 h with gentle agitation. After incubation, the solubilized homogenate was centrifuged at 16,000 × g for 35 min at 4 °C, and the resulting supernatant was used as the nuclear fraction. Protein concentration of the membrane, cytosol, and nuclei preparations was determined using the protein assay kit from Bio-Rad. For protein separation and detection, retinal protein preparations were subjected to SDS-PAGE and transferred onto polyvinylidene difluoride membranes. Following blocking in 5% nonfat milk at room temperature for 1 h, blots were incubated with primary antibody overnight at 4 °C (see Table 1 for antibody dilutions). After rinsing in Tris-buffered saline with 0.1% Tween 20, blots were incubated with HRP-conjugated secondary antibodies (1:20,000) for 1 h at room temperature. SuperSignal® West Dura Extended Duration chemiluminescent substrate (Pierce) was used to detect binding of the primary antibodies to their cognate antigens. HyBlot CL autoradiography films (Denville Scientific, Inc., Metuchen, NJ) were used to develop the target proteins, and Adobe Photoshop CS5 was used to analyze the signal density.
Results
Reduced Photoreceptor Apoptosis in Cnga3−/−/Nrl−/− Mice Treated with PKG Inhibitor
We previously showed that cones of Cnga3−/− and Cnga3−/−/Nrl−/− mice undergo early-onset apoptosis (8, 12). Retinas of these mice also showed a remarkable elevation of [cGMP] and increased PKG activity (12, 15, 22). Furthermore, we provided evidence that the cGMP accumulation contributed to cone degeneration (15). To determine whether the beneficial effects of cGMP reduction are associated with PKG signaling, we examined the effects of PKG inhibition on apoptotic cone death. We used two commonly used PKG inhibitors, KT5823 and (Rp)-8-Br-cGMPS (17–19) in our study. We found that treatment with PKG inhibitor significantly reduced photoreceptor apoptosis in Cnga3−/−/Nrl−/− mice. As shown in Fig. 1A, PKG activity in Cnga3−/−/Nrl−/− retinas was strongly reduced by PKG inhibitors. Subsequently, the increased TUNEL labeling was significantly reduced in KT5823-treated mice and was nearly completely abolished in (Rp)-8-Br-cGMPS-treated mice (Fig. 1, B and C). In addition, caspase-7 cleavage was abolished (Fig. 1D), and CHOP expression was reduced (Fig. 1E) in Cnga3−/−/Nrl−/− mice treated with PKG inhibitor, compared with vehicle-treated controls.
FIGURE 1.

Reduced photoreceptor apoptosis in Cnga3−/−/Nrl−/− mice treated with PKG inhibitor. A, reduced PKG activity in retinas of Cnga3−/−/Nrl−/− mice treated with KT5823 (left panel) or (Rp)-8-Br-cGMPS (right panel). The relative PKG activity in Cnga3−/−/Nrl−/− mice was normalized to the values in Nrl−/− mice. B and C, reduced TUNEL labeling on retinal sections of Cnga3−/−/Nrl−/− mice treated with KT5823 or (Rp)-8-Br-cGMPS. Shown are representative confocal images showing TUNEL-positive cells in Cnga3−/−/Nrl−/− mice treated with KT5823 (B, upper panels) or (Rp)-8-Br-cGMPS (B, lower panels), and corresponding quantitative analysis (C). D, reduced caspase-7 cleavage in retinas of Cnga3−/−/Nrl−/− mice treated with KT5823. Shown are representative images of the Western blot detection of caspase-7. E, reduced expression level of CHOP in retinas of Cnga3−/−/Nrl−/− mice treated with (Rp)-8-Br-cGMPS. Representative images of Western blot detection of CHOP are shown. IB, immunoblot. F, reduced phospho-CREB level in retinas of Cnga3−/−/Nrl−/− mice treated with (Rp)-8-Br-cGMPS. Shown are representative confocal images of the phospho-CREB immunofluorescence labeling. ONL, outer nuclear layer; INL, inner nuclear layer; RGC, retinal ganglion cell; Rp-8-Br, (Rp)-8-Br-cGMPS. Data are represented as mean ± S.E. from 3 to 4 assays using eyes/retinas prepared from 5 to 6 mice. Unpaired Student's t test was used to determine the significance of differences (*, p < 0.05; ***, p < 0.001).
We also examined the effects of PKG inhibition on the activity of cAMP-response element-binding protein (CREB), which is a known substrate of PKG. We found that the level of phospho-CREB was significantly reduced in Cnga3−/−/Nrl−/− mice treated with (Rp)-8-Br-cGMPS, compared with vehicle-treated controls (Fig. 1F), supporting the effectiveness of PKG inhibitor treatment.
Reduced cGMP Level, PKG Activity, and Photoreceptor Apoptosis in Cnga3−/−/Nrl−/−/Gucy2e−/− Mice
Cnga3−/−/Gucy2e−/− mice showed increased cone density and expression levels of cone proteins, compared with those in Cnga3−/− mice (15). This work examined whether deletion of RetGC1 reduces apoptotic cone death. We generated a Cnga3−/−/Nrl−/−/Gucy2e−/− mouse line to perform biochemical assays on a cone-dominant retina lacking a CNG channel and RetGC1. Retinal cGMP levels, PKG activity, and TUNEL labeling were examined in P15 Cnga3−/−/Nrl−/−/Gucy2e−/− mice and compared with those in age-matched Cnga3−/−/Nrl−/− and Nrl−/− mice. As shown in Fig. 2, the cGMP level, PKG activity, and TUNEL-positive cells in Cnga3−/−/Nrl−/− mice increased by about 70-, 2.5-, and 2.4-fold, respectively, compared with Nrl−/− mice. Deletion of retGC1 completely abolished cGMP (Fig. 2A) and nearly completely abolished elevated PKG activity (Fig. 2B) and TUNEL-positive labeling (Fig. 2, C and D).
FIGURE 2.

