

Background: The N-terminal fragment of amyloidogenic apoA-I mutants deposits as fibrils by unknown mechanisms.
Results: The G26R mutation partially prevents helix formation of the N-terminal fragment upon lipid binding, thereby facilitating β-transition and fibril formation.
Conclusion: Membrane binding modulates fibril formation of apoA-I through partially destabilized helical conformation.
Significance: The results reveal a new pathway for amyloid fibril formation by apoA-I.
Keywords: amyloid, apolipoprotein, fibril, lipid bilayer, membrane, apoA-I, helix
Abstract
The N-terminal amino acid 1–83 fragment of apolipoprotein A-I (apoA-I) has a strong propensity to form amyloid fibrils at physiological neutral pH. Because apoA-I has an ability to bind to lipid membranes, we examined the effects of the lipid environment on fibril-forming properties of the N-terminal fragment of apoA-I variants. Thioflavin T fluorescence assay as well as fluorescence and transmission microscopies revealed that upon lipid binding, fibril formation by apoA-I 1–83 is strongly inhibited, whereas the G26R mutant still retains the ability to form fibrils. Such distinct effects of lipid binding on fibril formation were also observed for the amyloidogenic prone region-containing peptides, apoA-I 8–33 and 8–33/G26R. This amyloidogenic region shifts from random coil to α-helical structure upon lipid binding. The G26R mutation appears to prevent this helix transition because lower helical propensity and more solvent-exposed conformation of the G26R variant upon lipid binding were observed in the apoA-I 1–83 fragment and 8–33 peptide. With a partially α-helical conformation induced by the presence of 2,2,2-trifluoroethanol, fibril formation by apoA-I 1–83 was strongly inhibited, whereas the G26R variant can form amyloid fibrils. These findings suggest a new possible pathway for amyloid fibril formation by the N-terminal fragment of apoA-I variants: the amyloidogenic mutations partially destabilize the α-helical structure formed upon association with lipid membranes, resulting in physiologically relevant conformations that allow fibril formation.
Introduction
Apolipoprotein A-I (apoA-I)2 is the major protein component of plasma high-density lipoprotein (HDL) and plays a central role in reverse cholesterol transport, a process by which excess cholesterol in peripheral cells is transferred to the liver for catabolism (1–3). In humans, the apoA-I molecule (243 residues) folds into two tertiary structure domains, comprising an N-terminal α-helix bundle spanning residues 1–187 and a separate less organized C-terminal region spanning the remainder of the molecule (4–6). Naturally occurring mutations in the N-terminal helix bundle domain of human apoA-I are known to affect its functionality mainly in two ways: mutations in the N terminus (residues 1–90) are associated with hereditary amyloidosis, whereas those within the central region (residues 140–170) are mostly associated with defective activation of lecithin-cholesterol acyltransferase (7, 8).
So far, there are about 20 known mutations in the APOA1 gene associated with hereditary systemic apoA-I amyloidosis (9, 10), among which G26R, the first and most common amyloidogenic mutation (11–13), is characterized by amyloid deposits in the peripheral nerves, kidneys, liver, and gastrointestinal tract (11, 14). Although the precise mechanism for the amyloidogenicity of variant apoA-I remains unclear, it is thought that an unstable N-terminal helix bundle conformation associated with amyloidogenic mutations promotes proteolytic cleavage of the full-length protein (10, 15, 16), which deposits in the extracellular space of target tissues (17). Indeed, we recently found that the G26R mutation destabilizes the helix bundle structure of full-length apoA-I (18), and also enhances the amyloid fibril formation of the N-terminal fragment (amino acids 1–83) at physiological neutral pH (19). This enhancing effect of the G26R mutation on fibril formation comes from the local effect on the first amyloidogenic core region (residues 14–22) in the N-terminal fragment of apoA-I (20). Alternatively, it is proposed that the mutation-induced structural perturbations in the amyloid-prone regions promote conformational conversions to the β-structure in the N-terminal segments (21, 22). In addition, it was reported that oxidative modification of apoA-I methionine residues induces transformation of full-length wild-type apoA-I into an amyloidogenic protein (23, 24).
Lipid membrane surfaces are well known to influence the folding, oligomerization, and fibril formation of a variety of amyloidogenic proteins (25–28). There are several possible mechanisms by which membrane binding can facilitate protein misfolding and aggregation. In some natively unfolded proteins such as islet amyloid polypeptide and α-synuclein, the formation of α-helical intermediate states of these proteins upon membrane binding promotes the aggregation of proteins through exposure of highly amyloidogenic sequences (29–32). Membrane binding also increases the local concentration of proteins, and reduces the dimensionality of their collision via diffusion, promoting inter-molecular interaction and fibrillization (33, 34). Most recently, lipid vesicles were shown to accelerate the nucleation step of α-synuclein (35) and amyloid-β peptide (36), triggering conversion into the aggregated state. Because apoA-I has a strong ability to bind to lipid membranes (37) and both the N- and C-terminal regions are involved in lipid binding (38), it is expected that membrane binding has a key role in the aggregation pathway of the N-terminal fragment of apoA-I. However, little is known about the effect of the lipid environment on the amyloidogenic property of apoA-I to date (39).
In the present study, we ask how membrane binding affects the fibril-forming properties of the amyloidogenic N-terminal 1–83 fragment of apoA-I variants. We also examine the role of partially α-helical conformation in the fibril-forming pathway of apoA-I. The results indicate that the G26R mutation prevents formation of the helical structure in the highly aggregation prone sequence upon membrane binding, facilitating the transition to the β-aggregate structure and fibril formation in the amyloidogenic N-terminal fragment of apoA-I.
Experimental Procedures
Preparation of ApoA-I Proteins and Peptides
ApoA-I 1–83 fragment and its engineered variants with substitutions G26R, Y18P/G26R, and L22C/G26R were expressed in Escherichia coli as thioredoxin fusion proteins and isolated and purified as described (19). Cleavage of the thioredoxin fusion protein with thrombin leaves the target apoA-I with two extra amino acids, Gly-Ser, at the amino terminus. The apoA-I preparations were at least 95% pure as assessed by SDS-PAGE. The apoA-I 8–33 and 8–33/G26R peptides were synthesized by the solid-phase method with Fmoc (N-(9-fluorenyl)methoxycarbonyl) chemistry. The amino and carboxyl termini were capped with an acetyl group and an amide group, respectively, to promote α-helix formation (40). Peptide purity was verified by analytical HPLC (>97%) and mass spectrometry. In all experiments, apoA-I variants and peptides were freshly dialyzed from 6 m guanidine hydrochloride and/or a 1% β-mercaptoethanol solution into the appropriate buffer before use. Labeling of cysteine-containing apoA-I variants with 6-acryloyl-2-dimethylaminonaphthalene (acrylodan) was employed as described previously (41).
Preparation of Small Unilamellar Vesicles (SUV)
SUV was prepared as described (37, 42). Briefly, a dried film of egg phosphatidylcholine (PC; Kewpie, Tokyo, Japan) was hydrated in 10 mm Tris buffer (150 mm NaCl, 0.02% NaN3, pH 7.4) and sonicated on ice under nitrogen. After removing titanium debris, the samples were centrifuged in a Beckman 70.1Ti rotor for 1.5 h at 15 °C at 40,000 rpm to separate any remaining large vesicles. The PC concentration of SUV was determined using an enzymatic assay kit from Wako Pure Chemicals (Osaka, Japan).
Circular Dichroism (CD) Spectroscopy
Far-UV CD spectra were recorded from 185 to 260 nm at 25 °C using a Jasco J-1500 spectropolarimeter. The apoA-I protein or peptide solutions of 50 μg/ml in 10 mm Tris buffer (pH 7.4) in the absence or presence of 2,2,2-trifluoroethanol (TFE) were subjected to CD measurements in a 2-mm quartz cuvette. For the mixture with egg PC SUV, the apoA-I protein or peptide was incubated with SUV for 1 h prior to the measurement. The results were corrected by subtracting the baseline for an appropriate blank sample. The α-helix content was derived from the molar ellipticity at 222 nm ([θ]222) using the equation: % α-helix = [(−[θ]222 + 3,000)/(36,000 + 3,000)] × 100 (43).
