

Abstract
The membrane protein complex between sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) and phospholamban (PLN) is a prime therapeutic target for reversing cardiac contractile dysfunctions caused by calcium mishandling. So far, however, efforts to develop drugs specific for this protein complex have failed. Here, we show that non-coding RNAs and single-stranded DNAs (ssDNAs) interact with and regulate the function of the SERCA/PLN complex in a tunable manner. Both in HEK cells expressing the SERCA/PLN complex, as well as in cardiac sarcoplasmic reticulum preparations, these short oligonucleotides bind and reverse PLN’s inhibitory effects on SERCA, increasing the ATPase’s apparent Ca2+ affinity. Solid-state NMR experiments revealed that ssDNA interacts with PLN specifically, shifting the conformational equilibrium of the SERCA/PLN complex from an inhibitory to a non-inhibitory state. Importantly, we achieved rheostatic control of SERCA function by modulating the length of ssDNAs. Since restoration of Ca2+ flux to physiological levels represents a viable therapeutic avenue for cardiomyopathies, our results suggest that oligonucleotide-based drugs could be used to fine-tune SERCA function to counterbalance the extent of the pathological insults.
Heart failure is the leading cause of death worldwide1. Despite important advances in pharmacological and device therapies, the incidence and the financial impact of this devastating condition is increasing1. Current drug therapies aim to manage and ameliorate patients’ symptoms rather than cure the disease, which has generated a growing emphasis on the development of alternative approaches, including gene therapy2,3. As heart failure is a multifactorial event, several different strategies can be undertaken to reverse the decline of cardiac function3. One possible approach is to improve cardiac contractility by targeting calcium-handling proteins2,4. An emerging target for gene therapy is the membrane protein complex between the sarco(endo)plasmic reticulum Ca2+ -ATPase (SERCA) and phospholamban (PLN). The SERCA/PLN complex is a central regulator of cardiac contractility, transporting approximately 70% of the Ca2+ ions in the human heart5,6. SERCA reuptakes Ca2+ ions from the cytosol into the sarcoplasmic reticulum lumen during diastole5. PLN, a 52 amino acid transmembrane protein, controls SERCA’s apparent Ca2+ affinity, reducing its ability to transport Ca2+ when unphosphorylated and augmenting it when phosphorylated at Ser16 by protein kinase A6. Aberrant interactions between SERCA and PLN mutants and concomitant Ca2+ mishandling have been correlated with dysfunctional contractility and heart disease7,8. As a result, several approaches have been pursued to reverse these conditions; not only SERCA gene transfer therapy4,9,10, but also siRNA11, miRNA inhibition12 and aptamers13,14, have shown promise as therapeutic avenues. In particular, SERCA-directed gene therapy is the most effective strategy to augment Ca2+ transport and muscle contractility, either using an adeno-associated virus to overexpress the ATPase in cardiomyocytes or targeting PLN to reverse SERCA inhibition10. While the former approach is being evaluated in clinical trials, the latter presents challenges. In fact, PLN-knockout mice15 and PLN-null mutations in humans16 progress to heart failure and lethal dilated cardiomyopathy, respectively. Although encouraging for large animal models17, gene transfer of PLNS16E, a pseudo-phosphorylated mutant, hampers β-adrenergic control of heart contractility as the mutant can no longer be phosphorylated. In addition, current gene therapy methods do not allow a controllable response to counteract the different degrees of heart disease manifestations4. Whereas phosphorylatable mutants mimicking the phosphorylated state of PLN are being developed18,19, there is a critical need to devise more direct, viable methods to target PLN.
Here, we report the unexpected discovery that small RNAs and single-stranded DNAs (ssDNAs) bind with low nanomolar dissociation constants (Kd) to the SERCA/PLN complex, regulating the ATPase’s apparent Ca2+ affinity in a tunable manner. Specifically, we found that both short RNAs and ssDNAs are able to reverse PLN’s inhibitory effects irrespective of their primary sequence. The functional effects are tunable by increasing or decreasing the oligonucleotides’ lengths, becoming significant for sizes encompassing typical endogenous miRNAs and reaching a plateau at 80 bases. Solid-state NMR and fluorescence spectroscopy data show that ssDNA binds PLN’s cytoplasmic domain specifically, but does not affect SERCA in the absence of the regulatory protein. In particular, NMR spectra show that ssDNA shifts the conformational equilibrium of the SERCA/PLN complex from inhibitory to a non-inhibitory state. Förster resonance energy transfer (FRET) experiments in HEK cells overexpressing the SERCA/PLN complex show that PLN remains bound to SERCA upon interacting with ssDNA. These functional effects, tested in membrane reconstituted systems, are reproducible in mammalian sarcoplasmic reticulum (SR) cardiac preparations. These findings indicate that chemically modified, non-coding RNA and single-stranded DNA templates with low off-target propensity can be exploited for developing compounds to target SERCA inhibition by PLN, thereby regulating Ca2+ transport in the SR. The rheostatic control of SERCA function achieved here opens up new possibilities for devising a graded response to varying extents of cardiac pathologies10.
Results
Small RNAs and ssDNAs bind PLN with low nanomolar dissociation constants, reversing its inhibitory effect on SERCA
Using affinity capillary electrophoresis (ACE), fluorescence polarization (FP), and native gel mobility shift assays, we found that short RNAs, of similar length to naturally occurring miRNA sequences, display low nanomolar dissociation constants for PLN. As an example, we report the dissociation constant of a random sequence RNA (50mer) to PLN in Fig. 1A and typical binding curves (80mer) in Fig. S1. Importantly, we found that these short RNAs not only bind, but also reverse PLN’s inhibition of SERCA. Fig. 1B shows the effect of the 50mer RNA on SERCA activity as monitored using coupled enzyme assays. The Ca2+ concentration at half maximal activity in the coupled assay curves indicates SERCA’s apparent affinity for Ca2+ ions (pKCa). When SERCA is bound to PLNWT, the ATPase activity decreases and the normalized curves show a concomitant reduction of the pKCa value. Upon binding the 50mer RNA, SERCA’s function is nearly restored (Fig. 1B), mimicking the effect of PLN phosphorylation at Ser1620. Since aptamers have been selected to target specific proteins without off-target effects on gene expression21, we tested the effects of a 50mer ssDNA random sequence. Indeed, we found that both short RNAs and ssDNAs have similar Kd values for PLN (Fig. 1A) and restoration of SERCA function (Fig. 1B).
Figure 1. Functional effect of RNA and ssDNA on the SERCA/PLN complex.

(a) Dissociation constants as measured by both fluorescence polarization and affinity capillary electrophoresis of both RNA and ssDNA. (b) pKCa of SERCA as measured by coupled enzyme assays. Dissociation constants versus ssDNA length obtained from fluorescence polarization (c) and affinity capillary electrophoresis (d). Sequences for (c) and (d) are found in Table 1.