Reduced cGMP level, PKG activity, and photoreceptor apoptosis in Cnga3−/−/Nrl−/−/Gucy2e−/− mice. A and B, reduced cGMP levels (A) and PKG activity (B) in Cnga3−/−/Nrl−/−/Gucy2e−/− retinas. The relative cGMP levels and PKG activity in Cnga3−/−/Nrl−/−/Gucy2e−/− mice were normalized to the values in Nrl−/− mice. C and D, reduced TUNEL labeling on the retinal section of Cnga3−/−/Nrl−/−/Gucy2e−/− mice. Shown are representative confocal images showing TUNEL-positive cells in Cnga3−/−/Nrl−/−/Gucy2e−/−, Cnga3−/−/Nrl−/−, and Nrl−/− mice (C), and corresponding quantitative analysis (D). ONL, outer nuclear layer; INL, inner nuclear layer. Data are represented as mean ± S.E. from 3 to 4 assays using retinas/eyes prepared from 6 to 8 mice. Unpaired Student's t test was used to evaluate the significances of differences (**, p < 0.01).
Increased Expression Levels of Cone Proteins in Cnga3−/−/Nrl−/− Mice Treated with PKG Inhibitor and in Cnga3−/−/Nrl−/−/Gucy2e−/− Mice
To evaluate whether suppressing PKG signaling improves cone survival, we examined expression levels of cone opsin in Cnga3−/−/Nrl−/− mice treated with PKG inhibitor. As shown in Fig. 3, A and B, M-opsin and S-opsin levels increased by about 28 and 23%, respectively, in Cnga3−/−/Nrl−/− mice treated with KT5823, compared with vehicle-treated controls. We also examined the expression levels of cone proteins in Cnga3−/−/Nrl−/−/Gucy2e−/− mice. Cone opsin and cone arrestin expression levels significantly increased in P60 Cnga3−/−/Nrl−/−/Gucy2e−/− mice, compared with those in age-matched Cnga3−/−/Nrl−/− mice (Fig. 3C).
FIGURE 3.

Increased expression levels of cone proteins in Cnga3−/−/Nrl−/− mice treated with PKG inhibitor and in Cnga3−/−/Nrl−/−/Gucy2e−/− mice. A and B, shown are representative images of the Western blot detection of M- and S-opsin (A) and corresponding densitometric analysis (B) in Cnga3−/−/Nrl−/− mice treated with KT5823. IB, immunoblot. C, Western blot image showing expression of M-opsin, S-opsin, and cone arrestin in Cnga3−/−/Nrl−/−/Gucy2e−/− mice at P60, compared with that in age-matched Cnga3−/−/Nrl−/− and Nrl−/− mice. Data are represented as means ± S.E. of measurements from 3 to 4 assays using retinas prepared from 4 to 5 mice. Unpaired Student's t test was used for determination of the significance of differences (*, p < 0.05).
Reduced Müller Glial Cell Activation in Cnga3−/−/Nrl−/− Mice Treated with PKG Inhibitor and in Cnga3−/−/Nrl−/−/Gucy2e−/− Mice
Müller glia are known to activate in response to retinal stress, including photoreceptor death and injury, by profound up-regulation of GFAP in intermediate filaments. GFAP immunolabeling is commonly used to assess Müller glial cell activation in retinal degeneration (23–25). We previously showed increased GFAP expression in Cnga3−/− retinas (8). In this study, we examined GFAP expression in Cnga3−/−/Nrl−/− mice treated with the PKG inhibitor to determine whether the overall retinal stress is reduced. Fig. 4A shows GFAP immunofluorescence labeling of retinal sections prepared from Cnga3−/−/Nrl−/− mice treated with (Rp)-8-Br-cGMPS or vehicle and the corresponding quantification of immunofluorescence intensity. The retinal sections from vehicle-treated mice showed strong up-regulation of GFAP across the retina, with immunostaining mainly detected in the inner and outer plexiform layers and the ganglionic layer (Fig. 4A, upper panels). In contrast, the retinal sections prepared from (Rp)-8-Br-cGMPS-treated mice showed significantly reduced GFAP immunoreactivity (Fig. 4A, lower panels). Similar results were obtained from mice treated with KT5823 (Fig. 4B). The reduction of Müller glial cell activation was also observed in Cnga3−/−/Nrl−/−/Gucy2e−/− mice, compared with that in Cnga3−/−/Nrl−/− mice (Fig. 4C).
FIGURE 4.

Reduced Müller glial cell activation in Cnga3−/−/Nrl−/− mice treated with PKG inhibitor and in Cnga3−/−/Nrl−/−/Gucy2e−/− mice. Shown are representative confocal images of GFAP immunofluorescence labeling on the peripheral, middle, and central regions of the retinal sections prepared from Cnga3−/−/Nrl−/− mice treated with (Rp)-8-Br-cGMPS (A) or KT5823 (B) or vehicle and from Cnga3−/−/Nrl−/−/Gucy2e−/− mice (C), and corresponding quantification of immunofluorescence intensity. ONL, outer nuclear layer; INL, inner nuclear layer; RGC, retinal ganglion cell. Data are represented as means ± S.E. of measurements from three assays using retinas prepared from 3 to 4 mice. Unpaired Student's t test was used for determination of the significance of differences (*, p < 0.05; **, p < 0.01).
Reduced ER Stress and Increased IP3R1 Phosphorylation in Retinas of Cnga3−/−/Nrl−/− Mice Treated with PKG Inhibitor
We previously showed that the levels of the ER stress marker proteins, including phospho-eIF2α, which represents a well characterized arm/pathway of ER stress (26, 27), were enhanced in Cnga3−/−/Nrl−/− retinas (12). In this study, we first investigated whether inhibition of PKG suppresses PERK/eIF2α pathway and ER stress. We found that PKG inhibitor treatment significantly reduced levels of phospho-eIF2α in Cnga3−/−/Nrl−/− retinas. The level of phospho-eIF2α was about 1.3-fold higher in Cnga3−/−/Nrl−/− mice than in Nrl−/− mice. Treatment with (Rp)-8-Br-cGMPS reduced the level of phospho-eIF2α by about 25%, compared with vehicle-treated Cnga3−/−/Nrl−/− controls (Fig. 5A), whereas treatment with KT5823 reduced it by about 40%, compared with vehicle-treated controls (Fig. 5B).
FIGURE 5.

