

Abstract
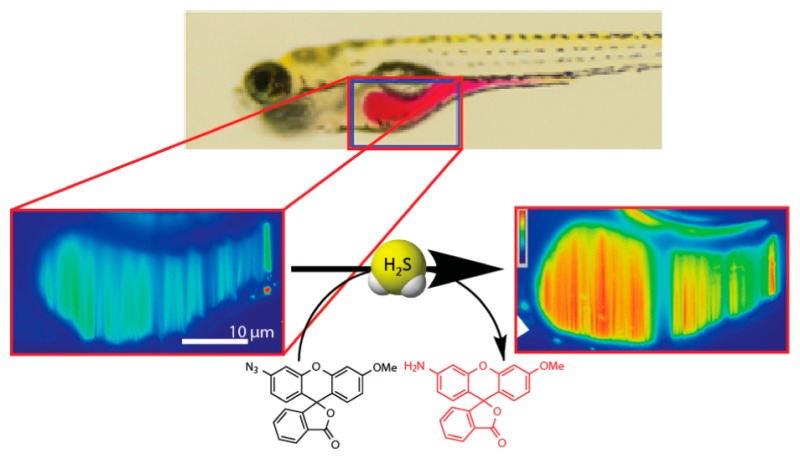
Hydrogen sulfide (H2S) is a critical gaseous signaling molecule emerging at the center of a rich field of chemical and biological research. As our understanding of the complexity of physiological H2S in signaling pathways evolves, advanced chemical and technological investigative tools are required to make sense of this interconnectivity. Toward this goal, we have developed an azide-functionalized O-methylrhodol fluorophore, MeRho-Az, which exhibits a rapid >1000-fold fluorescence response when treated with H2S, is selective for H2S over other biological analytes, and has a detection limit of 86 nM. Additionally, the MeRho-Az scaffold is less susceptible to photoactivation than other commonly used azide-based systems, increasing its potential application in imaging experiments. To demonstrate the efficacy of this probe for H2S detection, we demonstrate the ability of MeRho-Az to detect differences in H2S levels in C6 cells and those treated with AOAA, a common inhibitor of enzymatic H2S synthesis. Expanding the use of MeRho-Az to complex and heterogeneous biological settings, we used MeRho-Az in combination with light sheet fluorescence microscopy (LSFM) to visualize H2S in the intestinal tract of live zebrafish. This application provides the first demonstration of analyte-responsive 3D imaging with LSFM, highlighting the utility of combining new probes and live imaging methods for investigating chemical signaling in complex multicellular systems.
Introduction
The perception of hydrogen sulfide (H2S) in the scientific community has shifted dramatically in the 21st century.1 No longer viewed as merely a toxic geological and environmental pollutant, H2S is now at the center of a rich and expanding field focused on investigating its biological and physiological significance. Since 1996, when Abe and Kimura first suggested that H2S acts as a neuromodulator in hippocampal long-term potentiation,2 H2S has been recognized as an essential gasotransmitter that regulates many important physiological functions in the cardiovascular, nervous, endocrine, immune, and gastrointestinal systems.1,3−7 Biosynthesized by three main enzymes, cystathionine-β-synthase (CBS), cystathionine-γ-lyase (CSE), and 3-mercaptopyruvate sulfurtransferase (3-MST), H2S is generated enzymatically in the heart, brain, liver, and kidneys; however, localized H2S concentrations in the body are tissue-dependent, suggesting differential activation and action in various H2S-producing pathways.8 Once generated, H2S undergoes complex catabolism through its interactions with cellular oxidants, protein transition-metal centers, and reactive sulfur, oxygen, and nitrogen species (RSONs), all of which are sensitive to internal and external redox stimuli. For example, oxidative S-sulfhydration (or persulfidation) of protein cysteine residues is proposed to constitute a significant sulfide storage mechanism, which modifies protein function and the signaling activity of H2S.9−12 The intricacy of physiological H2S reactivity requires that researchers utilize advanced chemical and technological tools for H2S detection and imaging in order to gain a more detailed understanding of the interconnectivity of these networks.
In recent years chemists have answered the call for improved tools for H2S detection by developing small-molecule fluorescent probes and similar methods to investigate biological H2S.13,14 Historically the most widely utilized assay for H2S detection and quantification has been the methylene blue assay.15 This technique, however, requires sample homogenization and a harshly acidic workup that precludes real-time detection or live-animal imaging. These conditions also liberate sulfur from acid-labile sulfur pools and are thus not selective for H2S.16 By contrast, H2S quantification using monobromobimane (mBB) has better detection limits and enables separation of free, sulfane, and acid-labile sulfide pools.17 Although the mBB method offers a robust platform for H2S quantification, this technique still requires sample destruction and additional HPLC analysis. Similarly, the usefulness of other techniques including gas chromatography and sulfur-selective electrodes are limited by complex workups and/or insufficient sensitivity.18,19 Alternatively, reaction-based fluorescent probes offer the potential for in vivo compatibility, high sensitivity, and high spatiotemporal resolution while maintaining selectivity for H2S over other RSONs including free thiols, which are abundant in much higher concentrations than H2S in cellular milieu. On the basis of these requirements, three main reaction-based strategies for H2S imaging have been developed: using the dual-nucleophilicity of H2S to liberate ester-bound fluorophores with nearby reactive electrophilic sites,20−26 displacement of CuS from CuII-ligated fluorophores,27−29 and reduction of nitro- and azide-functionalized fluorophores.30−45 Among these strategies, H2S-mediated azide reduction has been the most broadly reported due to the plethora of amine-functionalized fluorophores available for modification and the ease of azide functional group installation. Azide reduction is often rapid and produces large (10–100-fold) fluorescence turn-ons, with the resultant probes exhibiting functional low micromolar detection limits and excellent selectivity profiles. One limitation of such methods, however, is the potential photoreduction of azides to amines, which can lead to unwanted photoactivation in long-term imaging experiments.39
Despite the rapidly advancing progress in H2S probe development, few examples of live-animal imaging and application of these tools in biological studies exist43,46−48 due to the substantial challenges associated with transitioning from cell culture to whole organisms. Additionally, the innate sensitivity of fluorophores to high energy excitation must be considered against tissue penetration requirements for in vivo imaging, thus complicating problems with azide photoactivation. Laser scanning confocal microscopy, a popular technique in three-dimensional fluorescence imaging, inherently sacrifices illumination efficiency in order to achieve highly resolved, focused images. This process often results in photobleaching (or photoactivation). An alternative imaging strategy with significantly reduced photobleaching and phototoxicity by comparison to confocal or even 2-photon illumination is light sheet fluorescence microscopy (LSFM), in which excitation light is confined to a thin sheet coinciding with the focal plane of a wide field imaging system.49−53 LSFM also provides access to significantly larger samples (>1 mm) than conventional confocal microscopy, while maintaining fast imaging times. LSFM of larval zebrafish, a useful model for many aspects of vertebrate development,54 enables the imaging of intestinal tract contents with three-dimensional spatial resolution of microns, temporal resolution of seconds, and durations of tens of hours. Such contents can include bacterial communities colonizing the gut of the fish, a particularly interesting target for future investigations of microbial sulfur metabolism and the role of H2S and associated sulfur-containing species in the gut microbiota.55,56 A key unmet step toward bridging this gap, however, is the utilization of analyte-responsive imaging tools in combination with LSFM. Combining these application-driven approaches would enable significant new avenues of investigation, such as small-molecule and secondary messenger trafficking, by providing access to real-time, analyte-responsive imaging in whole organisms. Toward this goal, we report herein the development of a bright fluorescent probe for selective H2S imaging and demonstrate for the first time analyte-responsive detection experiments in combination with LSFM in live zebrafish.