Isothermal Titration Calorimetry (ITC) Measurements
Heats of binding of apoA-I variants or peptides to SUV were measured with a VP-ITC instrument (GE Healthcare) at 25 °C. All solutions were degassed under vacuum before use. The SUV suspension was placed in the sample cell (1.33 ml) and titrated with 10-μl aliquots of the apoA-I with continual stirring at 400 rpm. Heats of dilution were determined in control experiments by injecting apoA-I solution into buffer, and the heats were subtracted from the heats determined in the corresponding apoA-I-SUV binding experiments to give the enthalpy of binding. The ITC results were fitted to the one-site binding model in Origin 7.1 (MicroCal) to obtain thermodynamic parameters of binding.
Fluorescence Measurements
Fluorescence measurements were carried out with a F-4500 or F-7000 fluorescence spectrophotometer (Hitachi) and an Fmax fluorescence plate reader (Molecular Devices) at 25 °C. To access the local environment of apoA-I single Trp variants or peptides, Trp emission fluorescence was recorded from 300 to 420 nm using a 290-nm excitation wavelength to avoid tyrosine fluorescence. In quenching experiments of Trp fluorescence, Trp emission spectra were recorded at increasing concentrations of KI (0–0.56 m) using a 5 m stock solution containing 1 mm Na2S2O3 to prevent the formation of iodine. Acrylodan emission fluorescence was collected from 380 to 600 nm using a 360-nm excitation wavelength.
Kinetics of aggregation and fibril formation of protein or peptide were monitored using the fluorescent dye, thioflavin T (ThT). ApoA-I 1–83 variants or 8–33 peptides (0.1 mg/ml) in 10 mm Tris buffer (pH 7.4) were incubated at 37 °C with agitation on an orbital rotator or microplate shaker in the presence of 10 μm ThT. ThT fluorescence was recorded at 485 nm with an excitation wavelength of 445 nm. The time-dependent increase in ThT fluorescence intensity was fitted to sigmoidal equation (44, 45),
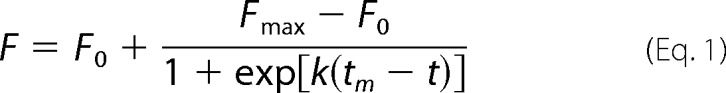 |
where F is the fluorescence intensity, F0 is the initial base line during the lag phase, and Fmax is the final base line after the growth phase has ended (maximal fluorescence). k is the apparent rate constant for the growth of fibrils and tm is the time to 50% of maximal fluorescence. The lag time is calculated as tm − 2/k.
Transmission Electron Microscopy (TEM)
A 2-μl droplet of the sample suspension was placed on a glow-discharged copper grid (300 mesh) coated with carbon, and then a 20-μl droplet of 2% phosphotungstic acid solution (adjusted at pH 7.4 with NaOH) was added. After excess staining solution was blotted with a filter paper after 1 min incubation, the grid was dried in vacuo. TEM images were obtained at 120 kV on an FEI Tecnai F20 transmission electron microscope.
Total Internal Reflection Fluorescence Microscopy (TIRFM)
To observe individual amyloid fibrils in sample solution (50 μm apoA-I), a fluorescence microscopic system based on an inverted microscope (IX70; Olympus, Tokyo, Japan) was used. ThT was excited using an argon laser (model 185F02-ADM; Spectra Physics, Mountain View, CA), and the fluorescent image was filtered with a bandpass filter (D490/30 Omega Optical, Brattleboro, VT) and visualized using an image intensifier (model VS4–1845; Video Scope International, Sterling, VA) coupled with a SIT camera (C2400-08; Hamamatsu Photonics, Shizuoka, Japan).
Results
Effects of Membrane Binding on Fibril Formation of ApoA-I 1–83 Variants
We first assessed the fibril-forming properties of apoA-I 1–83 variants in the absence or presence of egg PC SUV using the amyloidophilic dye, ThT. As shown in Fig. 1, A and B, the 1–83 variants of apoA-I in solution (0.1 mg/ml) exhibited significant increases in ThT fluorescence in a time-dependent manner, in which the G26R mutation greatly reduces the lag time for fibril formation (31 and 5.5 h for 1–83 and 1–83/G26R, respectively) (19). Such time-dependent development of ThT fluorescence for the apoA-I 1–83 gradually decreased with increasing concentrations of SUV, and that essentially no increase in ThT fluorescence was observed at PC to apoA-I weight ratio of 30 (Fig. 1A). Even in extensive incubations over 600 h, no significant increase in ThT fluorescence occurred for the 1–83 fragment at this PC/apoA-I ratio (data not shown). In contrast, the 1–83/G26R variant still exhibited significant increases in ThT fluorescence in the presence of SUV with increased lag phase (32 h at PC/apoA-I = 30, w/w) (Fig. 1B).
FIGURE 1.

Effects of membrane binding on fibril formation of apoA-I 1–83 variants. A and B, fibril formation was monitored by ThT fluorescence for apoA-I 1–83 (A) and 1–83/G26R (B) incubated at pH 7.4 in the absence or presence of increasing concentrations of egg PC SUV. The protein concentration was 0.1 mg/ml. a.u., arbitrary units. C, effects of protein concentration on the ThT fluorescence of apoA-I 1–83/G26R in the absence (dotted lines) or presence (dashed lines) of egg PC SUV. The PC/apoA-I weight ratio was 30. D, effects of the PC to apoA-I ratio on ThT fluorescence intensities for apoA-I 1–83/G26R incubated in the presence of egg PC SUV. Inset shows lag time of kinetics of ThT fluorescence intensities. The protein concentration was 0.05 mg/ml. N.D., not determined. E, TEM and TIRFM images of apoA-I 1–83 variants after 120 h incubation at pH 7.4 in the absence or presence of egg PC SUV. Scale bars of TEM and TIRFM represent 100 nm and 50 μm, respectively.
Fibril formation by the 1–83/G26R variant was significantly reduced by lowering the protein concentration to 50 μg/ml, whereas relatively small decreases in ThT fluorescence were observed for the 1–83/G26R on SUV (PC/apoA-I = 30, w/w) at the same protein concentration (Fig. 1C). The enhancing effect of SUV on fibril formation of apoA-I 1–83/G26R at 50 μg/ml depended upon the PC to apoA-I ratio, with there being an optimal SUV concentration for fibril formation (Fig. 1D), as reported for α-synuclein aggregation on SUV (35). At low PC/apoA-I ratios (≤10, w/w), a high local concentration of apoA-I on the SUV surface compared with that in bulk solution would promote protein aggregation and fibril formation. However, as the SUV concentration increases (PC/apoA-I ratios >10, w/w), fibril formation by the 1–83/G26R decreases likely due to dilution of apoA-I on the SUV surface as well as depletion of lipid-free apoA-I in solution. As a result, the lag time increased for low PC/apoA-I ratios (≤10, w/w) where nucleation occurs both on the SUV surface and in solution, and reached a maximal value when nucleation occurs only on the SUV surface (Fig. 1D, inset). Taken together, these results suggest that fibril formation by apoA-I 1–83/G26R on the SUV surface occurs by a different mechanism from that in bulk solution (35, 36).
Fig. 1E shows TEM and TIRFM images of apoA-I 1–83 variants after 120 h incubation in the absence or presence of SUV. Both apoA-I 1–83 and 1–83/G26R formed ThT active, straight fibrils in solution. In contrast, only the 1–83/G26R variant formed ThT active, straight fibrils in the presence of excess SUV. Interestingly, the 1–83 variant appeared to cause membrane budding or tubulation of vesicles after incubation instead of fibril formation, as reported for full-length apoA-I (46) and α-synuclein (32, 47). No fibrils were observed by TEM for the apoA-I 1–83 incubated with SUV over 240 h (data not shown). These results indicate that lipid binding strongly inhibits fibril formation by the amyloidogenic apoA-I 1–83 fragment (39), whereas the G26R mutant still retains the ability to form fibrils on lipid membranes.
