

Abstract
IL-15 is currently undergoing clinical trials to assess its efficacy for treatment of advanced cancers. The combination of IL-15 with soluble IL-15Rα generates a complex termed IL-15 superagonist (IL-15 SA) that possesses greater biological activity than IL-15 alone. IL-15 SA is considered an attractive antitumor and antiviral agent because of its ability to selectively expand NK and memory CD8+ T (mCD8+ T) lymphocytes. However, the adverse consequences of IL-15 SA treatment have not been defined. In this study, the effect of IL-15 SA on physiologic and immunologic functions of mice was evaluated. IL-15 SA caused dose- and time-dependent hypothermia, weight loss, liver injury, and mortality. NK (especially the proinflammatory NK subset), NKT, and mCD8+ T cells were preferentially expanded in spleen and liver upon IL-15 SA treatment. IL-15 SA caused NK cell activation as indicated by increased CD69 expression and IFN-γ, perforin, and granzyme B production, whereas NKT and mCD8+ T cells showed minimal, if any, activation. Cell depletion and adoptive transfer studies showed that the systemic toxicity of IL-15 SA was mediated by hyperproliferation of activated NK cells. Production of the proinflammatory cytokine IFN-γ, but not TNF-α or perforin, was essential to IL-15 SA–induced immunotoxicity. The toxicity and immunological alterations shown in this study are comparable to those reported in recent clinical trials of IL-15 in patients with refractory cancers and advance current knowledge by providing mechanistic insights into IL-15 SA–mediated immunotoxicity.
Introduction
Interleukin-15 is a four α-helix bundle cytokine produced constitutively by multiple cell types including dendritic cells, monocytes, macrophages, and epithelial cells of various origins (1, 2). IL-15 can be induced by stimulation with endotoxin, type I (IFN-α/β) and type II (IFN-γ) IFNs, dsRNA (3), and infection with viruses (4). It is a pluripotent cytokine that facilitates the generation, proliferation, and function of NK, NKT, and memory CD8+ T (mCD8+ T) cells as well as intestinal intraepithelial lymphocytes, as evidenced by the deficiency of those cells in IL-15−/− and IL-15Rα−/− mice (5, 6). Administration of exogenous IL-15 facilitates the expansion of both NK and CD8+ T cell populations, both of which play important roles in anticancer and antiviral immunosurveillance (6–9). The target cell specificity of IL-15 provides the possibility of it being superior to other cytokines as an agent to enhance antitumor and antiviral immunity (7, 9, 10). As such, IL-15 has been used to augment the efficacy of HIV vaccines and as an anticancer agent (7, 11, 12). Treatment with IL-15 alone, or as an adjuvant in antitumor vaccines, has shown efficacy in several experimental cancer models (13–16). Also, IL-15 administration has been shown to enhance bone marrow repopulation after allogeneic bone marrow transplantation (17). In cancer clinical trials, IL-15 has been administered alone and in combination with tumor-infiltrating lymphocytes (18). A recent first-in-human trial of recombinant human IL-15 in cancer patients showed clearance of lung lesions in patients with malignant melanoma (19). The toxicity profile for IL-15 was also defined and included fever, grade 3 hypotension, and liver injury. The authors reported expansion of peripheral blood NK cell numbers and a spike in plasma IFN-γ concentrations in patients receiving IL-15 treatment. However, the mechanisms by which IL-15 mediates toxicity were not provided and are difficult to determine in human models.
IL-15 uses a unique mechanism of action referred to as trans-presentation. As opposed to other cytokines that are secreted in soluble form, most IL-15 is transported to the surface of IL-15–producing cells (i.e., dendritic cells and monocytes) in complex with a unique high-affinity receptor, IL-15Rα. The IL-15/IL-15Rα complex is trans-presented to opposing NK, NKT, and mCD8+ T cells expressing IL-2/IL-15Rβ and the common γ-chain (20–24). In light of this mechanism of delivery, Rubinstein et al. (25) discovered that the combination of IL-15 with the IL-15Rα subunit in solution produced a compound with significantly longer half-life and greater biological activity than native IL-15. The resulting IL-15/IL-15Rα complex has been termed IL-15 superagonist (IL-15 SA). Recent preclinical studies showed that IL-15 SA is more effective than IL-15 in causing regression of established melanoma and pancreatic cancer in mice (26, 27). IL-15/IL-15Rα fusion proteins and IL-15 SA–expressing adenovirus expression systems have also been efficacious in experimental tumor models (28–30). These promising preclinical studies have generated robust interest in the application of IL-15 SA as an anticancer agent. However, little is known about the in vivo toxicity and dose limitations of IL-15 SA.
In the current study, the dose- and time-dependent effects of IL-15 SA treatment on physiological and immunological functions were examined in mice. Systemic administration of IL-15 SA caused significant hypothermia, weight loss, liver injury, and mortality. The immunotoxicity induced by IL-15 SA was mediated by systemic expansion of NK cells, which exhibited an activated phenotype characterized by upregulation of CD69, IFN-γ, granzyme B, and perforin. IL-15 SA–induced immunotoxicity was dependent on production of IFN-γ, a proinflammatory cytokine predominantly produced by NK cells after IL-15 SA treatment. This study provides new insights into the mechanisms of IL-15 SA–induced immunotoxicity that will be important to consider as IL-15 and IL-15 SA advance through clinical drug development.
Materials and Methods
Mice
Female, 8- to 12-wk-old C57BL/6Tac and Rag2/IL-2rg double knockout (KO) mice (Rag2−/−γc−/−) were purchased from Taconic Farms (Hudson, NY). Female, 8- to 12-wk-old homozygous CD8 null mice (B6.129S2-Cd8atm1Mak/J, CD8KO), homozygous IFN-γ null mice (B6.129S7-Ifngtm1Ts/J, IFN-γ KO), homozygous CD1d null mice (B6.129S6-Cd1d1/Cd1d2tm1Spb/J, CD1d KO), homozygous Perforin null mice (C57BL/6-Prf1tm1Sdz/J, Pfn KO), homozygous TNF-α null mice (B6.129S6-Tnftm1Gkl/J), and wild type C57BL/6J (WT) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). WT C57BL/6J mice served as controls in experiments using the KO strains. NK and NKT cells were depleted in mice by i.p. injection with anti-asialoGM1 IgG (50 μg/mouse; Cedarlane Laboratories, Hornby, ON, Canada) or anti-NK1.1 IgG (clone PK136, 50 μg/mouse; eBioscience, San Diego, CA) at 24 h prior to initiation of IL-15SA treatment. CD8 T cells were depleted by treatment with anti-CD8α IgG (clone 53-6.7, 50 μg/mouse; eBioscience) at 24 h prior to IL-15 SA treatment. Isotype-specific or nonspecific IgG served as control in all Ab-induced leukocyte depletion experiments. All studies were approved by the Institutional Animal Care and Use Committee at Vanderbilt University and complied with the National Institutes of Health’s Guide for the Care and Use of Experimental Animals.
IL-15 SA preparation
Recombinant mouse IL-15 was purchased from eBioscience (catalog number 34-8151-85). Mouse IL-15 Rα subunit Fc chimera (IL-15 Ra) was purchased from R&D Systems (Minneapolis, MN; catalog number 551-MR-100). For preparation of IL-15 SA, 20 μg IL-15 and 90 μg IL-15Ra were incubated in 400 μl sterile PBS at 37°C for 20 min to form the IL-15/IL-15 Ra complex. The complex was then diluted with sterile PBS to reach a concentration of 2 μg IL-15/ml, then aliquoted, and frozen. The IL-15 SA doses reported in the study are based on the amount of IL-15 present in the IL-15 SA complex.