We also assessed the affinity of different lengths of ssDNAs with sequences chosen at random that had no significant secondary structures, with the exception of the 30mer (Table 1 and Fig. 1C,D). We found sequence length to be the best indicator of affinity. Any sequence containing more than 10 bases had low nanomolar Kd values, while sequences containing 10 bases or less had significantly less affinity with higher Kd values. Surprisingly, all of the randomly chosen sequences containing more than 10 bases exhibited high affinity for PLN, suggesting that this is a sequence-independent interaction (Table 1). The 80mer used for the binding studies was synthesized as a completely randomized mixture, with a 25% probability of each base being present at every register position. This random library allowed affinity to be determined independently of a specific sequence. The high affinity (low nanomolar Kd) between the completely randomized sequence mixture of 80mer ssDNA and PLN confirms the sequence independence of this interaction. Among the longer sequences, the 30mer demonstrated slightly weaker affinity for PLN than predicted based on sequence length alone. This 30mer is the only sequence listed in Table 1 that exhibits significant secondary structure. The weaker than predicted affinity suggests a thermodynamic penalty to unfold the ssDNA before binding PLN. To analyze the nature of the intermolecular interactions, we carried out the binding assays at different salt concentrations (Fig. S2). Indeed, we found that the ssDNA/PLN interactions are persistent even at high salt concentrations and remain in the nanomolar range at NaCl concentrations up to 200 mM.
Table 1. Random ssDNA sequences used.
| Sequence Length | Sequence 5′⟶3′ |
|---|---|
| 5 | FAM-GCT TG |
| 10 | FAM-ATA GCT TGC A |
| 15 | FAM-AGT GAT AGC TAT GGT |
| 20 | FAM-AGC AGC ACA GAG GTC AGA TG |
| 30 | FAM-ACT GAG CAT GGG ATA ACC GTT CTC AGA CTT |
| 50 | FAM-AGC AGC ACA GAG GTC AGA TGC AGG TAG GGT CCT ATG CGT GCT ACC GTG AA |
| 80* | FAM-(N) 80 |
| 80** | AGC AGC ACA GAG GTC AGA TGC AGG TAG GGT CCT ATG CGT GCT ACC GTG AAAGC AGC ACA GAG GTC AGA TGATA GCT TGC A |
*A mixture of randomized 80-mer ssDNA sequences.
**80mer used for NMR.
We then performed ATPase assays with varying length oligonucleotides and found they give rise to a graded effect on ATPase activity. Figure 2A depicts the normalized SERCA/PLN activity curves upon addition of ssDNA at different lengths and varying concentrations of free calcium (pCa). In the absence of ssDNA, PLN binding results in decreased Ca2+ affinity of SERCA (lowest pKCa, with the brown curve shifted furthest to the right in Fig. 2A). Addition of ssDNA to the PLN/SERCA complex shifts the activity curve toward the higher pCa, indicating that the SERCA’s apparent affinity for Ca2+ ions is increasing; ssDNA reverses the inhibitory effect of PLN (Fig. 2A,B). The functional effect trends with sequence length, but not necessarily according to the relative affinity of the oligonucleotides (Kd values). This is evidenced by the 30mer, which displays a higher Kd than expected based on length alone, but still follows the length trend seen in the activity assays (Fig. 2A,B). It should be noted that all ATPase assays were performed at saturating ssDNA concentrations. Thus, the length trend (Fig. 2) observed is structural in origin and independent of the ssDNA concentration. Remarkably, longer oligonucleotides result in complete inhibition relief, an effect that mimics phosphorylation of PLN at Ser16 by protein kinase A22. With oligonucleotide sequences longer than 50 bases, we observed no further increase of SERCA activity beyond the physiological window, indicating that the effect is mediated by the direct interaction between ssDNA and PLN.
Figure 2. Tunable control of SERCA function using ssDNA of different lengths.

Coupled enzyme assays in DOPC:DOPE lipid vesicles. (a) SERCA activity normalized as a function of Ca2+ concentration. Black is SERCA alone, brown is SERCA and PLN; other colors represent addition of different lengths of ssDNA. (b) pKCa values for the different ssDNA lengths derived from the pCa values at half maximum activity of SERCA. (c) Effects of ssDNA on pig cardiac SR preparations.
To confirm the specificity of the ssDNA/PLN interactions, we incubated 80mer ssDNA with SERCA in the absence of PLN. Under these conditions, the apparent Ca2+ affinity was identical to that of SERCA alone, confirming that ssDNA does not have an activating effect without PLN (Fig. 2B). Additionally, both double-stranded DNA and a mixture of free deoxynucleotides (dNTP) show no effect on PLN’s inhibition of SERCA; the pKCa with either double-stranded DNA or free dNTPs is identical to the pKCa of the PLN inhibited pump (Fig. S3). Taken together, these data suggest that reversal of the inhibitory effect of ssDNAs is manifested in a length-dependent manner, with inhibition relief being progressively more effective with increasing sequence length and reaching a plateau at ~80 oligonucleotides.
ssDNA disrupts PLN inhibition without dissociating the SERCA/PLN complex
To probe the specific interactions between PLN and ssDNA both in the absence and presence of SERCA, we labeled SERCA with a FRET donor (AEDANS) at position Cys674 and PLN with a non-fluorescent acceptor (Dabcyl-SE) on Lys3. FRET sample preparation matched that of the coupled enzyme assay samples (see Methods). In the presence of the FRET pair, we detected a marked decrease in fluorescence signal upon formation of the complex between SERCA and PLN (Fig. 3B). Addition of an equimolar amount of 40mer ssDNA (Table 1) to the pre-formed SERCA/PLN complex under conditions similar to the activity assays did not change the FRET signal. These measurements were repeated with excess PLN, or excess ssDNA, and the results were similar to the FRET data with stoichiometrical amounts (Fig. 3B,C). The enhanced FRET and shift of the emission maximum seen with a 10-fold excess of PLN is indicative of more FRET between SERCA and PLN, resulting from saturating the PLN binding site in the ATPase (Fig. 3C). We have also recorded fluorescence of SERCAAEDANS without PLN, but in the presence of ssDNA. The fluorescence intensity was identical to that of SERCAAEDANS, illustrating that ssDNA does not interact with the ATPase alone (Fig. 3A). The FRET, ACE and fluorescence polarization data collectively demonstrate that the ssDNA interacts specifically with PLN irrespective of the presence of SERCA. Furthermore, ssDNA relieves PLN inhibition by causing a structural rearrangement of PLN without dissociating it from SERCA, in a manner resembling PLN phosphorylation at Ser16 – the endogenous mechanism for inhibition relief.
Figure 3. ssDNA does not dissociate PLN from SERCA.