Binding Properties of ApoA-I 1–83 Variants to Egg PC SUV
To compare the binding properties of apoA-I 1–83 variants to the SUV surface, we performed far-UV CD and ITC measurements. Dynamic light scattering measurements demonstrated that there was no change in size distribution of SUV upon binding of apoA-I (data not shown). Far-UV CD spectra of apoA-I 1–83/G26R with increasing concentrations of SUV (Fig. 2A) demonstrated a structural transition from predominantly random coil to α-helix upon SUV binding. The α-helix content derived from the CD spectra increases with increasing SUV PC to the apoA-I ratio; an increase in the α-helix content is much less for the 1–83/G26R variant than for the 1–83 variant, and even at high PC/apoA-I ratios, the α-helix contents of apoA-I 1–83/G26R are significantly smaller than those of apoA-I 1–83 (Fig. 2B). This suggests that the G26R mutation decreases the propensity of the apoA-I 1–83 fragment to form a α-helical structure upon lipid binding.
FIGURE 2.

Comparison of binding behaviors of apoA-I 1–83 variants to egg PC SUV. A, far-UV CD spectra of apoA-I 1–83/G26R bound to egg PC SUV. The protein concentration was 50 μg/ml. B, increases in α-helix content of apoA-I 1–83 (●) and 1–83/G26R (Δ) as a function of the weight ratio of PC to apoA-I. C, isothermal titration thermogram for binding of apoA-I 1–83/G26R to egg PC SUV. D, changes in the fraction % of apoA-I bound to SUV (● and ▴) and molar ratio of bound apoA-I to PC on the SUV surface (○ and Δ) for apoA-I 1–83 (dashed line) and 1–83/G26R (dotted line) with increasing weight ratio of PC to apoA-I. Fraction % of apoA-I bound to SUV were derived from Kd and Bmax values. Molar ratios of bound apoA-I to PC on the SUV surface were derived assuming that surface PC is located on the outer leaflet of SUV available for apoA-I binding is 67% of total PC.
Fig. 2C shows a typical titration curve for binding of apoA-I 1–83 variants to SUV. As listed in Table 1, the obtained parameters indicate that the 1–83 variants bind to SUV with high affinity similarly to full-length apoA-I (37), and the G26R mutation reduces the binding capacity. In addition, binding of the 1–83 variants to the SUV surface is an enthalpically favorable but entropically unfavorable process (37, 48).
TABLE 1.
Thermodynamic parameters of binding of apoA-I variants or peptides to egg PC SUV at 25 °C
| Kd | Bmax | ΔGa | ΔH | TΔSb | |
|---|---|---|---|---|---|
| μg/ml | amino acids/mol PC | kcal/mol | |||
| ApoA-I 1–83 | 3.8 ± 0.3 | 0.34 ± 0.01 | −11.1 ± 0.1 | −23.7 ± 1.9 | −12.6 ± 1.9 |
| ApoA-I 1–83/G26R | 1.7 ± 0.1 | 0.15 ± 0.01 | −11.6 ± 0.1 | −20.3 ± 0.8 | −8.7 ± 0.8 |
| ApoA-I 1–83/Y18P/G26R | 1.7 ± 0.2 | 0.083 ± 0.01 | −11.6 ± 0.1 | −19.8 ± 0.2 | −8.2 ± 0.2 |
| ApoA-I 8–33 | 2.1 ± 0.1 | 0.059 ± 0.01 | −10.8 ± 0.1 | −20.2 ± 0.4 | −9.4 ± 0.4 |
| ApoA-I 8–33/G26R | 2.7 ± 0.2 | 0.042 ± 0.01 | −10.7 ± 0.1 | −13.7 ± 0.1 | −3.0 ± 0.1 |
a Free energy was calculated according to ΔG = −RT ln 55.5(1/Kd).
b The entropy of binding was calculated from ΔG = ΔH − TΔS.
Using the dissociation constant, Kd, and the maximal binding capacity, Bmax, values for the SUV binding of the 1–83 variants listed in Table 1, we can estimate the fraction and amount of apoA-I bound to SUV at different SUV PC/apoA-I ratios. Fig. 2D shows percentages of apoA-I bound to SUV (closed circles and triangles) and surface concentrations of apoA-I on SUV (open circles and triangles) with increasing concentrations of SUV. At low PC/apoA-I ratios (≤10, w/w), SUV surfaces are saturated with apoA-I 1–83 variants and a significant population of lipid-free protein exists in solution in equilibrium with the SUV-bound state. In contrast, at high PC/apoA-I ratios (>30, w/w), both apoA-I 1–83 variants bind to SUV similarly in which the fraction of SUV-bound apoA-I becomes close to 100%. Thus, the distinct behaviors in the fibril formation of the 1–83 variants in the presence of SUV (Fig. 1, A and B) are not likely to be due to the differences in the amount of apoA-I bound to the SUV surface, but rather due to different conformations of the 1–83 variants bound to SUV.
Membrane-binding and Fibril-forming Properties of ApoA-I 8–33 and 8–33/G26R Peptides
To further evaluate the effects of the G26R mutation on the lipid-binding and fibril forming properties of the N-terminal residues of apoA-I, we used the apoA-I 8–33 fragment because this peptide contains a high lipid affinity region that forms α-helical structure upon lipid binding (49) as well as a strong fibril-forming region (20). The helical wheel diagram in Fig. 3A indicates that the G26R substitution places the strongly basic arginine residue into the nonpolar face of the amphipathic α-helix, destabilizing the helical structure (18, 19).
FIGURE 3.

Membrane-binding and fibril-forming properties of apoA-I 8–33 and 8–33/G26R peptides. A, helical wheel projection of residues 16–33 of apoA-I with the mutated position 26 on the nonpolar face of the amphipathic α-helix. B, isothermal titration thermogram for binding of apoA-I 8–33/G26R to egg PC SUV. C, far-UV CD spectra of apoA-I 8–33 and 8–33/G26R peptides in Tris buffer (a) or bound to egg PC SUV (PC = 3.0 mg/ml) (b and c). The peptide concentration was 50 μg/ml. D, α-helix contents of apoA-I 8–33 and 8–33/G26R bound to egg PC SUV at various weight ratios of PC to peptide. E and F, fibril formation was monitored by ThT fluorescence for apoA-I 8–33 (●) and 8–33/G26R (Δ) incubated at pH 7.4 in the absence (E) or presence (F) of egg PC SUV. a.u., arbitrary units. Peptide and PC concentrations were 0.1 and 6.0 mg/ml, respectively. G, TIRFM images of apoA-I 8–33 peptides after 120 h incubation at pH 7.4 in the absence or presence of egg PC SUV. Scale bars of TIRFM represent 50 μm.
ITC measurements of binding of apoA-I 8–33 peptides to SUV (Fig. 3B) indicated that both the 8–33 and 8–33/G26R peptides bind to SUV with similar affinity but reduced binding capacity, compared with the corresponding apoA-I 1–83 variants (Table 1). Significantly less exothermic heat in binding of the 8–33/G26R peptide compared with that of the 8–33 peptide suggests that the G26R mutation lowers the helical propensity of apoA-I 8–33 upon binding to SUV (37). Indeed, CD measurements demonstrated that the 8–33 and 8–33/G26R peptides undergo the conformational transition from random coil to α-helix upon binding to SUV, with the G26R mutation reducing the propensity to form α-helix (Fig. 3C). Comparison of the α-helical contents of the 8–33 and 8–33/G26R peptides with increasing ratios of SUV PC to peptide clearly shows that the 8–33/G26R peptide has less ability to form α-helix upon lipid binding compared with the 8–33 peptide (Fig. 3D). These results indicate that the G26R mutation impairs α-helical formation of 8–33 residues, perhaps resulting in the different helical conformation of the N-terminal region of apoA-I on the membrane surface. Such a different helical conformation is expected to modify fibril formation for amyloidogenic proteins (30, 31, 50).