Reduced levels of phospho-eIF2α and increased levels of phospho-IP3R1 in retinas of Cnga3−/−/Nrl−/− mice treated with PKG inhibitor. Shown are representative images of the Western blot detection of phospho-eIF2α and phospho-IP3R1 in retinas of Cnga3−/−/Nrl−/− mice treated with (Rp)-8-Br-cGMPS (A) or KT5823 (B) and corresponding densitometric analysis. The relative expression levels in Cnga3−/−/Nrl−/− mice were normalized to the values in Nrl−/− mice. Data are represented as means ± S.E. of measurements from four assays using retinas prepared from 4 to 5 mice. Unpaired Student's t test was used for determination of the significance of differences between the drug-treated and vehicle-treated mice (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
IP3R plays a critical role in the cellular Ca2+ regulation, including ER Ca2+ homeostasis and responses to cellular Ca2+ perturbation and stress. We have shown an increased IP3R1 phosphorylation in Cnga3−/−/Nrl−/− retinas (12). In this study, we examined the effects of PKG inhibitor on IP3R1 phosphorylation. The level of phospho-IP3R1 was about 30% higher in Cnga3−/−/Nrl−/− mice than in Nrl−/− mice (Fig. 5, A and B). Following treatment with the PKG inhibitor, the level of phospho-IP3R1 increased further. The level of phospho-IP3R1 in Cnga3−/−/Nrl−/− mice increased by about 33% following treatment with (Rp)-8-Br-cGMPS (Fig. 5A) and increased by about 12% following treatment with KT5823 (Fig. 5B), compared with vehicle-treated Cnga3−/−/Nrl−/− controls. Thus, inhibition of PKG activity reduced ER stress and increased IP3R1 phosphorylation in CNG channel-deficient mice.
In this work, we also examined the activity of the other arms/pathways of ER stress in CNG channel-deficient mice and the effects of PKG inhibition. These arms include the activating transcription factor 6 (ATF6) and the serine/threonine-protein kinase/endoribonuclease (IRE1) pathways. Activation of the ATF6 arm is characterized by cleavage of the pro-protein and production of the cleaved form. Using the anti-ATF6 antibody that recognizes both the pro-form and the cleaved form of the protein, our analysis showed the presence of the cleaved ATF6 in Cnga3−/−/Nrl−/− retinas, which was barely detected in Nrl−/− retinas (Fig. 6A). The cleaved form of ATF6 in Cnga3−/−/Nrl−/− retinas was increased by about 4.5-fold, compared with Nrl−/− retinas (Fig. 6A). Moreover, treatment with PKG inhibitor significantly suppressed cleavage of ATF6 (Fig. 6A). Activity of the IRE1 arm was evaluated by examining the expression levels of phospho-IRE1α using anti-phospho-IRE1α antibody. As shown in Fig. 6B, the level of phospho-IRE1α in Cnga3−/−/Nrl−/− retinas was increased by about 1.8-fold, compared with Nrl−/− retinas. Treatment with the PKG inhibitor reduced its phosphorylation, but the differences did not reach statistical significance (Fig. 6B).
FIGURE 6.

Increased ATF6 cleavage and increased phospho-IRE1 levels in Cnga3−/−/Nrl−/− retinas, compared with Nrl−/− mice, and the effects of treatment with PKG inhibitor. Shown are representative images of the Western blot detection of ATF6 (A) and phospho-IRE1 (B) in retinas of Nrl−/− and Cnga3−/−/Nrl−/− mice treated with vehicle or (Rp)-8-Br-cGMPS or KT5823, and the corresponding densitometric analyses. The relative expression levels in Cnga3−/−/Nrl−/− mice were normalized to the value in Nrl−/− mice. Data are represented as means ± S.E. of measurements from four assays using retinas prepared from 7 to 8 mice. Unpaired Student's t test was used for determination of the significance of differences (*, p < 0.05). IB, immunoblot.
Reduced ER Stress and Increased IP3R1 Phosphorylation in Cnga3−/−/Nrl−/−/Gucy2e−/− Retinas
We next examined whether ER stress and IP3R1 phosphorylation are altered in retinas of Cnga3−/−/Nrl−/−/Gucy2e−/− mice. Similar to our findings from the treatment with PKG inhibitor, the level of phospho-eIF2α was significantly reduced, and the level of phospho-IP3R1 was further increased in Cnga3−/−/Nrl−/−/Gucy2e−/− mice, compared with that in Cnga3−/−/Nrl−/− mice. As shown in Fig. 7, the level of phospho-eIF2α in Cnga3−/−/Nrl−/−/Gucy2e−/− mice decreased by about 37% and the level of phospho-IP3R1 increased by about 17%, compared with that in Cnga3−/−/Nrl−/− mice.
FIGURE 7.
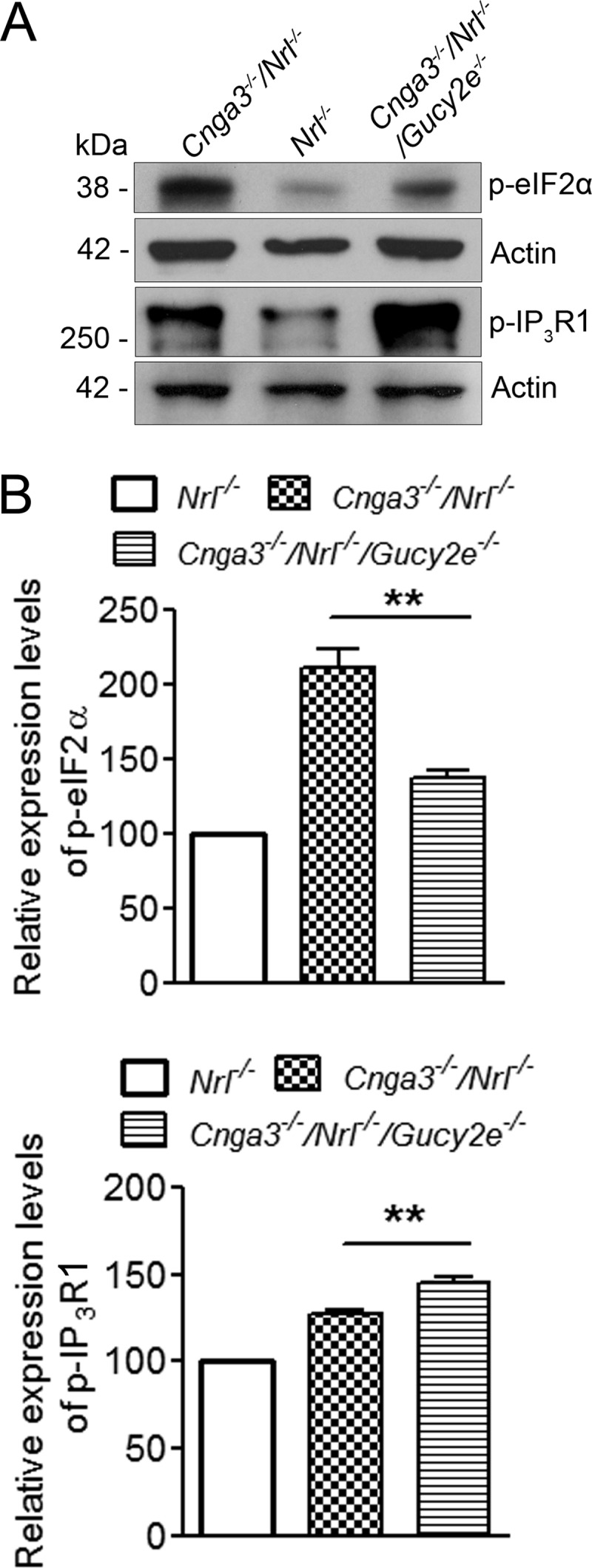
Reduced levels of phospho-eIF2α and increased levels of phospho-IP3R1 in Cnga3−/−/Nrl−/−/Gucy2e−/− retinas. Shown are representative images of the Western blot detection of phospho-eIF2α and phospho-IP3R1 in retinas of Cnga3−/−/Nrl−/−/Gucy2e−/−, Cnga3−/−/Nrl−/−, and Nrl−/− mice at P30 (A) and the corresponding densitometric analysis (B). The relative expression levels in Cnga3−/−/Nrl−/− and Cnga3−/−/Nrl−/−/Gucy2e−/−mice were normalized to the values in Nrl−/− mice. Data are represented as means ± S.E. of measurements from 3 to 4 assays using retinas prepared from 4 to 5 mice. Unpaired Student's t test was used for determination of the significance of differences between the drug-treated and vehicle-treated mice (**, p < 0.01).