IL-15 SA treatment protocols
In dose finding studies, mice received i.p. injections of IL-15 SA at doses of 0, 0.5, 1, or 2 μg for 4 d (days 0–3). Rectal temperature and body weight were measured at 24 h after the fourth injection. In time course studies, mice were treated with 2 μg IL-15 SA for 4 d. Rectal temperature and body weight were measured daily throughout the treatment period. On day 4, spleen and liver were harvested for measurement of leukocyte numbers and activation. Blood was obtained in heparinized syringes after carotid artery laceration in anesthetized mice breathing 2% isoflurane in oxygen by nose cone. Arterial blood gases were measured using i-Stat CG4+ cartridges (Abaxis, Union City, CA). For serum preparation, supernatant was obtained from nonheparinized whole blood after centrifugation (2000 × g for 10 min) to remove the blood clots. Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) concentrations were measured as indices of acute liver injury. Blood urea nitrogen (BUN) and creatinine concentrations were measured as indices of renal injury. ALT, AST, BUN, and creatinine concentrations were measured in the Translational Pathology Core Laboratory at Vanderbilt University using an ACE Alera Chemistry Analyzer (Alfa Wassermann West Caldwell, NJ).
Flow cytometry
Splenocytes and hepatic leukocytes were isolated as described previously. Briefly, spleens were harvested, placed in 35-mm dishes containing RPMI 1640 medium with 10% FBS, and homogenized by smashing with the plunger from a 10-ml syringe. The homogenate was passed through a 70-μm cell strainer, and erythrocytes were lysed with RBC Lysis Buffer (Sigma-Aldrich, St. Louis, MO). The remaining cells were counted using TC20 Automated Cell Counter (Bio-Rad, Hercules, CA) and centrifuged (300 × g for 5 min), and the cell pellet was resuspended in PBS. Livers were harvested after perfusion, which was achieved by cutting of the hepatic portal vein, insertion of a 25-g needle into the left ventricle of the heart and perfusion with 10 ml PBS. Harvested livers were smashed with the plunger from a 10-ml syringe and passed through a 70-μm cell strainer. The hepatic homogenate was washed, resuspended with 10 ml 37.5% Percoll Plus (GE Healthcare Life Sciences), and centrifuged (680 × g for 12 min at room temperature). The supernatant containing hepatocytes was discarded, erythrocytes were lysed, and the resulting mononuclear cells were counted using TC20 Automated Cell Counter (Bio-Rad).
For surface marker staining, cells were suspended in PBS (1 × 107 cells/ml) and incubated with anti-mouse CD16/32 (1 μl/ml; eBioscience) for 5 min to block nonspecific FcR-mediated Ab binding. One million cells were then transferred into polystyrene tubes. Fluorochrome-conjugated Abs or isotype controls (0.5 μg /tube) were added, incubated (4°C) for 30 min, and washed with 2 ml cold PBS. After centrifugation (300 × g for 5 min), cell pellets were fixed with 250 μl 1% paraformaldehyde. Abs used for surface marker labeling included CD3-Alexa Fluor 488, NK1.1-PE-Cy7, CD8-FITC, CD4-FITC, CD44-PE-Cy5, CD19-PE, CD69-PE, TCRγ-PE, NKG2D-PE, NKp46-PE, CD49a-PE, Trail-PE, DX5-allophycocyanin, CD11b-PE, and CD27-allophycocyanin (eBioscience; BD Biosciences, San Diego). CD1d/α-Galcer tetramers were provided from the National Institutes of Health Tetramer Core Facility (Emory University, Atlanta, GA). Appropriate isotype-specific Abs were used as controls.
For intracellular staining, spleens were harvested at 4 h after the last injection of IL-15 SA. Cell suspensions were labeled with fluorochrome-conjugated Abs to surface markers as described above. Cells were then fixed and permeabilized with Cytofix/Cytoperm Plus (BD Biosciences, 250 μl/tube) for 20 min at 4°C. After washing with BD Perm/Wash solution, anti–IFN-γ-PE (clone XMG1.2; eBioscience) was used to detect intracellular IFN-γ production. Intracellular granzyme B and perforin were stained with anti–granzyme B-PE (clone FGB12; Invitrogen) and anti–mouse perforin-PE (eBioscience). Fluorochrome-conjugated isotype-specific IgGs served as controls. All samples were analyzed using an Accuri C6 flow cytometer (BD Biosciences). Data were analyzed using Accuri C6 software.
Immunohistochemistry
Immunohistochemical (IHC) staining of NK, NKT, and T lymphocytes as well as macrophages in the marginal zone of spleen was performed on 5 μM cryosections of tissue. Primary Abs included polyclonal goat anti-mouse NKp46 (catalog number AF2225; R&D Systems), biotinylated rat anti-metallophilic macrophages Ab (clone MOMA-1; catalog number CL89149B; Cedarlane Laboratories) and biotinylated hamster anti-TCRβ Ab (clone H57-597, catalog number 553169; BD Biosciences). Isotype IgG Abs or donkey serum was used as controls to stain serial frozen sections. For primary biotinylated Ab staining, avidin-biotin complex alkaline phosphatase (ABC-AP Complex, catalog number AK-5000; Vector Laboratories, Burlingame, CA) was added for 1 h and then developed with Vector Red Kit containing Alkaline Phosphatase Substrate (catalog number SK-5100; Vector Laboratories). For goat anti-mouse NKp46 staining, the secondary Donkey anti-goat-biotin was added followed by incubation with avidin-biotin complex peroxidase (ABC-Standard Complex, catalog number PK-6100; Vector Laboratories) and development with DAB-Solution (SK-4100; Vector Laboratories). Sections were counterstained in Meyer’s hematoxylin, and slides were mounted with Permount.
Adoptive cell transfer
Splenic leukocytes were harvested as described previously. Splenic NK cells were isolated using mouse NK isolation kit II (Miltenyi Biotec). Briefly, splenocytes were incubated with a biotin–Ab mixture that binds macrophages/monocytes, dendritic cells, erythrocytes, B cells, and CD3+ T cells. Anti-biotin magnetic microbeads were added to bind Ab-bound cells. The suspension was passed through columns in a magnetic field that bind iron microbead Ab–laden cells and prevent their passage. NK cells, which do not bind the specified Abs, were washed through columns and collected. The enriched cell preparation was resuspended in sterile PBS followed by retro-orbital injection (1.0 × 106 cell/mouse) into Rag2−/−γc−/− mice.
Statistics
All data were analyzed using GraphPad Prism software. All values are presented as the mean ± SEM, except for body temperature, ALT and AST, for which median values are designated. A Student t test was used to examine the difference between paired vehicle and IL-15 SA groups. Data from multiple group experiments were analyzed using one-way ANOVA, followed by a post hoc Tukey’s test to compare groups. A p value < 0.05 was considered statistically significant for all experiments.