Results and Discusssion
Because of the small sample volume excited during LSFM experiments, a high dynamic range and a bright fluorophore are key probe requirements for analyte detection studies. Although fluorescein and rhodamine are common platforms in the design of reaction-based probes because of their excellent photophysical properties, including high extinction coefficients and quantum yields,57 their susceptibility to photobleaching and pH sensitivity can limit their versatility in certain biological environments. Additionally, although reaction-based probes for H2S detection based on azido-fluorescein or rhodamine conjugates have provided useful tools for investigations of H2S in context,30,42 the dynamic range of these scaffolds (typically <25-fold turn-on) remains insufficient for LSFM investigations. To overcome these limitations, we reasoned that the rhodol (or rhodafluor) family of fluorophores would provide an attractive platform well suited for LSFM with improved pH insensitivity and photostability, while retaining many of the key photophysical advantages of their parent structures.58 Additionally, O-alkylation of rhodols provides a potential handle for structural modification, and unlike fluorescein, O-alkylation typically does not appreciably mitigate the quantum yield. Consequently, rhodol derivatives have been adopted as sensors for hydrogen peroxide, hydrolase activity, nitroxyl, and thiols in recent years.59−62 We envisioned that an O-methylrhodol (MeRho) modified by an azide-functionalized xanthene core (MeRho-Az) would be locked in a nonfluorescent spirocyclic lactone tautomer. H2S-mediated azide reduction would unmask fluorophore fluorescence by regenerating the amine and unlocking the fluorescent open tautomer (Scheme 1). Given the inherent brightness of rhodols and the rapidity of H2S-mediated azide reduction, we reasoned that MeRho-Az would produce a strong fluorescent response to H2S when reduced, thus providing a high fidelity functional tool for studying H2S in vivo that is compatible with LSFM.
Scheme 1. Synthesis and H2S-Mediated Activation of MeRho-Az.
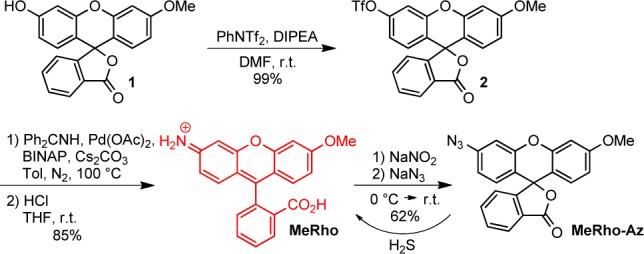
To test this design hypothesis, we first prepared the rhodol scaffold by adapting the modular rhodol synthesis reported by Yang and co-workers63 to convert methylfluorescein (1) to MeRho in two steps. Triflation of 1 with N-phenyl-bis(trifluoromethanesulfonamide) to form triflated methylfluorescein (2), and subsequent Buchwald-Hartwig amination with benzophenone imine as an ammonia equivalent, followed by acid hydrolysis, affords MeRho in 84% overall yield. During reaction optimization, we found that initially heating the amination reaction to 140 °C increased the yield significantly, presumably by either facilitating efficient formation of the L–Pd0 active catalyst or by accelerating oxidative addition of 2. Finally, diazotization of 2 and azidification under Sandmeyer conditions affords MeRho-Az.
With both MeRho and MeRho-Az in hand, the photophysical properties of each compound were characterized (Table 1). MeRho displays excellent solubility in aqueous buffer (50 mM PIPES, 100 mM KCl, pH 7.4), with absorption and fluorescence bands centered at 476 and 516 nm, respectively (Figure S1). As predicted, MeRho exhibits a high quantum yield (ΦMeRho = 0.57), whereas the quantum yield of the closed lactone form of MeRho-Az is essentially zero when excited at either the λmax (286 nm) or that of MeRho (476 nm). To establish the fidelity of the MeRho scaffold under physiological conditions, we investigated the pH-dependent fluorescence. By performing a pH titration and monitoring the fluorescence, we established that the MeRho fluorophore maintains a constant emission between pH 4.5 and 10 (Figure 1), with apparent pKa values of 3.3 and 12.2, making the biologically viable pH range superior to that of fluorescein (Figure S2). Additionally, the MeRho fluorophore maintains 75% of its maximum fluorescence under highly acidic conditions.
Table 1. Spectroscopic Properties of MeRho and MeRho-Az in PIPES Buffer (50 mM, 100 mM KCl, pH 7.4)a.
| λmax (nm) | ε (M–1 cm–1) | λem (nm) | ϕ | |
|---|---|---|---|---|
| MeRho | 476 | 30 800 | 516 | 0.57 |
| MeRho-Az | 286 | 13 900 | N/A | <0.01 |
Quantum yield are presented relative to fluorescein (0.1 M NaOH).
Figure 1.
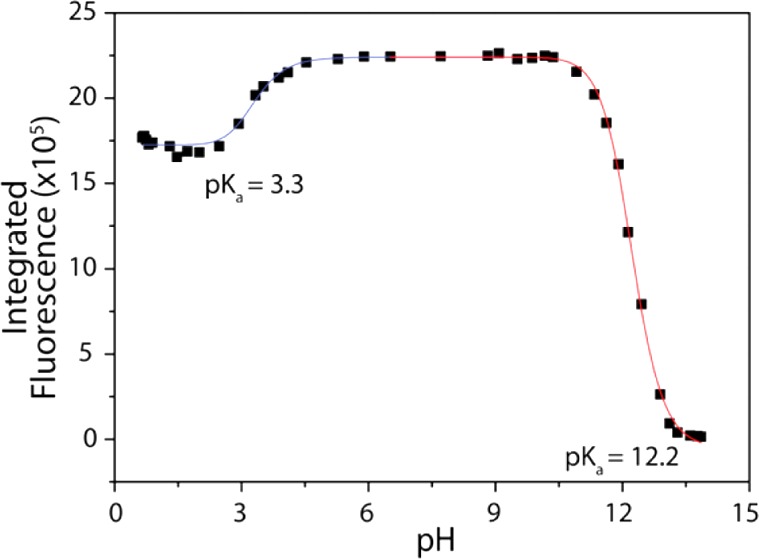
Integrated MeRho fluorescence (20 μM, λex = 476 nm, λem = 480–650 nm) in aqueous solution at various pH values (100 mM KCl).