Discussion
Contribution of cGMP/PKG Signaling to Apoptotic Cone Death
This study using a PKG inhibitor and deletion of retGC1, which effectively abolished PKG activation, establishes a role for PKG signaling in cone death. Suppressing cGMP production and inhibiting PKG activity significantly decreased apoptotic cone death, caspase-7 cleavage, and Müller glial cell activation in Cnga3−/−/Nrl−/− mice. The effectiveness of PKG inhibitor treatment was evident by the strong suppression of the enzyme's activity and by effective reduction of the activation of CREB (Fig. 1). Thus, our experimental data support the view that cGMP/PKG signaling activated in CNG channel deficiency contributes to cone death and that the toxicity of elevated cGMP is primarily mediated via PKG signaling.
The contribution of PKG signaling to photoreceptor death was previously shown in the rd1 mouse, a model of recessive retinitis pigmentosa caused by a nonsense mutation in the rod Pde6b gene. These mice also display ER stress-associated photoreceptor apoptosis (28, 29). cGMP accumulation has been shown to cause cell death in rd1 mice by overactivating the CNG channel, Ca2+ overload (30, 31), and by activating PKG signaling. Treatment of rd1 retina with PKG inhibitor reduced apoptotic photoreceptor death and improved rod survival (32). Thus, excessive activation of PKG signaling is harmful to photoreceptors, and our work provides insights into how cGMP/PKG signaling contributes to photoreceptor death.
Contribution of cGMP/PKG Signaling to ER Stress
CNG channel-deficient mice display early-onset ER stress, shown by studies at the protein and mRNA expression levels. The expression levels of phospho-eIF2α and CHOP were increased in CNG channel-deficient mice (12). Gene expression profiling studies demonstrated up-regulation of EIF2/ER stress pathways (13). In addition to examining the levels of phospho-eIF2α, we examined the activity of the other two arms/pathways of ER stress and detected increased cleavage of ATF6 and increased expression levels of phospho-IRE1α in Cnga3−/−/Nrl−/− retinas, compared with Nrl−/− retinas. The relevance of ER stress in cone degeneration is highlighted by a recent report that links mutations in ATF6 gene with achromatopsia (33). This is the first nonphotoreceptor-specific gene thus far identified to associate with achromatopsia. ER stress was also indirectly supported by increased levels of calpains, Bcl-2/Bcl-x, and caspase-12/caspase-7 cleavage (12). Thus, the activation of all three ER stress pathways and processing of the ER stress-associated caspases suggest that cone death in CNG channel deficiency is associated with ER stress. Experiments with the ER chemical chaperone TUDCA and the molecular chaperone 11-cis-retinal further support this view. Treatment with TUDCA effectively suppressed caspase-7 cleavage and phosphorylation of eIF2α and increased cone-specific protein expression levels (13). Treatment with 11-cis-retinal significantly improved cone survival, manifested as increased levels of cone proteins (data not shown). To understand the mechanism of ER stress and determine whether the elevated cGMP/PKG signaling contributes to ER stress, we examined the effects of PKG inhibitor and Gucy2e deletion. We found that inhibition of cGMP production and PKG activity effectively reduced ER stress in CNG channel-deficient mice, supporting a contribution of cGMP/PKG signaling to ER stress. Involvement of cGMP/PKG signaling in ER stress and apoptosis was previously shown in pancreatic β cells and myocardial cells (34, 35). This study for the first time demonstrated the regulatory role of cGMP/PKG signaling in ER stress in neural retinal cells.
Potential ER Ca2+ Dysregulation and Impaired ER Function in CNG Channel Deficiency
As a Ca2+ store, the ER is responsible for sequestering excess intracellular Ca2+ and releasing ER luminal Ca2+ stores when intracellular Ca2+ levels are low. ER Ca2+ homeostasis is crucial for cellular Ca2+ signaling and the ER's function as a site of cellular protein processing. ER requires high levels of luminal Ca2+ to maintain proper protein folding and exportation. Low luminal Ca2+ is known to cause an accumulation of incorrectly folded proteins and the subsequent unfolded protein response and ER stress (36–39). ER Ca2+ homeostasis is regulated by three ER Ca2+ channels/pumps as follows: the IP3R and ryanodine receptors for Ca2+ efflux out of the ER into the cytosol, and the sarco/endoplasmic reticulum Ca2+-ATPase for Ca2+ influx into the ER (Fig. 8). The activity of these channels/pumps is primarily regulated by cellular Ca2+ levels, their respective ligands, and numerous signaling molecules/pathways. As nonselective cation channels, CNG channels are the main source of the Ca2+ inward currents in the outer segments of photoreceptors and play a pivotal role in the light response/adaptation and cellular Ca2+ homeostasis. Cones lacking functional CNG channels likely suffer from cellular Ca2+ perturbation/cytosolic Ca2+ reduction. In mouse rods, shutting down the influx through the CNG channels in the light lowers free cytoplasmic Ca2+ nearly 10-fold (40). The lowered cytosolic Ca2+ level in CNG channel deficiency is also supported by the remarkable elevation in cellular cGMP levels, because cGMP production in photoreceptors is tightly negatively regulated by the cytosolic Ca2+ level via the guanylate cyclase-activating proteins/retinal guanylate cyclase regulatory axis (41). Cytosolic Ca2+ reduction would likely lead to a compensating increase in ER Ca2+ release via ER Ca2+-releasing channels (and probably a reduced ER Ca2+ influx via the sarco/endoplasmic reticulum Ca2+-ATPase pumps), which may subsequently lead to excess ER Ca2+ release/ER Ca2+ reduction and interfere with ER function. Nevertheless, the typical phenotypes of CNG channel deficiency are impaired opsin trafficking and opsin mislocalization (8, 20), reflecting impaired ER function/protein folding, and ER stress. It is worth mentioning that there are other Ca2+ channels in the inner segment membrane, including transient receptor potential channels, and these channels could also play a role in the cytosolic Ca2+ regulation (42, 43).
