

Background: The apurinic/apyrimidinic endonuclease 1 (APE1) endonuclease of base excision repair must access DNA damage within chromatin.
Results: Two natural variants of APE1 have a greater reduction of nuclease activity on nucleosomes.
Conclusion: Some amino acid residues in APE1 promote activity in the context of other proteins on DNA.
Significance: Knowing how enzymes tolerate protein obstructions on DNA is fundamental to understanding DNA repair in chromatin.
Keywords: base excision repair (BER), DNA damage, genetic polymorphism, nucleosome, protein-DNA interaction, AP endonuclease 1
Abstract
Non-coding apurinic/apyrimidinic (AP) sites are generated at high frequency in genomic DNA via spontaneous hydrolytic, damage-induced or enzyme-mediated base release. AP endonuclease 1 (APE1) is the predominant mammalian enzyme responsible for initiating removal of mutagenic and cytotoxic abasic lesions as part of the base excision repair (BER) pathway. We have examined here the ability of wild-type (WT) and a collection of variant/mutant APE1 proteins to cleave at an AP site within a nucleosome core particle. Our studies indicate that, in comparison to the WT protein and other variant/mutant enzymes, the incision activity of the tumor-associated variant R237C and the rare population variant G241R are uniquely hypersensitive to nucleosome complexes in the vicinity of the AP site. This defect appears to stem from an abnormal interaction of R237C and G241R with abasic DNA substrates, but is not simply due to a DNA binding defect, as the site-specific APE1 mutant Y128A, which displays markedly reduced AP-DNA complex stability, did not exhibit a similar hypersensitivity to nucleosome structures. Notably, this incision defect of R237C and G241R was observed on a pre-assembled DNA glycosylase·AP-DNA complex as well. Our results suggest that the BER enzyme, APE1, has acquired distinct surface residues that permit efficient processing of AP sites within the context of protein-DNA complexes independent of classic chromatin remodeling mechanisms.
Introduction
Apurinic/apyrimidinic (AP)2 sites are one of the most frequent lesions generated in DNA, arising via spontaneous hydrolysis at a rate of over 10,000 lesions per mammalian genome per day (1). AP sites are also created via damage-induced hydrolysis or DNA glycosylase-mediated cleavage of the N-glycosylic bond that connects the base to the sugar moiety in DNA. Glycosylases are damage-specific enzymes that initiate the process of base excision repair (BER), releasing a range of hydrolytic (e.g. uracil), alkylative (e.g. 3-methyladenine) and oxidative (e.g. 8-oxoguanine) base modifications (2). The resulting AP sites are non-instructional lesions, as they lack the information carried by the base moiety, and are therefore potentially mutagenic when replicated (3). Moreover, AP sites can arrest DNA and RNA polymerases, leading to the activation of responses that drive chromosomal rearrangements or cell death. Thus, an abasic site is a deleterious repair intermediate that can promote carcinogenic and degenerative outcomes, and their removal from DNA is essential for genome maintenance and stability.
The major enzyme responsible for initiating the repair of AP sites is AP endonuclease 1 (APE1 (4)). This multifunctional protein harbors a robust incision activity for AP sites, cleaving the DNA backbone immediately 5′ of the lesion. The resulting strand break is processed by the 5′-deoxyribose phosphate lyase and gap-filling polymerization activities of DNA polymerase β, prior to nick ligation, which make up the final steps of the BER response (5). Notably, the stepwise progression of BER occurs in a coordinated manner, such that the enzyme at each step of the process remains associated with its product and promotes the subsequent step, ensuring that the intermediates of repair (each more biologically detrimental than the initial base modification) are processed efficiently (6, 7). Not unexpectedly, the complete absence of APE1 leads to embryonic lethality in mice (8) and mammalian cell inviability in culture (9, 10), whereas reduced APE1 levels have been associated with disease susceptibility (11), underscoring the importance of this protein in upkeep of organismal health.
Importantly, the process of BER, like all DNA-templated processes in the cell, must function efficiently in the protein-packed context of chromatin. Chromatin entails the genomic DNA compacted and organized by an abundance of DNA-binding proteins, which collectively work to regulate DNA transcription, replication, and repair. The basic unit of chromatin compaction is that of the nucleosome core particle (NCP), consisting of 147 base pairs (bp) of DNA wrapped ∼1.7 times around an octamer of histones, generally comprised of two each of the four core histones: H2A, H2B, H3, and H4 (12). NCPs are separated by short stretches of DNA (“linker DNA”), ranging from 20 to 90 bp in length, which themselves are associated with a “linker histone” (H1 or H5) that directs higher levels of compaction among the nucleosome cores. Although chromatin is a dynamic structure, an essential trait for its role in regulating access to genomic information, the vast majority of DNA in the nucleus is associated with nucleosomes. Nucleosomes are generally inhibitory to every step of the BER process (13), including base damage recognition by DNA glycosylases (14–20), incision at AP sites by APE1 (21), and the DNA synthesis and ligation steps (20, 22). BER inhibition may be associated in part to the general physical constraints imposed on helical mobility within the nucleosome, but the greatest factor in impairment is likely the actual occlusion of the lesion by the histone proteins, as intervals along the DNA helix are in direct contact with the histone core, buried away from the solvent (23, 24).
The enzymatic activities of APE1 have been well characterized on a wide range of abasic DNA substrates, with a few studies examining its functionality in the context of nucleosomes (21, 22). APE1 incision activity is greatly reduced (∼10-fold) on nucleosomes at an AP site with its backbone sugar moiety in an “inward” orientation relative to an “outward” oriented AP site (21). Notably, APE1 activity on the outward-oriented AP site in nucleosomes was much lower than that on naked DNA (∼10-fold), highlighting the impact of the histone proteins on APE1, even when they are not directly occluding the lesion.
Haploinsufficiency of proteins in BER has been associated with increased disease susceptibility and reduced life expectancy (25), alluding to the possibility that even minor reductions in BER activity may have negative consequences to human health (5). In the human population there are a number of naturally occurring variants of APE1, many of which have been assessed for AP endonuclease activity on DNA substrates to determine whether they show reduced activities potentially indicative of deleterious health effects (26, 27). Although these variants have shown few differences in activity on short, naked DNA substrates, they have not been assessed for activities on a chromatin-relevant nucleosome, with which most DNA in the cell is associated. In the current study, we examined the nuclease activity of select APE1 variants/mutants at abasic sites on nucleosome DNA substrates.
We report within that the activity of the variants, like wild-type (WT) APE1, is reduced on nucleosomes relative to naked DNA, and that the orientation of the abasic site lesion relative to the histone proteins greatly impacts the activity of the enzyme. Notably, we find two variants (R237C and G241R), with amino acid substitutions in close proximity on the surface of the protein, to have significantly reduced activity on nucleosomes relative to WT APE1 and the other variants surveyed. This reduction in AP endonuclease activity is not seen on naked DNA, and corresponds to irregular binding of these variants to the nucleosome substrates. In addition, the reduced nuclease activity of these two variants was also apparent on AP-DNA preincubated with the alkyl adenine DNA glycosylase (AAG, also known as MPG), which binds abasic sites, highlighting the possibility that the reduced activity on the nucleosome substrates may be indicative of inhibition by DNA-bound proteins in general. Our results in total suggest that APE1 harbors particular surface region residues that facilitate access to AP sites in the context of nucleoprotein complexes. This finding may imply that BER proteins are equipped with a trait (termed “protein obstruction tolerance”) to cope with DNA damage embedded within protein-DNA complexes, alleviating the need for energy-driven processes (e.g. chromatin remodelers) for such high-frequency lesions.