Results
Treatment with 2 μg IL-15 SA for 4 d causes toxicity in mice
In a dose escalation study, WT mice were treated with 0, 0.5, 1, or 2 μg IL-15 SA for 4 consecutive days (days 0–3). Body temperature was measured on days 0–4. Significant hypothermia was observed on days 3 and 4 in mice that received 2 μg IL-15 SA but not in mice that received the 0-, 0.5-, or 1-μg doses (Fig. 1A). Mice treated with 2 μg IL-15 SA showed 20% mortality (8 of 10 surviving) on day 4 (Fig. 1A). There was significant weight loss in mice treated with 2 μg IL-15 SA, beginning on day 2 of treatment and progressing out to day 4 (Fig. 1B). IL-15 SA treatment also caused elevation of liver enzymes (ALT and AST) in serum, indicative of acute hepatocellular injury (Fig. 1C). However, IL-15 SA treatment did not trigger significant acute kidney injury or lung injury because plasma BUN and creatinine concentrations and blood PaO2/FiO2 ratios were not altered in IL-15 SA–treated mice (data not shown). Spleen weight more than doubled in IL-15 SA–treated mice compared with control (Fig. 1D). Liver weight was not different between groups (data not shown).
FIGURE 1.

IL-15 SA treatment causes systemic toxicity and the study of kinetics of IL-15 SA toxicity. (A) In dose escalation studies, WT C57BL/6 mice received i.p. injection of 0, 0.5, 1, and 2 μg IL-15 SA for 4 consecutive days (days 0–3). Body temperature and mortality were recorded daily on days 0–4. (B) To assess the temporal effect of IL-15 SA, WT mice received daily administration of 2 μg IL-15 SA for 4 d. Untreated WT mice served as control. Body weight was measured daily from days 0 to 4. Values represent body weight changes compared with the initial day 0 measurements. (C) Serum AST and ALT concentrations were measured at 24 h after the fourth i.p. injection of 2 μg IL-15 SA. Vehicle-treated mice served as control. (D) Upon 4 d of 2 μg IL-15 SA treatment, spleens were harvested from WT mice. Untreated WT mice were included as control. Spleen weights were measured in the two groups. (E) To study the kinetics of IL-15 SA toxicity, WT mice were treated with 2 μg IL-15 SA treatment for 4 d. Body temperature was measured at 24 and 48 h as well as 1, 2, 3, 4, and 7 wk after the last treatment of IL-15 SA. n = 8–10 mice/group. Data are representative of two to four separate experiments. *p < 0.05 compared with designated control.
The toxicity and lymphocyte expansion caused by IL-15 SA treatment was transient. Body temperature recovered to normal levels within 2 d after stopping treatment (day 5) (Fig. 1E).
IL-15 SA treatment expands NK, NKT, and mCD8 T cells in spleen and liver
Further studies were undertaken to fully characterize IL-15 SA–mediated lymphocyte expansion. After 4 d of 2 μg IL-15 SA treatment, splenic NK cells (CD3−NK1.1+) increased ∼16.3-fold; NKT cells (CD3+NK1.1+) expanded ∼13.1-fold; mCD8+ T cells (CD8+CD44high) were increased ∼11.5-fold, respectively, compared with vehicle-treated mice (Fig. 2A–C). However, naive CD8+ T (CD8+CD44low), CD4+ T, and B cells in spleen showed a decrease in percentage and no change in total numbers following IL-15 SA treatment (Fig. 2B, 2C). A similar expansion of hepatic NK, NKT, and mCD8+ T cells was observed in IL-15 SA–treated mice (Fig. 2D–F).
FIGURE 2.

IL-15 SA treatment elicits expansion of NK, NKT, and memory phenotype CD8+ T lymphocytes in vivo. WT mice were treated with vehicle or 2 μg IL-15 SA for 4 d. At 24 h following the last treatment, splenic and liver NK (CD3−NK1.1+), NKT (CD3+NK1.1+), and memory CD8+ T (CD8+CD44high) lymphocyte numbers were determined using flow cytometry. Dot plots (A and D) are representative of results obtained from 9 to 15 mice/group. The bar graphs show the relative (B and E) and total numbers (C and F) of splenic and hepatic NK, NKT, and memory CD8 T as well as naive CD8+ T (CD8+CD44low), CD4 (CD4+), and B (CD3−CD19+) lymphocytes in IL-15 SA–treated (▪) and vehicle-treated groups (□). Data are representative of three to four separate experiments. n = 9–15 mice/group. *p < 0.05 compared with vehicle control. WT mice received daily administration of vehicle or 2 μg IL-15 SA for 4 d. Spleens (G) and livers (H) were then harvested on the next day for H&E (top panels, original magnifications ×100 in spleen sections and ×400 in liver sections) and IHC (bottom panels, original magnifications ×100 in spleen sections and ×400 in liver sections) staining. In IHC-stained sections of spleen, NK and NKT cells were identified by NKp46 expression and are stained brown; macrophages in the marginal zone stained with anti–MOMA-1 and are shown in pink. In IHC staining of liver sections, NK (NKp46 positive, brown), T (TCR-β positive, pink), and NKT (NKp46 and TCR-β double positive, brown and pink) cells are shown, respectively. Olympus BX43 microscope and Olympus digital color camera DP73 were used for acquisition of the images.
H&E staining of spleen sections showed that the red pulp was filled with leukocytes in response to IL-15 SA treatment (Fig. 2G). IHC staining identified the majority of leukocytes in red pulp as NKp46+ (Fig. 2G). In liver, increased numbers of leukocytes were present in liver sinusoids and especially accumulated around the central vein in IL-15 SA–treated mice (Fig. 2H). IHC staining of NK (NKp46+), NKT (NKp46+TCRβ+), and T (TCRβ+) lymphocytes showed a substantial number of NK (brown), NKT (brown and pink), and T (pink) cells were located in the liver sinusoids and around the central vein in response to treatment with IL-15 SA (Fig. 2H). Of note, NK and NKT cell numbers decreased to the baseline level by 7 d after cessation of IL-15 SA treatment (Supplemental Fig. 1). mCD8+ T cells also declined after stopping IL-15 SA treatment but remained elevated at 7 wk after cessation of treatment (Supplemental Fig. 1).
IL-15 SA treatment promotes NK and NKT subset expansion and activation
NK cell subsets were characterized based on CD11b and CD27 expression and classified as immature (I, CD11lowCD27high), proinflammatory (II, CD11bhighCD27high), or cytotoxic (III, CD11bhighCD27low). IL-15 SA treatment promoted expansion of all subsets but preferentially expanded the proinflammatory CD11bhighCD27high subset (∼23.9-fold increase in spleen and ∼115-fold increase in liver) (Fig. 3A–F). In liver, there are two recently identified NK populations, classical NK cells (CD3−NK1.1+DX5+CD49a−Trail−), resembling those in spleen, and tissue-resident NK cells (CD3−NK1.1+DX5-CD49a+/Trail+) with a “memory-like” phenotype. In the experiment, a combination of anti-CD49a and anti-Trail Abs conjugated with the same fluorochrome was used to distinguish classical (double negative) and tissue-resident NK cells (CD49a or Trail positive). Both hepatic NK subsets were expanded by IL-15 SA treatment, but the classical hepatic NK cell population showed greater expansion than tissue-resident NK cells (Fig. 3G–I).
FIGURE 3.