FIGURE 8.

Mechanism of ER stress in CNG channel deficiency: potential involvement of the ER Ca2+ channels. In CNG channel deficiency, decreased cytosolic Ca2+ and increased cGMP/PKG signaling, which is a consequence of the reduced cytosolic Ca2+ level, activates ER Ca2+-releasing channels to remedy the cytosolic Ca2+ perturbation. As a consequence, the decreased ER Ca2+ level interferes with ER function/protein folding, which causes opsin mistrafficking/mislocalization and triggers unfolded protein response (UPR) and ER stress. Suppressing cGMP/PKG signaling by Gucy2e deletion or by PKG inhibitor inhibits IP3R, decreases ER Ca2+ release, and reduces ER stress. RyR, ryanodine receptor; SERCA, sarco/endoplasmic reticulum Ca2+-ATPase.
cGMP/PKG Signaling Contributes to ER Stress in CNG Channel Deficiency, Potential Involvement of the ER Ca2+ Channel IP3R1
In addition to impaired cytosolic Ca2+ homeostasis directly caused by lack of the functional CNG channels, the activated cGMP/PKG signaling may play a role in ER stress, via its regulation on ER Ca2+ channels. Our results showing increased IP3R1 phosphorylation in CNG channel-deficient mice on GC1 knock-out background or after treatment with the PKG inhibitor support a role of cGMP/PKG signaling in ER Ca2+ regulation. Expressed in all cell types, IP3R channels play a crucial role in cytosolic and ER Ca2+ homeostasis. Reduced cytosolic Ca2+ or increased inositol 1,4,5-trisphosphate levels are known to increase IP3R activity/release of Ca2+ from the ER (44–46). Receptor phosphorylation is also a known factor regulating the channel's activity. Although the reported data were controversial, IP3R phosphorylation has been shown to decrease the channel's activity and reduce Ca2+ release from the ER in a variety of experimental conditions (47–52). This work indicates increased IP3R1 phosphorylation by knocking out GC1 or inhibiting PKG, accompanied by reduced ER stress and apoptosis. The results suggest that cGMP/PKG signaling suppresses IP3R1 phosphorylation, promotes Ca2+ release from the ER, and subsequently causes ER Ca2+ reduction/ER malfunction. How cGMP/PKG signaling suppresses phosphorylation of IP3R1 in CNG channel-deficient cones remains to be determined and may involve complex mechanisms. Numerous molecules/pathways, including PKA (53, 54), PKC (49), PKG (47), AKT (48, 55), and ERK (52), have been reported to regulate IP3R phosphorylation. Furthermore, PKG has multiple targets, and cross-talks among pathways exist.
It is important to emphasize that the level of phospho-IP3R1 in Cnga3−/−/Nrl−/− mice always increased relative to Nrl−/− mice (Figs. 5 and 7) (12). A possible explanation for this observation is compensatory regulation. The Ca2+ release from the ER in CNG channel deficiency is likely increased (and Ca2+ entry is probably decreased) to compensate for the reduced cytosolic Ca2+ level, which could potentially lead to a reduction of ER Ca2+ level. Indeed, alteration of ryanodine receptors, including increased phosphorylation and expression level, can be detected in CNG channel-deficient retinas (data not shown). As a consequence, the function of IP3R could be suppressed to protect the ER from excess Ca2+ release/Ca2+ depletion. The suppressive regulation of IP3R has been shown to protect hippocampal neurons from apoptosis both in vitro and in an animal model of ER stress (56). The protective effects of cGMP/PKG signaling inhibition on ER stress and cone death, accompanied with increased IP3R phosphorylation, suggest that the regulatory suppression of IP3R1 in CNG channel deficiency is insufficient to compensate, likely due to the elevated cGMP/PKG signaling. Thus, the elevated cGMP/PKG signaling in CNG channel deficiency may interfere with the compensatory inhibition of IP3R1, leading to excess Ca2+ release from the ER and ER stress. Based on our findings, we postulate that the reduced cytosolic Ca2+ and increased cGMP/PKG signaling in CNG channel deficiency promotes Ca2+ release from the ER, leading to ER Ca2+ dysregulation/ER stress and apoptotic death (Fig. 8).
Presence of a Nonapoptotic cGMP/PKG-independent Death Mechanism in CNG Channel Deficiency
We found that TUNEL-positive staining was almost completely abolished in Gucy2e-deleted mice and in PKG inhibitor-treated Cnga3−/−/Nrl−/− mice (see Figs. 1 and 2). However, the cone rescue was only partial (Fig. 3) (15). These findings suggest that apoptosis may not be the only mechanism in cone death; a nonapoptotic, cGMP/PKG-independent mechanism may exist. The early-onset apoptotic cone death appears to be mediated mainly by cGMP/PKG signaling. The correlation between the time course of cGMP elevation and the TUNEL labeling (15) also supports this view.
In summary, this study shows that treatment with the PKG inhibitor or deletion of Gucy2e suppressed cone apoptosis and improved cone survival in CNG channel-deficient mice, demonstrating a role of cGMP/PKG signaling in cone death. ER stress was decreased by treatment with the PKG inhibitor and deletion of Gucy2e, indicating cGMP/PKG signaling dependence. Moreover, suppressing cGMP/PKG signaling increased IP3R1 phosphorylation, suggesting a role of ER Ca2+ channel regulation in ER stress and cone death. This work provides insights into ER Ca2+ channel regulation/ER stress in cone death and improves our understanding of the mechanism of cone degeneration in CNG channel deficiency.