Experimental Procedures
AP-DNA and Nucleosome Substrate Preparation
The 147-bp DNA substrates used in this study entailed the 601 nucleosome positioning sequence (28) and were created by PCR from a plasmid containing the 601 sequence using two different forward primers (each directing a different abasic site position), and a common reverse primer. The forward primers, “U −24” and “U −29,” each have a uracil residue positioned at 24 or 29 bases from the dyad (center) of the 601 sequence. These uracil locations dictate distinct orientations of the lesion relative to the histone octamer when reconstituted into nucleosomes, where U −24 is an inwardly oriented lesion (the backbone of the uridylate nucleotide is facing the histone octamer) and U −29 is an outwardly oriented lesion (the backbone of the uridylate nucleotide is facing away from the histone octamer, toward the solvent; the location of residues are shown as AP sites on nucleosome in Fig. 1A). DNAs were radiolabeled with 32P at the 5′ end of the strand containing the uracil lesion by incubation of the DNA with T4 polynucleotide kinase (Invitrogen) and [γ-32P]ATP (PerkinElmer Life Sciences) for 30 min at 37 °C. Notably, the reverse primer contains a 5′ biotin tag, preventing labeling of the strand without the uracil residues during radiolabeling. Uracil residues were subsequently converted to abasic (AP) sites as described previously (21), by treatment of the uracil DNAs with recombinant Escherichia coli UDG (New England Biolabs) for 30 min (reaction buffer: 25 mm HEPES (pH 7.5), 2 mm DTT, 0.2 mm EDTA, 100 μg/ml of BSA, 10% glycerol, 5 mm MgCl2, 4 mm ATP). Mononucleosomes (NCPs) were prepared by histone octamer transfer as described previously (18, 21). Three pmol of radiolabeled 147-bp DNA substrates were combined with 300 pmol of chicken erythrocyte core particles prepared from chicken erythrocytes (29) at high ionic strength, and reconstituted by subsequent incremental dialysis (30) (Fig. 1B).
FIGURE 1.
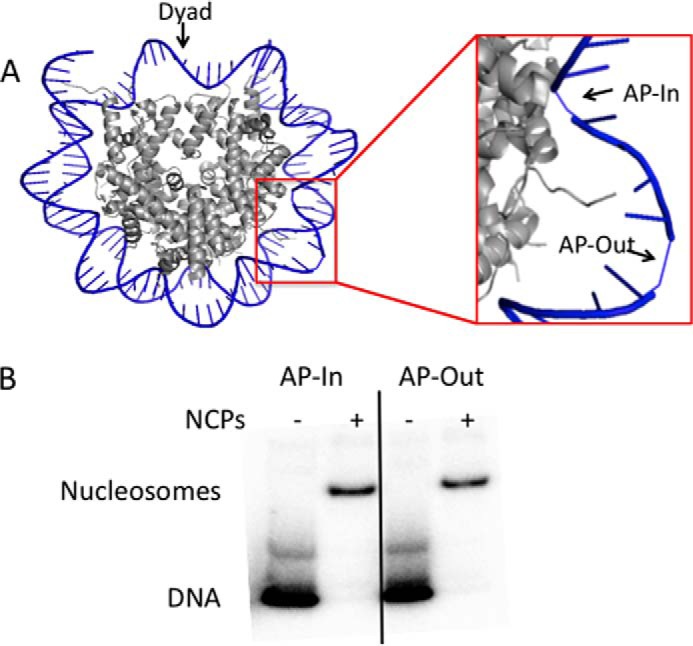
Reconstitution of AP-nucleosomes. A, image from Protein Data Bank structure 3LZ0 (orientation 1) (44) with AP-In (−24) and AP-Out (−29) sites shown. Note that only one strand of the DNA is shown, the opposing strand is hidden for clarity. B, nucleosomes were reconstituted by histone octamer transfer from nucleosome core particles isolated from chicken erythrocytes. Products were separated on 6% non-denaturing polyacrylamide gels. Lanes denoted with a − are DNA only, and lanes denoted with a + show the DNA reconstituted into nucleosome core particles. Note: the upper band in the lanes with DNA are caused by secondary structure of the 601 sequence DNA.
APE1 Proteins
Purified, recombinant, untagged human APE1 proteins used in this study were WT APE1, Q51H, I64V, P112L, D148E, R237C, G241R, P311S, and A317V described previously (26), and the APE1 mutants R237A (27) and Y128A (31). Four new mutants were created for assessment, i.e. E216A, E217A, P234A, and Q245R, by submission of their respective DNA sequences to GenScript (Piscataway, NJ). Synthesized DNAs were provided in pUC57, and the cDNAs were isolated following NdeI/BamHI digestion and transferred into the same restriction sites of pET11a using standard molecular biology techniques (32); we note that a silent nucleotide mutation was introduced into the internal NdeI site within the APE1 coding region. The proteins were expressed and purified as described previously (26).
AAG Protein
The AAG glycosylase expression vector was a generous gift from Dr. Leona Samson (MIT, Boston). The vector expresses an AAG fragment lacking residues 1–79 of the protein. Both the enzymatic activity and DNA binding specificity of this truncated AAG have been shown to be identical to the full-length AAG protein (33, 34). To express AAG from E. coli, BL21 cells were transformed with the recombinant plasmid pET19b-AAG and plated onto selective agar. A single colony was inoculated in 20 ml of LB/Amp media and grown overnight. The culture was diluted to 200 ml of fresh media and grown to an A600 of 0.6. AAG expression was induced with isopropyl 1-thio-β-d-galactopyranoside (0.8 mm) at 30 °C for 5 h. Cells were harvested by centrifugation and washed with cold water. The cell pellet was resuspended in 10 ml of cold E. coli lysis buffer (50 mm NaH2 PO4, 300 mm NaCl, 10 mm imidazole, pH 8.0), and cells were lysed by sonication. Cell lysate was centrifuged at 17,000 × g for 20 min at 4 °C and the His6-tagged AAG protein was purified from the supernatant with nickel-nitrilotriacetic acid-agarose beads according to the manufacturer's protocol (Qiagen).
AP Endonuclease Activity
For measurement of AP endonuclease activity, DNA and nucleosome substrates were incubated with APE1 proteins at 37 °C for 10 min, at the following concentrations: 5 nm APE1 on 1 pmol of naked DNA, 50 nm APE1 on 10 pmol of AP-Out NCPs, and 5 μm APE1 on 10 pmol of AP-In NCPs. The reaction buffer, consisting of 25 mm HEPES (pH 7.5), 2 mm DTT, 0.2 mm EDTA, 100 μg/ml of BSA, 10% glycerol, 5 mm MgCl2, and 4 mm ATP was identical to that used previously to assess APE1 activity on NCPs (21). To adjust for excess core particles present in reconstituted nucleosome samples, naked DNA samples had chicken erythrocyte core particles added to a final concentration of 300 pm. Reactions were terminated with addition of phenol:chloroform:isopropyl alcohol (20:19:1). DNAs were immediately treated with 1 m sodium borohydride (NaBH4) for 20 min on ice after APE1 reaction termination to reduce the remaining uncleaved AP site residues and prevent their breakage during sample boiling and electrophoresis. To resolve cleavage products after reaction termination, all samples were boiled and separated on 10% polyacrylamide (0.5% bisacrylamide), 7 m urea denaturing gels. Gels were run in 1× TBE buffer, exposed to PhosphorImager screens (GE Healthcare), screens (GE Healthcare), visualizedvisualized on a STORM 840 PhosphorImager (Amersham Biosciences), and images were analyzed with IMAGEQUANT software (GE Healthcare).