Having established that MeRho offers a bright, biocompatible fluorophore platform, we next investigated the viability of MeRho-Az as a fluorescent H2S sensor. MeRho-Az (5 μM) exhibits a rapid increase in fluorescence when treated with 50 equiv of NaHS (250 μM) in aqueous PIPES buffer (50 mM, 100 mM KCl, pH 7.4). Owing to the stark contrast in brightness between the azide- and amine-functionalized rhodol scaffolds, reduction of MeRho-Az to the parent amine produces a 1200-fold fluorescence turn on (F/F0) over 60 min (440-fold without any background correction) (Figure 2a). This represents one of the strongest fluorescent responses from H2S detection recorded to date. While the reaction of some probes with H2S may reach completion more quickly, the magnitude of response with MeRho-Az after only 5 min is significant. Furthermore, the fluorescence turn-on characteristics of MeRho-Az are faster and stronger than a recently reported nitro-reduction rhodol platform,64 which is consistent with previous findings from our group in which azide reduction on a naphthalimide scaffold proceeds faster and has a stronger turn-on than the corresponding nitro-functionalized analogue.65 After determining that MeRho-Az effectively reports on H2S, the sensitivity and detection limit of the probe was examined. A linear, concentration-dependent fluorescence relationship was observed between MeRho-Az fluorescence and increasing H2S concentrations (Figure 2b, Table S1). The detection limit was calculated to be the concentration at which the fluorescence equals that of [blank + 3σ] according to a linear regression fit of the data and determined to be 86 ± 7 nM. Supporting the validity of this detection limit, the MeRho-Az probe can differentiate between 1.0 and 0.10 μM H2S with a p value <0.01. Finally, to test the photostability of MeRho-Az, we prepared three common azide-based H2S detection probes HSN2, DNS-N3, and C7-Az,31,40,65 which are based on naphthalimide, dansyl, and coumarin fluorophores, respectively, and compared the photoactivation of each azide under identical conditions in the absence of H2S. As expected, the rhodol system in MeRho-Az exhibits significantly less photoactivation than the other azide-based systems (Figure 3c). Taken together, these data demonstrate the reactivity of MeRho-Az with H2S and highlights its sensitivity and potential for use in biological applications.
Figure 2.
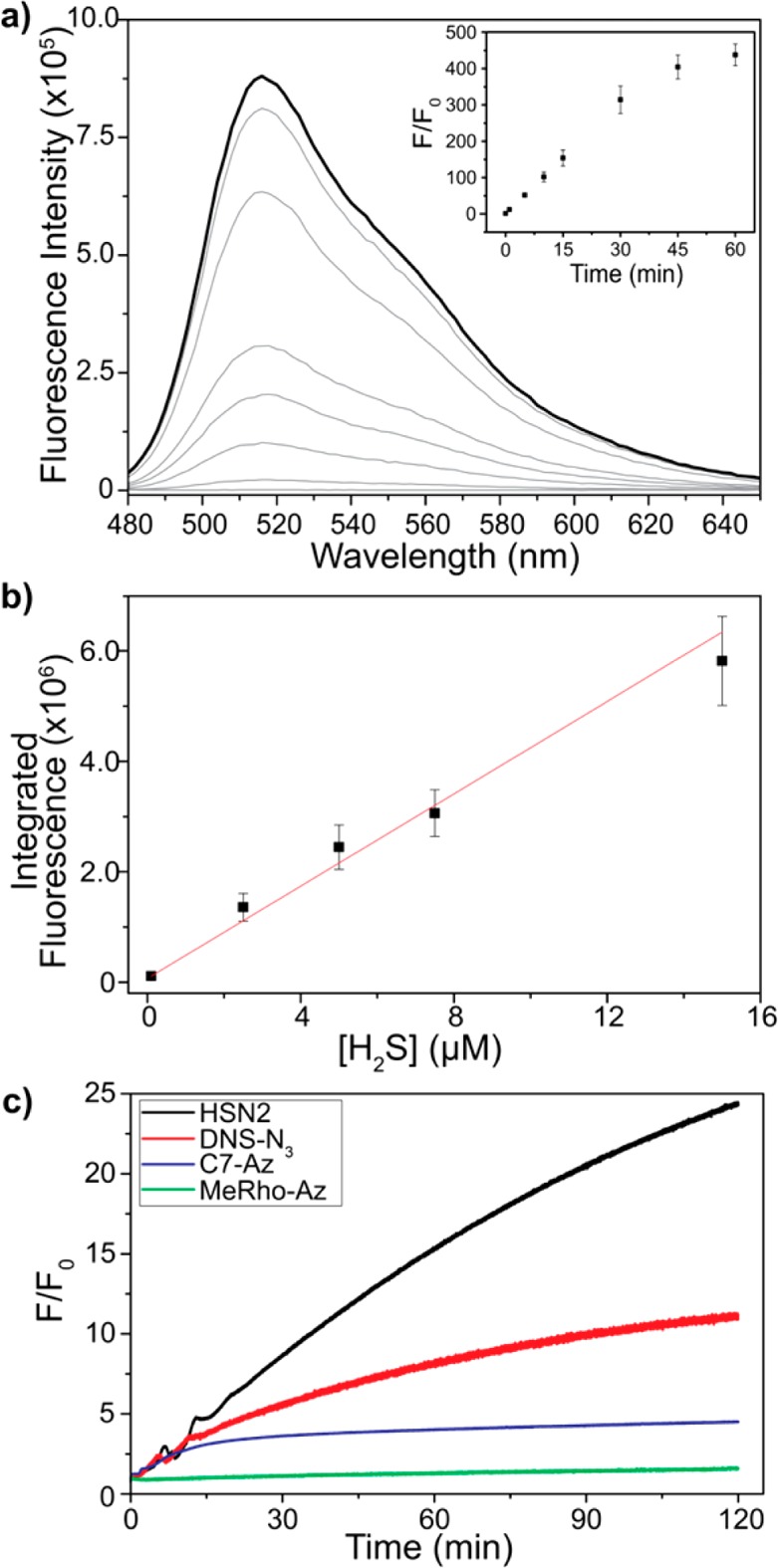
(a) Uncorrected fluorescent response of MeRho-Az to NaHS treatment over 60 min. Conditions: 5 μM MeRho-Az, 250 μM NaHS, PIPES buffer (50 mM, 100 mM KCl, pH 7.4), λex = 476 nm, λem = 480–650 nm, 37 °C. (b) Concentration-dependent fluorescence of MeRho-Az when treated with 0.10, 2.5, 5.0, 7.5, and 15 μM NaHS and incubation for 90 min at 37 °C. Each data point represents the average of at least three trials. Error bars were calculated as standard deviation. (c) Fluorescence photoactivation response of HSN2 (λex = 432 nm, λem = 542 nm), DNS-N3 (λex = 340 nm, λem = 550 nm), C7-Az (λex = 340 nm, λem = 445 nm), and MeRho-Az (λex = 476 nm, λem = 516 nm). Excitation slits: 2.6 nm. Data measured at 4 s–1.
Figure 3.
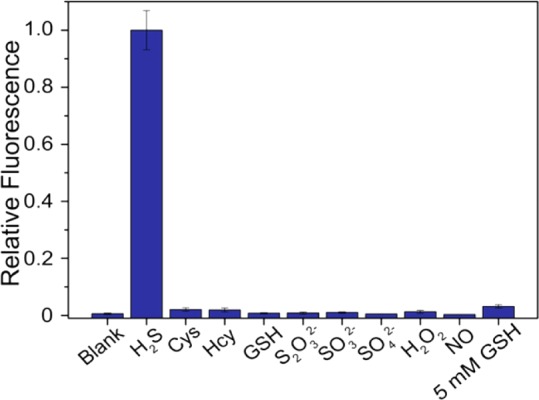
Selectivity profile of MeRho-Az toward reactive sulfur, oxygen, and nitrogen species. From left to right: blank, NaHS, l-cysteine, dl-homocysteine, glutathione, Na2S2O3, Na2SO3, Na2SO4, H2O2, and DEA NONOate. Conditions: 5 μM MeRho-Az, 250 μM RSONs, PIPES buffer (50 mM, 100 mM KCl, pH 7.4), λex = 476 nm, λem = 480–650 nm, 37 °C. Data were acquired after 60 min incubation at 37 °C.