Author Contributions
X. Q. D., H. M., and M. R. B. designed the study and wrote and revised the manuscript. S. M., M. B., and W. B. contributed to study design, manuscript writing, and revision. H. M., M. R. B., A. T., J. B., and F. Y. performed experiments and analyzed data. S. M. and M. B. provided Cnga3−/− mouse line and W. B. provided Gucy2e−/− mouse line. All authors approved the final version of the manuscript.
Acknowledgments
We thank Drs. Anand Swaroop, Cheryl M. Craft, and Muna I. Naash for providing the Nrl−/− mouse line and antibodies for M-opsin, cone arrestin, and S-opsin. We thank Melanie Mason for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants P30EY021725, T32EY023202, R01EY019490, R01EY08123, and R01EY019298. This work was also supported by the Deutsche Forschungsgemeinschaft. The authors declare that they have no conflicts of interest with the contents of this article.
- CNG
- cyclic nucleotide-gated
- ATF6
- activating transcription factor 6
- CHOP
- CCAAT/-enhancer-binding protein homologous protein
- CREB
- cAMP-response element-binding protein
- ER
- endoplasmic reticulum
- GFAP
- glial fibrillary acidic protein
- IP3R
- inositol 1,4,5-trisphosphate receptor
- IRE1
- serine/threonine-protein kinase/endoribonuclease
- UPR
- unfolded protein response
- TUDCA
- tauroursodeoxycholic acid
- P
- postnatal day.
References
- 1. Kaupp U. B., Seifert R. (2002) Cyclic nucleotide-gated ion channels. Physiol. Rev. 82, 769–824 [DOI] [PubMed] [Google Scholar]
- 2. Kohl S., Baumann B., Broghammer M., Jägle H., Sieving P., Kellner U., Spegal R., Anastasi M., Zrenner E., Sharpe L. T., Wissinger B. (2000) Mutations in the CNGB3 gene encoding the β-subunit of the cone photoreceptor cGMP-gated channel are responsible for achromatopsia (ACHM3) linked to chromosome 8q21. Hum. Mol. Genet. 9, 2107–2116 [DOI] [PubMed] [Google Scholar]
- 3. Nishiguchi K. M., Sandberg M. A., Gorji N., Berson E. L., Dryja T. P. (2005) Cone cGMP-gated channel mutations and clinical findings in patients with achromatopsia, macular degeneration, and other hereditary cone diseases. Hum. Mutat. 25, 248–258 [DOI] [PubMed] [Google Scholar]
- 4. Wissinger B., Gamer D., Jägle H., Giorda R., Marx T., Mayer S., Tippmann S., Broghammer M., Jurklies B., Rosenberg T., Jacobson S. G., Sener E. C., Tatlipinar S., Hoyng C. B., Castellan C., et al. (2001) CNGA3 mutations in hereditary cone photoreceptor disorders. Am. J. Hum. Genet. 69, 722–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Varsányi B., Somfai G. M., Lesch B., Vámos R., Farkas A. (2007) Optical coherence tomography of the macula in congenital achromatopsia. Invest. Ophthalmol. Vis. Sci. 48, 2249–2253 [DOI] [PubMed] [Google Scholar]
- 6. Thiadens A. A., Somervuo V., van den Born L. I., Roosing S., van Schooneveld M. J., Kuijpers R. W., van Moll-Ramirez N., Cremers F. P., Hoyng C. B., Klaver C. (2010) Progressive loss of cones in achromatopsia. An imaging study using spectral-domain optical coherence tomography. Invest. Ophthalmol. Vis. Sci. 51, 5952–5957 [DOI] [PubMed] [Google Scholar]
- 7. Genead M. A., Fishman G. A., Rha J., Dubis A. M., Bonci D. M., Dubra A., Stone E. M., Neitz M., Carroll J. (2011) Photoreceptor structure and function in patients with congenital achromatopsia. Invest. Ophthalmol. Vis. Sci. 52, 7298–7308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Michalakis S., Geiger H., Haverkamp S., Hofmann F., Gerstner A., Biel M. (2005) Impaired opsin targeting and cone photoreceptor migration in the retina of mice lacking the cyclic nucleotide-gated channel CNGA3. Invest. Ophthalmol. Vis. Sci. 46, 1516–1524 [DOI] [PubMed] [Google Scholar]
- 9. Biel M., Seeliger M., Pfeifer A., Kohler K., Gerstner A., Ludwig A., Jaissle G., Fauser S., Zrenner E., Hofmann F. (1999) Selective loss of cone function in mice lacking the cyclic nucleotide-gated channel CNG3. Proc. Natl. Acad. Sci. U.S.A. 96, 7553–7557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xu J., Morris L., Fliesler S. J., Sherry D. M., Ding X.-Q. (2011) Early-onset, slow progression of cone photoreceptor dysfunction and degeneration in CNG channel subunit CNGB3 deficiency. Invest. Ophthalmol. Vis. Sci. 52, 3557–3566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ding X. Q., Harry C. S., Umino Y., Matveev A. V., Fliesler S. J., Barlow R. B. (2009) Impaired cone function and cone degeneration resulting from CNGB3 deficiency: down-regulation of CNGA3 biosynthesis as a potential mechanism. Hum. Mol. Genet. 18, 4770–4780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thapa A., Morris L., Xu J., Ma H., Michalakis S., Biel M., Ding X. Q. (2012) Endoplasmic reticulum stress-associated cone photoreceptor degeneration in cyclic nucleotide-gated channel deficiency. J. Biol. Chem. 287, 18018–18029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ma H., Thapa A., Morris L. M., Michalakis S., Biel M., Frank M. B., Bebak M., Ding X. Q. (2013) Loss of cone cyclic nucleotide-gated channel leads to alterations in light response modulating system and cellular stress response pathways: a gene expression profiling study. Hum. Mol. Genet. 22, 3906–3919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mears A. J., Kondo M., Swain P. K., Takada Y., Bush R. A., Saunders T. L., Sieving P. A., Swaroop A. (2001) Nrl is required for rod photoreceptor development. Nat. Genet. 29, 447–452 [DOI] [PubMed] [Google Scholar]