Characterization of NK and NKT subsets following IL-15 SA treatment. Upon treatment with vehicle or 2 μg IL-15 SA for 4 d, spleens and livers were harvested on day 5. In the representative histograms, splenic (A) and hepatic (D) NK cells are divided into four subpopulations based on CD11b and CD27 expression, namely precursor (CD11blowCD27low), immature (I, CD11blowCD27high), mature proinflammatory (II, CD11bhighCD27high), and mature cytotoxic (III, CD11bhighCD27low) NK cells. The percentage and total numbers of NK cell subsets in spleen and liver are shown graphically in (B), (C), (E), and (F). n = 6–10 mice/group. In liver, NK cells are specifically classified into two separate subsets, namely classical (NK1.1+DX5+CD3−CD49a−Trail−) NK cells that resemble those in spleen and tissue-resident (NK1.1+DX5−CD3−CD49a+/Trail+) NK cells that mediate Ag-specific memory-like responses. Of note, liver tissue-resident DX5−CD49a+ NK cells express high levels of Trail, whereas classical DX5+CD49a− NK cells exhibit minimal expression of Trail. Thus, a combination of anti-CD49a and anti-Trail Abs conjugated with the same fluorochrome was used to distinguish classical (double negative) and tissue-resident NK cells (CD49a or Trail positive). The representative dot plot, percentage, and total numbers of NK cell subsets in liver are shown in (G)–(I). n = 5 mice/group. In representative dot plots, type I (CD3+NK1.1+/−α-Galcer-CD1d tetramer+) and type II NKT subsets (CD3+NK1.1+α-Galcer-CD1d tetramer−) in spleen (J) and liver (M) are shown in vehicle- and IL-15 SA–treated groups. The percentage (K and N) and total numbers (L and O) of each subset are shown in bar graphs. Data are representative of two to three separate experiments. *p < 0.05 compared with vehicle control.
NKT cells are also heterogeneous and composed of two major subsets: type I (invariant TCR, CD1d/α-Galcer reactive) and type II (variant TCR, not reactive to CD1d/α-Galcer). Both type I (CD3+NK1.1+/− α-Galcer-CD1d tetramer+) and type II NKT subsets (CD3+NK1.1+α-Galcer-CD1d tetramer−) were examined by flow cytometry. The treatment with IL-15 SA significantly increased the number of two NKT subsets in spleen and liver (Fig. 3J–O).
The impact of IL-15 SA treatment on NK cell activation and function was determined by examining expression of the early surface activation marker CD69 and production of IFN-γ, granzyme B, and perforin (Fig. 4A). Treatment with IL-15 SA induced increased expression of CD69 (Fig. 4A) on splenic NK cells. IL-15 SA treatment also elicited IFN-γ, granzyme B, and perforin production by NK cells (Fig. 4A). However, IL-15 SA did not upregulate CD69 expression or IFN-γ production by NKT cells (Fig. 4B). There is no augmented CD69 expression and slight increase in IFN-γ production by mCD8+ T cells upon IL-15 SA treatment (Fig. 4C).
FIGURE 4.
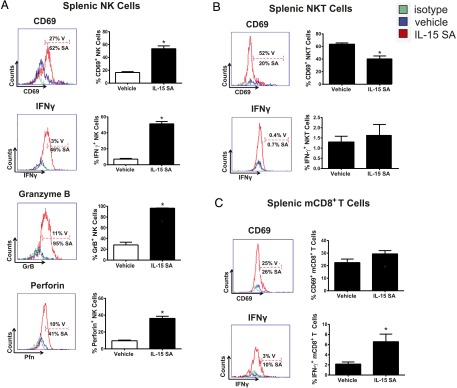
Characterization of NK, NKT and memory CD8+ T cell activation following IL-15 SA treatment. Representative histograms show the percentage of splenic NK cells (A) expressing CD69 on the surface as well as production of intracellular IFN-γ, granzyme B, and perforin from vehicle (blue)- and IL-15 SA (red)–treated mice. The gates were drawn based on isotype control staining (green). The bar graphs shown on the right quantify representative data with 9–10 mice per group. The activation status of NKT and memory CD8+ T cell upon IL-15 SA treatment was also analyzed. The representative histograms show expression of CD69 by splenic NKT (CD3+NK1.1+) (B) and mCD8+ (CD8+CD44high) T cells (C) from vehicle (blue)- and IL-15 SA (red)–treated mice. IFN-γ expression by NKT and mCD8+ T cells is also shown. Specific isotype staining (green) was determined as control. The percentage of CD69+ and IFN-γ+ cells in each group is shown in the figure. The bar graphs shown on the right quantify representative data with 8–10 mice/group. Data are representative of two to three separate experiments. *p < 0.05 compared with vehicle. SA, IL-15 SA; V, vehicle.
Depletion of NK cells reverses IL-15 SA–mediated toxicity in mice
Depletion of NK and NKT cells by treatment with anti-asialoGM1 or anti-NK1.1 prior to IL-15 SA treatment prevented development of hypothermia (Fig. 5A) but not the induction of liver injury evidenced by increased ALT and AST levels (Fig. 5B). To understand the specific contribution of invariant NKT cells to the toxicity of IL-15 SA, invariant NKT cell–deficient CD1d KO mice were treated with 2 μg IL-15 SA for 4 d. CD1d KO mice were not protected from IL-15 SA–induced hypothermia (Fig. 5C). Further studies were undertaken to determine the effect of CD8+ T cell depletion on IL-15 SA–induced toxicity. Total CD8+ T cells were depleted because it is not currently possible to selectively deplete mCD8+ T cells. Mice depleted of CD8+ T cells by treatment with anti-CD8α showed significantly more hypothermia in response to treatment with IL-15 SA compared with mice treated with nonspecific IgG (Fig. 5D). Likewise, CD8 KO mice showed significantly more hypothermia after IL-15 SA treatment than WT control mice (Fig. 5D). Upon IL-15 SA treatment, AST levels in WT mice treated with anti-CD8α were comparable to untreated mice and mice treated with nonspecific IgG. However, CD8 T cell–deficient mice showed an elevation of ALT levels in responses to IL-15 SA treatment, which was comparable to mice treated with IgG (Fig. 5E). Treatment of CD8 KO mice with anti-asialoGM1 reversed IL-15SA–induced hypothermia (Fig. 5F).
FIGURE 5.

Effect of lymphocyte depletion on IL-15 SA–induced toxicity. Mice were treated with 2 μg IL-15 SA for 4 d; rectal temperature was measured on day 5. (A) Mice were treated with anti-asialoGM1 or anti-NK1.1 IgG to deplete NK and NKT cells 1 d before initiation of 2 μg IL-15 SA treatment. (B) Serum AST and ALT concentrations in mice treated with anti-NK1.1 or nonspecific IgG were measured at 24 h after the fourth i.p. injection of 2 μg IL-15 SA. Vehicle-treated mice served as control. (C) CD1d KO mice were treated with 2 μg IL-15 SA. (D) CD8 T cells were depleted by treatment with anti-CD8α IgG at 24 h prior to IL-15 SA treatment or CD8 KO mice were treated with IL-15 SA. (E) Serum AST and ALT levels in mice treated with anti-CD8α IgG or nonspecific IgG were measured at 24 h after the last injection of IL-15 SA. (F) To deplete NK and NKT cells in CD8 KO mice, CD8 KO mice were treated with anti-asialoGM1 1 d before IL-15 SA treatment. Isotype-specific or nonspecific IgG served as control in Ab-induced leukocyte depletion experiments. WT mice served as control for experiments using genetically altered mice. NK (G and J), NKT (H and K), and mCD8+ T cells (I and L) from spleens of WT mice were characterized in mice treated with anti-asiaoGM1, anti-NK1.1, or anti-CD8α IgG as well as in CD8 KO mice. Untreated WT mice or WT mice treated with isotype matched IgG served as controls. n = 6–10 mice/group. Data are representative of two to three separate experiments. *p < 0.05 compared with untreated WT control, +p < 0.05 compared with IgG group or WT control, #p < 0.05 compared to CD8KO treated with IgG.