To test this idea, we investigated the effects of lipid binding on the fibril-forming properties of apoA-I 8–33 and 8–33/G26R peptides by ThT fluorescence assay. As shown in Fig. 3E, both 8–33 peptides exhibited significant increases in ThT fluorescence upon incubation in solution, with the G26R mutation enhancing fibril formation of apoA-I 8–33 peptide (20). Similarly to the case of apoA-I 1–83 variants, the increase in ThT fluorescence of the 8–33 peptide was strongly inhibited with binding to SUV, whereas the 8–33/G26R still exhibited a large increase in ThT fluorescence upon incubation with SUV (Fig. 3F). In addition, TIRFM measurements demonstrated that in the presence of an excess of SUV, fibril formation was observed only for the 8–33/G26R peptide despite both 8–33 and 8–33/G26R peptides having a strong ability to form fibrils in solution (Fig. 3G). These results indicate that the enhancing effect of the G26R mutation on fibril formation by apoA-I on lipid membranes comes from the local perturbation of the amyloidogenic region located in residues 8–33 of apoA-I.
Evaluation of Membrane-bound States of ApoA-I 1–83 and 8–33 Variants Using Trp or Acrylodan Fluorescence
To probe the conformational differences of amyloidogenic regions in the N-terminal residues of apoA-I on lipid membranes, we monitored Trp or acrylodan fluorescence of apoA-I 1–83 variants. In single Trp variants, W@8, W@50, and W@72 of the apoA-I 1–83 fragment, intrinsic Trp residues (Trp8, Trp50, and Trp72) were substituted to Phe to generate single Trp variants (19). For attachment of acrylodan to apoA-I 1–83 variants, we introduced a Cys mutation, L22C, because Leu22 is located in the most aggregation prone segment of the N-terminal 1–83 residues of apoA-I (20, 21, 51).
Fig. 4A shows Trp fluorescence emission spectra of apoA-I 1–83 W@8 in buffer solution or bound to SUV. A significant blue shift in wavelength of maximum fluorescence (WMF) with increased fluorescence intensity upon SUV binding is consistent with the transfer of Trp8 into a more hydrophobic lipid environment. Comparison of WMF among three single Trp mutants of apoA-I 1–83/G26R (Fig. 4B) indicates that Trp50 is also involved in the conformational transition upon binding to SUV like Trp8, whereas the region around Trp72 is unlikely to contribute to the lipid binding because of no change in WMF. We also examined the effects of the G26R mutation on Trp environments of Trp8 and Trp50 using apoA-I 1–83 single Trp variants bound to SUV. Higher WMF values in both the W@8 and W@50 variants of apoA-I 1–83/G26R than those for the corresponding 1–83 variants on the SUV surface (Fig. 4C) indicate that Trp residues at positions 8 and 50 in the G26R variant are more exposed to the solvent compared with the 1–83 variant. Consistent with this finding, a higher WMF value and KI quenching ratio (parameters of solvent accessibility of Trp8) for the G26R variant were also seen in apoA-I 8–33 peptides (Fig. 4D).
FIGURE 4.

Effects of membrane binding on Trp or acrylodan fluorescence of apoA-I 1–83 variants and 8–33 peptides. A, Trp fluorescence spectra of apoA-I 1–83 W@8 (50 μg/ml) in the absence (solid line) or presence (dashed line) of SUV (1.5 mg/ml of PC). The excitation wavelength was 290 nm. a.u., arbitrary units. B, change in WMF of apoA-I 1–83/G26R single Trp variants by binding to SUV. C, comparison of WMF of W@8 and W@50 variants of apoA-I 1–83 bound to SUV. D, WMF and quenching ratio at 0.56 m KI as a parameter of solvent accessibility for apoA-I 8–33 and 8–33/G26R peptides bound to SUV. E, acrylodan fluorescence spectra of apoA-I 1–83/L22C-Ac (acrylodan) (50 μg/ml) in the absence (solid line) or presence (dashed line) of SUV (1.5 mg/ml PC). F, changes in WMF of apoA-I 1–83/L22C-acrylodan and 1–83/L22C-acrylodan/G26R by binding to SUV.
Fig. 4E shows acrylodan fluorescence emission spectra of apoA-I 1–83/L22C-acrylodan in buffer solution and bound to SUV. Similarly to Trp fluorescence, a significant blue shift in WMF with increased fluorescence intensity upon SUV binding was observed, indicating the transfer of the acrylodan molecule into a more hydrophobic lipid environment (41, 52). Comparison of WMF of the 1–83 L22C-acrylodan variants (Fig. 4F) further indicates that the acrylodan molecule attached at position 22 in the G26R variant is more exposed to the solvent compared with the corresponding 1–83 variant. Taken together, these results suggest that the G26R mutation induces more solvent-exposed conformation of the amyloidogenic regions in the N-terminal fragment of apoA-I on lipid membranes.
Effects of TFE on Fibril Formation of ApoA-I 1–83 Variants
Formation of α-helical structure has been reported to promote fibril formation of a number of amyloidogenic proteins (31, 53). Interestingly, a partially folded, helical intermediate structure contributes to the acceleration of fibril growth of protein, whereas over-stabilization of a helical structure prevents fibril formation (54–56). Besides membrane binding, TFE is also known to induce partially helical intermediate conformations of protein, thereby promoting amyloid fibril formation (57–59). Consequently, we examined the fibril-forming properties of apoA-I 1–83 variants in the presence of TFE, mimicking membrane-induced conformational changes to some extent.
Far-UV CD spectra of apoA-I 1–83/G26R in the presence of increasing concentrations of TFE demonstrate that TFE induces the transition of the secondary structure of the 1–83/G26R from a random coil to α-helical structure (Fig. 5A). Such effects of TFE in inducing the helical transition were similar between the 1–83 and 1–83/G26R variants (Fig. 5B). It should be noted that the absence of a well defined isodichroic point in CD spectra (Fig. 5A) suggests a non-two-state transition of the secondary structure across the range of TFE concentrations, consistent with the results of thermal unfolding of full-length apoA-I (60). By plotting the mean residue ellipticities at 198 nm against 222 nm from CD spectra, we constructed transition diagrams for the 1–83 variants (Fig. 5C) to identify the helical intermediate conformation (57, 58). The observation of nonlinearity reflects multiple secondary structure transitions; the two intersecting lines shown in Fig. 5C indicate that the 1–83 variants adopt unfolded (0% TFE), well folded (30–40% TFE), and intermediate conformations (around 10–14% TFE).
FIGURE 5.

Effects of TFE on fibril formation by apoA-I 1–83 variants. A, far-UV CD spectra of apoA-I 1–83/G26R in 0–40% TFE. The protein concentration was 50 μg/ml. B, increases in α-helix content of apoA-I 1–83 (●) and 1–83/G26R (Δ) as a function of TFE %. C, transition diagram obtained from the ellipticity values at 222 and 198 nm in the far-UV CD spectra for apoA-I 1–83/G26R. D, effect of TFE concentration on ThT fluorescence of apoA-I 1–83 variants. ApoA-I 1–83 in 10% TFE (○), apoA-I 1–83/G26R in 10% TFE (Δ) or 30% TFE (▾). The protein concentration was 0.1 mg/ml. a.u., arbitrary units.
Next, we evaluated the kinetics of fibril formation of the 1–83 variants under different secondary structure conformations (at 10 or 30% TFE) using ThT fluorescence (Fig. 5D). At a intermediate helical conformation (10% TFE), fibril formation of the 1–83 fragment was strongly inhibited, whereas there were great increases in ThT fluorescence for the 1–83/G26R variant. With a well folded helical conformation (30% TFE), in contrast, the 1–83/G26R variant exhibited no significant increase in ThT fluorescence over time. These results indicate that although the TFE-induced helix formation strongly inhibits β-transition and fibril formation by the 1–83 variants of apoA-I, the G26R variant has an ability to form fibrils when it has a partially helical intermediate conformation.