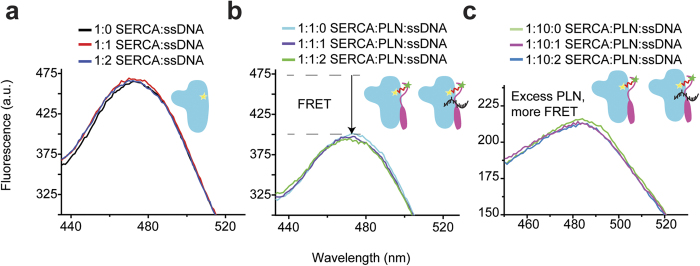
(a) Fluorescence spectra of SERCA alone and upon addition of ssDNA. (b) FRET of SERCA-PLN complex (cyan) in the absence and presence of ssDNA. Upon addition of ssDNA (purple and green) the FRET signal does not change, indicating no structural changes within the range of the FRET probes. (c) SERCA-PLN FRET at a 10:1 PLN:SERCA ratio (green).
ssDNA interacts with the SERCA/PLN complex in living cells
The SERCA/PLN interaction studies were also carried out in HEK cells. Specifically, FRET in-cell assays were used to investigate the interactions of ssDNA with free PLN and in complex with SERCA23,24. We utilized PLNAFA, a pentamer-destabilizing mutant with full inhibitory activity to minimize PLN oligomerization and detect small changes in binding affinity. Although it runs as a monomer on SDS-PAGE, PLNAFA has a slight tendency to form pentamers in membranes and exhibits a significant intra-pentameric FRET when overexpressed in living cells. A cell-by-cell comparison of FRET with PLN expression level revealed a hyperbolic dependence of FRET efficiency on protein concentration for a population of HEK cells expressing the fluorescent proteins Cer-PLNAFA and YFP-PLNAFA, fused at the N-terminus of PLN (Fig. S4). From this curve, we calculated the apparent dissociation constant for oligomerization (KD1) as well as the intrinsic FRET efficiency of the PLN oligomer (FRETmax), reporting on the inter-protomer binding affinity and quaternary structure changes, respectively. In the absence of ssDNA, we found that PLNAFA is able to form pentamers, though with reduced propensity compared to PLNWT. In contrast, addition of the 50-mer ssDNA to the cells containing plasmids encoding for Cer-PLNAFA and YFP-PLNAFA induced a notable increase in the apparent PLN-PLN affinity, representing an approximately 20% increase in PLN oligomerization (Fig. S4). A significant decrease in FRETmax was observed, which corresponds to an increase of the donor-acceptor distance (i.e., increased interprotomer affinity) as previously observed for PLNR9C and its phosphomimetic mutants25,26. For the SERCA/PLN complex, cell transfection with 50-mer ssDNA had striking effects both on the binding affinity and the structure of the complex. Unlike the in vitro fluorescence experiments in which the AEDANS donor probe was attached at Cys674 in the SERCA’s P-domain, the Cer probe placed on the A-domain of SERCA is able to detect a four-fold increase of PLN affinity for SERCA upon oligonucleotide addition, with a concomitant decrease of SERCA/PLN FRETmax (Fig. 4). Taken with the in vitro ATPase activity assays, these data demonstrate that ssDNA binding to PLN mimics both the structural and functional effects of phosphorylation on the SERCA/PLN regulatory complex. The decrease of FRETmax upon complex formation is suggestive of a structural rearrangement of PLN within the complex; rather than the dissociation of PLN from the ATPase.
Figure 4. FRET experiments in living cells.
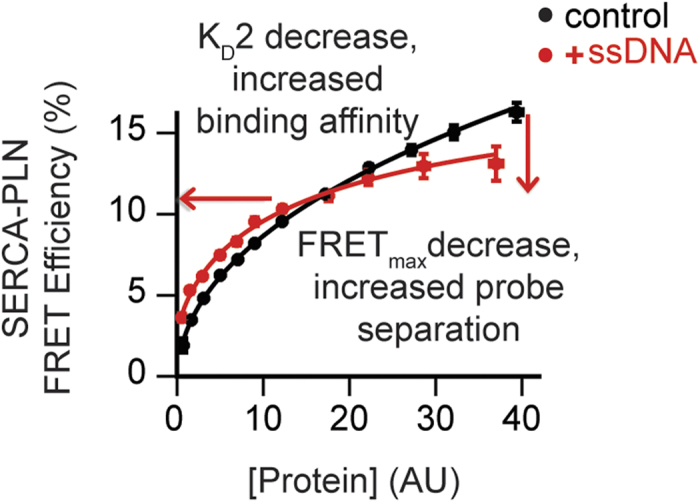
ssDNA increased the affinity of the SERCA/PLN interaction, and decreased FRETmax. ssDNA induces a conformational change of the complex, increasing fluorescent probe separation distance.
ssDNA reverses PLN’s inhibition of SERCA in cardiac SR preparations
To assess the reproducibility of the ssDNA effects observed in the reconstituted systems in more biologically relevant conditions, we performed the activity assays using crude cardiac SR preparations isolated from pig ventricles, containing the SERCA2a isoform of the ATPase (see Material and Methods). Since PLN is endogenously expressed in ventricles, direct addition of ssDNA to the heavy SR preparations containing the SERCA2a/PLN complex would be expected to increase the ATPase apparent Ca2+ affinity. Indeed, addition of ssDNA (1 μM 80mer) to the pig crude SR vesicles augmented the pKCa by 0.15 (Fig. 2C). While the effect of ssDNA on SERCA activity in pig crude SR is somewhat lower than in the reconstituted system; this slight discrepancy is likely due to the challenges in determining the exact amounts of the SERCA/PLN complex in these native preparations, as well as their molar ratio and phosphorylation state. Overall, these data confirm the efficacy of ssDNA in relieving PLN inhibition of the mammalian SERCA2a/PLN complex under native conditions.
NMR mapping of ssDNA binding epitope on PLN
To identify the specific residues of PLN interacting with the ssDNA, we used solid-state NMR (ssNMR) spectroscopy. We reconstituted U-13C/15N labeled PLN in deuterated DMPC lipid vesicles and monitored the chemical shifts of the backbone and side chain 13C resonances in the presence and absence of ssDNA (80mer). To detect the dynamic cytoplasmic domain of PLN, we used the refocused [1H, 13C]-RINEPT experiment27, which is well suited for protein domains undergoing fast reorientation (cytoplasmic domain of PLN) and insensitive to rigid domains on the NMR time scale (PLN’s transmembrane domain)28. In the free form of PLN, the resonances corresponding to the cytoplasmic region are all detectable. Addition of ssDNA to PLN at a 1:1 molar ratio causes the intensities of several amino acids peaks in the [1H, 13C]-RINEPT spectrum to decrease, with several peaks becoming broadened beyond detection (Fig. 5). The latter indicates the rigidification of PLN’s cytoplasmic domain and an increase in rotational correlation time upon ssDNA binding. In contrast, natural abundance 13C lipid signals are not affected by ssDNA and remain essentially unchanged (Fig. 5). The most affected resonances of PLN are located at the N-terminal portion of domain Ia (i.e., Lys3, Val4, Leu7, Thr8/17, Arg9/13/14, Ala/0/11/15, Ile12/18, and Glu2/19). To confirm that the ssDNA targets the cytoplasmic domain resonances, we compared the ssNMR data with solution NMR experiments carried out on PLN reconstituted in isotropic bicelles (q = 0.33). Due to the large size of the bicelle/PLN complex, solution NMR [1H, 15N]-HSQC experiments do not detect the transmembrane residues29, but provides visualization of the cytoplasmic domain resonances. Addition of 15mer ssDNA causes extensive line broadening (residues Glu2, Thr8, Ala11, Ile12/18 Arg13/14, and Met20 - Fig. S5), which is in qualitative agreement with ssNMR experiments. Taken with the FRET data, the NMR experiments indicate that the transmembrane domain of PLN remains essentially unperturbed and attached to the ATPase, while ssDNA primarily targets the cytoplasmic domain of PLN interfering with its regulatory function of SERCA.