Naked and Nucleosome AP-DNA Binding
APE1 protein (at concentrations ranging from 0 to 20 nm) was incubated in 20-μl reactions with 1 pmol of naked AP-Out DNA for 15 min on ice in the enzymatic reaction buffer used in the endonuclease activity assays, except with 0.4 mm EDTA and lacking the ATP and MgCl2 components. APE1 binding was assessed on AP-Out nucleosome substrates under the same conditions, but on 3 pmol of NCPs over an APE1 concentration range of 0–0.8 μm. Binding to uracil-containing substrate controls (U-Out DNA and U-Out NCPs) were done under the same conditions and protein concentrations as those of the corresponding AP-DNAs and NCPs. DNA/NCP binding was resolved on non-denaturing 8% polyacrylamide gels, run on ice at 100 V for 60 (DNA) or 140 min (NCPs). Substrates were visualized and quantified as above.
Thermal Stabilities of APE1 Proteins
To determine the relative heat-associated stabilities, the variants in this study were tested for AP endonuclease activity on naked AP-DNA (as performed above for endonuclease activity measurements) after preincubation at different temperatures. Initially, all variants were incubated at 50 °C for 15 min prior to endonuclease activity assessment. The variants that still showed activity after the 50 °C incubation (WT, Q51H, I64V, and D148E) were tested again, this time with a preincubation at 52 °C for 15 min prior to endonuclease assessment. Variants that showed no activity after the 50 °C incubation (R237C, G241R, P311S, and A317V) were tested for activity after a 15-min incubation at 45 °C.
AAG Binding to AP-DNA and Effect on APE1 Activity
To determine the binding of AAG to AP-DNA, different amounts of purified, recombinant AAG (0–120 μm) were incubated with 1 pmol of naked AP-Out DNA for 15 min on ice using the same binding buffer used for APE1 and visualized in the same manner (above). Impact of AAG on WT APE1 endonuclease activity was performed by preincubation of 1 pmol of naked AP-Out DNA with different concentrations of AAG (0–120 μm) at 37 °C for 10 min prior to addition of APE1 at a concentration of 5 nm. Endonucleolytic activity measurements were subsequently performed as noted above for naked DNA. Heat inactivation of AAG was done by incubation of AAG at 65 °C for 20 min.
Results
Specific APE1 Variants Exhibit Impaired Endonuclease Activity on Nucleosome Substrates
The population- and tumor-associated variants characterized here (Q51H, I64V, P112L, D148E, R237C, G241R, P311S, and A317V) have been shown previously to exhibit similar AP endonuclease activity on short, naked DNAs harboring the abasic site analog tetrahydrofuran (26). In addition, WT APE1 activity has been previously reported to be reduced on nucleosomes relative to naked DNA, with the reduction being much greater when the AP site is inwardly oriented, with the backbone of the lesion facing toward the histone octamer (21). Considering the high proportion of nuclear DNA associated with histones, we examined the possibility that the APE1 variants might exhibit different activities on substrate DNAs associated with NCPs.
Natural (UDG-generated) AP site-containing DNAs of 147 bp in length, naked or assembled in nucleosomes, were incubated with each APE1 protein (5 nm for naked DNAs, 50 nm for AP-Out nucleosomes, and 10 μm for AP-In nucleosomes) for 10 min at 37 °C, and the radiolabeled reaction products were subsequently assessed by denaturing gel electrophoresis (Fig. 2A); the quantification of the nuclease activities of each variant on the different 147-bp substrates is summarized in Fig. 2B. Also assessed for comparative purposes was activity of the APE1 variant R237A, a previously characterized protein with reduced AP site incision activity (27). Similar amounts of cleavage were observed on the naked DNA substrate for all of the APE1 variants (Fig. 2), including R237A (Fig. 2A), which did not appear to have a substantial decrease in activity in the reaction conditions employed here. However, in comparison to the WT protein and the other variants tested, there was a distinct ∼2–3-fold reduction in the nuclease activity of the two variants, R237C and G241R, as well as the R237A mutant, on the nucleosome substrates. Moreover, the reduced activity for these variants/mutants was seen on both the inward (AP-In) and outward (AP-Out) abasic site orientations (Fig. 2), despite the significant difference in APE1 protein concentration being required to detect product formation for each of the constructs (see above).
FIGURE 2.
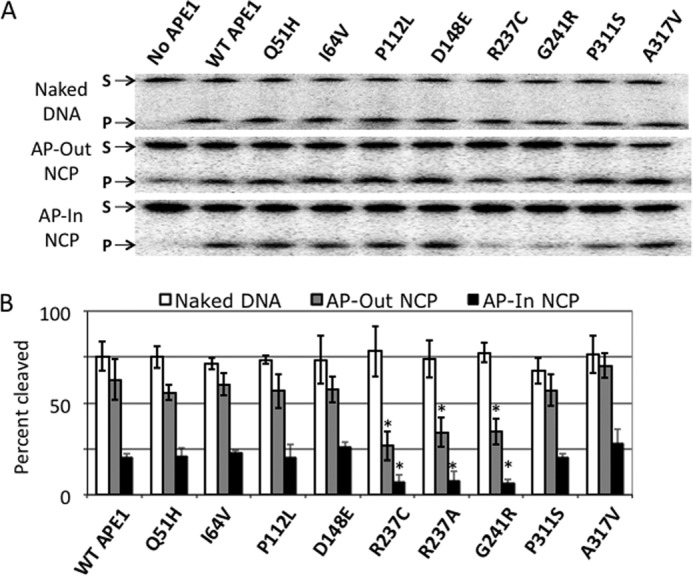
Nuclease activity of APE1 variants on naked DNA and nucleosome substrates. A, denaturing polyacrylamide gels separating the full-length 147-bp substrate (S, upper band) from the APE1 cleavage product (P, lower band) for naked AP-DNA (top gel), AP-Out nucleosomes (middle gel), and AP-In nucleosomes (bottom gel). B, chart showing percent cleavage of substrates after 10 min incubation with each APE1 protein. Bars represent the average of at least 3 independent measurements, error bars represent standard deviations. Asterisks mark significant difference (as determined by t test) between the measurement of the variant and the corresponding measurement of WT APE1, with p values as follows: R237C AP-Out NCPs, p < 5 × 10−5; R237C AP-In NCP, p < 0.004; R237A AP-Out NCPs, p < 0.005; R237A AP-In NCP, p < 0.02; G241R AP-Out NCPs, p < 2 × 10−4; G241R AP-In NCP, p < 0.005.
Unusual Binding of the R237C and G241R Variants to Naked and Nucleosome DNA Substrates
As shown previously, reduced nuclease activity of APE1 on nucleosome substrates correlates closely with a reduced affinity of the protein for AP-DNA (21). This is unsurprising, as the presence of the histones likely occludes the enzyme from recognizing the lesion, particularly when the AP site is in an inward orientation. Indeed, the presence of histones on the DNA appears to have a general inhibitory effect on the APE1-substrate interaction, as the endonuclease has a marked reduction in activity (∼10-fold) on nucleosomes with outward oriented AP sites compared with naked DNA (see Ref. 21 and Fig. 2B).