Analysis of splenic lymphocyte populations showed that treatment with anti-asialoGM1 or anti-NK1.1 nearly ablated IL-15 SA–induced NK and NKT cell expansion whereas mCD8 T cell expansion was unaffected (anti-asialoGM1) or enhanced (anti-NK1.1) (Fig. 5G–I). Treatment with anti-CD8α greatly attenuated IL-15 SA–induced expansion of CD8+ T cells and CD8+ T cells were absent in CD8 KO mice (Fig. 5L). Expansion of NK or NKT cells was not affected by anti-CD8α treatment and in CD8 KO mice (Fig. 5J, 5K).
Adoptive transfer of NK cells into Rag2−/−γc−/− mice re-establishes IL-15 SA toxicity
The contribution of NK cells to IL-15 SA–mediated toxicity was further demonstrated by adoptive transfer of NK cells from WT mice into Rag2−/−γc−/− mice (Fig. 6). Treatment of Rag2−/−γc−/− mice with IL-15 SA after adoptive transfer of WT NK cells elicited significant hypothermia and weight loss and showed 22% mortality (seven of nine surviving) (Fig. 6A, 6B). Rag2−/−γc−/− mice that were treated with IL-15 SA but did not receive NK cell adoptive transfer did not develop hypothermia or lose weight (Fig. 6A, 6B). Likewise, Rag2−/−γc−/− mice that received WT NK cell adoptive transfer and were treated with vehicle did not develop hypothermia or weight loss (Fig. 6A, 6B).
FIGURE 6.

Toxicity of IL-15 SA in Rag2−/−γc−/− mice receiving adoptive transfer of WT NK cells. WT NK cells (1.0 × 106 cells/mouse) were injected i.v. into Rag2−/−γc−/−mice 3 h before the initiation of IL-15 SA (2 μg) treatment. Rag2−/−γc−/− mice that received NK cell transfer and vehicle treatment or IL-15 SA treatment without NK cell transfer served as control. (A) Body temperature was measured at 8 d after NK cell transfer and IL-15 SA treatment. (B) Body weight was measured at the indicated time points. (C) Liver gross morphology and H&E-stained liver sections (original magnification ×400) at 8 d after treatment. Olympus BX43 microscope and Olympus digital color camera DP73 were used for acquisition of the H&E-stained images. (D) ALT and AST concentrations in serum at 8 d after adoptive transfer and IL-15 SA treatment. Splenocytes and hepatic leukocytes were harvested at 8 d after NK cell transfer and IL-15 SA treatment. The numbers of splenic and hepatic NK cells (E and H) and CD69 expression by NK cells (F, G, I, and J) was determined using flow cytometry. n = 6–9 mice/group. Data are representative of two separate experiments. *p < 0.05 compared with mice that did not receive NK cell transfer and were treated with IL-15 SA, +p < 0.05 compared with mice that received NK cell transfer but not IL-15 SA treatment.
Livers from Rag2−/−γc−/− mice that received adoptive transfer of NK cells and IL-15 SA treatment exhibited altered gross morphology and histology (Fig. 6C). The livers of Rag2−/−γc−/− mice that were treated with IL-15 SA but did not receive NK cell adoptive transfer as well as Rag2−/−γc−/− mice that received NK cell transfer and vehicle treatment had normal hepatic architecture (Fig. 6C). IL-15 SA–treated Rag2−/−γc−/− mice that were engrafted with NK cells exhibited hepatic inflammation and H&E-stained liver sections showed accumulation of leukocytes and destruction of normal liver sinusoidal architecture (Fig. 6C). Hepatocellular injury was present in those mice as evidenced by elevation of serum ALT and AST levels (Fig. 6D).
The number of NK cells in spleens and livers of engrafted Rag2−/−γc−/− mice significantly increased upon IL-15 SA treatment when compared with the other experimental groups (Fig. 6E, 6H). IL-15 SA treatment induced activation of engrafted NK cells as indicated by increased CD69 expression (Fig. 6F, 6G, 6I, 6J).
Ablation of IFN-γ reverses IL-15 SA–induced immunotoxicity
To determine the functional importance of IFN-γ, TNF-α and perforin/granzyme in the pathogenesis of IL-15 SA–induced immunotoxicity, IL-15 SA–induced hypothermia was examined in IFN-γ–, TNF-α–, and perforin-deficient mice (Fig. 7). IFN-γ KO mice did not develop significant hypothermia in response to IL-15 SA treatment compared with WT control mice (Fig. 7A). Treatment of TNF-α and Pfn KO mice with IL-15 SA induced hypothermia at a level that was not significantly different from WT mice (Fig. 7B, 7C).
FIGURE 7.
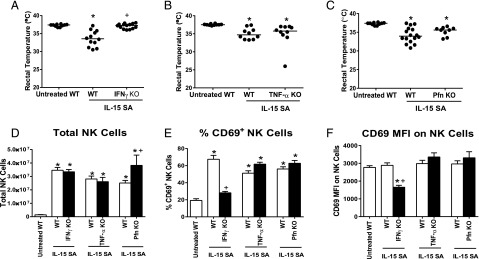
Effect of IFN-γ, TNF-α, or perforin deficiency on IL-15 SA–mediated immunotoxicity. IFN-γ KO, TNF-α KO, Pfn KO mice, and WT controls received 2 μg IL-15 SA treatment for 4 d. (A–C) Body temperature was measured at 24 h after the last IL-15 SA treatment. Untreated WT mice were included as control. Splenic NK numbers (D) and CD69 expression (E and F) were measured using flow cytometry. n = 9–16 mice/group. Data are representative of two to three separate experiments. *p < 0.05 compared with untreated WT control, +p < 0.05 compared with IL-15 SA–treated WT mice.
Compared with WT control mice, there was no difference in NK cell number in the spleens of IFN-γ and TNF-α KO but more NK cells in Pfn KO mice upon IL-15 SA treatment (Fig. 7D). However, NK cells failed to be activated by IL-15 SA stimulation in IFN-γ KO mice as indicated by the absence of increased CD69 expression (Fig. 7E, 7F). However, NK cell CD69 expression by NK cells from TNF-α and Pfn KO mice treated with IL-15 SA was increased and not different from WT treated with IL-15 SA (Fig. 7E, 7F).
NKT cell numbers were higher in IFN-γ and TNF-α KO mice and not different in Pfn KO mice in response to IL-15 SA treatment compared with WT mice. Upon IL-15 SA treatment, mCD8+ T cell numbers were higher in in IFN-γ KO mice and not different in TNF-α and Pfn KO mice in comparison with WT mice (Supplemental Fig. 2A, 2D, 2G).