Figure 5. Mapping of PLN residues involved in ssDNA binding.

[1H, 13C] RINEPT spectra of PLN in DMPC lipid vesicles in the absence (black) and presence (red) of ssDNA (80mer). Blue asterisks indicate lipid signals; green asterisks indicate a change in peak shape. Dotted circles indicate missing peaks.
Previously, we found that PLN bound to SERCA undergoes conformational transitions between three major states (Fig. 6b): an inhibitory T state, with the transmembrane (TM) domain bound to SERCA and the cytoplasmic domain associated to the membrane; an inhibitory R state, with the TM domain bound to SERCA and the cytoplasmic domain unfolded and dissociated from the membrane; and a sparsely populated, non-inhibitory B or bound state, with both the TM and cytoplasmic domains interacting with SERCA28. PLN phosphorylation at Ser16 shifts the equilibrium toward the B state, undergoing a local structural rearrangement and effectively reversing PLN’s inhibition of SERCA. To assess the effect of ssDNA on the PLN conformational equilibrium in the presence of SERCA, we utilized PLNAFA, which simplifies the interpretation of the ssDNA binding on the SERCA/PLN complex by eliminating the monomer/pentamer equilibrium. To resolve the resonances in the aliphatic region of the spectrum, the cytosolic domain Ia of PLNAFA was selectively labeled at 6 different sites (Val4, Leu7, Ala11, Ala15, Ile12, Ile18). These cytosolic residues (in the absence of SERCA) display chemical shift perturbations upon the addition of ssDNA, indicating binding between ssDNA and PLNAFA (Fig. S6). Dipolar assisted rotational resonance (DARR) MAS [13C, 13C] experiments of U-13C/15N labeled PLN in complex with unlabeled SERCA were performed in the absence and presence of ssDNA (Fig. 6). Specifically, the DARR peaks corresponding to the Cα/Cβ correlations of the two Ala residues are sensitive to the conformational equilibrium between the different states and can be used as reporter residues (Fig. 6). In the absence of SERCA at 20 °C, these two residues populate a major conformational state (T state) and a sparsely populated R state. Upon addition of SERCA, there is a population shift toward the bound state that is augmented upon phosphorylation of PLN. Upon binding ssDNA, we observe a progressive shift of the two Ala residues of PLNAFA toward lower fields, similar to what is observed with the non-inhibitory phosphorylated form of PLN. These results strongly support the fluorescence studies and show that ssDNA does not detach PLN from SERCA; rather it shifts the conformational equilibrium toward a non-inhibitory or D (DNA bound) state in a manner similar to the phosphorylated state. A hypothetical model of the conformational equilibrium of the SERCA/PLN complex and the effects of ssDNA is reported in Fig. 6B.
Figure 6.

Effects of ssDNA on the conformational equilibrium of the SERCA/PLN complex (a) Portions of the [13C, 13C]-DARR spectra indicating the conformational equilibrium between the different states of PLN as probed by the alanine residues in the cytoplasmic domain. A small population of AFA PLN exists in the B (bound) state, upon phosphorylation, a higher population transitions to the B state (with a slight shift upon the phosphorylation), a further shift occurs upon addition of ssDNA to the D (DNA) bound state. (b) Proposed regulatory model of SERCA by PLN and effects of ssDNA. ssDNA shifts the monomer to pentamer equilibrium toward the oligomeric state. Phosphorylation of Ser16 shifts the equilibrium toward the non-inhibitory bound state (B-state). ssDNA is proposed to bind phospholamban and mimic the phosphorylated state, shifting the equilibrium toward a non-inhibitory D-state.
Discussion
Homeostatic regulation of Ca2+ ions in muscle is crucial to proper contractility5. SERCA plays a central role in this delicate equilibrium and its function is regulated within a physiological window by PLN28. Super-inhibition of SERCA by PLN mutants or hyper-phosphorylation of PLN tip the homeostatic balance and lead to cardiomyopathies2,30. So far, there have been no drugs developed with a direct effect on the SERCA/PLN complex. However, the complexity of Ca2+ transport dysregulation in heart failure calls for therapeutic approaches that parallel the extent of the pathology, which is yet to be achieved.
Our study demonstrates that short RNAs and ssDNAs are able to regulate SERCA activity in a tunable manner by binding the cytoplasmic domain of PLN with nanomolar dissociation constants. Binding of ssDNA to the SERCA/PLN complex induces structural changes and promotes inhibition relief. Due to the high affinity of ssDNA for PLN, the tunability of the effects is not concentration-dependent, but rather length-dependent, with a range from 5 nucleotides, exhibiting minimal effects on SERCA activity, to 80 nucleotides, conferring a nearly complete relief of inhibition. The binding of the ssDNA is likely driven by the electrostatic interactions between the negatively charged phosphate groups of the oligonucleotide and the positively charged Lys and Arg side-chains of PLN. Such charge-charge interactions have been found in sequence-independent ssDNA binding proteins, such as T4 gp32, adeno-DBP and multiple others31,32,33. However, electrostatic interactions alone do not fully justify the unique binding seen between ssDNA and PLN. Single chain nucleic acids are essential for PLN binding, since neither double-stranded DNA nor a mixture of free dNTPs are able to reverse PLN inhibition of SERCA. ssDNA binds PLN without detaching it from the ATPase; rather, it shifts its conformational equilibrium toward a non-inhibitory state18,28. Previously, we found that PLN bound to SERCA undergoes conformational transitions between three major states: an inhibitory T state, with the transmembrane (TM) domain bound to SERCA and the cytoplasmic domain associated to the membrane; an inhibitory R state, with the TM domain bound to SERCA and the cytoplasmic domain unfolded and dissociated from the membrane; and a sparsely populated, non-inhibitory B or bound state, with both the TM and cytoplasmic domains interacting with SERCA28. By tracing the population of the SERCA-bound state of PLN, we found its conformational equilibrium is driven by ssDNA toward a new, distinct state (D state) (Fig. 6), which is non-inhibitory. Since FRET experiments show no detachments of PLN from SERCA, we surmise that the ssDNA may adopt a PLN-bound conformation with the phosphate backbone mimicking the effects of Ser16 and/or Thr17 phosphorylation and restoring SERCA’s apparent affinity for Ca2+ ions.