To determine whether the reduced nuclease activity of the R237C and G241R variants on nucleosome substrates is due to an altered substrate interaction relative to WT APE1, binding equilibria were determined by EMSA (Fig. 3). Naked AP-Out DNA (Fig. 3A) or AP-Out nucleosome substrates (Fig. 3B) were incubated with increasing concentrations of select APE1 variants for 15 min at 4 °C. In Fig. 3A, WT APE1 and the variant A317V, which showed similar endonuclease activity to that of WT on nucleosomes, exhibited a similar, characteristic, concentration-dependent naked AP-Out substrate binding profile. The R237C and G241R variants, however, although generating the anticipated APE1-DNA band at quantitatively similar levels as WT and A317V (∼40% of substrate bound at 20 nm protein), produced more DNA “smearing” between the bound and free DNA bands (indicated by the arrow; Fig. 3A), suggesting an irregular affinity for the AP site substrate that results in dissociation of the protein-DNA complex during the electrophoresis process. We note that the binding of APE1 is specific for the presence of the abasic site, as the WT protein at 20 nm shows no complex formation with the 147-bp naked DNA harboring a uracil (U-DNA) at the same location as the AP site (Fig. 3A).
FIGURE 3.
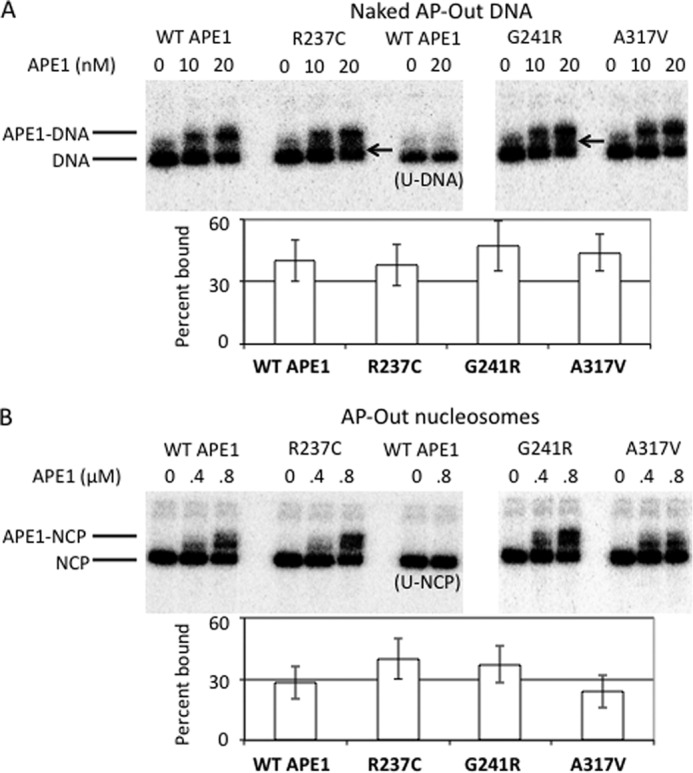
APE1 variant binding to AP sites in naked and nucleosome AP-Out DNA. A, assessment of electrophoretic mobility shifts of naked 147-bp AP-Out DNA substrates (lower band) to APE1-DNA complexes (upper band) on non-denaturing polyacrylamide gels associated with different concentrations of select APE1 variants. The DNA substrate with a uracil in place of the AP site is used as a binding specificity control, labeled as “U-DNA.” Arrows mark smearing in gel, lacking clear separation between the bands associated with the DNA and APE1-DNA complexes. Percent naked DNA bound by 20 nm of each variant after a 15-min incubation at 4 °C (bottom), as determined by the fraction of DNA in the gel residing above the substrate bands in the gel image (top). B, same as A, with AP-Out nucleosomes substrates, the lower band representing the NCPs and the upper band representing the APE1-NCP complex. As with the naked DNAs, a nucleosome substrate with a uracil in place of the AP site was used as a binding specificity control, labeled as U-NCP. Percent AP-Out nucleosomes bound by 0.8 μm of each protein after a 15-min incubation on ice (bottom), as determined by the fraction of DNA in the gel residing above the substrate bands in the gel image (top). Bars in graphs represent the average of at least 3 independent measurements, error bars represent standard deviations.
Like the binding to naked AP-DNA, the variants exhibited stable complex formation with the outwardly oriented nucleosome substrate (Fig. 3B). Unexpectedly although, the R237C and G241R variants did not show an obvious decrease in affinity for the AP-nucleosomes compared with WT APE1 or the A317V variant, and indeed showed binding to the substrate that was trending greater quantitatively. However, both R237C and G241R did appear to retard the nucleosome AP-Out substrates differently than WT and A317V, producing a broader band that might represent multiple protein binding events or multiple binding states, indicating an interaction of R237C and G241R with AP-Out nucleosome DNA that is distinct from WT APE1.
The DNA Binding Mutant Y128A Exhibits Unaffected Nuclease Activity on Nucleosome Substrates
Although the APE1 variants R237C and G241R did not show a quantitatively decreased affinity for the 147-bp “601 sequence” naked or nucleosome substrates (Fig. 3), we considered the possibility that a general decrease in DNA or nucleosome substrate binding (below the measurable detection threshold of EMSA) might lead to a pronounced defect in enzymatic activity. To test this possibility, we determined the endonuclease activity on nucleosome substrates of the APE1 Y128A mutant, which has previously been shown to exhibit an ∼4-fold decrease in cleavage activity mainly due to reduced DNA-substrate binding capacity (31). Consistent with our prior studies using short DNA substrates, the Y128A mutant showed reduced activity on the 147-bp naked DNA substrate relative to WT APE1 at 5 nm protein (Fig. 4, A and B). When the concentration of Y128A was increased 4-fold (20 nm), its nuclease activity on naked DNA roughly matched that of the other variants in the study. Moreover, when the concentration of Y128A was increased 4-fold in the nucleosome cleavage assay, from 50 (like that of WT APE1) to 200 nm, its cleavage activity was nearly equal to that of the WT protein (Fig. 4, A and C); thus the Y128A mutant did not show the proportionally reduced activity seen for the R237C and G241R variants on NCP-associated DNA. This finding argues that diminished DNA-binding activity does not necessarily lead to reduced activity on nucleosome substrates, and implies a unique role for the Arg-237 and Gly-241 residues in accessing lesions within protein-DNA complexes.
FIGURE 4.
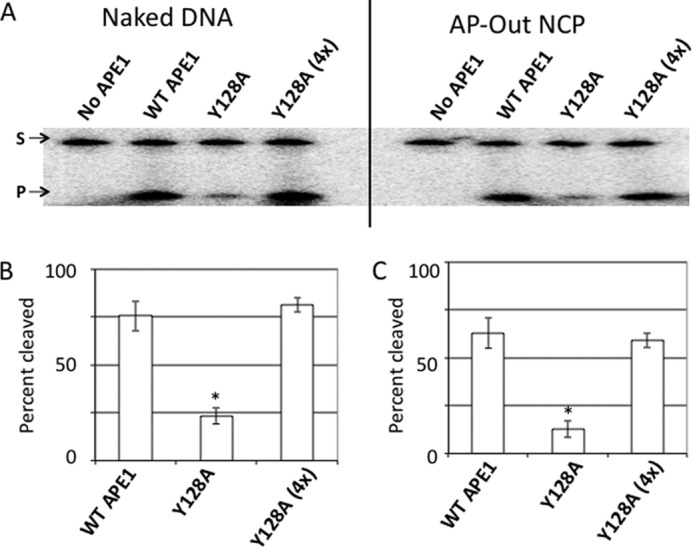
Activity of binding-defective APE1 mutant Y128A on nucleosome substrates. A, denaturing polyacrylamide gels separating the full-length 147-bp substrate (S, upper band) from the APE1 cleavage product (P, lower band) for the naked DNA (first four lanes), and the AP-Out nucleosome substrates (last four lanes). On naked DNA, the APE1 mutant Y128A was used at concentrations of 5 nm (as done for WT APE1), and at 20 nm (as Y128A (4x)). On AP-Out NCP substrates, WT APE1 and Y128A concentrations were 50 nm, and the Y128A (4x) was performed at 200 nm. B and C, charts showing percent cleavage of substrates (naked or AP-Out NCP DNAs) after a 10-min incubation with WT APE1 and Y128A at 5 and 20 nm (4x). Bars represent the average of at least 3 independent measurements, error bars represent standard deviations. Asterisks mark significant difference (as determined by t test) between the measurement of the variant and the corresponding measurement of the WT APE1, with p values as follows: Y128A naked DNA, p < 2 × 10−4; Y128A AP-Out Nucleosomes, p < 2 × 10−5.