- 15. Xu J., Morris L., Thapa A., Ma H., Michalakis S., Biel M., Baehr W., Peshenko I. V., Dizhoor A. M., Ding X. Q. (2013) cGMP accumulation causes photoreceptor degeneration in CNG channel deficiency: evidence of cGMP cytotoxicity independently of enhanced CNG channel function. J. Neurosci. 33, 14939–14948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang R. B., Robinson S. W., Xiong W. H., Yau K. W., Birch D. G., Garbers D. L. (1999) Disruption of a retinal guanylyl cyclase gene leads to cone-specific dystrophy and paradoxical rod behavior. J. Neurosci. 19, 5889–5897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Michel M., Green C. L., Eskin A., Lyons L. C. (2011) PKG-mediated MAPK signaling is necessary for long-term operant memory in Aplysia. Learn. Mem. 18, 108–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fallahian F., Karami-Tehrani F., Salami S., Aghaei M. (2011) Cyclic GMP induced apoptosis via protein kinase G in oestrogen receptor-positive and -negative breast cancer cell lines. FEBS J. 278, 3360–3369 [DOI] [PubMed] [Google Scholar]
- 19. Tegeder I., Del Turco D., Schmidtko A., Sausbier M., Feil R., Hofmann F., Deller T., Ruth P., Geisslinger G. (2004) Reduced inflammatory hyperalgesia with preservation of acute thermal nociception in mice lacking cGMP-dependent protein kinase I. Proc. Natl. Acad. Sci. U.S.A. 101, 3253–3257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Carvalho L. S., Xu J., Pearson R. A., Smith A. J., Bainbridge J. W., Morris L. M., Fliesler S. J., Ding X. Q., Ali R. R. (2011) Long-term and age-dependent restoration of visual function in a mouse model of CNGB3-associated achromatopsia following gene therapy. Hum. Mol. Genet. 20, 3161–3175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Matveev A. V., Fitzgerald J. B., Xu J., Malykhina A. P., Rodgers K. K., Ding X. Q. (2010) The disease-causing mutations in the carboxyl terminus of the cone cyclic nucleotide-gated channel CNGA3 subunit alter the local secondary structure and interfere with the channel active conformational change. Biochemistry 49, 1628–1639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Michalakis S., Mühlfriedel R., Tanimoto N., Krishnamoorthy V., Koch S., Fischer M. D., Becirovic E., Bai L., Huber G., Beck S. C., Fahl E., Büning H., Paquet-Durand F., Zong X., Gollisch T., et al. (2010) Restoration of cone vision in the CNGA3−/− mouse model of congenital complete lack of cone photoreceptor function. Mol. Ther. 18, 2057–2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. de Raad S., Szczesny P. J., Munz K., Remé C. E. (1996) Light damage in the rat retina: glial fibrillary acidic protein accumulates in Muller cells in correlation with photoreceptor damage. Ophthalmic Res. 28, 99–107 [DOI] [PubMed] [Google Scholar]
- 24. Bringmann A., Reichenbach A. (2001) Role of Muller cells in retinal degenerations. Front. Biosci. 6, E72–E92 [DOI] [PubMed] [Google Scholar]
- 25. Zhao T. T., Tian C. Y., Yin Z. Q. (2010) Activation of Muller cells occurs during retinal degeneration in RCS rats. Adv. Exp. Med. Biol. 664, 575–583 [DOI] [PubMed] [Google Scholar]
- 26. Szegezdi E., Logue S. E., Gorman A. M., Samali A. (2006) Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 7, 880–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim I., Xu W., Reed J. C. (2008) Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 7, 1013–1030 [DOI] [PubMed] [Google Scholar]
- 28. Sanges D., Comitato A., Tammaro R., Marigo V. (2006) Apoptosis in retinal degeneration involves cross-talk between apoptosis-inducing factor (AIF) and caspase-12 and is blocked by calpain inhibitors. Proc. Natl. Acad. Sci. U.S.A. 103, 17366–17371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sanges D., Marigo V. (2006) Cross-talk between two apoptotic pathways activated by endoplasmic reticulum stress: differential contribution of caspase-12 and AIF. Apoptosis 11, 1629–1641 [DOI] [PubMed] [Google Scholar]
- 30. Paquet-Durand F., Beck S., Michalakis S., Goldmann T., Huber G., Mühlfriedel R., Trifunović D., Fischer M. D., Fahl E., Duetsch G., Becirovic E., Wolfrum U., van Veen T., Biel M., Tanimoto N., Seeliger M. W. (2011) A key role for cyclic nucleotide gated (CNG) channels in cGMP-related retinitis pigmentosa. Hum. Mol. Genet. 20, 941–947 [DOI] [PubMed] [Google Scholar]
- 31. Tosi J., Davis R. J., Wang N. K., Naumann M., Lin C. S., Tsang S. H. (2011) shRNA knockdown of guanylate cyclase 2e or cyclic nucleotide gated channel α1 increases photoreceptor survival in a cGMP phosphodiesterase mouse model of retinitis pigmentosa. J. Cell. Mol. Med. 15, 1778–1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Paquet-Durand F., Hauck S. M., van Veen T., Ueffing M., Ekström P. (2009) PKG activity causes photoreceptor cell death in two retinitis pigmentosa models. J. Neurochem. 108, 796–810 [DOI] [PubMed] [Google Scholar]
- 33. Kohl S., Zobor D., Chiang W. C., Weisschuh N., Staller J., Menendez I. G., Chang S., Beck S. C., Garcia Garrido M., Sothilingam V., Seeliger M. W., Stanzial F., Benedicenti F., Inzana F., Héon E., et al. (2015) Mutations in the unfolded protein response regulator ATF6 cause the cone dysfunction disorder achromatopsia. Nat. Genet. 47, 757–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Takimoto E., Champion H. C., Li M., Belardi D., Ren S., Rodriguez E. R., Bedja D., Gabrielson K. L., Wang Y., Kass D. A. (2005) Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat. Med. 11, 214–222 [DOI] [PubMed] [Google Scholar]