An important corollary to our study is the possibility of direct interactions of endogenous short RNA sequences (such as miRNA) with the SERCA/PLN complex. In the heart, miRNAs are involved both in cardiogenesis and disease including: hypertrophy, ischemia, and electrical remodeling34,35,36,37. miRNAs are up- or down-regulated in response to stress during cardiac disorders37 and they are notorious for their pleiotropic action, affecting multiple genes in different tissues37. The latter has prevented the progression of miRNA regulation to therapeutic applications38,39. Based on the high, non-sequence specific affinity of short RNAs for PLN, it is possible that endogenous miRNAs may have a role in cardiac pathophysiology that goes beyond gene regulation. As miRNAs of different lengths and sequences are produced for both protective and pathological roles34,36,40, the up-regulation of miRNAs under cardiac remodeling and heart failure may favor their direct physical interaction with the SERCA/PLN complex, affecting Ca2+ regulation and cardiac contractility.
Irrespective of the possible role of the endogenous miRNAs toward the SERCA/PLN complex in cardiomyocytes, the nanomolar affinity of ssDNA for PLN and its ability to reverse its inhibitory effects on SERCA constitutes a unique opportunity to exploit oligonucleotides as scaffolds for the design of small molecules to target Ca2+ regulation. While DNA aptamers or miRNA constructs have been successfully used to target and regulate the PLN gene11,41, silencing or ablation of the PLN gene has resulted in cardiomyopathies. In contrast, tunable regulation of PLN inhibitory function of SERCA by oligonucleotide-based drugs may represent a more promising therapeutic avenue in which the extent of SERCA activation can be fine-tuned to match the pathological insult.
Methods
Wild type phospholamban expression and purification was achieved through the previously published methods42. Isotope labeling was accomplished via 13C-enriched glucose and 15N-enriched ammonium chloride. Calcium ATPase (SERCA1a isoform) was extracted from rabbit skeletal muscle according to previously reported procedure and purified by affinity chromatography using Reactive Red43. Pig cardiac SR vesicles (Lindenfelser’s Meats, Monticello, MN, USA) were prepared as reported previously44. Lipids were obtained from Avanti Polar Lipids (Alabaster, AL). Single stranded DNA (ssDNA) sequences were purchased from Integrated DNA Technologies, Inc. (Coralville, IA). Other chemicals were from Sigma Aldrich (St. Louis, MO).
SERCA Activity Assays
Recombinant PLN was co-reconstituted with SERCA in multilamellar vesicles of 1,2-dioleyl-sn-glycero-3-phosphocholine (DOPC) and 1,2-dioleyl-sn-glycero-3-phosphoethanolamine (DOPE) at 4:1 DOPC:DOPE molar ratio. A solution of ssDNA in deionized water was added directly to the vesicles and incubated for 20 minutes at 25 °C prior to starting the assay. The molar ratio of the components were 700:10:1:1 (lipid : PLN : SERCA : DNA). Calcium dependence of the SERCA ATPase activity was measured at 25 °C using a coupled enzyme assay45 utilizing λ340 absorbance (NADH) as an analytical signal using a Spectromax microplate reader (Molecular Devices). The initial rate was measured as a function of calcium concentration, and the data were fit using the Hill equation (1):
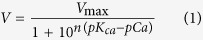 |
where: V is the initial rate; Vmax is the maximum rate; pCa is the negative logarithm of calcium concentration; pKCa is the pCa value where V = Vmax/2, and n is Hill coefficient. For comparison, each data set was normalized to Vmax after fitting. Inhibitory effects are expressed as changes in pKCa relative to SERCA alone.
Activity assays of SERCA2a were performed directly with pig cardiac SR, containing ~20 μg of total protein (bicinchoninic acid assay). Cardiac SR vesicles were diluted to 100 μl with buffer and incubated with 1 μM of ssDNA (80mer) for 30 min prior to measurements.
NMR sample preparation
Magic angle spinning (MAS) NMR samples were prepared by co-dissolving 10 mg acyl chain deuterated 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC-d54) and 2 mg of recombinant [U-13C/15N]-PLN. The solvent was removed under a stream of nitrogen gas and desiccated overnight. The lipid/protein film was rehydrated with 1 ml of 20 mM MOPS, 100 mM NaCl (pH 7.0). The suspension was vortexed, briefly sonicated, lyophilized, re-suspended in 10 μL of ddH2O and transferred to a 3.2 mm thin wall MAS rotor. The final samples contained approximately 50% H2O (w/w) with a lipid:PLN ratio of 40:1. Magic angle spinning samples with PLN and SERCA were prepared by co-mixing the proteins at a 1:1 molar ratio in 1% C12E8, and adding them to 10 mg of DMPC-d54 solubilized with 1% C12E8. Next, ssDNA suspended in ddH2O was added and the detergent was removed by 3 hr incubation with BioBeads SM2 at a 30:1 Biobeads:C12E8 ratio at room temperature. The sample was diluted to 50 mL with buffer, pelleted by centrifugation at 4 °C (100,000 × g, 30 minutes) and the pellet was packed in the MAS rotor. Isotropic bicelle samples for solution NMR were prepared by dissolving 1 mg of [U-13C, 15N] PLN in 20 mM MOPS, 100 mM NaCl, and 5% D2O containing 174 mg/ml of 1,2-dihexanoyl-sn-glycero-3-phosphocholine (DHPC). The mixture was transferred to lyophilized DMPC (21.9 mg, corresponding to q = 0.33), vortexed, sonicated and subjected to several freeze/thaw cycles, until a clear solution was formed.
NMR experiments
All NMR experiments were performed on a VNMRS spectrometer operating at 1H frequency of 600 MHz. Magic angle spinning (MAS) experiments were performed at 8 kHz spinning rate and at 25 °C, utilizing 1H/13C MAS probe (Varian). Pulse widths were 5.5 μsec (13C), 2.5 μsec (1H) and a 100 kHz 1H decoupling field strength was used. Two dimensional experiments were refocused Insensitive Nuclei Enhanced by Polarization Transfer (RINEPT) and Dipolar Assisted Rotational Resonance (DARR)27,46. A contact time of 1 ms was utilized for cross-polarization. Spectral widths were 100 kHz (direct dimension), 3.33 kHz (indirect dimension, RINEPT) and 40 kHz (indirect dimension, DARR). Chemical shifts were referenced to the 13CH2 signal of adamantane (40.48 ppm). Typically 640 scans with 30 increments in the indirect dimension were acquired. Data were processed using NMRPipe and analyzed with Sparky47.