Disparate Heat Sensitivity among the APE1 Variants
Although the APE1 endonuclease reactions in this study were run at 37 °C for only 10 min, we considered the possibility that among the APE1 variants there may be a range of temperature-associated instabilities, and that the reduced incision activity and abnormal binding of the R237C and G241R variants may be associated with decreased stability under the reaction conditions employed. To test this possibility, we measured the nuclease activity of the variants on naked AP-Out DNA, after preincubation of each protein at a defined temperature for 15 min. Initially, all variants were incubated at 50 °C (Fig. 5A), and under these conditions, a subset (WT, Q51H, I64V, P112L, and D148E) remained basically fully active, whereas others displayed either some (I64V and P112L) or essentially complete diminishment of AP endonuclease activity (R237C, G241R, P311S, and A317V). It is noteworthy that the subset of variants that completely lost functionality after the 50 °C preincubation was the four most C-terminal variants in the set tested.
FIGURE 5.
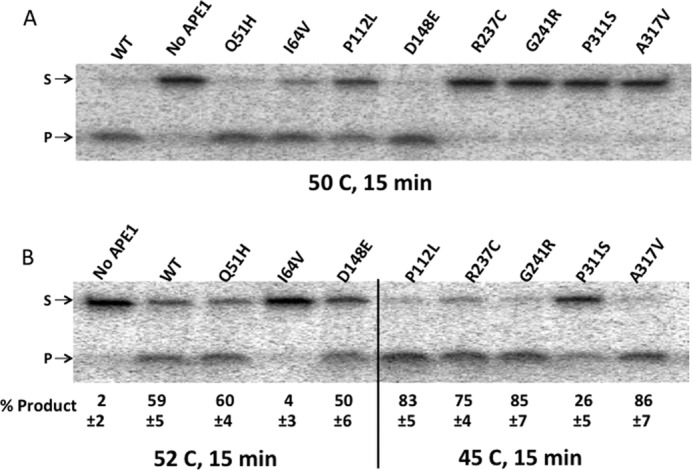
Relative heat inactivation temperatures for the APE1 variants. A, denaturing polyacrylamide gels separating the full-length 147-bp substrate (S, upper band) from the APE1 cleavage product (P, lower band) for the naked DNA after a 10-min incubation with APE1 following a 15-min preincubation of each variant at 50 °C. B, as in A, with variants functional after 50 °C (WT, Q51H, I64V, and D148E) retested for activity after a 15-min preincubation at 52 °C, and variants that lost most or all activity at 50 °C (P112L, R237C, G241R, P311S, and A317V) retested for activity after a 15-min preincubation at 45 °C. Numbers below the gel represent the average percentage of product of three independent experiments, and the errors represent standard deviations. Significance was determined by t test for each variant within the 52 and 45 °C assessments: WT versus Q51H, p = 0.4; WT versus I64V, p < 0.02; WT versus D148E, p = 0.2; A317V versus P112L, p = 0.4; A317V versus R237C, p = 0.18; A317V versus G241R, p = 0.3; A317V versus P311S, p < 0.01. Variants were considered equal if their cleavage measurements, at the same temperature, were not significantly different. Variants were ranked as greater/lesser if their cleavage measurements were significantly different from other variants within the same temperature.
For better resolution in determining the relative temperature sensitivities of the variants, the three variants that remained most active after the 50 °C preincubation (Q51H, I64V, and D148E) were retested with WT after a 15-min incubation at 52 °C, whereas the remaining variants were retested after preincubation at 45 °C (Fig. 5B). There is a clear range of temperature sensitivities among the variants, with the most stable proteins (WT, Q51H, and D148E) showing near full activity after the 15-min preincubation at a temperature that is 15 °C above the temperature used to assess activity (37 °C), whereas the most sensitive variant, P311S, was negatively affected even after a 15-min preincubation at a temperature only 8 °C above 37 °C. Based on the results of Fig. 5, the variants can be ranked as follows in terms of heat inactivation tolerance: WT, Q51H, D148E > I64V > P112L, G241R, A317V, R237C > P311S. Importantly, the two variants in the study that show reduced activity on nucleosomes (R237C and G241R) were not the most sensitive to heat exposure, showing “intermediate” activity that is not significantly different from the A317V variant used in the comparative studies above. Thus, the reduction in activity of R237C and G241R on nucleosome substrates is unlikely related to a decrease in stability under the reaction conditions employed.
Normal Activity of APE1 Mutants Created in the Region of Arg-237 and Gly-241
The two APE1 variants (R237C and G241R) showing decreased endonuclease activity on nucleosome substrates have residue substitutions that occur in close proximity to one another, specifically within the same helix on the surface of the protein. We wanted to test the possibility that other residues in this region might adversely affect enzyme activity in the context of nucleosomes. We selected 4 candidate residues for targeted mutagenesis, because they span the region harboring R237C and G241R: Glu-216, Glu-217, Pro-234, and Gln-245 (shown visually in Fig. 6A). Glu-216 and Glu-217 were substituted with alanine to interfere with any potential charge-associated functions they might provide, whereas the alanine substitution of Pro-234 was chosen to disrupt the structural role of the proline. Gln-245 was substituted with arginine, also in an attempt to interfere with ionic interactions associated with that glutamine.
FIGURE 6.
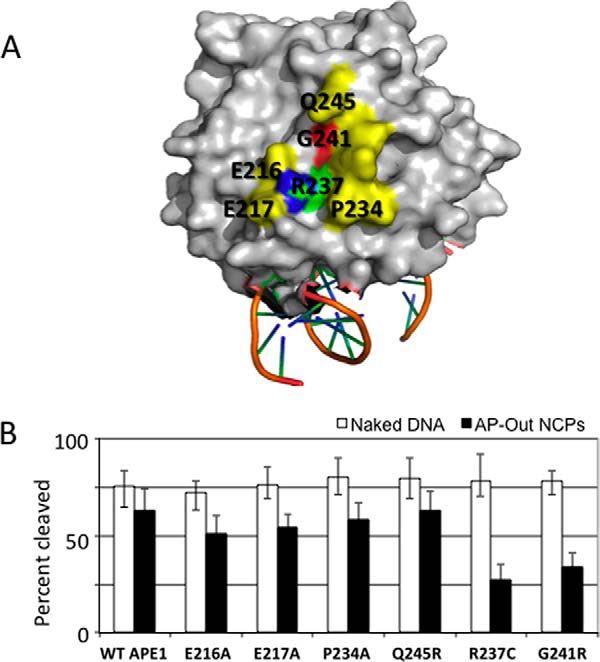
Nuclease activities of APE1 mutants in close proximity to Arg-237 and Gly-241. A, image of the surface of APE1 (41) on which the Arg-237 and Gly-241 residues reside, in addition to the locations of specific amino acids in close proximity that are mutated for testing. B, percent cleavage of naked AP-Out DNA and AP-Out nucleosome substrates after a 10-min incubation with newly created APE1 mutants E216A, E217A, P234A, and Q245R. WT APE1, R237C, and G241R data from Fig. 1 were added for comparison. Bars represent the average of at least 3 independent measurements, error bars represent standard deviations.