Effects of Y18P Mutation on Fibril-forming Property of ApoA-I 1–83/G26R
To probe the contribution of the first amyloidogenic region (residues 14–22) in the N-terminal 1–83 fragment of apoA-I to the fibril-forming ability of the 1–83/G26R variant on SUV, we introduced a Y18P point mutation. Tyr-18 is located at the center of the amyloidogenic segment containing very hydrophobic amino acids (14LATVYVDVL22), and the Y18P substitution was shown to cause strong inhibition of fibril formation by this region in solution (20).
Far-UV CD measurements demonstrated that the Y18P mutation does not significantly alter the helix-forming properties of apoA-I 1–83/G26R upon binding to SUV (Fig. 6A) and in 10% TFE solution (Fig. 6B). ITC measurements of binding of apoA-I 1–83/Y18P/G26R to SUV indicated that this variant can avidly bind to SUV with similar affinity to the 1–83/G26R variant, although the Y18P mutation reduces the binding capacity of apoA-I 1–83/G26R (Table 1). As shown in Fig. 6C, the kinetics of fibril formation of the 1–83/Y18P/G26R variant assessed by ThT fluorescence revealed that this variant forms fibrils in solution with markedly increased lag time (61 h) compared with that for the 1–83/G26R variant (5.5 h). SUV binding strongly inhibited fibril formation by apoA-I 1–83/Y18P/G26R even at a low PC/apoA-I ratio. Such inhibitory effects of the Y18P mutation on the fibril-forming properties of apoA-I 1–83/G26R were also seen in 10% TFE solution (Fig. 6D). These results indicate that the amyloidogenic region around Tyr18 is involved in the fibril-forming ability of apoA-I 1–83/G26R on lipid membranes.
FIGURE 6.

Effects of Y18P/G26R double mutation on fibril formation by apoA-I 1–83. A, increases in α-helix content of apoA-I 1–83/Y18P/G26R (○) as a function of the weight ratio of PC to apoA-I in comparison with 1–83 (●) and 1–83/G26R (Δ). B, effect of TFE on α-helix contents of apoA-I 1–83, 1–83/G26R, and 1–83/Y18P/G26R in solution. C, fibril formation was monitored by ThT fluorescence for apoA-I 1–83/Y18P/G26R incubated at pH 7.4 in the absence or presence of increasing concentrations of egg PC SUV. The data of apoA-I 1–83/G26R in solution (dotted line) is also shown for comparison. PC/apoA-I weight ratios were 0 (○), 10 (Δ), and 30 (■). The protein concentration was 0.1 mg/ml. a.u., arbitrary units. D, ThT fluorescence of apoA-I 1–83/Y18P/G26R in 10% TFE (●). The data of apoA-I 1–83/G26R (dotted line) are also shown for comparison. The protein concentration was 0.1 mg/ml.
Discussion
The results of the present study demonstrate that lipid binding plays a crucial role in fibril formation by the N-terminal fragment of apoA-I. In the lipid-free state, apoA-I 1–83 has a predominantly random coil structure that is highly susceptible to a transition to β-aggregate structure (19). In contrast, lipid binding stabilizes the α-helical structure thereby strongly inhibiting fibril formation by the protein (Figs. 1 and 2). Similar inhibitory effects of lipid environments on fibril formation were also reported in an apoA-I fragment (39) or peptides (61) and apolipoprotein C-II (62); in these cases binding to phospholipid membranes induces the transformation of unfolded protein to α-helix structure, thereby preventing the fibrillogenic process. As with other natively unstructured amyloidogenic proteins such as islet amyloid polypeptide (56) and α-synuclein (54), it is conceivable that stabilization of the α-helical state upon membrane binding generates a kinetic trap in transition to the β-sheet structure, thereby inhibiting amyloid fibril formation.
A significant finding of this study is that the amyloidogenic G26R mutation promotes fibril formation by the amyloidogenic region in the N-terminal fragment of apoA-I on the membrane surface. This effect occurs because partially destabilized α-helical conformations on lipid membranes are precursors of protein aggregation (30, 31). Far-UV CD analyses of apoA-I 1–83 fragments (Fig. 2, A and B) and 8–33 peptides (Fig. 3, C and D) indicate that the G26R mutation impairs the α-helical transition of the N-terminal lipid-binding region of apoA-I. Significant reduction of the exothermic heats of apoA-I upon lipid binding by the G26R mutation, especially seen in apoA-I 8–33 peptides (Table 1) further demonstrates the lower helical propensity of the G26R variants of apoA-I. Similar acceleration of β-aggregation via helical intermediate states was reported in the N-terminal 1–93 fragment of apoA-I under acidic conditions (15, 63). In many amyloidogenic proteins, it is known that membrane-mediated misfolding and promotion of fibril formation proceed via partially folded, helical intermediate states (64–67). Thus, it is likely that the transformation of transient α-helical intermediates to the β-sheet structure is a general pathway in the membrane-induced fibril formation of natively unfolded amyloidogenic proteins (31, 53). The balance of whether the membrane binding favors or disfavors fibril formation appears to depend on the level of stabilization of the α-helical structure (55, 56, 68).
Many amyloidogenic mutations including G26R are located in α-helical segments of the N-terminal region of apoA-I that place proline or charged residues in the nonpolar face of an amphipathic α-helix (10). Thus, it is thought that these mutations destabilize the helix bundle structure of apoA-I (18, 19), thereby promoting proteolysis. In addition to such destabilizing effects, we speculate that the amyloidogenic mutation in apoA-I may accelerate fibril formation of the proteolytic N-terminal fragment bound to cell membranes by promoting partially α-helical states. Consistent with this concept, although the TFE-induced helix formation strongly inhibits β-transition and fibril formation by the 1–83 variants of apoA-I, the G26R variant has an ability to form fibrils at a partially helical intermediate conformation (Fig. 5). The mechanism by which the 1–83/G26R variant forms fibrils at the TFE-induced partially helical conformations is unclear, but it is plausible that the G26R mutation promotes fibril formation by the apoA-I 1–83 fragment not only by inducing partially destabilized α-helical conformation but also by enhancing the nucleation step of fibril formation (19). Interestingly, it was reported that TFE-induced oligomerization and fibrillization behavior varied significantly between PD mutants of α-synuclein despite their secondary structural conformations being similar (58). In addition, it was recently shown that certain familial α-synuclein mutations cause impaired membrane interactions and an exposed, membrane-bound conformer of α-synuclein, leading to a greater propensity to undergo the membrane-induced formation of neurotoxic aggregates (69).
Estimation of the β-aggregation propensity (70) of the N-terminal 1–83 residues in apoA-I based on amino acid sequence indicates that residues 14–22 and 49–57 are predicted to be the most aggregation prone regions (21, 51, 71). Combined with the secondary structure examination of the apoA-I G26R variant (13) and a finding that a peptide comprising residues 46–59 of apoA-I aggregates to form amyloid-like fibrils (72), it is considered that residues 14–31 and 46–59 in the apoA-I G26R variant are highly amyloidogenic regions that undergo the transition to the β-strand structure. Interestingly, the first amyloidogenic region overlaps with a very hydrophobic segment in the hydropathy plot (Fig. 7A), implying the high lipid-binding ability of this region (73).
FIGURE 7.

Model of fibril formation of the N-terminal fragment of apoA-I mutant on lipid membranes. A, β-aggregation propensity and hydropathy of the N-terminal 1–83 residues of apoA-I. Residues 14–31 (13) and 46–59 (72), which are predicted to be β-aggregation prone regions, are shown as arrows. B, I, apoA-I 1–83/G26R has a predominantly random coil structure in solution. II, membrane binding induces the formation of α-helical structure probably in two regions including Trp8 and Trp50, respectively. α-Helix is depicted as cylinders. The possible formation of the α-helical structure around residue 26 (dotted cylinder) is inhibited by the G26R mutation. III and IV, association of apoA-I 1–83/G26R in partially helical states induces a high local concentration of the highly amyloidogenic region including Arg26, leading to the transition to β-strand structure and acceleration of the intermolecular aggregation. The β-structure is shown as arrows.