Affinity Capillary Electrophoresis (ACE)
The experiments were performed on a commercial CE system (P/ACE MDQ, Beckman Coulter, Inc., Fullerton, CA) with laser induced fluorescence (LIF) detection (λex = 488 nm, λem = 520 nm). Binding and separation buffers for ACE and fluorescence polarization were made in nuclease free water (Coralville) and filtered through a 0.2 μm membrane filter (Millipore) before use. Separation buffer consisted of 20 mM MOPS and 0.1% (w/v) octaethylene glycol monododecyl ether (C12E8) at pH 7.0. Binding buffer additionally contained 0.25 mM DTT, 1 mM MgCl2, 1 mM KCl, and 5 mM CaCl2. Fluorescently labeled ssDNA samples in binding buffer (2.5 nM) were titrated with PLN and injected into a 50 cm × 50 μm fused silica capillary (Polymicro Technologies, Phoenix, AZ) using hydrodynamic injection (1 psi for 4 s). Separations were performed at reversed polarity, 30 kV for 10 min. Electropherograms were analyzed using Cutter 7.0; peak heights of the free ssDNA peaks were used to calculate the bound fractions according to the equation (2)48:
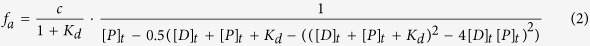 |
Where: fa is the bound fraction; c is maximum bound fraction; [P]t and [D]t are total concentrations of PLN and DNA, respectively. Note that denominator represents the free concentration of PLN.
Fluorescence Polarization (FP)
The experiments were performed on a SynergyTM 2 Microplate Reader (BioTek Instruments, Inc., Winooski, VT) with similar filter settings (λex = 485 ± 20 nm, λem = 528 ± 20 nm). Sample prepation was similar to the ACE experiments; 15 μL aliquots of mixtures were loaded into a corning 3540 microplate (Corning Incorporated, Corning, NY). Polarization values were calculated from the parallel and perpendicular fluorescence intensities. Calibration was done using the g factor, calculated in the Gen 5TM software (BioTek Instruments, Inc., Winooski, VT). Bound fractions were determined using equation (3):
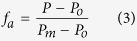 |
where: fa is the bound fraction; P, Pm and Po are measured polarizations of a sample, complex, and free ssDNA, respectively. The polarization of the complex (Pm) was taken as the plateau with an excess of PLN. The overall fluorescence intensity of the samples with increasing PLN concentrations, was monitored and bound fraction was modified if the overall fluorescence intensity was positively or negatively biased according to the published method49.
Native Gel Mobility Shift Assay
Native 10% Tris-Borate-EDTA (TBE) gels were prepared according to the published procedure50. Phospholamban was dissolved in binding buffer (0.1% C12E8, 20 mM Tris-HCl, 1 mM MgCl2, pH 7.0) and incubated with fluorescein-labeled ssDNA for 20 min at 25 °C. Samples were mixed at a 1:1 (v:v) ratio with ×2 loading buffer (Bromophenol blue (20% w/v), glycerol (50% v/v), 25 mM Tris-HCl, pH 7.0). Samples were run at 100 V at 4 °C and imaged by exposure to UV light. Signals were integrated using ImageJ densitometry software (National Institute of Mental Health, Bethesda, Maryland, USA). The Kd was determined from the non-linear fitting of the signal intensities versus PLN concentration51.
FRET Experiments in reconstituted systems
SERCA was incubated with 1,5-I-AEDANS (Invitrogen), which specifically interacts with Cys 674 of the ATPase. The acceptor, Dabcyl-SE, was added at a 20-fold excess to WT PLN in 1% SDS, 100 mM NaHCO3, pH 9.0. The reaction was performed for 16 hours, and the labeled protein was purified using reverse phase HPLC. The steady-state fluorescence wavelengths were 350 nm (excitation) and 400–600 nm (emission).
FRET in-cell Binding Assay
N-termini of canine SERCA2a and canine PLN were fused with either mCerulean (Cer), or enhanced yellow fluorescent protein (YFP)23. Phospholamban oligomerization, quaternary structure, and SERCA binding were measured by fluorescence resonance energy transfer in living cells (line AAV-293). Cells were cultured in complete DMEM growth medium with 10% fetal bovine serum, 1% L-glutamine and incubated at 37 °C under 5% CO2. The cultured cells were subjected to transient transfection using the MBS mammalian transfection kit (Stratagene, La Jolla, CA). Cells were co-transfected with plasmids encoding Cer-PLN-AFA and YFP-PLN-AFA, or Cer-SERCA and YFP-PLN-WT with a molar ratio of 1:5 or 1:20 respectively26, either in the presence or absence of 1 μM unlabeled 50-mer ssDNA. Following transfection, the cells were mildly trypsinized, resuspended in DMEM growth medium, plated on poly-D-lysine-coated glass bottom dishes, and allowed to adhere for 2 hours before imaging, as described previously24. The effect of ssDNA on PLN oligomerization and interaction with SERCA was quantified in live cells using wide-field fluorescence microscopy by acceptor sensitization FRET (EFRET) as described previously24,52. MetaMorph software was used to acquire a montage of 48 images using 40X objective having a numerical aperture of 0.75 and a motorized stage (Prior, Rockland, MA). Focus was automatically maintained by an optical feedback system (Perfect Focus System, Nikon). The exposure time was 150 ms for each channel: Cer, YFP, and FRET (Cer excitation/YFP emission). FRET efficiency was calculated according to the equation (4):
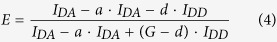 |
where: IDD and IAA are the fluorescence emission intensities of the donor channel (472/30 nm) with excitation of 427/10 nm, and the acceptor channel (542/27 nm) with excitation of 504/12 nm, respectively; IDA is the fluorescence emission intensity of the FRET channel (542/27 nm) with excitation of 427/10 nm. The constants a and d are cross-talk coefficients determined from acceptor-only or donor-only control samples respectively. G represents the ratio of the sensitized emission to the corresponding amount of donor recovery. We obtained values of 0.085, 0.737 and 4.6 for a, d, and G ratio respectively.
The effect of ssDNA on parameters related to structure and binding affinity of PLN oligomer and SERCA-PLN regulatory complex were quantified by performing an ‘in-cell’ binding assay as described previously24. The FRET efficiency of individual cells co-expressing Cer-PLN-AFA and YFP-PLN-AFA, or Cer-SERCA and YFP-PLN-WT was plotted against relative protein concentration, quantified from the observed YFP fluorescence intensities. The cell-by-cell concentration dependence of FRET was fit to a hyperbolic curve to obtain the values of FRETmax, KD1, and KD2. The maximal FRET (FRETmax) is the intrinsic FRET of the protein complex, providing an estimate of average distances between the binding partners. KD is the dissociation constant of the protein complex, providing an estimate of the apparent binding affinity. KD1 is the apparent dissociation constant of the PLN-PLN oligomer and KD2 is the apparent dissociation constant of the PLN-SERCA regulatory complex. For visualization, data from individual cells were pooled and the pooled data were fit by a hyperbolic or Hill function.
Statistical Analysis
The data is pooled from 8 independent experiments for PLB-PLB FRET and 4 independent experiments for SERCA-PLB FRET using approximately 400–1200 cells per sample for each experiment. Errors are reported as standard error of the mean and statistical significance was evaluated using Student’s T test, where p < 0.05 was considered significant.