After expression and purification of these four mutant proteins, they were tested for endonuclease activity on the naked AP-Out DNA and AP-Out nucleosomes relative to WT APE1 (Fig. 6B). All four of the selected mutants functioned similarly to that of WT on both naked and nucleosome-associated AP-DNAs. The E216A mutant had the lowest relative activity on nucleosomes among all of the proteins tested (besides R237A, R237C, and G241R), yet its incision efficiency was not significantly different from WT APE1. Although these amino acid substitutions are not exhaustive for all residues within the region of interest, the results suggest that the observed protein obstruction tolerance defects are particular to the Arg-237 and Gly-241 residues, and presumably not to a more generalizable domain of the APE1 protein.
Reduced Nuclease Activity of R237C and G241R Variants in the Presence of the AAG Glycosylase
Nucleosomes may interfere with DNA-interacting proteins due to direct occlusion by steric interference or due to the inherently reduced dynamics of the nucleosome DNA itself. Because we have found that APE1 has reduced activity on nucleosomes relative to naked DNA, we wanted to test the possibility that another DNA-associated protein, one not expected to impart DNA torsional constraints, might affect APE1 activity, particularly the two variants exhibiting decreased activity on NCP-substrates. We therefore determined the activity of APE1 in the context of excess AAG, a DNA glycosylase known to stably bind abasic sites and to bind AP-DNA nonspecifically as well (35). We found that WT APE1 (5 nm) cleaved the AP-DNA substrate (50 nm) normally in the presence of AAG up to a concentration of 60 μm, at which point APE1 activity was modestly reduced (Fig. 7A). We also found that there were likely multiple nonspecific AAG-DNA complexes formed with the 147-bp AP-DNA, and that at the concentration at which WT APE1 showed reduced activity (60 μm AAG) there was no unbound DNA (Fig. 7B).
FIGURE 7.
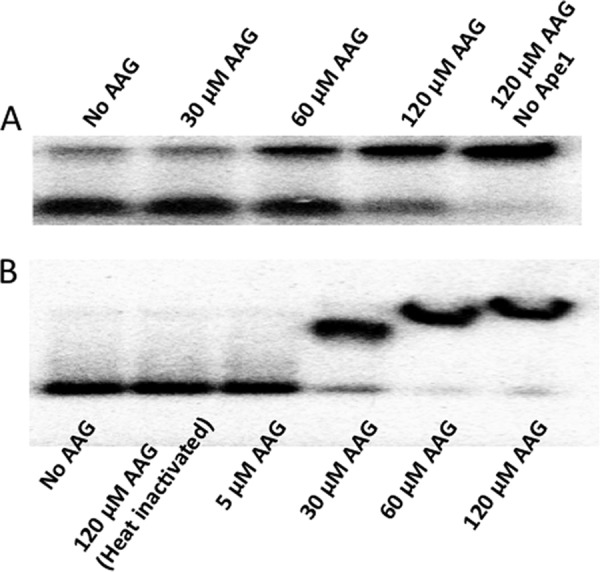
AAG-associated APE1 activity inhibition and AP-DNA binding. A, WT APE1 endonuclease activity on naked AP-Out DNA in the presence of different concentrations of AAG glycosylase. Upper bands are the DNA substrate, lower bands are the endonuclease cleavage product. B, non-denaturing polyacrylamide gel showing binding of AAG to naked AP-Out DNA at different concentrations.
Using the 60 μm concentration of AAG, we determined the effect of the glycosylase on AP endonuclease activity among the variants. Notably, whereas WT and most of the variants showed nearly equivalent endonuclease activity in the presence of AAG or heat-inactivated AAG (which does not bind to the AP-DNA (Fig. 7B)), the R237C and G241R proteins displayed an ∼2-fold decreased activity on DNA strictly in the presence of excess active AAG (Fig. 8). The G241R seemed to be less affected by AAG than R237C (although the difference was not significant), a pattern similar to what was seen for the two variants on AP-nucleosomes (Fig. 2B). Thus, the reduction of nuclease activity of the two outlying variants, which was prevalent on NCP-associated DNA, is not specific for interference by histones, and may represent a more general impairment in activity in the context of nucleoprotein complexes.
FIGURE 8.
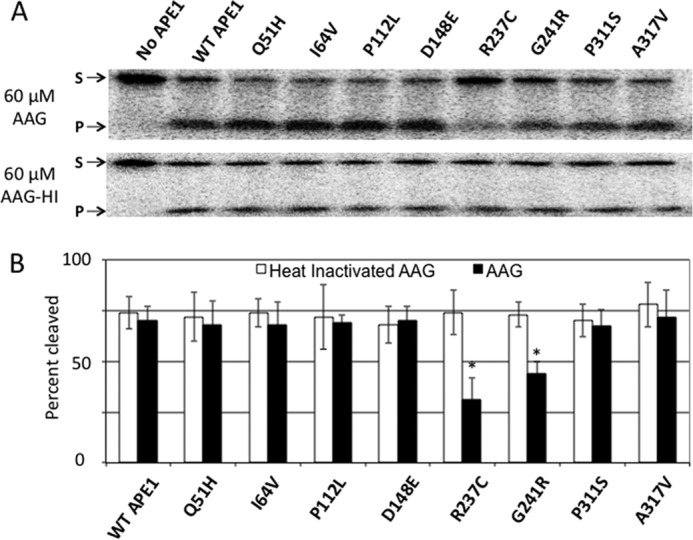
Effect of excess AAG glycosylase on APE1 variant nuclease activities. A, denaturing polyacrylamide gels separating the full-length 147-bp substrate (S, upper band) from the APE1 cleavage product (P, lower band) for the naked AP-Out DNA after a 10-min incubation with APE1 proteins in the presence of 60 μm AAG. B, percent cleavage of naked AP-Out DNA in the presence of heat-inactivated or active AAG. Bars represent the average of at least 3 independent measurements, error bars represent standard deviations. Asterisks mark significant difference (as determined by t test) between the measurement of the variant and the corresponding measurement of the WT APE1, with p values as follows: R237C, p < 5 × 10−4; G241R, p < 0.01.
Discussion
We set out to examine whether APE1 variants found within the population or reported to be associated with cancer might exhibit differential activity on AP site lesions in the context of nucleosomes. Indeed, we report herein that the rare population variant, G241R, and the tumor-associated variant, R237C, show a reduced capacity to incise at an abasic site in the context of nucleosomes. This impaired activity with respect to the WT enzyme was independent of the orientation of the AP lesion relative to the underlying histone proteins, being observed on both inward and outward AP sites. Our results also indicate that the more common APE1 population variants (in terms of frequency of appearance, i.e. Q51H, I64V, and D148E) exhibit normal enzymatic activity, consistent with recent experimental evidence (26, 27, 36), although defects in RNA processing have been reported (37). Overall, we find some rare APE1 variants do indeed have reduced activity on AP sites within biologically relevant DNA substrates, and thus could potentially contribute to disease susceptibility by leading to an overall reduced BER capacity.