Fig. 7B presents a proposed model of fibril formation by the N-terminal 1–83/G26R fragment of apoA-I on lipid membranes, which is reminiscent of the membrane-mediated aggregation model for human islet amyloid polypeptide (65, 66). ApoA-I 1–83/G26R has predominantly a random coil structure in solution (step I), but membrane binding induces the formation of α-helical structure probably in two regions including Trp8 and Trp50, respectively (5, 74) (step II). In this step, formation of the α-helical structure around residue 26 is inhibited by the G26R mutation (18). Association of apoA-I 1–83/G26R in partially α-helical conformations induces high local concentrations of the highly amyloidogenic region including Arg26 (step III), leading to the transition to β-strand structure and acceleration of the intermolecular aggregation (step IV), and subsequent formation of mature amyloid fibrils. Consistent with this model, the finding that substitution of Tyr18 located at the center of this highly amyloidogenic as well as hydrophobic region with a proline residue strongly inhibited fibril formation of apoA-I 1–83/G26R on SUV (Fig. 6C) implies the important role of the first amyloidogenic region on fibril formation by apoA-I 1–83/G26R on lipid membranes.
In summary, we have demonstrated for the first time that the amyloidogenic G26R mutation facilitates fibril formation of the N-terminal fragment of apoA-I on membrane surfaces through partially helical conformations. Because many other amyloidogenic mutations in apoA-I are located in α-helical segments of the N-terminal region (10), it is likely that partially α-helical conformations that form upon association with lipid membranes are physiologically relevant conformations of mutated apoA-I in fibril formation. Biological membranes represent an abundant surface with the potential to serve as a template for amyloid formation (28). The present findings suggest a new pathway involving cell membrane or lipoprotein surfaces for amyloid fibril formation by the N-terminal fragment of amyloidogenic apoA-I variants.
Author Contributions
C. M. and F. O. performed the experiments. T. B. and T. S. recorded the TEM and TIRFM images, respectively. S. M., K. T., A. S., and A. O. designed and synthesized peptides. C. M., K. O., and H. S. designed the study, and C. M. and H. S. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
TEM observations were performed by Drs. K. Yan and K. Hirose (EM team, Tsukuba Innovation Arena, AIST, Tsukuba), partly supported by IBEC Innovation Platform, AIST. We are indebted to Dr. Michael C. Phillips (University of Pennsylvania) and Dr. Galyna Gorbenko (Kharkov National University) for valuable advice.
This work was supported in part by Grant-in-Aid for Scientific Research 25293006 and 25670014 (to H. S.) from Japan Society for the Promotion of Science, and the research program for development of intelligent Tokushima artificial exosome (iTEX) from Tokushima University. The authors declare that they have no conflicts of interest with the contents of this article.
- apoA-I
- apolipoprotein A-I
- ITC
- isothermal titration calorimetry
- SUV
- small unilamellar vesicle
- TEM
- transmission electron microscopy
- TFE
- 2,2,2-trifluoroethanol
- ThT
- thioflavin T
- TIRFM
- total internal reflection fluorescence microscopy
- PC
- phosphatidylcholine
- WMF
- wavelength of maximum fluorescence.
References
- 1. Rosenson R. S., Brewer H. B., Jr., Davidson W. S., Fayad Z. A., Fuster V., Goldstein J., Hellerstein M., Jiang X. C., Phillips M. C., Rader D. J., Remaley A. T., Rothblat G. H., Tall A. R., Yvan-Charvet L. (2012) Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation 125, 1905–1919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rader D. J., Alexander E. T., Weibel G. L., Billheimer J., Rothblat G. H. (2009) The role of reverse cholesterol transport in animals and humans and relationship to atherosclerosis. J. Lipid Res. 50, S189–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Phillips M. C. (2014) Molecular mechanisms of cellular cholesterol efflux. J. Biol. Chem. 289, 24020–24029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Davidson W. S., Hazlett T., Mantulin W. W., Jonas A. (1996) The role of apolipoprotein AI domains in lipid binding. Proc. Natl. Acad. Sci. U.S.A. 93, 13605–13610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Saito H., Dhanasekaran P., Nguyen D., Holvoet P., Lund-Katz S., Phillips M. C. (2003) Domain structure and lipid interaction in human apolipoproteins A-I and E, a general model. J. Biol. Chem. 278, 23227–23232 [DOI] [PubMed] [Google Scholar]
- 6. Mei X., Atkinson D. (2011) Crystal structure of C-terminal truncated apolipoprotein A-I reveals the assembly of high density lipoprotein (HDL) by dimerization. J. Biol. Chem. 286, 38570–38582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sorci-Thomas M. G., Thomas M. J. (2002) The effects of altered apolipoprotein A-I structure on plasma HDL concentration. Trends Cardiovasc. Med. 12, 121–128 [DOI] [PubMed] [Google Scholar]
- 8. Frank P. G., Marcel Y. L. (2000) Apolipoprotein A-I: structure-function relationships. J. Lipid Res. 41, 853–872 [PubMed] [Google Scholar]
- 9. Rowczenio D., Dogan A., Theis J. D., Vrana J. A., Lachmann H. J., Wechalekar A. D., Gilbertson J. A., Hunt T., Gibbs S. D., Sattianayagam P. T., Pinney J. H., Hawkins P. N., Gillmore J. D. (2011) Amyloidogenicity and clinical phenotype associated with five novel mutations in apolipoprotein A-I. Am. J. Pathol. 179, 1978–1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gursky O., Mei X., Atkinson D. (2012) The crystal structure of the C-terminal truncated apolipoprotein A-I sheds new light on amyloid formation by the N-terminal fragment. Biochemistry 51, 10–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nichols W. C., Dwulet F. E., Liepnieks J., Benson M. D. (1988) Variant apolipoprotein AI as a major constituent of a human hereditary amyloid. Biochem. Biophys. Res. Commun. 156, 762–768 [DOI] [PubMed] [Google Scholar]
- 12. Rader D. J., Gregg R. E., Meng M. S., Schaefer J. R., Zech L. A., Benson M. D., Brewer H. B., Jr. (1992) In vivo metabolism of a mutant apolipoprotein, apoA-IIowa, associated with hypoalphalipoproteinemia and hereditary systemic amyloidosis. J. Lipid Res. 33, 755–763 [PubMed] [Google Scholar]
- 13. Lagerstedt J. O., Cavigiolio G., Roberts L. M., Hong H. S., Jin L. W., Fitzgerald P. G., Oda M. N., Voss J. C. (2007) Mapping the structural transition in an amyloidogenic apolipoprotein A-I. Biochemistry 46, 9693–9699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Joy T., Wang J., Hahn A., Hegele R. A. (2003) APOA1 related amyloidosis: a case report and literature review. Clin. Biochem. 36, 641–645 [DOI] [PubMed] [Google Scholar]
- 15. Andreola A., Bellotti V., Giorgetti S., Mangione P., Obici L., Stoppini M., Torres J., Monzani E., Merlini G., Sunde M. (2003) Conformational switching and fibrillogenesis in the amyloidogenic fragment of apolipoprotein A-I. J. Biol. Chem. 278, 2444–2451 [DOI] [PubMed] [Google Scholar]
- 16. Obici L., Franceschini G., Calabresi L., Giorgetti S., Stoppini M., Merlini G., Bellotti V. (2006) Structure, function and amyloidogenic propensity of apolipoprotein A-I. Amyloid 13, 191–205 [DOI] [PubMed] [Google Scholar]