NKT and mCD8 T cells in IFN-γ KO mice failed to be activated by IL-15 SA stimulation as indicated by lack of CD69 upregulation whereas activation of NKT and mCD8 T cells from TNF-α and perforin-deficient mice was similar to that observed in WT mice after IL-15 SA treatment (Supplemental Fig. 2B, 2C, 2E, 2F, 2H, 2I).
Discussion
Cancer immunotherapy is a rapidly evolving field of basic and clinical research. Recent advances show that deinhibition of lymphocytes by blockade of coinhibitory receptors such as PD-1 is effective in treating advanced cancers, which recently resulted in fast track approval of pembrolizumab for the treatment of metastatic melanoma (31–33). Cytokine-based treatments also hold promise for the treatment of cancer and viral diseases. IL-15 is an attractive drug candidate because of its selective actions on NK and mCD8+ T cells, both of which mediate anticancer and antiviral immunity (7, 9). Several papers have demonstrated the efficacy of IL-15 in experimental models of melanoma, pancreatic cancer, and multiple myeloma (29, 34). The recent paper by Conlon et al. (19) reported clearance of pulmonary lesions in patients with malignant melanoma that were treated with IL-15. However, IL-15 administration may also have untoward consequences. It has been implicated in the pathogenesis of acute lymphoblastic leukemia (35) and large granular lymphocyte leukemia (36). The administration of IL-15 also exacerbates graft-versus-host disease after allogeneic bone marrow transplantation (17). Thus, as IL-15 moves through drug development, it is important to understand its toxicity profile. IL-15 is known to cause considerable toxicity in nonhuman primates at higher doses. Adverse side effects included weight loss and skin rash (37–39). Most recently, Conlon et al. (19) reported hypotension, thrombocytopenia, liver injury, fever, and rigors in cancer patients receiving IL-15. Unlike IL-2, IL-15 did not cause vascular leak syndrome (40). In the current study, IL-15 SA treatment caused marked hypothermia and weight loss and a modest increase in liver enzymes in WT mice. Thus, the toxicity profile of IL-15 family immunobiologicals is similar among species. However, the cellular and molecular mechanisms by which IL-15 and its analogs mediate systemic toxicity had not been previously defined. The present study advances current knowledge by showing that NK cells and IFN-γ are central to IL-15 SA–mediated immunotoxicity.
Conlon et al. (19) reported expansion of NK and mCD8+ T cells in blood and markedly increased plasma IFN-γ concentrations in cancer patient receiving IL-15 treatment. However, a cause and effect relationship between increased NK cell numbers and IFN-γ production with IL-15–associated toxicity was not established. The present study provides a direct link between NK cell expansion and IFN-γ production with the adverse responses caused by IL-15 SA in mice. Of note, the dose of IL-15 SA applied to mice in our study is higher than the highest dose of IL-15 administered to patients in the cancer treatment trial (19). However, Conlon and colleagues administered IL-15 to patients for 12 consecutive days, whereas mice were treated for a period of 4 d in our study. Thus, we administered a higher dose but over a shorter period of time. Nevertheless, IL-15 SA induced physiologic dysfunction, weight loss, and liver injury in mice, a toxicity profile that was similar to that reported in humans. In addition, the toxicity observed in mice was more severe and characterized by 20% mortality. Thus, although species differences in sensitivity to IL-15 SA–induced toxicity may exist, we were able to provide mechanistic insights into IL-15 SA–mediated immunotoxicity that cannot be addressed in human studies.
Rubinstein et al. (25) used a dose of IL-15 SA (1.5 μg) in mice that was similar to the dose used in our study and was administered for 2 d rather than 4 d. Rubinstein et al. (25) observed NK and mCD8 T cell expansion that was similar to that reported in our study but did not mention toxic signs in mice under their treatment regimen. In our dose escalation study, we did not observe toxicity when administering IL-15 SA at 1 μg/day but observed toxicity at the 2-μg dose. Thus, it is likely that Rubinstein et al. (25) were using a dose that was slightly less than the toxic dose observed in our study and was administered over a shorter period of time (2 versus 4 d). These observations indicate that IL-15 SA–induced immunotoxicity is dose and time dependent.
IL-15 SA preferentially expanded the proinflammatory (CD11bhighCD27high) NK subset, which produces large amounts of cytokines, such as IFN-γ and TNF-α, and also possesses significant cytotoxic functions (41). NK cell expansion and activation appears to contribute to IL-15 SA-induced immunotoxicity since NK cell depletion ablated IL-15 SA-induced hypothermia and weight loss but did not modify the modest increase in liver enzymes observed in wild type mice. Interestingly, adoptive transfer of NK cells into Rag2γc KO mice followed by treatment with IL-15 SA caused significant hepatotoxicity characterized by gross pathology, marked hepatic leukocyte infiltration and liver enzyme elevations. The more profound liver injury observed in that model may reflect loss of the attenuating influence of CD8+ T cells, and possibly other lymphocyte populations, that are absent in Rag2γC KO mice. Our results show that CD8-deficient mice have an increased sensitivity to IL-15 SA-induced hypothermia, which suggests that CD8+ cells are able to temper IL-15 SA-induced injury.
IFN-γ–deficient, but not TNF-α– and perforin-deficient, mice were resistant to IL-15 SA–induced hypothermia and weight loss. Moreover, IFN-γ played an essential role in IL-15 SA–mediated NK cell activation as indicated by attenuation of NK cell activation in IFN-γ–deficient mice. Although TNF-α is also an important proinflammatory cytokine that is secreted by NK cells, it does not appear to play an important role in the immunotoxicity caused by IL-15 SA. That contention is further supported by our observation that IL-15 SA–mediated NK cell activation is independent of the presence of TNF-α. Taken together; our results indicate that NK cell–derived IFN-γ is an important mediator of IL-15 SA–induced immunotoxicity. IL-15, in conjunction with the monokines IL-12 and IL-18, is a potent inducer of IFN-γ secretion by NK cells (42). Previous studies have described a toxic role for IFN-γ during endotoxin-induced shock and polymicrobial sepsis (43, 44). NK cells facilitate the activation of macrophages, dendritic cells, and other APC populations to amplify proinflammatory responses through positive feedback mechanisms (45). The enhanced inflammatory response results in heightened cytokine secretion, leukocyte recruitment, and cytotoxic activity. However, the cellular interactions underlying IL-15 SA–induced toxicity remain to be fully defined.
The recent discovery of IL-15 SA advances the potential of IL-15–based therapy. IL-15 SA has more potent and prolonged action than native IL-15 and preliminary studies suggest enhanced efficacy in experimental models of cancer (25, 28). The mechanisms of IL-15 SA–mediated immunotoxicity and antitumor efficacy may be mediated through different cellular mechanisms. Although IL-15 SA–induced toxicity is mediated through expansion and activation of NK cells, the antitumor efficacy is reportedly dependent on expansion and activation of tumor resident mCD8+ T cells. Cheng et al. (30) reported that the efficacy of IL-15 SA in models of liver cancer is mediated through activation of tumor specific CD8+ T cell responses. Xu et al. (28) demonstrated the importance of mCD8+ T cells in the antitumor efficacy of an IL-15/IL-15Rα fusion protein in a murine model of multiple myeloma. Chang et al. (29) delivered IL-15 SA–expressing adenovirus in a model of hepatocellular carcinoma. However, they concluded that the antitumor effect of the IL-15 SA–adenovirus construct was mediated by NK cells. Thus, there is some controversy in the field. Last, the toxicity of IL-15 SA is transient. NK and NKT cell numbers declined rapidly after conclusion of IL-15 SA treatment and returned to baseline value by 1 wk after IL-15 SA treatment. Although mCD8 T cell numbers also declined after cessation of IL-15 SA treatment, levels of mCD8 T cells remained above baseline at 7 wk after completion of IL-15 SA treatment. Taken together, these findings provide new information regarding the cellular and molecular mechanisms underlying IL-15 SA–induced toxicity in vivo.