After establishing the concentration-dependent reactivity for MeRho-Az with H2S, we examined the reactivity of various RSONs toward the probe to establish a selectivity profile (Figure 3). No fluorescence was observed upon introduction of 50 equiv (250 μM) of biological thiols cysteine (Cys), homocysteine (Hcy), or glutathione (GSH) over 60 min. Sulfur anions thiosulfate, (S2O32–), sulfite (SO32–), and sulfate (SO42–), as well as hydrogen peroxide (H2O2) and nitric oxide (NO) also all proved to be chemically inert toward the probe. Additionally, MeRho-Az exhibits 32-fold preferential reactivity with H2S relative to 5 mM GSH loading, thus reinforcing the excellent selectivity of the H2S-mediated azide-reduction mechanism.
On the basis of the excellent H2S sensing properties of MeRho-Az, we sought to establish the efficacy of MeRho-Az for detecting endogenously produced H2S in cells. After incubation of C6 rat glial cells, which express the H2S-producing CBS enzyme, with 5 μM MeRho-Az for 45 min, the cells were fixed and imaged using a fluorescence microscope (Figure 4). We then compared this fluorescence response with cells that had been pretreated with either a slow-releasing H2S donor (AP39, 100 nM)66 or a common CBS inhibitor (aminooxyacetic acid, AOAA, 20 μM).67 We observed a significant reduction in fluorescence in cells treated with AOAA by contrast to untreated cells, suggesting that MeRho-Az is sufficiently sensitive to detect endogenous levels of enzymatically produced H2S. Furthermore, cells treated with low concentrations of AP39 showed enhanced fluorescence, highlighting the sensitivity of the system. These results demonstrate the applicability of the MeRho-Az platform in cellular environments, which can likely be extended to assays involving biological fluids such as serum, blood, or tissue homogenates.
Figure 4.
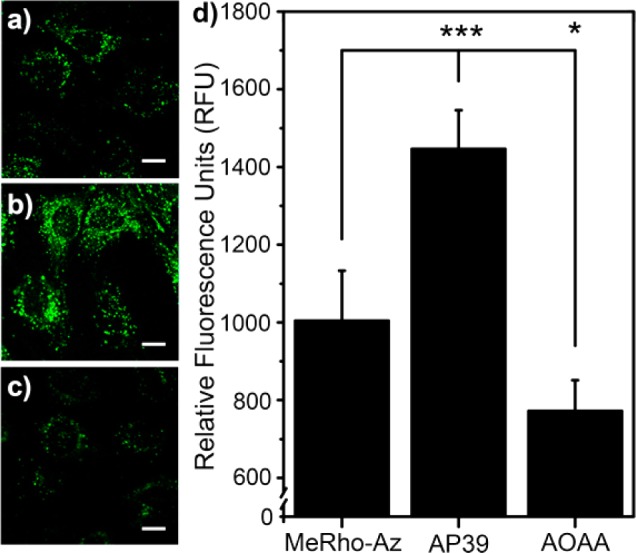
Fluorescence imaging of H2S in C6 cells. Cells were imaged after incubation with 5 μM MeRho-Az for 45 min after pretreatment with (a) no pretreatment, (b) 100 nM AP39 for 60 min, or (c) 20 μM AOAA for 45 min. Scale bars = 5 μm. (d) Quantified cellular fluorescence after reaction of MeRho-Az with endogenous H2S (MeRho-Az, N = 24 cells), after addition of exogenous H2S (AP39, N = 24 cells), and after inhibition of enzymatic H2S production (AOAA, N = 24 cells).
To further establish MeRho-Az as an in vivo H2S reporter, we next examined its biocompatibility using LSFM. Because little is known about endogenous sulfide dynamics in developing zebrafish, we focused our initial efforts on H2S release from a commonly used slow-releasing H2S donor, diallyl trisulfide (DATS). To confirm, as previously reported, that a thiol such as GSH is required to achieve H2S release from DATS,68 we used MeRho-Az with DATS to detect liberated H2S and observed a dose-dependent release of H2S in response to [GSH] (Figure S3). To expand on the use of MeRho-Az and to establish its validity for use with LSFM in live organisms, we chose to use larval (7 days post fertilization) zebrafish for imaging studies. At this stage in their development, zebrafish are approximately 3 mm in length and maintain a high level of transparency. Also, a key benefit of LSFM is that the collection of illuminated sheets that make up the three-dimensional images is obtained on a time scale (∼10 s in total) significantly faster than the time scale of gut peristalsis (∼1 min), which allows for direct analysis of the actual gut volume with minimal artifacts from translational movement. We first tested the toxicity of MeRho-Az in larval zebrafish by orally gavaging69 7 nL of buffered solutions (50 mM PIPES, 100 mM KCl, pH 7.4) containing 5 μM MeRho-Az and monitored the fish over time. No toxicity was observed for 20 h, and although this cannot completely rule out unwanted biological effects in longer term experiments, it suggests the safe use of MeRho-Az as a viable in vivo fluorescent reporter over the time scale of hours. To test the ability of MeRho-Az to provide an analyte-responsive signal toward H2S, larval zebrafish were orally gavaged with buffered solutions containing: buffer only, 5 μM MeRho-Az, 5 μM MeRho-Az + 250 μM DATS, 5 μM MeRho-Az + 250 μM DATS + 250 μM GSH, or 5 μM MeRho. After 60 min of recovery time, a three-dimensional image of the intestinal bulb for each fish was acquired using LSFM (Figure 5).
Figure 5.
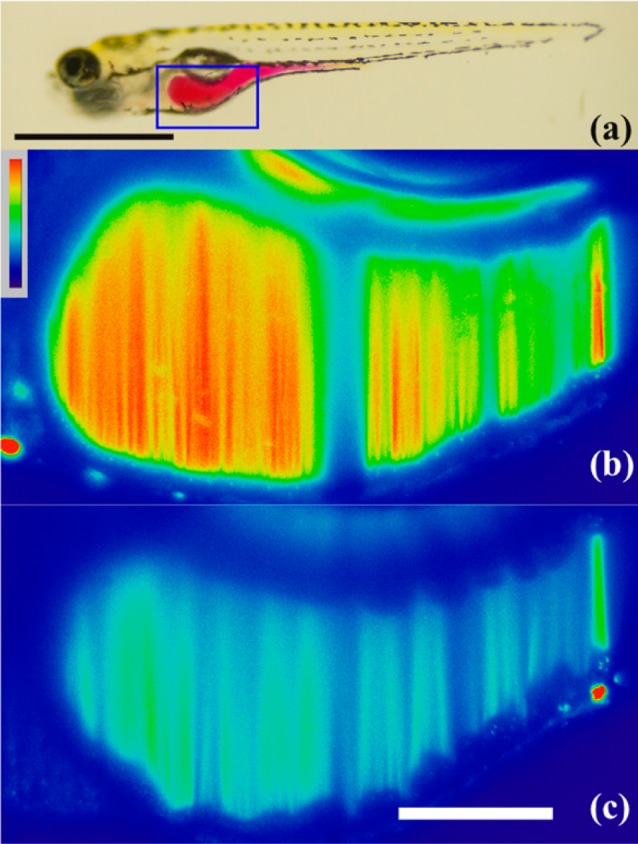
2D slices of LSFM images of live zebrafish 60 min after gavage. (a) Larval zebrafish (7 dpf) gavaged with phenol red to highlight the intestine (scale bar = 1 mm). The boxed region corresponds to the intestinal bulb expanded below in (b,c). Zebrafish gavaged with (b) 5 μM MeRho-Az + 250 μM DATS + 250 μM GSH or (c) 5 μM MeRho-Az. Scale bar in (b) and (c) = 10 μm.