Additional Information
How to cite this article: Soller, K. J. et al. Rheostatic Regulation of the SERCA/Phospholamban Membrane Protein Complex Using Non-Coding RNA and Single-Stranded DNA oligonucleotides. Sci. Rep. 5, 13000; doi: 10.1038/srep13000 (2015).
Supplementary Material
Acknowledgments
The authors would like to acknowledge Drs. T. Gopinath and K.M. Mote for technical assistance. The NMR experiments were performed at the Minnesota NMR Center. This work was supported by the NIH (GM064742 and GM072701 to G.V.; GM063533 to M.B.) and AHA (13POST14670054 to V.V and 13PRE16950023 to K.S).
Footnotes
GV, MB, MJ, and RV have filed a US patent no. US20140187615 A1 for the use of XNAs as therapeutics for heart failure. The remaining authors declare no competing interest.
Author Contributions G.V., M.T.B. and S.L.R. conceived, directed and analyzed all experimental research; G.V., M.T.B., S.L.R., V.V.V. and K.J.S. prepared the manuscript. K.J.S., R.V. and N.W. prepared NMR samples; K.J.S. and R.V. performed NMR spectroscopy experiments and activity assays. K.J.S. and V.V.V. analyzed NMR data. K.J.S., M.J. and J.Y. prepared and performed CE and FP binding experiments. N.A. performed and analyzed FRET experiments.
References
- Go A. S. et al. Heart disease and stroke statistics—2014 update: a report from the American Heart Association. Circulation 129, e28–e292, 10.1161/01.cir.0000441139.02102.80 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kho C., Lee A. & Hajjar R. J. Altered sarcoplasmic reticulum calcium cycling—targets for heart failure therapy. Nature reviews. Cardiology 9, 717–733, 10.1038/nrcardio.2012.145 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J. et al. Designing heart performance by gene transfer. Physiological reviews 88, 1567–1651, 10.1152/physrev.00039.2007 (2008). [DOI] [PubMed] [Google Scholar]
- Gwathmey J. K., Yerevanian A. & Hajjar R. J. Targeting sarcoplasmic reticulum calcium ATPase by gene therapy. Human gene therapy 24, 937–947, 10.1089/hum.2013.2512 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers D. M. Cardiac excitation-contraction coupling. Nature 415, 198–205, 10.1038/415198a (2002). [DOI] [PubMed] [Google Scholar]
- MacLennan D. H. & Kranias E. G. Phospholamban: a crucial regulator of cardiac contractility. Nature reviews. Molecular cell biology 4, 566–577, 10.1038/nrm1151 (2003). [DOI] [PubMed] [Google Scholar]
- Haghighi K., Gregory K. N. & Kranias E. G. Sarcoplasmic reticulum Ca-ATPase-phospholamban interactions and dilated cardiomyopathy. Biochemical and biophysical research communications 322, 1214–1222, 10.1016/j.bbrc.2004.07.164 (2004). [DOI] [PubMed] [Google Scholar]
- Schmitt J. P. et al. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science (New York, N.Y.) 299, 1410–1413, 10.1126/science.1081578 (2003). [DOI] [PubMed] [Google Scholar]
- Greenberg B. et al. Design of a phase 2b trial of intracoronary administration of AAV1/SERCA2a in patients with advanced heart failure: the CUPID 2 trial (calcium up-regulation by percutaneous administration of gene therapy in cardiac disease phase 2b). JACC. Heart failure 2, 84–92, 10.1016/j.jchf.2013.09.008 (2014). [DOI] [PubMed] [Google Scholar]
- Kairouz V., Lipskaia L., Hajjar R. J. & Chemaly E. R. Molecular targets in heart failure gene therapy: current controversies and translational perspectives. Annals of the New York Academy of Sciences 1254, 42–50, 10.1111/j.1749-6632.2012.06520.x (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bish L. T. et al. Cardiac Gene Transfer of Short Hairpin RNA Directed Against Phospholamban Effectively Knocks Down Gene Expression but Causes Cellular Toxicity in Canines. Human gene therapy 22, 969–977, 10.1089/hum.2011.035 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlquist C. et al. Inhibition of miR-25 improves cardiac contractility in the failing heart. Nature 508, 531–535, 10.1038/nature13073 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y., Honda T., Matsuura K., Kimura Y. & Inui M. In vitro selection and characterization of DNA aptamers specific for phospholamban. The Journal of pharmacology and experimental therapeutics 329, 57–63, 10.1124/jpet.108.149526 (2009). [DOI] [PubMed] [Google Scholar]
- Sakai H. et al. A cell-penetrating phospholamban-specific RNA aptamer enhances Ca transients and contractile function in cardiomyocytes. Journal of molecular and cellular cardiology 76C, 177–185, 10.1016/j.yjmcc.2014.09.006 (2014). [DOI] [PubMed] [Google Scholar]
- Kiriazis H. et al. Hypertrophy and functional alterations in hyperdynamic phospholamban-knockout mouse hearts under chronic aortic stenosis. Cardiovascular research 53, 372–381 (2002). [DOI] [PubMed] [Google Scholar]
- Haghighi K. et al. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. The Journal of clinical investigation 111, 869–876, 10.1172/JCI17892 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshijima M. et al. Chronic suppression of heart-failure progression by a pseudophosphorylated mutant of phospholamban via in vivo cardiac rAAV gene delivery. Nature medicine 8, 864–871, 10.1038/nm739 (2002). [DOI] [PubMed] [Google Scholar]
- Ha K. N., Gustavsson M. & Veglia G. Tuning the structural coupling between the transmembrane and cytoplasmic domains of phospholamban to control sarcoplasmic reticulum Ca(2+)-ATPase (SERCA) function. Journal of muscle research and cell motility 33, 485–492, 10.1007/s10974-012-9319-4 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha K. N. et al. Controlling the inhibition of the sarcoplasmic Ca2+-ATPase by tuning phospholamban structural dynamics. The Journal of biological chemistry 282, 37205–37214, 10.1074/jbc.M704056200 (2007). [DOI] [PubMed] [Google Scholar]
- Simmerman H. K., Collins J. H., Theibert J. L., Wegener A. D. & Jones L. R. Sequence analysis of phospholamban. Identification of phosphorylation sites and two major structural domains. The Journal of biological chemistry 261, 13333–13341 (1986). [PubMed] [Google Scholar]
- Keefe A. D., Pai S. & Ellington A. Aptamers as therapeutics. Nature reviews. Drug discovery 9, 537–550, 10.1038/nrd3141 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmerman H. K. & Jones L. R. Phospholamban: protein structure, mechanism of action, and role in cardiac function. Physiological reviews 78, 921–947 (1998). [DOI] [PubMed] [Google Scholar]