- 17. Arciello A., De Marco N., Del Giudice R., Guglielmi F., Pucci P., Relini A., Monti D. M., Piccoli R. (2011) Insights into the fate of the N-terminal amyloidogenic polypeptide of ApoA-I in cultured target cells. J. Cell Mol. Med. 15, 2652–2663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chetty P. S., Ohshiro M., Saito H., Dhanasekaran P., Lund-Katz S., Mayne L., Englander W., Phillips M. C. (2012) Effects of the iowa and milano mutations on apolipoprotein A-I structure and dynamics determined by hydrogen exchange and mass spectrometry. Biochemistry 51, 8993–9001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Adachi E., Nakajima H., Mizuguchi C., Dhanasekaran P., Kawashima H., Nagao K., Akaji K., Lund-Katz S., Phillips M. C., Saito H. (2013) Dual role of an N-terminal amyloidogenic mutation in apolipoprotein A-I: destabilization of helix bundle and enhancement of fibril formation. J. Biol. Chem. 288, 2848–2856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Adachi E., Kosaka A., Tsuji K., Mizuguchi C., Kawashima H., Shigenaga A., Nagao K., Akaji K., Otaka A., Saito H. (2014) The extreme N-terminal region of human apolipoprotein A-I has a strong propensity to form amyloid fibrils. FEBS Lett. 588, 389–394 [DOI] [PubMed] [Google Scholar]
- 21. Das M., Mei X., Jayaraman S., Atkinson D., Gursky O. (2014) Amyloidogenic mutations in human apolipoprotein A-I are not necessarily destabilizing - a common mechanism of apolipoprotein A-I misfolding in familial amyloidosis and atherosclerosis. FEBS J. 281, 2525–2542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Petrlova J., Bhattacherjee A., Boomsma W., Wallin S., Lagerstedt J. O., Irbäck A. (2014) Conformational and aggregation properties of the 1–93 fragment of apolipoprotein A-I. Protein Sci 23, 1559–1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wong Y. Q., Binger K. J., Howlett G. J., Griffin M. D. (2010) Methionine oxidation induces amyloid fibril formation by full-length apolipoprotein A-I. Proc. Natl. Acad. Sci. U.S.A. 107, 1977–1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chan G. K., Witkowski A., Gantz D. L., Zhang T. O., Zanni M. T., Jayaraman S., Cavigiolio G. (2015) Myeloperoxidase-mediated methionine oxidation promotes an amyloidogenic outcome for apolipoprotein A-I. J. Biol. Chem. 290, 10958–10971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gorbenko G. P., Kinnunen P. K. (2006) The role of lipid-protein interactions in amyloid-type protein fibril formation. Chem. Phys. Lipids 141, 72–82 [DOI] [PubMed] [Google Scholar]
- 26. Butterfield S. M., Lashuel H. A. (2010) Amyloidogenic protein-membrane interactions: mechanistic insight from model systems. Angew. Chem. Int. Ed. Engl. 49, 5628–5654 [DOI] [PubMed] [Google Scholar]
- 27. Axelsen P. H., Komatsu H., Murray I. V. (2011) Oxidative stress and cell membranes in the pathogenesis of Alzheimer's disease. Physiology 26, 54–69 [DOI] [PubMed] [Google Scholar]
- 28. Bucciantini M., Rigacci S., Stefani M. (2014) Amyloid aggregation: role of biological membranes and the aggregate-membrane system. J. Phys. Chem. Lett. 5, 517–527 [DOI] [PubMed] [Google Scholar]
- 29. Jayasinghe S. A., Langen R. (2007) Membrane interaction of islet amyloid polypeptide. Biochim. Biophys. Acta 1768, 2002–2009 [DOI] [PubMed] [Google Scholar]
- 30. Abedini A., Raleigh D. P. (2009) A critical assessment of the role of helical intermediates in amyloid formation by natively unfolded proteins and polypeptides. Protein Eng. Des. Sel. 22, 453–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hebda J. A., Miranker A. D. (2009) The interplay of catalysis and toxicity by amyloid intermediates on lipid bilayers: insights from type II diabetes. Annu. Rev. Biophys. 38, 125–152 [DOI] [PubMed] [Google Scholar]
- 32. Jiang Z., de Messieres M., Lee J. C. (2013) Membrane remodeling by α-synuclein and effects on amyloid formation. J. Am. Chem. Soc. 135, 15970–15973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Byström R., Aisenbrey C., Borowik T., Bokvist M., Lindström F., Sani M. A., Olofsson A., Gröbner G. (2008) Disordered proteins: biological membranes as two-dimensional aggregation matrices. Cell. Biochem. Biophys. 52, 175–189 [DOI] [PubMed] [Google Scholar]
- 34. Shen L., Adachi T., Vanden Bout D., Zhu X. Y. (2012) A mobile precursor determines amyloid-β peptide fibril formation at interfaces. J. Am. Chem. Soc. 134, 14172–14178 [DOI] [PubMed] [Google Scholar]
- 35. Galvagnion C., Buell A. K., Meisl G., Michaels T. C., Vendruscolo M., Knowles T. P., Dobson C. M. (2015) Lipid vesicles trigger α-synuclein aggregation by stimulating primary nucleation. Nat. Chem. Biol. 11, 229–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Terakawa M. S., Yagi H., Adachi M., Lee Y. H., Goto Y. (2015) Small liposomes accelerate the fibrillation of amyloid β(1–40). J. Biol. Chem. 290, 815–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Saito H., Dhanasekaran P., Nguyen D., Deridder E., Holvoet P., Lund-Katz S., Phillips M. C. (2004) α-Helix formation is required for high affinity binding of human apolipoprotein A-I to lipids. J. Biol. Chem. 279, 20974–20981 [DOI] [PubMed] [Google Scholar]
- 38. Tanaka M., Dhanasekaran P., Nguyen D., Ohta S., Lund-Katz S., Phillips M. C., Saito H. (2006) Contributions of the N- and C-terminal helical segments to the lipid-free structure and lipid interaction of apolipoprotein A-I. Biochemistry 45, 10351–10358 [DOI] [PubMed] [Google Scholar]
- 39. Monti D. M., Guglielmi F., Monti M., Cozzolino F., Torrassa S., Relini A., Pucci P., Arciello A., Piccoli R. (2010) Effects of a lipid environment on the fibrillogenic pathway of the N-terminal polypeptide of human apolipoprotein A-I, responsible for in vivo amyloid fibril formation. Eur. Biophys. J. 39, 1289–1299 [DOI] [PubMed] [Google Scholar]
- 40. Venkatachalapathi Y. V., Phillips M. C., Epand R. M., Epand R. F., Tytler E. M., Segrest J. P., Anantharamaiah G. M. (1993) Effect of end group blockage on the properties of a class A amphipathic helical peptide. Proteins 15, 349–359 [DOI] [PubMed] [Google Scholar]
- 41. Koyama M., Tanaka M., Dhanasekaran P., Lund-Katz S., Phillips M. C., Saito H. (2009) Interaction between the N- and C-terminal domains modulates the stability and lipid binding of apolipoprotein A-I. Biochemistry 48, 2529–2537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mizuguchi C., Hata M., Dhanasekaran P., Nickel M., Phillips M. C., Lund-Katz S., Saito H. (2012) Fluorescence analysis of the lipid binding-induced conformational change of apolipoprotein E4. Biochemistry 51, 5580–5588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sparks D. L., Lund-Katz S., Phillips M. C. (1992) The charge and structural stability of apolipoprotein A-I in discoidal and spherical recombinant high density lipoprotein particles. J. Biol. Chem. 267, 25839–25847 [PubMed] [Google Scholar]
- 44. Nielsen L., Khurana R., Coats A., Frokjaer S., Brange J., Vyas S., Uversky V. N., Fink A. L. (2001) Effect of environmental factors on the kinetics of insulin fibril formation: elucidation of the molecular mechanism. Biochemistry 40, 6036–6046 [DOI] [PubMed] [Google Scholar]