No difference in fluorescence was observed when comparing the signal between a vehicle-gavaged control group and fish gavaged with either MeRho-Az alone or MeRho-Az + DATS, confirming that LSFM was not causing photoactivation of the azide and that H2S release from DATS was GSH-dependent (Figure 6).70 In contrast, fish gavaged with both MeRho-Az and DATS/GSH were measurably brighter than the vehicle or MeRho-Az alone (Figure 6), confirming that H2S was being captured by MeRho-Az and visualized using LSFM (Figure 5, see Supporting Information for a link to video compiling 2D image slices into a 3D representation). To compare the relative intensity of the fully activated probe, we also gavaged fish with the fluorophore, MeRho, alone, which resulted in an identical intensity to that observed with MeRho-Az with DATS/GSH, which is consistent with efficient H2S-mediated activation of MeRho-Az in the zebrafish gut. To the best of our knowledge, these data demonstrate the first use of LSFM for live-animal imaging of analyte-responsive reaction-based probes, thus opening the door for new investigations of whole-organism imaging in the context of reactive small molecule analytes. In a broader context, the three-dimensional imaging capability afforded by LSFM is crucial for accurately determining fluorescence intensity in a whole organism due to the heterogeneity of basal autofluorescence, reflection, and absorption of various tissues and organs. Differentiation and separation of these different signals would not be possible without the 3D intensity map afforded by LSFM imaging.
Figure 6.

Average fluorescence intensity in zebrafish intestinal bulb, normalized to the mean of the buffer-gavaged set. Each dot represents one fish, each of which provided ∼107 intensity measurements. Boxes extend to the first and third quartile; whiskers enclose data within 1.5 times the interquartile range. Solid lines denote median, and dashed lines denote mean values. Shown are measurements for fish orally gavaged with 6.9 nL of buffered solutions (50 mM PIPES, pH 7.4): buffer (N = 5), 5 μM MeRho-Az (N = 9), 5 μM MeRho-Az + 250 μM DATS (N = 8), 5 μM MeRho-Az + 250 μM DATS + 250 μM GSH (N = 8), and 5 μM MeRho (N = 6).
Conclusions
In summary, motivated by enabling new whole-animal imaging techniques for H2S, we have developed a bright, selective fluorescent probe for H2S detection based on a rhodol platform and demonstrated its application both in cells and in LSFM experiments with live zebrafish. This new application of LSFM for use with reaction-based analyte-responsive probes is enabled by the large dynamic range, high photostability, and excellent selectivity afforded by MeRho-Az and would not have been possible with previously reported H2S sensing systems. On the basis of the broad importance of sulfide in gastrointestinal biology, we are currently exploring this, as well as other techniques to investigate sulfide genesis and action in developing gut microbiota.
Experimental Section
Materials and Methods
Reagents were purchased from Sigma-Aldrich or Tokyo Chemical Industry (TCI) and used as received. Methylfluorescein (1), HSN2, DNS-N3, and C7-Az, and AP39 were synthesized as reported previously.31,40,65,66,71 Deuterated solvents were purchased from Cambridge Isotope Laboratories and used as received. Silica gel (SiliaFlash F60, Silicycle, 230–400 mesh) was used for column chromatography. Preparatory chromatography was performed on Silicycle SiliaPlates (1 mm thickness). 1H and 13C{1H} NMR spectra were recorded on a Varian INOVA 500 MHz NMR instrument. Chemical shifts are reported in ppm relative to residual protic solvent resonances. UV–visible spectra were acquired on a Cary 100 spectrometer equipped with a Quantum Northwest TLC-42 dual cuvette temperature controller at 37.00 ± 0.05 °C. Fluorescence spectra were obtained on a Quanta Master 40 spectrofluorometer (Photon Technology International) equipped with a Quantum Northwest TLC-50 temperature controller at 37.0 ± 0.05 °C.
Spectroscopic Materials and Methods
Piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES) and potassium chloride (99.999%) were used to make buffered solutions (50 mM PIPES, 100 mM KCl, pH 7.4) in Millipore water. Buffer solutions were sparged with N2 to remove dissolved oxygen. Anhydrous sodium hydrosulfide (NaHS) was purchased from Strem Chemicals and handled under nitrogen. DEA NONOate (used to generate NO) was purchased from Cayman. Stock solutions of MeRho-Az were prepared in an N2-filled glovebox and stored at −25 °C until immediately before use. Aqueous stock solutions of l-cysteine, homocysteine, glutathione, NaS2O3, Na2SO3, Na2SO4, and H2O2 were freshly prepared in an N2-filled glovebox prior to use. Stock solutions of DEA NONOate were prepared in degassed 0.01 M NaOH immediately prior to use. Spectroscopic measurements were obtained under anaerobic conditions using septum-sealed cuvettes obtained from Starna Scientific.
General Procedure for Fluorescence and Selectivity Measurements
A septum-sealed cuvette was charged with 3.00 mL of buffer (50 mM PIPES, 100 mM KCl, pH 7.4) in a glovebox. After injection of a MeRho-Az (15 μL, 1.0 mM in DMSO) stock solution via syringe, an initial fluorescence spectrum was recorded (λex = 476 nm, λem = 480–650 nm). A NaHS stock solution (15 μL, 50 mM in PIPES buffer) was then injected via syringe, and the fluorescence was recorded after 1, 5, 10, 15 30, 45, and 60 min. The reaction cuvette was incubated at 37 °C during the experiment.
pKa Determination
An aqueous MeRho solution (20 μM, 100 mM KCl, 10 mL) was prepared in a centrifuge tube and acidified to pH 0.656 using 12.1 M HCl. After transferring 3.00 mL of this solution to a cuvette, the MeRho fluorescence was recorded (λex = 476 nm, λem = 480–650 nm). The solution in the cuvette was then returned to the centrifuge tube, and the MeRho solution was basified incrementally to pH 13.859 using stock solutions of KOH at various concentrations (10, 5, 1, 0.1 M). A fluorescence spectrum was recorded at each pH increment.
Determination of Detection Limit
The fluorescence of seven blank cuvettes containing MeRho-Az (5 μM, λex = 476 nm, λem = 480–650 nm) was recorded after incubation at 37 °C for 90 min in PIPES buffer (50 mM, 100 mM KCL, pH 7.4). Then MeRho-Az was treated with NaHS at various concentrations (0.10, 2.5, 5.0, 7.5, 15 μM), and the fluorescence spectra were measured after incubation for 90 min at 37 °C. Each data point represents at least three trials. A linear regression was constructed using the background-corrected fluorescence measurements, and the detection limit was determined to be concentration at which the fluorescence equals that of [blank + 3σ].