- 35. Oyadomari S., Takeda K., Takiguchi M., Gotoh T., Matsumoto M., Wada I., Akira S., Araki E., Mori M. (2001) Nitric oxide-induced apoptosis in pancreatic beta cells is mediated by the endoplasmic reticulum stress pathway. Proc. Natl. Acad. Sci. U.S.A. 98, 10845–10850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Benali-Furet N. L., Chami M., Houel L., De Giorgi F., Vernejoul F., Lagorce D., Buscail L., Bartenschlager R., Ichas F., Rizzuto R., Paterlini-Bréchot P. (2005) Hepatitis C virus core triggers apoptosis in liver cells by inducing ER stress and ER calcium depletion. Oncogene 24, 4921–4933 [DOI] [PubMed] [Google Scholar]
- 37. Mekahli D., Bultynck G., Parys J. B., De Smedt H., Missiaen L. (2011) Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb. Perspect. Biol. 3, a004317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Luciani D. S., Gwiazda K. S., Yang T. L., Kalynyak T. B., Bychkivska Y., Frey M. H., Jeffrey K. D., Sampaio A. V., Underhill T. M., Johnson J. D. (2009) Roles of IP3R and RyR Ca2+ channels in endoplasmic reticulum stress and beta-cell death. Diabetes 58, 422–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chao C. C., Huang C. C., Lu D. Y., Wong K. L., Chen Y. R., Cheng T. H., Leung Y. M. (2012) Ca2+ store depletion and endoplasmic reticulum stress are involved in P2X7 receptor-mediated neurotoxicity in differentiated NG108–15 cells. J. Cell Biochem. 113, 1377–1385 [DOI] [PubMed] [Google Scholar]
- 40. Woodruff M. L., Olshevskaya E. V., Savchenko A. B., Peshenko I. V., Barrett R., Bush R. A., Sieving P. A., Fain G. L., Dizhoor A. M. (2007) Constitutive excitation by Gly90Asp rhodopsin rescues rods from degeneration caused by elevated production of cGMP in the dark. J. Neurosci. 27, 8805–8815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Polans A., Baehr W., Palczewski K. (1996) Turned on by Ca2+! The physiology and pathology of Ca2+-binding proteins in the retina. Trends Neurosci. 19, 547–554 [DOI] [PubMed] [Google Scholar]
- 42. Szikra T., Cusato K., Thoreson W. B., Barabas P., Bartoletti T. M., Krizaj D. (2008) Depletion of calcium stores regulates calcium influx and signal transmission in rod photoreceptors. J. Physiol. 586, 4859–4875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Baldridge W. H., Kurennyi D. E., Barnes S. (1998) Calcium-sensitive calcium influx in photoreceptor inner segments. J. Neurophysiol. 79, 3012–3018 [DOI] [PubMed] [Google Scholar]
- 44. Finch E. A., Turner T. J., Goldin S. M. (1991) Calcium as a coagonist of inositol 1,4,5-trisphosphate-induced calcium release. Science 252, 443–446 [DOI] [PubMed] [Google Scholar]
- 45. Miyawaki A., Furuichi T., Maeda N., Mikoshiba K. (1990) Expressed cerebellar-type inositol 1,4,5-trisphosphate receptor, P400, has calcium release activity in a fibroblast L cell line. Neuron 5, 11–18 [DOI] [PubMed] [Google Scholar]
- 46. Sienaert I., De Smedt H., Parys J. B., Missiaen L., Vanlingen S., Sipma H., Casteels R. (1996) Characterization of a cytosolic and a luminal Ca2+-binding site in the type I inositol 1,4,5-trisphosphate receptor. J. Biol. Chem. 271, 27005–27012 [DOI] [PubMed] [Google Scholar]
- 47. Murthy K. S., Zhou H. (2003) Selective phosphorylation of the IP3R-I in vivo by cGMP-dependent protein kinase in smooth muscle. Am. J. Physiol. Gastrointest. Liver Physiol. 284, G221–G230 [DOI] [PubMed] [Google Scholar]
- 48. Szado T., Vanderheyden V., Parys J. B., De Smedt H., Rietdorf K., Kotelevets L., Chastre E., Khan F., Landegren U., Söderberg O., Bootman M. D., Roderick H. L. (2008) Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc. Natl. Acad. Sci. U.S.A. 105, 2427–2432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Arguin G., Regimbald-Dumas Y., Fregeau M. O., Caron A. Z., Guillemette G. (2007) Protein kinase C phosphorylates the inositol 1,4,5-trisphosphate receptor type 2 and decreases the mobilization of Ca2+ in pancreatoma AR4–2J cells. J. Endocrinol. 192, 659–668 [DOI] [PubMed] [Google Scholar]
- 50. Caron A. Z., Chaloux B., Arguin G., Guillemette G. (2007) Protein kinase C decreases the apparent affinity of the inositol 1,4,5-trisphosphate receptor type 3 in RINm5F cells. Cell Calcium 42, 323–331 [DOI] [PubMed] [Google Scholar]
- 51. Marchi S., Marinello M., Bononi A., Bonora M., Giorgi C., Rimessi A., Pinton P. (2012) Selective modulation of subtype III IP(3)R by Akt regulates ER Ca2+ release and apoptosis. Cell Death Dis. 3, e304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bai G. R., Yang L. H., Huang X. Y., Sun F. Z. (2006) Inositol 1,4,5-trisphosphate receptor type 1 phosphorylation and regulation by extracellular signal-regulated kinase. Biochem. Biophys. Res. Commun. 348, 1319–1327 [DOI] [PubMed] [Google Scholar]
- 53. Soulsby M. D., Wojcikiewicz R. J. (2005) The type III inositol 1,4,5-trisphosphate receptor is phosphorylated by cAMP-dependent protein kinase at three sites. Biochem. J. 392, 493–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Haug L. S., Jensen V., Hvalby O., Walaas S. I., Ostvold A. C. (1999) Phosphorylation of the inositol 1,4,5-trisphosphate receptor by cyclic nucleotide-dependent kinases in vitro and in rat cerebellar slices in situ. J. Biol. Chem. 274, 7467–7473 [DOI] [PubMed] [Google Scholar]
- 55. Khan M. T., Wagner L., 2nd, Yule D. I., Bhanumathy C., Joseph S. K. (2006) Akt kinase phosphorylation of inositol 1,4,5-trisphosphate receptors. J. Biol. Chem. 281, 3731–3737 [DOI] [PubMed] [Google Scholar]
- 56. Feng X., Krogh K. A., Wu C. Y., Lin Y. W., Tsai H. C., Thayer S. A., Wei L. N. (2014) Receptor-interacting protein 140 attenuates endoplasmic reticulum stress in neurons and protects against cell death. Nat. Commun. 5, 4487. [DOI] [PMC free article] [PubMed] [Google Scholar]