In conclusion, the current study demonstrates time- and dose-dependent toxicity of IL-15 SA in mice. Toxicity is characterized by systemic physiologic dysfunction reflected by the development of hypothermia and weight loss. Acute liver injury was also observed in IL-15 SA–treated mice. The toxic effects of IL-15 SA appear to be mediated primarily by expansion and activation of NK cells and are dependent on the presence of IFN-γ. Thus, the current study provides significant characterization of specific systemic, cellular, and molecular alterations caused by IL-15 SA treatment.
Supplementary Material
Acknowledgments
We thank Wu Lan in Luc Van Kaer’s laboratory for the provision of CD1d/α-Galcer tetramers and Michael Hill and Pavlo Gilchuk in Sebastian Joyce’s laboratory for assistance with NKT cell staining.
This work was supported by National Institutes of Health Grant R01 GM66885 and National Institute of Health Tetramer Core Facility (Contract HHSN272201300006C) for provision of CD1d/α–Galcer tetramers.
The online version of this article contains supplemental material.
- ALT
- alanine aminotransferase
- AST
- aspartate aminotransferase
- BUN
- blood urea nitrogen
- IHC
- immunohistochemical
- KO
- knockout
- mCD8+ T
- memory CD8+ T
- SA
- superagonist
- WT
- wild-type.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Fehniger T. A., Caligiuri M. A. 2001. Interleukin 15: biology and relevance to human disease. Blood 97: 14–32. [DOI] [PubMed] [Google Scholar]
- 2.Ma A., Koka R., Burkett P. 2006. Diverse functions of IL-2, IL-15, and IL-7 in lymphoid homeostasis. Annu. Rev. Immunol. 24: 657–679. [DOI] [PubMed] [Google Scholar]
- 3.Mattei F., Schiavoni G., Belardelli F., Tough D. F. 2001. IL-15 is expressed by dendritic cells in response to type I IFN, double-stranded RNA, or lipopolysaccharide and promotes dendritic cell activation. J. Immunol. 167: 1179–1187. [DOI] [PubMed] [Google Scholar]
- 4.Ahmad R., Ennaciri J., Cordeiro P., El Bassam S., Menezes J. 2007. Herpes simplex virus-1 up-regulates IL-15 gene expression in monocytic cells through the activation of protein tyrosine kinase and PKC ζ/λ signaling pathways. J. Mol. Biol. 367: 25–35. [DOI] [PubMed] [Google Scholar]
- 5.Kennedy M. K., Glaccum M., Brown S. N., Butz E. A., Viney J. L., Embers M., Matsuki N., Charrier K., Sedger L., Willis C. R., et al. 2000. Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15-deficient mice. J. Exp. Med. 191: 771–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lodolce J., Burkett P., Koka R., Boone D., Chien M., Chan F., Madonia M., Chai S., Ma A. 2002. Interleukin-15 and the regulation of lymphoid homeostasis. Mol. Immunol. 39: 537–544. [DOI] [PubMed] [Google Scholar]
- 7.Ahmad A., Ahmad R., Iannello A., Toma E., Morisset R., Sindhu S. T. 2005. IL-15 and HIV infection: lessons for immunotherapy and vaccination. Curr. HIV Res. 3: 261–270. [DOI] [PubMed] [Google Scholar]
- 8.Zanoni I., Spreafico R., Bodio C., Di Gioia M., Cigni C., Broggi A., Gorletta T., Caccia M., Chirico G., Sironi L., et al. 2013. IL-15 cis presentation is required for optimal NK cell activation in lipopolysaccharide-mediated inflammatory conditions. Cell Reports 4: 1235–1249. [DOI] [PubMed] [Google Scholar]
- 9.Bachanova V., Miller J. S. 2014. NK cells in therapy of cancer. Crit. Rev. Oncog. 19: 133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diab A., Cohen A. D., Alpdogan O., Perales M. A. 2005. IL-15: targeting CD8+ T cells for immunotherapy. Cytotherapy 7: 23–35. [DOI] [PubMed] [Google Scholar]
- 11.Cha E., Graham L., Manjili M. H., Bear H. D. 2010. IL-7 + IL-15 are superior to IL-2 for the ex vivo expansion of 4T1 mammary carcinoma-specific T cells with greater efficacy against tumors in vivo. Breast Cancer Res. Treat. 122: 359–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suck G., Oei V. Y., Linn Y. C., Ho S. H., Chu S., Choong A., Niam M., Koh M. B. 2011. Interleukin-15 supports generation of highly potent clinical-grade natural killer cells in long-term cultures for targeting hematological malignancies. Exp. Hematol. 39: 904–914. [DOI] [PubMed] [Google Scholar]
- 13.Klebanoff C. A., Gattinoni L., Palmer D. C., Muranski P., Ji Y., Hinrichs C. S., Borman Z. A., Kerkar S. P., Scott C. D., Finkelstein S. E., et al. 2011. Determinants of successful CD8+ T-cell adoptive immunotherapy for large established tumors in mice. Clin. Cancer Res. 17: 5343–5352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu P., Steel J. C., Zhang M., Morris J. C., Waldmann T. A. 2010. Simultaneous blockade of multiple immune system inhibitory checkpoints enhances antitumor activity mediated by interleukin-15 in a murine metastatic colon carcinoma model. Clin. Cancer Res. 16: 6019–6028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang M., Yao Z., Dubois S., Ju W., Muller J. R., Waldmann T. A. 2009. Interleukin-15 combined with an anti-CD40 antibody provides enhanced therapeutic efficacy for murine models of colon cancer. Proc. Natl. Acad. Sci. USA 106: 7513–7518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Capitini C. M., Fry T. J., Mackall C. L. 2009. Cytokines as adjuvants for vaccine and cellular therapies for cancer. Am. J. Immunol. 5: 65–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alpdogan O., Eng J. M., Muriglan S. J., Willis L. M., Hubbard V. M., Tjoe K. H., Terwey T. H., Kochman A., van den Brink M. R. 2005. Interleukin-15 enhances immune reconstitution after allogeneic bone marrow transplantation. Blood 105: 865–873. [DOI] [PubMed] [Google Scholar]
- 18.Forget M. A., Malu S., Liu H., Toth C., Maiti S., Kale C., Haymaker C., Bernatchez C., Huls H., Wang E., et al. 2014. Activation and propagation of tumor-infiltrating lymphocytes on clinical-grade designer artificial antigen-presenting cells for adoptive immunotherapy of melanoma. J. Immunother. 37: 448–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Conlon K. C., Lugli E., Welles H. C., Rosenberg S. A., Fojo A. T., Morris J. C., Fleisher T. A., Dubois S. P., Perera L. P., Stewart D. M., et al. 2015. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J. Clin. Oncol. 33: 74‑82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blaser B. W., Schwind N. R., Karol S., Chang D., Shin S., Roychowdhury S., Becknell B., Ferketich A. K., Kusewitt D. F., Blazar B. R., Caligiuri M. A. 2006. Trans-presentation of donor-derived interleukin 15 is necessary for the rapid onset of acute graft-versus-host disease but not for graft-versus-tumor activity. Blood 108: 2463–2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chapoval A. I., Fuller J. A., Kremlev S. G., Kamdar S. J., Evans R. 1998. Combination chemotherapy and IL-15 administration induce permanent tumor regression in a mouse lung tumor model: NK and T cell‑mediated effects antagonized by B cells. J. Immunol. 161: 6977–6984. [PubMed] [Google Scholar]