- Kelly E. M., Hou Z., Bossuyt J., Bers D. M. & Robia S. L. Phospholamban Oligomerization, Quaternary Structure, and Sarco(endo)plasmic Reticulum Calcium ATPase Binding Measured by Fluorescence Resonance Energy Transfer in Living Cells. Journal of Biological Chemistry 283, 12202–12211, 10.1074/jbc.M707590200 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidwell P., Blackwell D. J., Hou Z., Zima A. V. & Robia S. L. Phospholamban Binds with Differential Affinity to Calcium Pump Conformers. Journal of Biological Chemistry 286, 35044–35050, 10.1074/jbc.M111.266759 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha K. N. et al. Lethal Arg9Cys phospholamban mutation hinders Ca2+-ATPase regulation and phosphorylation by protein kinase A. Proceedings of the National Academy of Sciences of the United States of America 108, 2735–2740, 10.1073/pnas.1013987108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Z., Kelly E. M. & Robia S. L. Phosphomimetic Mutations Increase Phospholamban Oligomerization and Alter the Structure of Its Regulatory Complex. Journal of Biological Chemistry 283, 28996–29003, 10.1074/jbc.M804782200 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D. M., Bendall M. R., Pegg D. T., Doddrell D. M. & Field J. Two-dimensional 13C•1H polarization transfer J spectroscopy. Journal of Magnetic Resonance (1969) 42, 298–306, http://dx.doi.org/10.1016/0022-2364(81)90219-5 (1981). [Google Scholar]
- Gustavsson M. et al. Allosteric regulation of SERCA by phosphorylation-mediated conformational shift of phospholamban. Proceedings of the National Academy of Sciences of the United States of America, 10.1073/pnas.1303006110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son W. S. et al. ‘q-Titration’ of long-chain and short-chain lipids differentiates between structured and mobile residues of membrane proteins studied in bicelles by solution NMR spectroscopy. Journal of Magnetic Resonance 214, 111–118, http://dx.doi.org/10.1016/j.jmr.2011.10.011 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranias E. G. & Hajjar R. J. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circulation research 110, 1646–1660, 10.1161/CIRCRESAHA.111.259754 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamoo Y., Friedman A. M., Parsons M. R., Konigsberg W. H. & Steitz T. A. Crystal structure of a replication fork single-stranded DNA binding protein (T4 gp32) complexed to DNA. Nature 376, 362–366 (1995). [DOI] [PubMed] [Google Scholar]
- Revzin A. The Biology of Nonspecific DNA-Protein Interactions. (CRC Press, 1990). [Google Scholar]
- Dickey T. H., Altschuler S. E. & Wuttke D. S. Single-Stranded DNA-Binding Proteins: Multiple Domains for Multiple Functions. Structure 21, 1074–1084, http://dx.doi.org/10.1016/j.str.2013.05.013 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumarswamy R. & Thum T. Non-coding RNAs in cardiac remodeling and heart failure. Circulation research 113, 676–689, 10.1161/CIRCRESAHA.113.300226 (2013). [DOI] [PubMed] [Google Scholar]
- Bronze-da-Rocha E. MicroRNAs Expression Profiles in Cardiovascular Diseases. BioMed Research International. 2014, 23, 10.1155/2014/985408 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan L., Xiong X., Liu Y. & Wang J. miRNA-1: functional roles and dysregulation in heart disease. Molecular BioSystems, 10.1039/C4MB00338A (2014). [DOI] [PubMed] [Google Scholar]
- Winbanks C. E., Ooi J. Y. Y., Nguyen S. S., McMullen J. R. & Bernardo B. C. MicroRNAs differentially regulated in cardiac and skeletal muscle in health and disease: Potential drug targets? Clinical and Experimental Pharmacology and Physiology, n/a-n/a, 10.1111/1440-1681.12281 (2014). [DOI] [PubMed] [Google Scholar]
- van Rooij E. & Olson E. N. MicroRNA therapeutics for cardiovascular disease: opportunities and obstacles. Nature reviews. Drug discovery 11, 860–872, 10.1038/nrd3864 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dangwal S. & Thum T. microRNA therapeutics in cardiovascular disease models. Annual review of pharmacology and toxicology 54, 185–203, 10.1146/annurev-pharmtox-011613-135957 (2014). [DOI] [PubMed] [Google Scholar]
- Bao M.-H. et al. Let-7 in cardiovascular diseases, heart development and cardiovascular differentiation from stem cells. International journal of molecular sciences 14, 23086–23102, 10.3390/ijms141123086 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gröβl T. et al. A Novel Artificial MicroRNA Expressing AAV Vector for Phospholamban Silencing in Cardiomyocytes Improves Ca2+ Uptake into the Sarcoplasmic Reticulum. PloS one 9, e92188, 10.1371/journal.pone.0092188 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck B. et al. Overexpression, purification, and characterization of recombinant Ca-ATPase regulators for high-resolution solution and solid-state NMR studies. Protein expression and purification 30, 253–261 (2003). [DOI] [PubMed] [Google Scholar]
- Stokes D. L. & Green N. M. Three-dimensional crystals of CaATPase from sarcoplasmic reticulum. Symmetry and molecular packing. Biophysical Journal 57, 1–14 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ablorh N.-A. et al. Accurate quantitation of phospholamban phosphorylation by immunoblot. Analytical Biochemistry 425, 68–75, http://dx.doi.org/10.1016/j.ab.2012.01.028 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden T. D., Chapman D. & Quinn P. J. Cholesterol modulates activity of calcium-dependant ATPase of the sarcoplasmic reticulum. Nature 279, 538–541 (1979). [DOI] [PubMed] [Google Scholar]
- Takegoshi K., Nakamura S. & Terao T. in Chemical Physics Letters Vol. 344 631–637 (2001). [Google Scholar]
- Delaglio F. et al. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. Journal of Biomolecular NMR 6, 277–293, 10.1007/BF00197809 (1995). [DOI] [PubMed] [Google Scholar]
- Bowser M. T. & Chen D. D. Y. Monte Carlo Simulation of Error Propagation in the Determination of Binding Constants from Rectangular Hyperbolae. 2. Effect of the Maximum-Response Range. The Journal of Physical Chemistry A 103, 197–202, 10.1021/jp982917e (1999). [DOI] [Google Scholar]
- Wei a. P. & Herron J. N. Use of synthetic peptides as tracer antigens in fluorescence polarization immunoassays of high molecular weight analytes. Analytical chemistry 65, 3372–3377 (1993). [DOI] [PubMed] [Google Scholar]
- Onizuka T., Endo S., Hirano M., Kanai S. & Akiyama H. Design of a Fluorescent Electrophoretic Mobility Shift Assay Improved for the Quantitative and Multiple Analysis of Protein-DNA Complexes. Bioscience, Biotechnology, and Biochemistry 66, 2732–2734, 10.1271/bbb.66.2732 (2002). [DOI] [PubMed] [Google Scholar]
- Jing M. & Bowser M. T. Isolation of DNA aptamers using micro free flow electrophoresis. Lab on a chip 11, 3703–3709, 10.1039/c1lc20461k (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zal T. & Gascoigne N. R. J. Photobleaching-Corrected FRET Efficiency Imaging of Live Cells. Biophysical Journal 86, 3923–3939, http://dx.doi.org/10.1529/biophysj.103.022087 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.