Consistent with our current observations, we reported previously that WT APE1 has greatly reduced (∼10-fold) endonuclease activity on AP site nucleosomes with outwardly oriented lesions relative to naked DNA (21). In fact, the sensitivity of APE1 to other proteins on DNA may have been essential for detecting differences in activity among the variants, as naked DNA substrates provide little challenge to the incision process. Interestingly, unlike APE1, the human uracil DNA glycosylase, UNG1, displays only modest differences in activity on uracil moieties in identical nucleosome substrates relative to the naked DNA controls (18). Although both proteins associate with the minor groove and are capable of translocating processively along the DNA to search for their respective substrates, APE1 makes distinct contacts with both strands of the DNA during catalysis, whereas UNG1 interacts with a single strand of DNA (38–42). In addition, APE1 induces a greater bend in the DNA helix for enzymatic activity (6), requiring more substrate flexibility for efficient catalysis. Thus, the requirement to interact with both strands and the increased kinking of the DNA are both likely contributors to the reduced activity of APE1 on nucleosomes.
One possible explanation for the reduced activity of the two errant APE1 variants in this study is a defect in their ability to tolerate the reduced flexibility of nucleosome-associated DNA, thus reducing their substrate interaction efficiency. However, this is not supported by our nucleosome binding assessment (Fig. 3), which showed no quantitative difference between the R237C, G241R, and WT APE1 proteins, although their interactions appeared characteristically different. In addition, the R237C and G241R variants showed a relative reduction in activity on AP-DNA bound by the glycosylase AAG (Fig. 8), a mobile protein on DNA (43) that does not impose the limited dynamics associated with nucleosomes, arguing against the conclusion that these two variants are strictly sensitive to reduced substrate tractability. Nevertheless, it is clear from our work that, whereas the R237C and G241R proteins do not exhibit a major AP-DNA binding defect, the abnormal interactions observed with substrate DNA are likely reflective of an impaired interaction that contributes to the reduced efficiency on nucleosome substrates.
Given their close proximity on the APE1 surface, it is noteworthy that similarly reduced incision capacities were measured for R237C (as well as R237A) and G241R on nucleosome substrates. The observation that the R237C variant exhibits an impaired AP-DNA binding capacity on 147-bp substrates (Fig. 3) is consistent with our prior studies using shorter oligonucleotide DNAs (26). Because Arg-237 forms a hydrogen bonding network with neighboring glutamate residues (Glu-216 and Glu-217), it seems likely that mutation of this position could result in altered protein integrity and reduced functionality (27). Although the E216A and E217A mutants showed some reduction in APE1 activity on nucleosomes (Fig. 6B), it is perhaps not surprising that they did not have the significant impact seen in R237C, as each individual glutamate mutation would only contribute to disruption of part of the hydrogen bonding network. Significantly, the difference in DNA binding of the R237C and G241R variants does not fully explain their reduced activities on protein-associated AP-DNAs, because the APE1 mutant Y128A, with a severe DNA binding defect, does not show hypersensitivity to AP sites in nucleosome substrates (Fig. 4).
An unexpected, yet exciting, result of the work described herein was the identification of residues within APE1 that appear to facilitate recognition of AP sites within the context of pre-assembled protein-DNA complexes. Importantly, the incision defects observed for R237C and G241R were recapitulated with both histone- and glycosylase-bound DNA substrates, suggesting a more generalizable role for these two amino acids in damage recognition. It is tempting to speculate that APE1 has evolved a protein surface region to facilitate AP site identification within protein-DNA complexes, because (i) such lesions are frequent products within the genome and are likely to exist in some form of nucleoprotein configuration and (ii) without a “simple” mechanism, there would be an enormous cellular energy demand to regularly carry out chromatin remodeling around the damage to facilitate repair. It will be interesting going forward to determine whether other components of BER, which similarly must deal with high-frequency lesions, have evolved comparable protein obstruction tolerance regions or residues to aid in efficient execution of the repair response within the vast sea of chromatin.
Author Contributions
J. M. H. coordinated the study, performed the experiments shown, and co-wrote the paper. P. M. provided technical assistance and contributed to preparation of Figs. 7 and 8. D. R. M. provided technical assistance and prepared the APE1 variants used throughout the study. D. M. W. conceived the study, provided feedback during study development, and co-wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Robert Brosh (NIA, National Institutes of Health) for editorial feedback, Dr. Mingrui Duan (Washington State University) for help with figure assembly, and Dr. Takashi Tadokoro (NIA, National Institutes of Health) for creating the APE1 protein structure image.
This work was supported, in whole or in part, by National Institutes of Health Grants ES020955 (to J. M. H.) and ES004106 and ES002614 (to M. Smerdon, Washington State University)), from the National Institute of Environmental Health Sciences, and the Intramural Research Program at the National Institutes of Health, NIA. The authors declare that they have no conflicts of interest with the contents of this article.
- AP
- apurinic/apyrimidinic
- APE1
- AP endonuclease 1
- BER
- base excision repair
- NCP
- nucleosome core particle
- AAG
- alkyl adenine DNA glycosylase.