Cell Culture
C6 cells were obtained from ATTC and cultured in Dulbecco’s Eagle Medium (DMEM, GIBCO) supplemented with 10% fetal bovine serum (FBS, HyClone) and 1% penicillin/streptomycin. Cells were seeded on a 22 mm diameter glass coverslip at ∼2.3 × 106 cells per well in a six-well culture dish and allowed to adhere for 24 h in 2.0 mL DMEM (37 °C, 5% CO2) prior to experiment. Cells were then washed with 1x Dulbecco’s Phosphate Buffered Saline (1× DPBS, 3×) and treated with 2.0 mL of DMEM containing either 100 nM AP39 or 20 μM AOAA. After incubation for 1 h or 45 min, respectively, the cells were washed with 1x DPBS (3×) and treated with 2.0 mL of DMEM containing 5 μM MeRho-Az and incubated at 37 °C at 5% CO2 for an additional 45 min. As a blank, cells were treated with 5 μM MeRho-Az and incubated in DMEM media for 45 min. After incubation, cells were washed with 1× DPBS (3×) and fixed in 3.7% paraformaldehyde in 1× DPBS at 37 °C for 15 min followed by two rinses and 1 wash with 1× DPBS. Coverslips containing fixed cells were mounted in Vectashield Hardset Mounting Medium (Vector Laboratories).
Fluorescence Microscopy and Statistical Analysis
Images were acquired on a confocal microscope (Olympus Fluoview 1000) using an oil-immersion 60× (1.4 NA) objective. All images were processed with ImageJ.72 Fluorescent data was analyzed using ImageJ software and all statistical comparisons were performed using Prism. (One-way ANOVA with Dunnett’s post-test was performed using GraphPad Prism version 7.0 for Windows; GrahPad Software: San Diego, CA www.graphpad.cpm)
Light Sheet Fluorescence Microscopy
Light sheet fluorescence microscopy was performed using a home-built instrument similar in design to that of Keller et al.50 and described previously.55 In brief: fluorescence excitation illumination was provided by a 488 nm Coherent sapphire laser (Coherent, Santa Clara, California), shaped into a thin sheet by a mirror galvanometer (Cambridge Technology, Bedford, MA) and telecentric scan lens (Sill Optics). Detection was performed with a Zeiss W Plan-Apochromat 40×/1.0 DIC objective lens and a pco.Edge scientific CMOS camera (PCO, Kelheim, Germany). This LSFM setup can image the volume containing the intestinal bulb used in this study (400 × 350 × 300 μm3), with 1 μm steps between planes, in approximately 10 s, leading to images unblurred by gut peristalsis. All microscope control, image acquisition, and analysis software were custom-written in MATLAB, C++, and Python.
Specimen Mounting and Imaging Protocols
Larval zebrafish were mounted for imaging as described previously.56 In brief: specimens were held in 0.5% agarose gel, and suspended in a temperature controlled specimen chamber containing embryo medium, held at 28 °C. All experiments involving zebrafish were performed according to protocols approved by the University of Oregon Institutional Animal Care and Use Committee (protocol #12–18RR).
Zebrafish Imaging
Larval zebrafish (7 days post fertilization) were orally gavaged as described69 with either vehicle (50 mM PIPES, pH 7.4), 5 μM MeRho-Az, 5 μM MeRho-Az + 250 μM DATS + 250 μM GSH, or 5 μM MeRho with a total injection volume of 6.9 nL. This injection volume is sufficient to fill the intestinal space. After 60 min of recovery time, a three-dimensional image (z spacing of 1 μm) of the intestinal bulb of each fish was acquired. Fish in all experimental groups remained alive and healthy for the duration of the experiment, with no indication of toxicity for 20 h afterward. Excitation light was provided by a 488 nm laser, delivering 10 mW of power to the sample. Emission light was filtered through a 525/50 nm bandpass filter and collected on a sCMOS camera.
Data Analysis
For each three-dimensional data set, the average background intensity was measured in a region of tissue outside of the intestinal tract and used to provide a minimum threshold value for pixel intensity. Next, a 20 μm thick section within the intestinal bulb was selected at a depth where the bulb was visible in its fullest extent. Voxels from these sections with an average voxel intensity greater than the background threshold were used to measure the integrated gut brightness.
Synthesis of 2
Compound 1 (0.400 g, 1.15 mmol) was combined with N-phenyl-bis(trifluoromethanesulfonamide) (0.412 g, 1.15 mmol) in DMF (3 mL) and stirred at room temperature for 15 h. The reaction mixture was then diluted with water and extracted into EtOAc. The organic phase was washed with brine and dried using Na2SO4. After removal of the solvent under reduced pressure, the crude product was purified using column chromatography (100% DCM) to afford the pure product 2 as a white crystalline solid (0.550 g, 99% yield). 1H NMR (500 MHz, CDCl3) δ (ppm): 8.04 (d, J = 7.6 Hz, 1H), 7.66 (m, 2H), 7.24 (d, J = 4.7 Hz, 1H), 7.16 (d, J = 7.5 Hz, 1H), 6.94 (d, J = 8.8 Hz, 1H), 6.89 (d, J = 8.8 Hz, 1H), 6.79 (s, 1H), 6.71 (d, J = 8.8 Hz, 1H), 6.65 (d, J = 9.7 Hz, 1H), 3.84 (s, 3H). 13C{1H} NMR (125 MHz, CDCl3) δ (ppm): 169.1, 161.8, 152.8, 152.2, 152.2, 135.5, 130.3, 130.2, 129.1, 126.4, 125.5, 124.0, 120.1, 120.0, 117.5, 116.8, 112.6, 110.7, 110.6, 101.1, 81.8, 55.8.
Synthesis of MeRho
In a glovebox, compound 2 (0.368 g, 0.769 mmol), Pd(OAc)2 (17 mg, 77 μmol), BINAP (72 mg, 120 μmol), and Cs2CO3 (0.752 g, 2.31 mmol) were dissolved in toluene (20 mL) in a three-neck flask fitted with a reflux condenser. After the reaction vessel was sealed under N2 and removed from the glovebox, benzophenone imine was added via syringe (0.152 mL, 0.923 mmol). The reaction was heated and stirred at 140 °C for 5 min, and then the temperature was reduced to 100 °C for an additional 8 h. After heating, the reaction mixture was allowed to cool to room temperature and was filtered through a plug of Celite. After removal of the solvent under reduced pressure, the residue was dissolved in a solution of THF (20 mL) and 1 M HCl (2 mL) and stirred at room temperature overnight. The THF was removed under reduced pressure, the crude product was diluted with water, and the pH was neutralized. The aqueous solution was extracted into EtOAc, and the organic phase was washed with brine and dried using Na2SO4. The crude product was purified using column chromatography (EtOAc:hexanes gradient from 1:4 to 4:1) to afford MeRho as a pale orange powder (0.226 g, 85% yield). 1H NMR (500 MHz, DMSO) δ (ppm): 7.97 (d, J = 7.7 Hz, 1H), 7.78 (t, J = 8.0 Hz, 1H), 7.70 (t, J = 7.6 Hz, 1H), 7.24 (d, J = 7.6 Hz, 1H), 6.90 (d, J = 2.5 Hz, 1H), 6.66 (dd, J = 8.8, 2.5 Hz, 1H), 6.60 (d, J = 8.8 Hz, 1H), 6.44 (d, J = 2.0 Hz, 1H), 6.37 (d, J = 8.5 Hz, 1H), 6.33 (dd, J = 8.6, 2.0 Hz, 1H), 5.66 (s, 2H), 3.80 (s, 3H). 13C{1H} NMR (125 MHz, DMSO) δ (ppm): 168.7, 160.9, 152.5, 152.1, 151.9, 151.3, 135.5, 129.9, 128.8, 128.5, 126.4, 124.5, 124.0, 111.5, 111.3, 111.2, 105.2, 100.7, 99.0, 83.6, 55.6. HRMS (m/z): [M + H]+ calcd for [C21H16NO4]+ 346.1079, found 346.1096.