- 45. Zhou Z., Fan J. B., Zhu H. L., Shewmaker F., Yan X., Chen X., Chen J., Xiao G. F., Guo L., Liang Y. (2009) Crowded cell-like environment accelerates the nucleation step of amyloidogenic protein misfolding. J. Biol. Chem. 284, 30148–30158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Varkey J., Isas J. M., Mizuno N., Jensen M. B., Bhatia V. K., Jao C. C., Petrlova J., Voss J. C., Stamou D. G., Steven A. C., Langen R. (2010) Membrane curvature induction and tubulation are common features of synucleins and apolipoproteins. J. Biol. Chem. 285, 32486–32493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lee J. H., Hong C. S., Lee S., Yang J. E., Park Y. I., Lee D., Hyeon T., Jung S., Paik S. R. (2012) Radiating amyloid fibril formation on the surface of lipid membranes through unit-assembly of oligomeric species of α-synuclein. PLoS ONE 7, e47580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Arnulphi C., Jin L., Tricerri M. A., Jonas A. (2004) Enthalpy-driven apolipoprotein A-I and lipid bilayer interaction indicating protein penetration upon lipid binding. Biochemistry 43, 12258–12264 [DOI] [PubMed] [Google Scholar]
- 49. Tanaka M., Tanaka T., Ohta S., Kawakami T., Konno H., Akaji K., Aimoto S., Saito H. (2009) Evaluation of lipid-binding properties of the N-terminal helical segments in human apolipoprotein A-I using fragment peptides. J. Pept. Sci. 15, 36–42 [DOI] [PubMed] [Google Scholar]
- 50. Yanagi K., Ashizaki M., Yagi H., Sakurai K., Lee Y. H., Goto Y. (2011) Hexafluoroisopropanol induces amyloid fibrils of islet amyloid polypeptide by enhancing both hydrophobic and electrostatic interactions. J. Biol. Chem. 286, 23959–23966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Girych M., Gorbenko G., Trusova V., Adachi E., Mizuguchi C., Nagao K., Kawashima H., Akaji K., Lund-Katz S., Phillips M. C., Saito H. (2014) Interaction of thioflavin T with amyloid fibrils of apolipoprotein A-I N-terminal fragment: resonance energy transfer study. J. Struct. Biol. 185, 116–124 [DOI] [PubMed] [Google Scholar]
- 52. Tricerri M. A., Behling Agree A. K., Sanchez S. A., Jonas A. (2000) Characterization of apolipoprotein A-I structure using a cysteine-specific fluorescence probe. Biochemistry 39, 14682–14691 [DOI] [PubMed] [Google Scholar]
- 53. Abedini A., Raleigh D. P. (2009) A role for helical intermediates in amyloid formation by natively unfolded polypeptides? Phys. Biol. 6, 015005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhu M., Fink A. L. (2003) Lipid binding inhibits α-synuclein fibril formation. J. Biol. Chem. 278, 16873–16877 [DOI] [PubMed] [Google Scholar]
- 55. Jayasinghe S. A., Langen R. (2005) Lipid membranes modulate the structure of islet amyloid polypeptide. Biochemistry 44, 12113–12119 [DOI] [PubMed] [Google Scholar]
- 56. Williamson J. A., Loria J. P., Miranker A. D. (2009) Helix stabilization precedes aqueous and bilayer-catalyzed fiber formation in islet amyloid polypeptide. J. Mol. Biol. 393, 383–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Munishkina L. A., Phelan C., Uversky V. N., Fink A. L. (2003) Conformational behavior and aggregation of alpha-synuclein in organic solvents: modeling the effects of membranes. Biochemistry 42, 2720–2730 [DOI] [PubMed] [Google Scholar]
- 58. Anderson V. L., Ramlall T. F., Rospigliosi C. C., Webb W. W., Eliezer D. (2010) Identification of a helical intermediate in trifluoroethanol-induced α-synuclein aggregation. Proc. Natl. Acad. Sci. U.S.A. 107, 18850–18855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yamaguchi K., Naiki H., Goto Y. (2006) Mechanism by which the amyloid-like fibrils of a β2-microglobulin fragment are induced by fluorine-substituted alcohols. J. Mol. Biol. 363, 279–288 [DOI] [PubMed] [Google Scholar]
- 60. Gursky O., Atkinson D. (1996) Thermal unfolding of human high-density apolipoprotein A-1: implications for a lipid-free molten globular state. Proc. Natl. Acad. Sci. U.S.A. 93, 2991–2995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mendoza-Espinosa P., Montalvan-Sorrosa D., García-González V., Moreno A., Castillo R., Mas-Oliva J. (2014) Microenvironmentally controlled secondary structure motifs of apolipoprotein A-I-derived peptides. Mol. Cell. Biochem. 393, 99–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Griffin M. D., Mok M. L., Wilson L. M., Pham C. L., Waddington L. J., Perugini M. A., Howlett G. J. (2008) Phospholipid interaction induces molecular-level polymorphism in apolipoprotein C-II amyloid fibrils via alternative assembly pathways. J. Mol. Biol. 375, 240–256 [DOI] [PubMed] [Google Scholar]
- 63. Di Gaetano S., Guglielmi F., Arciello A., Mangione P., Monti M., Pagnozzi D., Raimondi S., Giorgetti S., Orrù S., Canale C., Pucci P., Dobson C. M., Bellotti V., Piccoli R. (2006) Recombinant amyloidogenic domain of apoA-I: analysis of its fibrillogenic potential. Biochem. Biophys. Res. Commun. 351, 223–228 [DOI] [PubMed] [Google Scholar]
- 64. Okada T., Wakabayashi M., Ikeda K., Matsuzaki K. (2007) Formation of toxic fibrils of Alzheimer's amyloid β-protein-(1–40) by monosialoganglioside GM1, a neuronal membrane component. J. Mol. Biol. 371, 481–489 [DOI] [PubMed] [Google Scholar]
- 65. Knight J. D., Hebda J. A., Miranker A. D. (2006) Conserved and cooperative assembly of membrane-bound α-helical states of islet amyloid polypeptide. Biochemistry 45, 9496–9508 [DOI] [PubMed] [Google Scholar]
- 66. Apostolidou M., Jayasinghe S. A., Langen R. (2008) Structure of α-helical membrane-bound human islet amyloid polypeptide and its implications for membrane-mediated misfolding. J. Biol. Chem. 283, 17205–17210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Georgieva E. R., Ramlall T. F., Borbat P. P., Freed J. H., Eliezer D. (2010) The lipid-binding domain of wild type and mutant α-synuclein: compactness and interconversion between the broken and extended helix forms. J. Biol. Chem. 285, 28261–28274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Guo C., Côté S., Mousseau N., Wei G. (2015) Distinct helix propensities and membrane interactions of human and rat IAPP(1–19) monomers in anionic lipid bilayers. J. Phys. Chem. B 119, 3366–3376 [DOI] [PubMed] [Google Scholar]
- 69. Ysselstein D., Joshi M., Mishra V., Griggs A. M., Asiago J. M., McCabe G. P., Stanciu L. A., Post C. B., Rochet J. C. (2015) Effects of impaired membrane interactions on alpha-synuclein aggregation and neurotoxicity. Neurobiol. Dis. 79, 150–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tartaglia G. G., Pawar A. P., Campioni S., Dobson C. M., Chiti F., Vendruscolo M. (2008) Prediction of aggregation-prone regions in structured proteins. J. Mol. Biol. 380, 425–436 [DOI] [PubMed] [Google Scholar]
- 71. Raimondi S., Guglielmi F., Giorgetti S., Di Gaetano S., Arciello A., Monti D. M., Relini A., Nichino D., Doglia S. M., Natalello A., Pucci P., Mangione P., Obici L., Merlini G., Stoppini M., Robustelli P., Tartaglia G. G., Vendruscolo M., Dobson C. M., Piccoli R., Bellotti V. (2011) Effects of the known pathogenic mutations on the aggregation pathway of the amyloidogenic peptide of apolipoprotein A-I. J. Mol. Biol. 407, 465–476 [DOI] [PubMed] [Google Scholar]
- 72. Wong Y. Q., Binger K. J., Howlett G. J., Griffin M. D. (2012) Identification of an amyloid fibril forming peptide comprising residues 46–59 of apolipoprotein A-I. FEBS Lett. 586, 1754–1758 [DOI] [PubMed] [Google Scholar]
- 73. Saito H., Lund-Katz S., Phillips M. C. (2004) Contributions of domain structure and lipid interaction to the functionality of exchangeable human apolipoproteins. Prog. Lipid Res. 43, 350–380 [DOI] [PubMed] [Google Scholar]
- 74. Chetty P. S., Mayne L., Lund-Katz S., Stranz D., Englander S. W., Phillips M. C. (2009) Helical structure and stability in human apolipoprotein A-I by hydrogen exchange and mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 106, 19005–19010 [DOI] [PMC free article] [PubMed] [Google Scholar]