- 22.Castillo E. F., Stonier S. W., Frasca L., Schluns K. S. 2009. Dendritic cells support the in vivo development and maintenance of NK cells via IL-15 trans-presentation. J. Immunol. 183: 4948–4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han K. P., Zhu X., Liu B., Jeng E., Kong L., Yovandich J. L., Vyas V. V., Marcus W. D., Chavaillaz P. A., Romero C. A., et al. 2011. IL-15:IL-15 receptor α superagonist complex: high-level co-expression in recombinant mammalian cells, purification and characterization. Cytokine 56: 804–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mortier E., Woo T., Advincula R., Gozalo S., Ma A. 2008. IL-15Rα chaperones IL-15 to stable dendritic cell membrane complexes that activate NK cells via trans presentation. J. Exp. Med. 205: 1213–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rubinstein M. P., Kovar M., Purton J. F., Cho J. H., Boyman O., Surh C. D., Sprent J. 2006. Converting IL-15 to a superagonist by binding to soluble IL-15Rα. Proc. Natl. Acad. Sci. USA 103: 9166–9171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stoklasek T. A., Schluns K. S., Lefrancois L. 2006. Combined IL-15/IL-15Rα immunotherapy maximizes IL-15 activity in vivo. J. Immunol. 177: 6072–6080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Epardaud M., Elpek K. G., Rubinstein M. P., Yonekura A. R., Bellemare-Pelletier A., Bronson R., Hamerman J. A., Goldrath A. W., Turley S. J. 2008. Interleukin-15/interleukin-15Rα complexes promote destruction of established tumors by reviving tumor-resident CD8+ T cells. Cancer Res. 68: 2972–2983. [DOI] [PubMed] [Google Scholar]
- 28.Xu W., Jones M., Liu B., Zhu X., Johnson C. B., Edwards A. C., Kong L., Jeng E. K., Han K., Marcus W. D., et al. 2013. Efficacy and mechanism-of-action of a novel superagonist interleukin-15: interleukin-15 receptor αSu/Fc fusion complex in syngeneic murine models of multiple myeloma. Cancer Res. 73: 3075–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang C. M., Lo C. H., Shih Y. M., Chen Y., Wu P. Y., Tsuneyama K., Roffler S. R., Tao M. H. 2010. Treatment of hepatocellular carcinoma with adeno-associated virus encoding interleukin-15 superagonist. Hum. Gene Ther. 21: 611–621. [DOI] [PubMed] [Google Scholar]
- 30.Cheng L., Du X., Wang Z., Ju J., Jia M., Huang Q., Xing Q., Xu M., Tan Y., Liu M., et al. 2014. Hyper-IL-15 suppresses metastatic and autochthonous liver cancers by promoting tumor-specific CD8 T cell responses. J. Hepatol. 61: 1297‑1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Callahan M. K., Wolchok J. D. 2013. At the bedside: CTLA-4- and PD-1-blocking antibodies in cancer immunotherapy. J. Leukoc. Biol. 94: 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Curran M. A., Montalvo W., Yagita H., Allison J. P. 2010. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. USA 107: 4275–4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.2014. PD-1 inhibitor approved for melanoma. Cancer Discov. 4: 1249. [DOI] [PubMed] [Google Scholar]
- 34.Fujisaki H., Kakuda H., Shimasaki N., Imai C., Ma J., Lockey T., Eldridge P., Leung W. H., Campana D. 2009. Expansion of highly cytotoxic human natural killer cells for cancer cell therapy. Cancer Res. 69: 4010–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Williams M. T., Yousafzai Y., Cox C., Blair A., Carmody R., Sai S., Chapman K. E., McAndrew R., Thomas A., Spence A., et al. 2014. Interleukin-15 enhances cellular proliferation and upregulates CNS homing molecules in pre-B acute lymphoblastic leukemia. Blood 123: 3116–3127. [DOI] [PubMed] [Google Scholar]
- 36.Mishra A., Liu S., Sams G. H., Curphey D. P., Santhanam R., Rush L. J., Schaefer D., Falkenberg L. G., Sullivan L., Jaroncyk L., et al. 2012. Aberrant overexpression of IL-15 initiates large granular lymphocyte leukemia through chromosomal instability and DNA hypermethylation. Cancer Cell 22: 645–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berger C., Berger M., Hackman R. C., Gough M., Elliott C., Jensen M. C., Riddell S. R. 2009. Safety and immunologic effects of IL-15 administration in nonhuman primates. Blood 114: 2417–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Waldmann T. A., Lugli E., Roederer M., Perera L. P., Smedley J. V., Macallister R. P., Goldman C. K., Bryant B. R., Decker J. M., Fleisher T. A., et al. 2011. Safety (toxicity), pharmacokinetics, immunogenicity, and impact on elements of the normal immune system of recombinant human IL-15 in rhesus macaques. Blood 117: 4787–4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perera P. Y., Lichy J. H., Waldmann T. A., Perera L. P. 2012. The role of interleukin-15 in inflammation and immune responses to infection: implications for its therapeutic use. Microbes Infect. 14: 247‑261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwartz R. N., Stover L., Dutcher J. 2002. Managing toxicities of high-dose interleukin-2. Oncology 16: 11–20. [PubMed] [Google Scholar]
- 41.Hayakawa Y., Huntington N. D., Nutt S. L., Smyth M. J. 2006. Functional subsets of mouse natural killer cells. Immunol. Rev. 214: 47–55. [DOI] [PubMed] [Google Scholar]
- 42.de Rham C., Ferrari-Lacraz S., Jendly S., Schneiter G., Dayer J. M., Villard J. 2007. The proinflammatory cytokines IL-2, IL-15 and IL-21 modulate the repertoire of mature human natural killer cell receptors. Arthritis Res. Ther. 9: R125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blank C., Luz A., Bendigs S., Erdmann A., Wagner H., Heeg K. 1997. Superantigen and endotoxin synergize in the induction of lethal shock. Eur. J. Immunol. 27: 825–833. [DOI] [PubMed] [Google Scholar]
- 44.Ito H., Koide N., Hassan F., Islam S., Tumurkhuu G., Mori I., Yoshida T., Kakumu S., Moriwaki H., Yokochi T. 2006. Lethal endotoxic shock using α-galactosylceramide sensitization as a new experimental model of septic shock. Lab. Invest. 86: 254–261. [DOI] [PubMed] [Google Scholar]
- 45.Sherwood E. R., Toliver-Kinsky T. 2004. Mechanisms of the inflammatory response. Best Pract. Res. Clin. Anaesthesiol. 18: 385–405. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.