Synthesis of MeRho-Az
A solution of NaNO2 (31 mg, 0.45 mmol) in water (2.5 mL) was chilled in an ice bath under foil. A suspension of MeRho (0.100 g, 0.289 mmol) in 6 M HCl (2 mL) was added dropwise, and the reaction was stirred at 0 °C for 30 min. A solution of NaN3 (58 mg, 0.89 mmol) in water (2 mL) was then added dropwise, and the reaction was allowed to warm to room temperature and stir for 4 h. While taking care to shield the crude material from light, water was added, and the pH was neutralized. The crude product was extracted into EtOAc, and the organic phase was washed with brine and dried using Na2SO4. Purification of the crude product by preparatory chromatography (3:2 hexanes:EtOAc) afforded pure MeRho-Az as a white solid (67 mg, 62% yield). 1H NMR (600 MHz, CDCl3) δ (ppm): 8.05 (d, J = 7.6 Hz, 1H), 7.68 (m, 2H), 7.17 (d, J = 7.6 Hz, 1H), 6.97 (d, J = 2.2 Hz, 1H), 6.80 (m, 2H), 6.73 (m, 2H), 6.65 (dd, J = 8.8, 2.5 Hz, 1H), 3.87 (s, 3H). 13C{1H} NMR (125 MHz, CDCl3) δ (ppm): 169.3, 161.5, 153.0, 152.3, 152.2, 142.5, 135.1, 129.9, 129.7, 129.1, 126.6, 125.2, 123.8, 115.8, 114.9, 112.0, 110.9, 107.2, 100.9, 82.4, 55.6. FTIR (ATR, cm–1): 2110 (s), 1761 (s), 1608 (s), 1260 (m), 1495 (s), 1420 (s), 1282 (m), 1250 (m), 1215 (s), 1161 (m), 1097 (s), 1080 (s), 1030 (m), 828 (s), 757 (s), 691 (m). HRMS (m/z): [M + H]+ calcd for [C21H14N3O4]+ 372.0984, found 372.0975.
Acknowledgments
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number 1P50GM098911 and R00GM092970 (to MDP). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The NMR facilities at the University of Oregon are supported by NSF/ARRA CHE-0923589. The Biomolecular Mass Spectrometry Core of the Environmental Health Sciences Core Center at Oregon State University is supported, in part, by the NIEHS (P30ES000210) and the NIH. The University of Oregon Fish Facility is supported, in part, by the NIH under award number P01 HD022486.
Supporting Information Available
Supplemental figures, NMR spectra, 3D video of imaging experiments. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b04196.
The authors declare no competing financial interest.
Supplementary Material
References
- Wang R. Physiol. Rev. 2012, 92, 791–896. [DOI] [PubMed] [Google Scholar]
- Abe K.; Kimura H. J. Neurosci. 1996, 16, 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. FASEB J. 2002, 16, 1792–1798. [DOI] [PubMed] [Google Scholar]
- Kimura H. Mol. Neurobiol. 2002, 26, 13–19. [DOI] [PubMed] [Google Scholar]
- Yang W.; Yang G.; Jia X.; Wu L.; Wang R. J. Physiol. 2005, 569, 519–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde A.; Bhatia M. Inflammation Allergy: Drug Targets 2011, 10, 118–122. [DOI] [PubMed] [Google Scholar]
- Siebert N.; Cantre D.; Eipel C.; Vollmar B. Am. J. Physiol.: Gastrointest. Liver Physiol. 2008, 295, G1266–1273. [DOI] [PubMed] [Google Scholar]
- Wilinski B.; Wilinski J.; Somogyi E.; Piotrowska J.; Opoka W. Pharmacol. Rep. 2013, 65, 737–742. [DOI] [PubMed] [Google Scholar]
- Mustafa A. K.; Gadalla M. M.; Sen N.; Kim S.; Mu W.; Gazi S. K.; Barrow R. K.; Yang G.; Wang R.; Snyder S. H. Sci. Signaling 2009, 2, ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul B. D.; Snyder S. H. Nat. Rev. Mol. Cell Biol. 2012, 13, 499–507. [DOI] [PubMed] [Google Scholar]
- Bailey T. S.; Zakharov L. N.; Pluth M. D. J. Am. Chem. Soc. 2014, 136, 10573–10576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishigami M.; Hiraki K.; Umemura K.; Ogasawara Y.; Ishii K.; Kimura H. Antioxid. Redox Signaling 2009, 11, 205–214. [DOI] [PubMed] [Google Scholar]
- Lin V. S.; Chang C. J. Curr. Opin. Chem. Biol. 2012, 16, 595–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xuan W.; Sheng C.; Cao Y.; He W.; Wang W. Angew. Chem., Int. Ed. 2012, 51, 2282–2284. [DOI] [PubMed] [Google Scholar]
- Fogo J. K.; Popowsky M. Anal. Chem. 1949, 21, 732–734. [Google Scholar]
- Shen X.; Peter E. A.; Bir S.; Wang R.; Kevil C. G. Free Radical Biol. Med. 2012, 52, 2276–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X.; Pattillo C. B.; Pardue S.; Bir S. C.; Wang R.; Kevil C. G. Free Radical Biol. Med. 2011, 50, 1021–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardencki W. J. Chromatogr. A 1998, 793, 1–19. [Google Scholar]
- García-Calzada M.; Marbán G.; Fuertes A. B. Anal. Chim. Acta 1999, 380, 39–45. [Google Scholar]
- Liu C.; Pan J.; Li S.; Zhao Y.; Wu L. Y.; Berkman C. E.; Whorton A. R.; Xian M. Angew. Chem., Int. Ed. 2011, 50, 10327–10329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Y.; Karpus J.; Kabil O.; Zhang S.-Y.; Zhu H.-L.; Banerjee R.; Zhao J.; He C. Nat. Commun. 2011, 2, 495. [DOI] [PubMed] [Google Scholar]
- Liu C.; Peng B.; Li S.; Park C.-M.; Whorton A. R.; Xian M. Org. Lett. 2012, 14, 2184–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z.; Xu L.; Zhou J.; Xu Y.; Zhu W.; Qian X. Chem. Commun. 2012, 48, 10871–10873. [DOI] [PubMed] [Google Scholar]
- Qian Y.; Zhang L.; Ding S.; Deng X.; He C.; Zheng X. E.; Zhu H.-L.; Zhao J. Chem. Sci. 2012, 3, 2920–2923. [Google Scholar]
- Zhang J.; Sun Y.-Q.; Liu J.; Shi Y.; Guo W. Chem. Commun. 2013, 49, 11305–11307. [DOI] [PubMed] [Google Scholar]
- Wei C.; Zhu Q.; Liu W.; Chen W.; Xi Z.; Yi L. Org. Biomol. Chem. 2014, 12, 479–485. [DOI] [PubMed] [Google Scholar]
- Sasakura K.; Hanaoka K.; Shibuya N.; Mikami Y.; Kimura Y.; Komatsu T.; Ueno T.; Terai T.; Kimura H.; Nagano T. J. Am. Chem. Soc. 2011, 133, 18003–18005. [DOI] [PubMed] [Google Scholar]
- Gu X.; Liu C.; Zhu Y.-C.; Zhu Y.-Z. Tetrahedron Lett. 2011, 52, 5000–5003. [Google Scholar]
- Qu X.; Li C.; Chen H.; Mack J.; Guo Z.; Shen Z. Chem. Commun. 2013, 49, 7510–7512. [DOI] [PubMed] [Google Scholar]
- Lippert A. R.; New E. J.; Chang C. J. J. Am. Chem. Soc. 2011, 133, 10078–10080. [DOI] [PubMed] [Google Scholar]