References
- 1. Lindahl T. (1993) Instability and decay of the primary structure of DNA. Nature 362, 709–715 [DOI] [PubMed] [Google Scholar]
- 2. Brooks S. C., Adhikary S., Rubinson E. H., Eichman B. F. (2013) Recent advances in the structural mechanisms of DNA glycosylases. Biochim. Biophys. Acta 1834, 247–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Loeb L. A., Preston B. D. (1986) Mutagenesis by apurinic/apyrimidinic sites. Annu. Rev. Genet. 20, 201–230 [DOI] [PubMed] [Google Scholar]
- 4. Li M., Wilson D. M., 3rd. (2014) Human apurinic/apyrimidinic endonuclease 1. Antioxid. Redox Signal. 20, 678–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brenerman B. M., Illuzzi J. L., Wilson D. M., 3rd. (2014) Base excision repair capacity in informing healthspan. Carcinogenesis 35, 2643–2652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wilson S. H., Kunkel T. A. (2000) Passing the baton in base excision repair. Nat. Struct. Biol. 7, 176–178 [DOI] [PubMed] [Google Scholar]
- 7. Dianov G. L., Parsons J. L. (2007) Co-ordination of DNA single strand break repair. DNA Repair 6, 454–460 [DOI] [PubMed] [Google Scholar]
- 8. Xanthoudakis S., Smeyne R. J., Wallace J. D., Curran T. (1996) The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc. Natl. Acad. Sci. U.S.A. 93, 8919–8923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fung H., Demple B. (2005) A vital role for Ape1/Ref1 protein in repairing spontaneous DNA damage in human cells. Mol. Cell 17, 463–470 [DOI] [PubMed] [Google Scholar]
- 10. Izumi T., Brown D. B., Naidu C. V., Bhakat K. K., Macinnes M. A., Saito H., Chen D. J., Mitra S. (2005) Two essential but distinct functions of the mammalian abasic endonuclease. Proc. Natl. Acad. Sci. U.S.A. 102, 5739–5743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Meira L. B., Devaraj S., Kisby G. E., Burns D. K., Daniel R. L., Hammer R. E., Grundy S., Jialal I., Friedberg E. C. (2001) Heterozygosity for the mouse Apex gene results in phenotypes associated with oxidative stress. Cancer Res. 61, 5552–5557 [PubMed] [Google Scholar]
- 12. Luger K., Mäder A. W., Richmond R. K., Sargent D. F., Richmond T. J. (1997) Crystal structure of the nucleosome core particle at 2.8-Å resolution. Nature 389, 251–260 [DOI] [PubMed] [Google Scholar]
- 13. Odell I. D., Wallace S. S., Pederson D. S. (2013) Rules of engagement for base excision repair in chromatin. J. Cell. Physiol. 228, 258–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nilsen H., Lindahl T., Verreault A. (2002) DNA base excision repair of uracil residues in reconstituted nucleosome core particles. EMBO J. 21, 5943–5952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Beard B. C., Wilson S. H., Smerdon M. J. (2003) Suppressed catalytic activity of base excision repair enzymes on rotationally positioned uracil in nucleosomes. Proc. Natl. Acad. Sci. U.S.A. 100, 7465–7470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Prasad A., Wallace S. S., Pederson D. S. (2007) Initiation of base excision repair of oxidative lesions in nucleosomes by the human, bifunctional DNA glycosylase NTH1. Mol. Cell. Biol. 27, 8442–8453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cole H. A., Tabor-Godwin J. M., Hayes J. J. (2010) Uracil DNA glycosylase activity on nucleosomal DNA depends on rotational orientation of targets. J. Biol. Chem. 285, 2876–2885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hinz J. M., Rodriguez Y., Smerdon M. J. (2010) Rotational dynamics of DNA on the nucleosome surface markedly impact accessibility to a DNA repair enzyme. Proc. Natl. Acad. Sci. U.S.A. 107, 4646–4651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Menoni H., Shukla M. S., Gerson V., Dimitrov S., Angelov D. (2012) Base excision repair of 8-oxoG in dinucleosomes. Nucleic Acids Res. 40, 692–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rodriguez Y., Smerdon M. J. (2013) The structural location of DNA lesions in nucleosome core particles determines accessibility by base excision repair enzymes. J. Biol. Chem. 288, 13863–13875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hinz J. M. (2014) Impact of abasic site orientation within nucleosomes on human APE1 endonuclease activity. Mutat. Res. 766, 19–24 [DOI] [PubMed] [Google Scholar]
- 22. Odell I. D., Barbour J. E., Murphy D. L., Della-Maria J. A., Sweasy J. B., Tomkinson A. E., Wallace S. S., Pederson D. S. (2011) Nucleosome disruption by DNA ligase III-XRCC1 promotes efficient base excision repair. Mol. Cell. Biol. 31, 4623–4632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li Q., Wrange O. (1993) Translational positioning of a nucleosomal glucocorticoid response element modulates glucocorticoid receptor affinity. Genes Dev. 7, 2471–2482 [DOI] [PubMed] [Google Scholar]
- 24. Li Q., Wrange O. (1995) Accessibility of a glucocorticoid response element in a nucleosome depends on its rotational positioning. Mol. Cell. Biol. 15, 4375–4384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cabelof D. C. (2012) Haploinsufficiency in mouse models of DNA repair deficiency: modifiers of penetrance. Cell. Mol. Life Sci 69, 727–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Illuzzi J. L., Harris N. A., Manvilla B. A., Kim D., Li M., Drohat A. C., Wilson D. M., 3rd. (2013) Functional assessment of population and tumor-associated APE1 protein variants. PloS One 8, e65922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hadi M. Z., Coleman M. A., Fidelis K., Mohrenweiser H. W., Wilson D. M., 3rd. (2000) Functional characterization of Ape1 variants identified in the human population. Nucleic Acids Res. 28, 3871–3879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lowary P. T., Widom J. (1998) New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 276, 19–42 [DOI] [PubMed] [Google Scholar]
- 29. Fernandez A. G., Anderson J. N. (2007) Nucleosome positioning determinants. J. Mol. Biol. 371, 649–668 [DOI] [PubMed] [Google Scholar]
- 30. Libertini L. J., Small E. W. (1980) Salt induced transitions of chromatin core particles studied by tyrosine fluorescence anisotropy. Nucleic Acids Res. 8, 3517–3534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nguyen L. H., Barsky D., Erzberger J. P., Wilson D. M., 3rd. (2000) Mapping the protein-DNA interface and the metal-binding site of the major human apurinic/apyrimidinic endonuclease. J. Mol. Biol. 298, 447–459 [DOI] [PubMed] [Google Scholar]
- 32. Erzberger J. P., Barsky D., Schärer O. D., Colvin M. E., Wilson D. M., 3rd. (1998) Elements in abasic site recognition by the major human and Escherichia coli apurinic/apyrimidinic endonucleases. Nucleic Acids Res. 26, 2771–2778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. O'Connor T. R. (1993) Purification and characterization of human 3-methyladenine-DNA glycosylase. Nucleic Acids Res. 21, 5561–5569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Roy R., Biswas T., Hazra T. K., Roy G., Grabowski D. T., Izumi T., Srinivasan G., Mitra S. (1998) Specific interaction of wild-type and truncated mouse N-methylpurine-DNA glycosylase with ethenoadenine-containing DNA. Biochemistry 37, 580–589 [DOI] [PubMed] [Google Scholar]
- 35. Baldwin M. R., O'Brien P. J. (2010) Nonspecific DNA binding and coordination of the first two steps of base excision repair. Biochemistry 49, 7879–7891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim W. C., Ma C., Li W. M., Chohan M., Wilson D. M., 3rd, Lee C. H. (2014) Altered endoribonuclease activity of apurinic/apyrimidinic endonuclease 1 variants identified in the human population. PloS One 9, e90837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chohan M., Mackedenski S., Li W. M., Lee C. H. (2015) Human apurinic/apyrimidinic endonuclease 1 (APE1) has 3′ RNA phosphatase and 3′ exoribonuclease activities. J. Mol. Biol. 427, 298–311 [DOI] [PubMed] [Google Scholar]
- 38. Parikh S. S., Mol C. D., Slupphaug G., Bharati S., Krokan H. E., Tainer J. A. (1998) Base excision repair initiation revealed by crystal structures and binding kinetics of human uracil-DNA glycosylase with DNA. EMBO J. 17, 5214–5226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Parikh S. S., Putnam C. D., Tainer J. A. (2000) Lessons learned from structural results on uracil-DNA glycosylase. Mutat. Res. 460, 183–199 [DOI] [PubMed] [Google Scholar]
- 40. Carey D. C., Strauss P. R. (1999) Human apurinic/apyrimidinic endonuclease is processive. Biochemistry 38, 16553–16560 [DOI] [PubMed] [Google Scholar]
- 41. Mol C. D., Izumi T., Mitra S., Tainer J. A. (2000) DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination (corrected). Nature 403, 451–456 [DOI] [PubMed] [Google Scholar]
- 42. Zharkov D. O., Mechetin G. V., Nevinsky G. A. (2010) Uracil-DNA glycosylase: structural, thermodynamic and kinetic aspects of lesion search and recognition. Mutat. Res. 685, 11–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hedglin M., O'Brien P. J. (2008) Human alkyladenine DNA glycosylase employs a processive search for DNA damage. Biochemistry 47, 11434–11445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vasudevan D., Chua E. Y., Davey C. A. (2010) Crystal structure of nucleosome core particle composed of the widom 601 DNA sequence. J. Mol. Biol. 403, 1–10 [DOI] [PubMed] [Google Scholar]