- Peng H.; Cheng Y.; Dai C.; King A. L.; Predmore B. L.; Lefer D. J.; Wang B. Angew. Chem., Int. Ed. 2011, 50, 9672–9675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X.; Lin W.; Zheng K.; He L. Chem. Commun. 2012, 48, 10529–10531. [DOI] [PubMed] [Google Scholar]
- Chen B.; Lv C.; Tang X. Anal. Bioanal. Chem. 2012, 404, 1919–1923. [DOI] [PubMed] [Google Scholar]
- Chen S.; Chen Z.-J.; Ren W.; Ai H.-W. J. Am. Chem. Soc. 2012, 134, 9589–9592. [DOI] [PubMed] [Google Scholar]
- Das S. K.; Lim C. S.; Yang S. Y.; Han J. H.; Cho B. R. Chem. Commun. 2012, 48, 8395–8397. [DOI] [PubMed] [Google Scholar]
- Hartman M. C. T.; Dcona M. M. Analyst 2012, 137, 4910–4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R.; Yu F.; Chen L.; Chen H.; Wang L.; Zhang W. Chem. Commun. 2012, 48, 11757–11759. [DOI] [PubMed] [Google Scholar]
- Zheng K.; Lin W.; Tan L. Org. Biomol. Chem. 2012, 10, 9683–9688. [DOI] [PubMed] [Google Scholar]
- Bailey T. S.; Pluth M. D. J. Am. Chem. Soc. 2013, 135, 16697–16704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B.; Li W.; Lv C.; Zhao M.; Jin H.; Jin H.; Du J.; Zhang L.; Tang X. Analyst 2013, 138, 946–951. [DOI] [PubMed] [Google Scholar]
- Li W.; Sun W.; Yu X.; Du L.; Li M. J. Fluoresc. 2013, 23, 181–186. [DOI] [PubMed] [Google Scholar]
- Lin V. S.; Lippert A. R.; Chang C. J. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 7131–7135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W.; Fan J.; Hu C.; Cao J.; Zhang H.; Xiong X.; Wang J.; Cui S.; Sun S.; Peng X. Chem. Commun. 2013, 49, 3890–3892. [DOI] [PubMed] [Google Scholar]
- Zhou G.; Wang H.; Ma Y.; Chen X. Tetrahedron 2013, 69, 867–870. [Google Scholar]
- Santos-Figueroa L. E.; de laTorre C.; El Sayed S.; Sancenón F.; Martínez-Máñez R.; Costero A. M.; Gil S.; Parra M. Eur. J. Org. Chem. 2014, 2014, 1848–1854. [Google Scholar]
- Cao J.; Lopez R.; Thacker J. M.; Moon J. Y.; Jiang C.; Morris S. N. S.; Bauer J. H.; Tao P.; Mason R. P.; Lippert A. R. Chem. Sci. 2015, 6, 1979–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Q. Q.; Song Y. C.; Li Z.; Gao X. H.; Ma H. M. Chem. Commun. 2013, 49, 502–504. [DOI] [PubMed] [Google Scholar]
- Dufton N.; Natividad J.; Verdu E. F.; Wallace J. L. Sci. Rep. 2012, 2, 499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisken J.; Stainier D. Y. Development 2009, 136, 1963–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller P. J.; Schmidt A. D.; Wittbrodt J.; Stelzer E. H. Science 2008, 322, 1065–1069. [DOI] [PubMed] [Google Scholar]
- Huisken J. BioEssays 2012, 34, 406–411. [DOI] [PubMed] [Google Scholar]
- Jemielita M.; Taormina M. J.; Delaurier A.; Kimmel C. B.; Parthasarathy R. J. Biophotonics 2013, 6, 920–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santi P. A. J. Histochem. Cytochem. 2011, 59, 129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunwald D. J.; Eisen J. S. Nat. Rev. Genet. 2002, 3, 717–724. [DOI] [PubMed] [Google Scholar]
- Taormina M. J.; Jemielita M.; Stephens W. Z.; Burns A. R.; Troll J. V.; Parthasarathy R.; Guillemin K. Biol. Bull. 2012, 223, 7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemielita M.; Taormina M. J.; Burns A. R.; Hampton J. S.; Rolig A. S.; Guillemin K.; Parthasarathy R. mBio 2014, 5, e01751-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J.; Dodani S. C.; Chang C. J. Nat. Chem. 2012, 4, 973–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker J. E.; Haugland R. P.; Ryan D.; Hewitt P. C.; Haugland R. P.; Prendergast F. G. Anal. Biochem. 1992, 207, 267–279. [DOI] [PubMed] [Google Scholar]
- Dickinson B. C.; Huynh C.; Chang C. J. J. Am. Chem. Soc. 2010, 132, 5906–5915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya M.; Asanuma D.; Kuranaga E.; Takeishi A.; Sakabe M.; Miura M.; Nagano T.; Urano Y. J. Am. Chem. Soc. 2011, 133, 12960–12963. [DOI] [PubMed] [Google Scholar]
- Kawai K.; Ieda N.; Aizawa K.; Suzuki T.; Miyata N.; Nakagawa H. J. Am. Chem. Soc. 2013, 135, 12690–12696. [DOI] [PubMed] [Google Scholar]
- Yang X.-F.; Huang Q.; Zhong Y.; Li Z.; Li H.; Lowry M.; Escobedo J. O.; Strongin R. M. Chem. Sci. 2014, 5, 2177–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng T.; Yang D. Org. Lett. 2010, 12, 496–499. [DOI] [PubMed] [Google Scholar]
- Zhu D.; Xue L.; Li G.; Che Y.; Jiang H. Org. Chem. Front. 2014, 1, 501–505. [Google Scholar]
- Montoya L. A.; Pluth M. D. Chem. Commun. 2012, 48, 4767–4769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczesny B.; Módis K.; Yanagi K.; Coletta C.; Le Trionnaire S.; Perry A.; Wood M. E.; Whiteman M.; Szabo C. Nitric Oxide 2014, 41, 120–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asimakopoulou A.; Panopoulos P.; Chasapis C. T.; Coletta C.; Zhou Z.; Cirino G.; Giannis A.; Szabo C.; Spyroulias G. A.; Papapetropoulos A. Br. J. Pharmacol. 2013, 169, 922–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benavides G. A.; Squadrito G. L.; Mills R. W.; Patel H. D.; Isbell T. S.; Patel R. P.; Darley-Usmar V. M.; Doeller J. E.; Kraus D. W. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 17977–17982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocchiaro J. L.; Rawls J. F. J. Visualized Exp. 2013, e4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- By utilizing a control of buffer-gavaged fish instead of ungavaged fish, the relative strength and variability of mucosal autofluorescence is suppressed. Additionally, any morphological changes to the gut cavity upon gavage are accounted for in buffer-gavaged fish.
- Mugherli L.; Burchak O. N.; Chatelain F.; Balakirev M. Y. Bioorg. Med. Chem. Lett. 2006, 16, 4488–4491. [DOI] [PubMed] [Google Scholar]
- Schneider C. A.; Rasband W. S.; Eliceiri K. W. Nat. Methods 2012, 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.